

Abstract
Both deficiencies and excesses of iron represent major public health problems throughout the world. Understanding the cellular and organismal processes controlling iron homeostasis is critical for identifying iron-related diseases and in advancing the clinical treatments for such disorders of iron metabolism. Iron regulatory proteins (IRPs) 1 and 2 are key regulators of vertebrate iron metabolism. These RNA binding proteins post-transcriptionally control the stability or translation of mRNAs encoding proteins involved in iron homeostasis thereby controlling the uptake, utilization, storage or export of iron. Recent evidence provides insight into how IRPs selectively control the translation or stability of target mRNAs, how IRP RNA binding activity is controlled by iron-dependent and iron-independent effectors, and the pathological consequences of dysregulation of the IRP system.
Keywords: Iron, Iron regulatory protein, Iron responsive element, Translational control, RNA stability, Protein degradation, Iron-sulfur protein, Phosphorylation, Protein kinase C
1. Introduction
1.1. Necessity and toxicity of iron
Iron is considered a micronutrient because the adequate daily intake is in the low milligram range [1]. Despite this relatively low dietary requirement, iron deficiency is a major global health problem due, in part, to the low abundance of iron in many foods coupled with reduced bioavailability of non-heme iron, a common dietary iron source. Impairment of health caused by nutritional iron deficiency affects more than 2 billion people worldwide resulting in significant negative social and economic impacts [2,3]. In fact, iron deficiency is listed by the World Health Organization as one of the top ten leading risk factors for significant health impairment or death [4]. While most affected individuals live in developing countries, even industrialized areas such as the United States have significant numbers of affected individuals [5]. The nutritional requirement for iron in vertebrates arises because it is an essential component of proteins that perform redox or non-redox roles in many critical cellular functions including respiration and cell division. In addition to anemia, iron deficiency impairs muscle, immune and cognitive function and can increase the incidence of low birthweight and preterm delivery [6–11]. It is worth noting that iron deficiency can impair health in the absence of anemia and a significant fraction of the two billion individuals suffering pathological consequences of iron deficiency fall into this category [12]. Given the significant impact of iron deficiency on human health, there is much interest in accurately quantifying its occurrence, reducing its prevalence, as well as understanding how cells and tissues respond to deficiencies or excesses of this critical nutrient [2,3,13,14].
Dysregulation of iron metabolism associated with hemochromatosis and other iron overload disorders is also a significant health concern [15–19]. Iron’s toxicity arises due to iron-induced formation of reactive oxygen species (ROS) that damage cellular structures [20]. In hemochromatosis, iron overload damages the liver, pancreas, heart and other tissues. Furthermore, iron overload is causative of, or has been associated with the development of, several neurodegenerative diseases including Parkinson’s, Frie-dreich’s ataxia, Aceruloplasminemia, Pantothenate kinase deficiency and others [20–26]. The molecular basis for diseases of iron overload includes improper control of fundamental pathways of iron metabolism such as iron absorption or transport, iron–sulfur protein biogenesis, iron storage, or, in an animal model, the general control of iron metabolism by iron regulatory protein 2 (IRP2), all of which can lead to degenerative processes in specific tissues [27–31]. In order to maintain optimal health, the concentration and distribution of iron must be tightly controlled to provide enough to meet cellular requirements while avoiding excessive levels that are toxic. Understanding the tissue and cell-type specific processes underlying the control of iron homeostasis remains a critical factor in identifying iron-related diseases and in advancing the clinical treatments for such disorders of iron metabolism [19].
2. Brief overview of whole body and cellular iron homeostasis
2.1. Dietary iron absorption
Multiple physiological processes are used to maintain iron homeostasis in diverse organisms. These include the absorption, use, storage and export of iron. In mammals, absorption of dietary iron represents the only means to regulate body iron content [32–34]. Identification of the transporters and other proteins functioning in the absorption of dietary non-heme and heme iron has greatly enhanced the understanding of this process that is central to the control of body iron content. Iron can be absorbed as non-heme iron or heme iron with the latter being more efficiently taken up [1]. A candidate for the long sought intestinal transporter required for heme absorption was recently identified [35,36]. Non-heme iron is absorbed through the action of divalent metal transporter 1 (DMT1; also called nRAMP2) and DCYTB, a ferrireductase, present at the apical membrane in the proximal small intestine (Fig. 1) [37–39]. DMT1 is required for absorption of dietary non-heme iron after birth but is not essential in all tissues [40]. Two isoforms of DMT1 protein exist that differ at their C-terminus, have tissue specific roles in iron absorption, and exhibit different subcellular trafficking [41]. Mutation of DMT1 impairs iron absorption and can cause microcytic anemia in animals and humans [37,42–44]. Iron controls the abundance of DMT1 mRNA as well as the subcellular localization of the transporter itself [37,38,45]. DCYTB was identified as a gene up-regulated in duodenum in response to iron deficiency and other scenarios causing enhanced iron absorption [39]. DCYTB is believed to donate electrons for reduction of ferric iron to the ferrous state for transport by DMT1. However, recent work has questioned the essential role of DCYTB, at least in mice [46]. Once internalized, iron traverses the intestinal mucosal cell but the mechanism is not well understood, although it may involve transcytosis [47]. Heme absorbed from the diet is presumably degraded by heme oxygenase and the iron released enters the intracellular non-heme iron pool. Export of iron into the circulation requires the iron exporter ferroportin (FPN; also called IREG1 and MTP1) [48] and a ferroxidase, to oxidize iron so it can be bound by transferrin (TF). Ferroportin is essential for iron export from enterocytes, macrophages and hepatocytes [48]. Until recently the ferroxidase involved in oxidizing iron exported by FPN has been presumed to be hephaestin, a multicopper oxidase related to the long known serum protein, ceruloplasmin [34]. Mutation of hephaestin, in the sex-linked anemia mouse, causes microcytic anemia and illustrates the importance of hephaestin in iron absorption [34,49]. However, a recent study demonstrates a clear role for ceruloplasmin in intestinal iron absorption under some circumstances such as in response to acute phlebotomy [50]. In brain, a novel form of ceruloplasmin directly interacts with FPN suggesting channeling of iron between the transporter and ferroxidase; a similar situation exists in yeast [51,52]. An analogous interaction between FPN and either ceruloplasmin or hephaestin may occur in relation to the process of dietary iron absorption. How apoTf directly acquires iron is not clear. Major factors influencing the rate of intestinal iron absorption are the size of body iron stores, the rate of erythropoiesis and inflammation [32]. Dietary factors including the type of iron (heme versus non-heme iron) and the presence of inhibitors or enhancers of iron absorption also influence intestinal iron absorption [1]. The recently discovered iron regulatory hormone hepcidin has a critical role in controlling iron absorption through its ability to bind to and control the cell surface expression of FPN [53,54]. Hepcidin has a major role in the control of body iron homeostasis in response to changes in dietary iron intake and in pathological situations such as the anemia of chronic disease.
Fig. 1.

Control of mammalian iron homeostasis by the IRE/IRP regulatory network. A generic mammalian cell depicting the various roles of mammalian proteins encoded by IRE-containing mRNAs (red lettering). Tf-Fe3+ binds to Tf R on the cell surface where the TF-Fe3+/Tf R complex is endocytosed. Acidification of the endosome causes the release of Fe3+ (red balls) from TF where it is reduced to Fe2+ (yellow balls) by the ferrireductase Steap3 before export from the endosome by DMT1 (divalent metal transporter 1). ApoTf/Tf R complex is then returned to the cell surface where it dissociates and initiates another round of iron uptake. TF-mediated iron uptake is ubiquitous, but it is especially important in erythroid precursor cells where it is the primary source of iron for utilization in heme synthesis. DMT1 is also localized on the apical membrane of duodenal enterocytes where it transports Fe2+ reduced possibly by the membrane reductase DCYTB or a member of a Steap family of metalloreductases [300]. Iron taken up by the Tf R enters a cytosolic free iron pool thought to consist of Fe2+ bound to small molecular weight molecules. IRPs sense iron in this pool and regulate the translation of 5′ IRE-containing mRNAs (H- and L-ferritin subunits, eALAS (erythroid aminolevulinate synthase), m-aconitase, ferroportin) or the stability of 3′ IRE-containing mRNAs (Tf R and possibly DMT1). eALAS is a mitochondrial enzyme and the rate limiting enzyme in heme synthesis in precursor erythroid cells. Mitochondrial aconitase is an enzyme in the tricarboxylic acid cycle that requires a [4Fe–4S] cluster for activity. Iron that is not utilized or stored in ferritin is exported by ferroportin. Ferroportin is expressed in duodenal enterocytes, hepatocytes, placenta and macrophages. Export of iron is coupled to the oxidation of iron possibly by the membrane bound protein hephaestin (HP) or the serum multicopper oxidase ceruloplasmin (CP).
2.2. Interorgan transport and cellular metabolism of iron
One or two milligrams of iron is absorbed by men and women, respectively, every day. However, on a daily basis, the majority of iron flux in the body (i.e. ~25 mg), flows through a tightly controlled cycle between the erythron and the reticuloendothelial (RE) system [1,55]. In this process, senescent red cells are engulfed by macrophages of the RE, such as liver Kupffer cells [55]. Heme from the destroyed red cells is degraded and exported through the action of ferroportin and a ferroxidase, presumably ceruloplasmin, although recent evidence suggests another ferroxidase may be involved [50]. The ferric iron so-formed binds to serum apoTf forming holoTF. TF binds two iron atoms, in the ferric oxidation state, with extremely high affinity. HoloTF can then travel to the bone marrow and be used for formation of new erythrocytes, which completes the red cell cycle.
Transferrin-mediated iron uptake represents the primary means for uptake of non-heme iron by most cell types. HoloTF binds to cell surface transferrin receptor 1 (Tf R1) and the Tf R1/ TF complex is internalized through receptor-mediated endocytosis (Fig. 1) [56,57]. The process of iron release to the cytosol requires endosomal acidification, reduction of iron by a recently identified ferrireductase, Steap3 [58], and ferrous iron export to the cytosol through the action of DMT1 [42]. Release of iron from TF is stimulated by the interaction of TF with Tf R1 (reviewed in [56,57,59]). The complex of Tf R1 and apoTF is returned to the cell surface, completing the TF/Tf R1 cycle (Fig. 1), and apoTF is released to be re-charged with iron (Fig. 1). Iron delivered to the cytosol can be used for metabolic utilization and formation of essential iron-containing proteins in both the mitochondria and cytosol (Fig. 1). Mitochondria have a particularly important role in the formation of iron-containing proteins since the majority of iron–sulfur (Fe–S) proteins are made in this organelle, cytosolic Fe–S cluster formation depends on mitochondrial function, and several critical steps in heme formation take place in the mit-ochondrial matrix where 5-aminolevulinate synthase (ALAS) and ferrochelatase are found [60–62]. In the cytosol, excess iron can be stored in ferritin, a large roughly spherical molecule composed of twenty-four subunits of two types, heavy- (H) or light-chain (L) ferritin which can store several thousand iron atoms [63], or be exported by FPN. A critical aspect of the maintenance of cellular iron homeostasis is the control of the expression of genes encoding proteins required for the uptake (Tf R1, DMT1), utilization (5-aminolevulinate synthase (ALAS) and erythroid ALAS (eALAS)), storage (H- and L-ferritin) or export (FPN) of iron [19]. In order to coordinate these processes, sensing of cellular iron status is required if organisms are to use iron-containing proteins in essential metabolic pathways while simultaneously avoiding the deleterious consequences of iron overload. Iron regulatory proteins (IRPs) are central components of a sensory and regulatory system required for the maintenance of iron homeostasis in vertebrates.
2.3. IRPs are key iron sensors in vertebrates
Since its discovery in 1937 [64] the intriguing properties of ferritin have stimulated many insightful studies that have helped elucidate the major mechanisms for sensing and controlling cellular and organismal iron homeostasis in vertebrates. Initial studies by Granick and others demonstrated that ferritin abundance and synthesis were iron-responsive [65–67]. Munro and colleagues, as well as others, made the critical observation that mammalian cells contained a cytosolic iron sensor that controlled ferritin synthesis [68–70]. In a seminal study, Munro’s laboratory then demonstrated that iron stimulated ferritin synthesis by activating the translation of ferritin mRNAs [71]. The next critical step in studies of ferritin expression involved the identification of an evolutionarily conserved 28 nucleotide (nt) sequence, termed the iron responsive element (IRE), which was present in the 5′ untranslated region (UTR) of both H- and L-ferritin mRNAs. The IRE proved to be necessary and sufficient for iron-dependent control of ferritin mRNA translation [72–74]. These observations occurred concurrently with the discovery of cytosolic iron-regulated RNA binding proteins, IRP1 and IRP2, which recognize the IRE in a sequence and structure specific manner [75,76]. The first suggestion of a general post-transcriptional regulatory network controlling mammalian iron metabolism arose from a comparison of the findings concerning ferritin regulation with the observations of Kühn and associates, who showed that Tf R1 mRNA stability was iron-regulated and that the regulatory element responsible was present in the 3′ UTR [77,78]. In this manner, Harford and colleagues then established that IRE-dependent post-transcriptional control was central to the regulation of cellular iron metabolism in vertebrates [79,80].
Since the discovery of IREs in H- and L-ferritin and Tf R1 mRNAs, IRE or IRE-like elements have been identified in at least ten animal cell mRNAs (Fig. 2) (reviewed in [10,19]). These include the mRNAs encoding proteins involved in heme formation in erythroid cells (eALAS); iron uptake, DMT1; iron export, FPN as well as two tricarboxylic acid cycle enzymes, mitochondrial aconitase (m-acon) and the iron-protein subunit of succinate dehydrogenase [81]. These studies have established the central role of the IRE-IRP system in controlling iron metabolism in vertebrates and some invertebrates, and have served as impetus for the discovery of other post-transcriptional regulatory networks with similar physiological roles in lower eukaryotes and prokaryotes [82–86]. Putative IREs have been observed in three novel mRNAs. Glycolyate oxidase (GOX) mRNA was found to contain a putative IRE in its 5′ UTR [87]. More recently a single putative IRE was discovered in the 3′ UTR of myotonic dystrophy kinase-related Cdc42-binding kinase α (MRCK α) and in one splice variant of CDC14A mRNAs [88,301]. These observations point towards an expanded role for IRP in cell physiology. The presence of a putative IRE in MRCK α mRNA suggests a role for IRPs in cytoskeletal dynamics, which may possibly be related to endocytosis of TF, while the putative IRE in CDC14A suggests new roles for iron in cell division. Whether IRPs have a physiological role in controlling GOX, CDC14A and MRCK α expression remains to be elucidated.
Fig. 2.
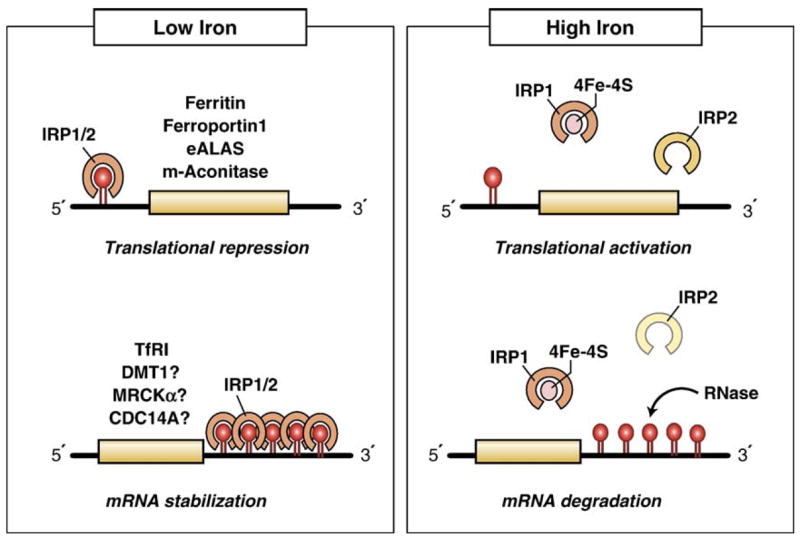
Cellular regulation of mammalian iron homeostasis by the IRPs. IRPs bind to IREs located in either the 5′- or 3′-UTRs of specific mRNAs. During low iron conditions, IRP1 and IRP2 bind with high affinity to 5′ IREs and to the five 3′ IREs in Tf R mRNA, resulting in the translational repression of 5′ IRE-containing mRNAs and the stabilization of the Tf R mRNA. During high iron conditions, IRPs lose their affinity for IREs, increasing translation of 5′ IRE-containing mRNAs and mediating degradation of the Tf R mRNA. Increased iron levels result in the conversion of the IRP1 RNA binding form into the [4Fe–4S] cluster c-acon form, while increased iron and/or heme levels mediate IRP2 proteasomal degradation. Whether the 3′ IRE in DMT1 mRNA is functional is not clear.
2.4. IRPs control mRNA translation or stability
IRP1 and IRP2 are key cytosolic iron sensors in mammalian cells. When cells are iron-deficient, IRPs bind IREs with high affinity (KD ~20–100 pM), inhibiting the translation of ferritin and other mRNAs containing 5′ IREs while stabilizing mRNAs containing 3′ IREs, such as Tf R1 mRNA (Fig. 2). When cells are iron-sufficient, IRPs lose their high affinity RNA binding capacity and fail to bind to IREs. Consequently, ferritin synthesis is activated while Tf R1 mRNA is degraded. The post-transcriptional regulation of ferritin and Tf R1 mRNAs by IRPs allows for rapid alterations in gene expression in response to fluctuations in iron concentrations, and ensures that cells acquire sufficient iron for their needs while preventing iron toxicity.
Although IRP1 and IRP2 share 64% amino acid sequence identity the regulation of their RNA binding activity occurs through different mechanisms. Both IRPs are members of the aconitase family of proteins [89,90]. IRP1 is a dual function protein that exhibits two mutually exclusive activities depending on cellular iron status. IRP1 binds the IRE with very high affinity when cells are iron-deficient. When iron levels rise, a [4Fe–4S] cluster is assembled in IRP1 resulting in the loss of high affinity RNA binding capacity and the acquisition of aconitase activity. In fact, in the iron-replete form, IRP1 is the cytosolic isoform of aconitase (c-acon) [91–95]. Aconitases are ancient enzymes that interconvert citrate to isocitrate through a stereospecific dehydration/rehydration mechanism requiring direct binding of substrate to the [4Fe–4S] cluster [96,97]. Formation and complete loss of the [4Fe–4S] cluster is required for functional interconversion of IRP1 and c-acon. IRP2 is regulated primarily through protein degradation [98,99]. One notable difference between IRP1 and IRP2 is the presence of a unique 73-amino acid (73-aa) insertion near the N-terminus of IRP2. Unlike IRP1, IRP2 cannot form a [4Fe–4S] cluster, and hence does not exhibit aconitase activity [100].
How do the IRPs recognize IREs? The recently solved structure of the c-acon form of IRP1 provides evidence supporting the prediction that the IRE binds within a cleft present between domains 1–3 and domain 4 [101–103]. Similar to mitochondrial aconitase, the [4Fe–4S] cluster sits deep in the solvent accessible cleft of c-acon, although the cleft is deeper in c-acon [102]. In the absence of the [4Fe–4S] cluster, the cleft is predicted to open allowing RNA recognition [101,103]. Both the [4Fe–4S] cluster and the IRE influence the conformation of IRP1 [104–106]. Since the IRPs do not contain classical RNA recognition motifs, understanding how they bind RNA remains a major unresolved issue. Studies indicate that IRPs require a stem and loop structure in which the latter usually has the sequence CAGUGX where the sixth nucleotide (nt) is usually U or C [107] (Fig. 3). The stem, or RNA helix, contains an unpaired or “bulged” C residue five basepairs 5′ of the loop. In the case of the H- and L-ferritin IREs, this bulge is part of larger internal loop/bulge region while all other 5′ or 3′ IREs are predicted to contain a single C-bulge (Fig. 3) [107,108]. The sequence of the IRE loop and the presence of the bulge nucleotide region are critical for IRP binding. Differences in structure and sequence of the IRE RNA helix contribute to differential binding of IRPs to specific IREs (see below).
Fig. 3.
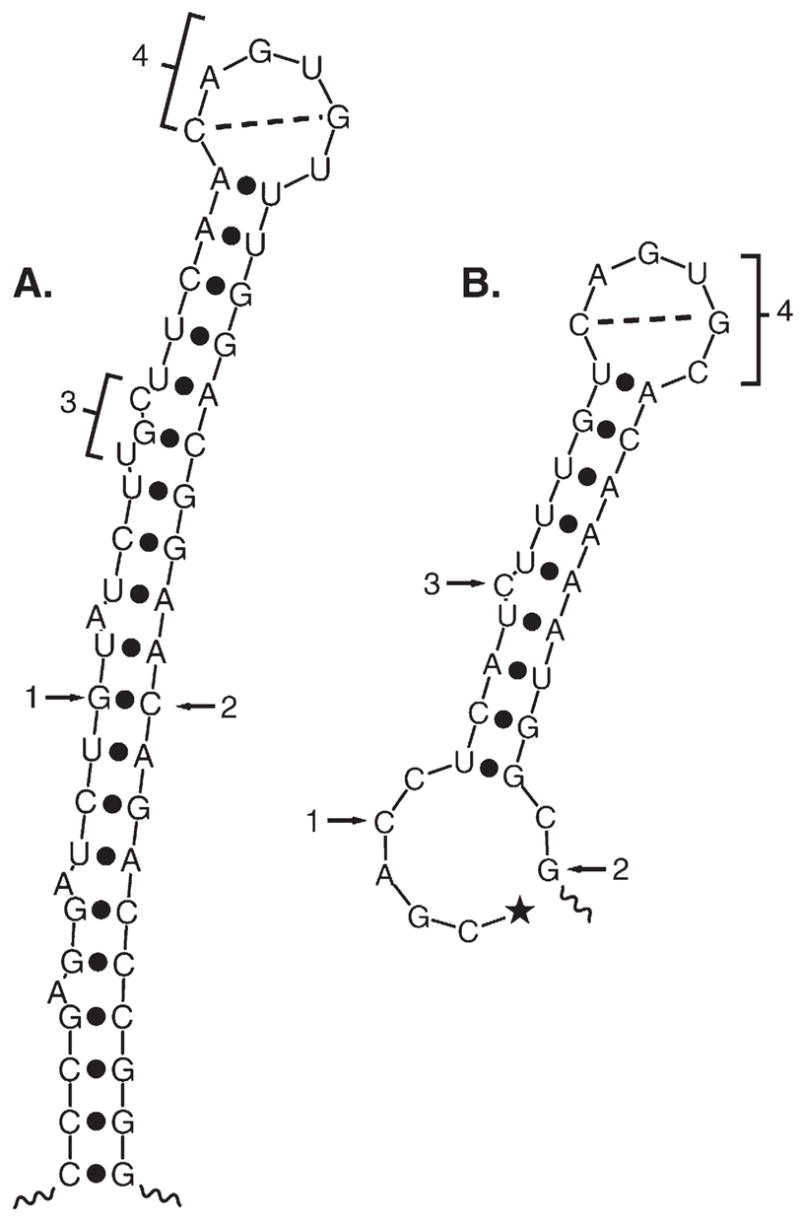
Predicted structure of ferritin and m-acon IRE regions. (A) The rat L-ferritin IRE plus flanking sequences (IRE region). Note that ferritin IRE is the central part of a large stem loop structure stabilized by sequences flanking the IRE. This structure has been confirmed in solution [143,150]. The 5′ and 3′ ends of the ferritin 28 nt IRE are indicated by arrows 1 and 2. The internal loop/bulge is indicated (bracket 3) as is the terminal loop (bracket 4). Potential basepairing between loop nucleotides 1 and 5 is shown. (B) m-acon 5′ UTR structure using the best example of an IRE-like fold obtained from computer predictions. In contrast to the ferritin and Fpn IRE regions, the m-acon IRE is not predicted to form as frequently. The highly conserved RNA sequence flanking [134] the m-acon IRE lacks extensive basepairing since, in contrast to ferritin and Fpn, the m-acon (and eALAS) IRE is <10 nt from the 5′ end. The 5′ CAP is shown for m-acon (★); ferritin cap is 28 nt from 5′ end of IRE. Structures were predicted by M-fold version 3.2.
2.5. Regulation of IRE-containing mRNAs by IRPs
While iron controls ferritin expression transcriptionally and through changes in protein stability [109,110], regulation of ferritin mRNA translation is generally viewed as the primary means for controlling cellular iron storage capacity. The increase in IRE RNA binding activity in iron-deficient cells concordant with the reduction in ferritin synthesis [75,76] argued that IRPs act as repressors of mRNA translation. This was confirmed using cell-free translation systems [111]. Examination of the 5′ UTR of ferritin, as well as other IRE-containing mRNAs, revealed that in most cases the IRE lies within approximately 70 nt of the 5′ methylated cap present at the 5′end of mRNAs, suggesting that cap proximity is important for IRP action. When a functional IRE was present more than 70 nt 3′ of the cap, IRP no longer repressed mRNA translation [112,113]. These studies established the “position effect” requirement for IRP function. Further analysis revealed that IRP1 binding to the ferritin IRE failed to block interaction of the cap binding complex eIF4F to the mRNA 5′end [114]. However, IRP1 did block the recruitment of the small ribosomal subunit and associated factors to ferritin mRNA, a step requiring interaction with mRNA-bound eIF4F. Future studies of IRP-dependent translational regulation will likely focus on understanding how some IRE-containing mRNAs largely evade IRP action (e.g., eALAS and m-acon mRNAs) and on the mechanism(s) underlying derepression of IRE-containing mRNAs [115].
Iron regulation of Tf R1 expression is largely controlled through changes in RNA degradation [77,116] although Tf R1 gene transcription and possibly protein stability can serve important roles [116–119]. Iron-dependent control of Tf R1 mRNA stability depends on the presence of five IREs and a rapid turnover determinant in the 3′ UTR, which provides a site for an endonuclease [78–80,120,121]. One unanswered question is why multiple IREs are needed for regulation? Recent analysis of the 3′ UTR of Tf R1 mRNA when IRPs are bound indicates that structural changes in the IRE helix occur in the context of the five IREs. These structural changes facilitate IRP2 binding and do not occur when Tf R1 IREs are examined as isolated elements. Unlike IRP2, IRP1 binds well to single Tf R1 IREs [122]. These studies show that alterations in the structure of the Tf R1 IRE region allows for binding of either IRP1 or IRP2, further enhancing the stability of Tf R1 mRNA. Further work on the identification of the nuclease(s) involved in Tf R1 mRNA degradation and determining how this nuclease(s) functions in relation to IRP binding is needed. A second Tf R isoform has been identified that binds serum TF and has a role in sensing extracellular iron [123–125]. However, Tf R2 is not controlled by IRPs.
In addition to Tf R1, DMT1, MRCKα and CDC14A mRNA appear to contain an IRE or IRE-like structure in the 3′ UTR. The IRE-like structure in DMT1 mRNA binds IRPs in vitro [126]. However, the function of this putative IRE is unclear. While DMT1 mRNA levels can be iron-regulated [37,38,127], in other scenarios the IRE-like structure appears to be non-functional [128–130]. Furthermore, multiple alternatively spliced forms of DMT1 mRNA are observed [127]. However, not all contain the IRE-like structure and some of the non-IRE containing variants are iron-responsive [127,128]. Whether the IRE-like structure in DMT1 mRNA is functional remains to be determined. The putative IRE in MRCKα mRNA binds to IRP and MRCKα mRNA half-life is iron-regulated [88]. Future studies should determine if the putative IRE in MRCKα is required for iron regulation of MRCKα mRNA degradation. The function of the putative IRE in one splice variant of CDC14A mRNA remains to be confirmed by direct analysis of the regulation of CDC14A mRNA expression by IRPs.
2.6. Evidence of combinatorial control by IRPs
The presence of IREs in mRNAs encoding proteins involved in multiple aspects of iron metabolism or tricarboxylic acid cycle function argue that these mRNAs are not identically regulated by IRPs. This suggests that the IRE-IRP system likely uses combinatorial regulation to control iron and energy metabolism [107]. Combinatorial control provides a mechanism where the outputs (e.g., translation rate of specific mRNAs) from a system of interest can be varied over a wide range in response to the same signal. Evidence of combinatorial control by IRPs comes from studies demonstrating the hierarchical translational regulation of IRE-containing mRNAs [107]. Several studies have shown that ferritin mRNA is more potently regulated than m-acon or eALAS mRNAs [131–139]. In animal and cell culture studies, ferritin protein abundance and synthesis rate vary over a 50-fold range while m-acon varies by only 2-fold in response to the same change in iron availability [134,137]. These studies suggest that the IRE region in ferritin mRNA evolved to promote optimal translational regulation. A key question arising from these studies is how do some mRNAs with 5′ IREs largely evade IRP-dependent regulation while others are strongly repressed? The answer appears to lie, at least in part, in RNA (i.e., IRE) target site diversity, and differences in the individual structural requirements of IRP1 versus IRP2 for RNA recognition.
The general proof-of-principle that IRP1 and IRP2 have overlapping but not identical requirements for RNA recognition comes from SELEX experiments where novel IREs were identified that bound preferentially to either IRP1 or IRP2 [140–142]. A similar stem–loop secondary structure in all IREs is recognized by IRPs, however, sequence and structural differences in the helix of naturally occurring IREs can alter IRP binding [108,143,144]. As noted earlier, all natural IREs are generally composed of ~28 nt forming a stem–loop usually with the loop sequence CAGUGX where X is usually a U or C. Base pairing between the first and fifth nt of the loop has been observed in NMR studies of the IRE, and is essential for IRP1 binding [141,145–147]. Sequences that constitute the stem region of the canonical 28 nt IRE, and regions flanking it, are much better conserved for the same mRNA (>95%) compared to the same region in IREs of other mRNAs (~30–80%) [107,148]. All IREs contain an unpaired or bulged C residue in the stem five nucleotides from the 5′ terminus of the loop. However, in the ferritin IRE, the unpaired C is part of a larger internal loop, the so-called internal loop/bulge (IL/B) region (Fig. 3) [108,143,149,150]. The size of bulged nucleotide or internal loop regions affects linking of RNA helices and influences accessibility of the major groove to protein contacts [151–155]. IRP2 prefers the ferritin type IL/B IRE while IRP1 binds either the single C bulge or the IL/B type IRE [108,156]. As noted above, the Tf R1 IREs, though predicted to exhibit a single C-bulge, appear to form a structure similar to the ferritin IL/B when present in their native sequence context [122]. Sequences outside or flanking the canonical 28 nt IRE sequence can also affect IRE function [150,157]. For example, the ferritin IRE is typically flanked by sequences that extend the RNA helix such that the IRE is a central part of a larger and more stable structure compared to the m-acon or eALAS IREs (Fig. 3). The evidence suggests that the flanking sequences are structurally integrated with the IRE in ferritin mRNA facilitating high affinity binding of IRP1 and IRP2 [150]. Mutations in the IRE region, which are linked to several human diseases, can decrease or increase binding of IRPs [158–161].
These studies indicate that IRP1 and IRP2 have different requirements for high affinity RNA binding in vitro and provide a potential mechanistic basis for the differential regulation of IRE-containing mRNAs in vivo [108]. Further support for this concept comes from studies of mRNAs containing non-natural IRE sequences that show IRP1 and IRP2 can selectively control the translation of mRNA in cells [162]. However, studies of natural IRE-containing mRNAs suggest that the correlation between in vitro binding of IRPs to specific IREs and mRNA regulation by IRPs in vivo has not been fully elucidated. Of particular note is the finding that loss of IRP2, but less so IRP1, leads to dysregulation of ferritin, Tf R1 and eALAS expression in mice [163–165]. These studies suggest that during normal physiological conditions IRP1 cannot fully compensate for IRP2. These results, however, appear to stand in contrast with studies of IRP binding to specific IREs where IRP1 was found to bind all IREs as well as, or better than, IRP2 and the finding that IRP2 prefers the ferritin IRE [108,166]. This apparent discrepancy may involve tissue specific differences in the abundance of each IRP or of individual IRE-containing mRNAs and will hopefully be resolved in future studies. The use of animal models will be key to understanding how IRE sequence and structure affect IRP function and how the selective actions of these RNA binding proteins ultimately control cellular and organismal metabolism.
3. Recent advances concerning the regulation of IRP function
In recent years significant progress has been made in defining many aspects of IRP1 and IRP2 regulation and function. For IRP1 these include determining: (a) the physiological processes and some participants underlying the assembly and disassembly of its Fe–S cluster; (b) the role of phosphorylation in controlling the aconitase function and mechanism of iron-regulation of RNA binding; and (c) the consequences of dysregulation of IRP1 function either through loss of its expression or its unregulated activation. Advancements concerning the function and regulation of IRP2 include: (a) identifying additional iron- and heme-mediated regulatory mechanisms for controlling RNA binding; and (b) the dysregulation of iron metabolism and neurodegenerative events that occur when IRP2 is ablated.
3.1. Linkages between IRP1 and pathways of Fe–S cluster assembly
The presence of the [4Fe–4S] cluster in the c-acon form of IRP1 has unique consequences for the regulation of IRP1 function. First, it provides a connection between the regulation of cellular iron metabolism by IRP1 and a central pathway of iron consumption, the assembly of Fe–S proteins. Second, the solvent accessibility of the [4Fe–4S] cluster in aconitases renders them sensitive to cluster perturbants including reactive oxygen (superoxide anion, O2−) and nitrogen species (nitric oxide, NO; peroxynitrite, ONOO−) that promote disassembly of the cluster. This makes IRP1 particularly responsive to oxidative or inflammatory stresses or to environmental agents (e.g., paraquat) that are capable of inducing formation of specific cluster perturbants. Third, the action of such perturbants can be controlled by phosphorylation of IRP1/c-acon or by the presence of aconitase substrates (e.g., citrate) or inhibitors [167,168] that can impair perturbant action by binding the Fe–S cluster.
Studies in yeast have provided insight into the metabolic relationships between pathways of iron metabolism and the sensing and regulation of iron homeostasis [169–171]. These studies indicate that the flux of iron through the mitochondrial Fe–S assembly pathway, and not the absolute concentration of cellular iron, serves as a key indicator of cellular iron status. Changes in Fe–S cluster assembly are sensed by the critical transcriptional regulators of yeast iron metabolism, Aft1 and Aft2 [171]. Analysis of the pathways of Fe–S cluster assembly in mammalian cells lags behind what is known in yeast but recent studies have begun to identify the mitochondrial and cytosolic participants. Frataxin, a mitochondrial matrix protein defective in Friedreich’s ataxia (FA), has a role in the formation of mito-chondrial and cytosolic Fe–S proteins but its precise function is still being elucidated [60,62,172,173]. The mitochondrial ABC half-transporter Abcb7 appears to be required for maturation of cytosolic Fe–S proteins as is its yeast orthologue, Atm1p, and is defective in X-linked sideroblastic anemia with ataxia (XLSA/A) [60,62,174]. Using a genetic approach in yeast, Walden and colleagues [175] identified Cfd1 as a cytosolic factor that promotes cluster assembly in IRP1. Cfd1 appears to be a P-loop type ATPase required for cytosolic Fe–S cluster assembly. Lill and coworkers have identified additional yeast cytosolic proteins that likely function together with Cfd1 to form the so-called CIA, or cytosolic iron–sulfur cluster assembly, machinery (reviewed in [60]). Mammalian orthologues of these proteins exist, which suggests that the CIA machinery may play a role in iron-sensing by IRP1 in vivo. Recent identification of cytosolic members of the iron sulfur cluster (Isc) assembly gene family in mammalian cells suggest that multiple systems may promote Fe–S cluster assembly or Fe–S cluster repair in the cytosol [62,176,177].
Recent evidence supports a role of mitochondria in the conversion of IRP1 to c-acon [178–183] and are consistent with results in yeast wherein this organelle has an essential role in cytosolic Fe–S cluster assembly [60]. The recent studies in mammalian cells show that impairment of mitochondrial [178–183] and cytosolic [177] Fe–S cluster assembly pathways leads to activation of IRP1 RNA binding activity and a decrease in cytosolic aconitase activity. Disruption of these pathways of Fe–S biogenesis can result in maladaptive changes in iron metabolism. For instance, a mutant mitochondrial glutaredoxin involved in iron sulfur cluster biogenesis was recently found to cause anemia in zebrafish due to defects in IRP1, but not IRP2; over-activation of IRP1 leads to excessive repression of eALAS and hence, heme formation [183]. Studies of the etiology of the human diseases FA and XLSA/A have provided further insight concerning the relationships between Fe–S biogenesis and iron metabolism. While FA appears to primarily be a disease involving altered mito-chondrial function, including Fe–S biogenesis, recent evidence demonstrates that an inappropriate activation of IRP1 may contribute to pathological changes in iron metabolism [181,182]. Furthermore, disruption of Abcb7, also impacts IRP1. Abcb7, like its yeast counterpart Atm1p [184], is proposed to transport mi-tochondrially derived metabolites to the cytosol for cluster biogenesis. Loss of Abcb7 in liver results in a several-fold activation of IRP1 RNA binding [180]. A smaller activation of IRP2 was also observed. This response of IRPs is associated with increased TfR1 mRNA abundance and increased iron accumulation. Hence, it appears that like yeast, cellular iron sensors in mammalian cells respond not to the total level of iron but to the size of a regulatory iron pool or perhaps to the flux of iron though specific pathways (e.g., Fe–S assembly pathways). Furthermore, it is apparent that there is significant interplay between mitochondrial and cytosolic pathways of iron metabolism in eukaryotes from yeast to mammals. Additional work is needed to further define the link between cellular Fe–S biogenesis and the regulation of iron metabolism in mammalian cells including how dysregulation of IRP1 contributes to maladaptive changes in cellular iron metabolism in human disease.
3.2. Pathways of Fe–S cluster disassembly affect IRP1 function
Much interest has focused on how the Fe–S cluster in c-acon is disassembled in order to generate IRP1 RNA binding activity (Fig. 4). This is important not only in response to oxidative or inflammatory stresses but is also relevant to our understanding of how IRP1 is activated merely in response to iron deficiency. The Fe–S cluster of c-acon does not spontaneously disassemble when iron levels are low, at least on a physiological time scale. Instead, the disassembly process must be initiated by factors extrinsic to c-acon. Several studies have examined the role of cluster perturbants such as O2 and O2 metabolites as well as reactive nitrogen species such as nitric oxide (NO) or peroxy-nitrite (ONOO−) (Fig. 4) that will be discussed here. It is important to note, however, that protein-mediated removal of the [4Fe–4S] cluster of c-acon has not been ruled out. Low molecular weight cluster perturbants (e.g., O2−) promote loss of the Fe–S cluster and increase RNA binding activity or in some cases damage the IRP1 apoprotein leading to its inactivation (reviewed in [10,185,186]). There are several situations where oxygen or ROS likely influence the balance between c-acon and IRP1. These include the constant presence of oxygen or ROS in cells and the changes in their level in response to hypoxia or oxidative stress such as during ischemia/reperfusion injury, in the action of specialized cells (e.g., macrophages) [186–188] or in response to certain drugs [185,189,190] or toxicants [191,192].
Fig. 4.
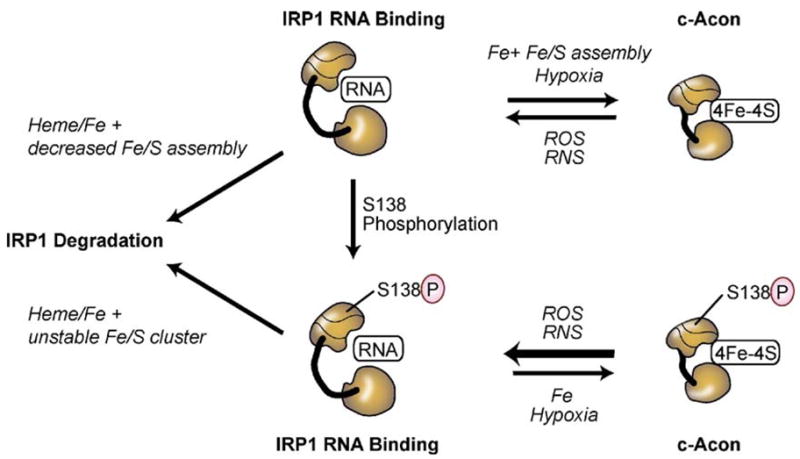
Pathways for regulation of IRP1: IRP1 can be regulated through mechanisms dependent or independent of the [4Fe–4S] cluster. In the cluster dependent pathway IRP1 has dual roles either as a high affinity IRE RNA binding protein when devoid of the [4Fe–4S] cluster or it is the cytosolic isoform of aconitase (c-acon) when the [4Fe–4S] cluster is assembled. The c-acon form does not bind RNAwith high affinity. The [4Fe–4S] cluster is accessible to low molecular cluster perturbants including reactive oxygen species (ROS) such as O2− and H2O2 or reactive nitrogen species such as NO or ONOO−. Hypoxia stabilizes the c-acon form by reducing the level of oxygen or ROS. IRP1 can be phosphorylated at S138 and S711; both sites are not accessible in the c-acon form but are readily phosphorylated in the RNA binding form. Studies with phosphomimetic mutants of IRP1 indicate that the Fe–S cluster can be assembled and the protein exhibits robust aconitase activity. However, the Fe–S cluster is much more sensitive to disruption by cluster perturbants including oxygen and hydrogen peroxide. Iron stimulates the degradation of S138 phosphomimetic mutants of IRP1 as well as S138 phosphorylated IRP1. The non-phosphorylated IRP1 apoprotein is also subject to iron induced protein degradation when the Fe–S cluster biogenesis pathway is impaired.
ROS can influence IRP1 function though multiple mechanisms and produce different responses depending on the amount and species of ROS examined. IRP1 RNA binding activity can be increased by oxygen and reactive oxygen species (ROS) [179,193–197]. Interestingly, the level of oxygen or ROS influence iron-regulation of IRP1 in animals and cultured cells [179,194–196]. The reduction in IRP1 RNA binding activity in response to hypoxia is antagonized by iron chelation [194], suggesting that iron and cluster perturbants have opposing effects on the interconversion of IRP1/c-acon [179,194,195]. In contrast, reversal of the effect of hypoxia on IRP1 by re-oxygenation is dependent on new protein synthesis suggesting inefficient conversion of c-acon to IRP1 or that cluster removal is protein-mediated in some cases [194]. Recent evidence demonstrates that the response of IRP1 to hypoxia and reoxygenation can vary in a cell type specific manner, depending on whether cells use additional levels of gene regulation to control ferritin expression [198]. IRP1 also can be inactivated by high levels ROS. Under such circumstances IRP1 is inactivated presumably through oxidative damage [190,192] and/or degradation [179,197,199]. The loss of IRP1 in response to high levels of ROS and other cluster perturbants may represent a means to prevent over accumulation of RNA binding activity. Cluster removal from c-acon is regulated by the action of H2O2, rapidly activating IRP1 through a membrane-initiated signaling pathway [188,196]. Direct treatment of c-acon with H2O2 does not generate RNA binding activity indicating that additional proteins, and/or altered sensitivity of c-acon to cluster perturbants, must be involved in IRP1 activation when intact cells are treated with H2O2 [200]. The action of H2O2 on IRP1/c-acon can be antagonized by other oxidants in cultured cells [201]. Finally, it is of interest to note that aconitases can vary in the stability of their Fe–S cluster [202], and in the case of IRP1/c-acon the stability of the cluster is regulated through phosphorylation of IRP1 at S138 [203,204]. S138 phosphomimetic mutants of c-acon display enhanced sensitivity to oxygen and H2O2 [203,204]. Future studies are needed to address issues including the mechanism and efficiency of conversion of c-acon to IRP1 and the extent to which dead-end inactive forms of the protein are produced in response to oxidants [204]. Finally, additional animal models are needed in order to more clearly establish the roles of ROS in controlling IRP function and iron homeostasis in vivo.
Much interest has focused on determining the role of NO and other reactive nitrogen species (RNS) such as peroxynitrite (ONOO−) in influencing iron metabolism because the Fe–S cluster of aconitases has long been known to be a target of these agents [205]. Modulation of IRP function by RNS has been suggested to have important roles in both the inflammatory response and in aspects of cell-mediated immunity[185,206, 207]. Numerous studies using cultured cells and more importantly, purified IRP1/c-acon protein, have established that under suitable conditions NO and ONOO−increase IRP1 RNA binding by directly targeting the Fe–S cluster, and in the case of NO, promoting iron loss from cells [208–215]. Future studies need to resolve a number of critical issues concerning the role of RNS in controlling the function of IRP1. First, what are the biologically relevant RNS that act on c-acon to convert it to IRP1? Second, how do these reactive species modify c-acon and what species of IRP1 are created?
It is now generally accepted that the Fe–S cluster of aconitases is a direct target of NO [209,210,216–219]. This was demonstrated by loss of aconitase activity, loss of cluster iron and spectroscopic analysis. Together these results indicate that NO directly attacks, and promotes complete removal of the Fe–S cluster in aconitases with generation of high affinity RNA binding activity. NO treatment of c-acon can directly generate RNA binding activity without a need for additional proteins or other compounds (e.g., reductants) [219]. However, exposure to NO also leads to the generation of other forms of IRP1 such as dinitrosyl iron specie(s) or possibly S-nitroso-IRP1, that lack aconitase activity [210,219,220]. Future studies should determine the efficiency of NO-mediated conversion of c-acon to the RNA binding form or to other species. It will also be necessary to determine whether NO-generated forms of IRP1 (e.g., the dinitrosyl iron form of IRP1) are functional pools of the protein or are degraded within the cell.
Because NO reacts very rapidly with superoxide anion to produce the strong oxidant ONOO−, many investigations have proposed that ONOO−elicits the biological response [208,216, 221–223]. In the case of c-acon, there are differing views concerning the mechanism of action and efficacy of ONOO−in enhancing accumulation of IRP1 RNA binding activity. The commonality in these in vitro studies is the increased efficiency of ONOO−, compared to NO, in inactivating c-acon [210,216, 219,220,222,223]. Critical unresolved issues are what are the species of IRP1 produced after disruption of the Fe–S cluster by ONOO−, are they observed in vivo, and what are their fate? These species include the [3Fe–4S] form of c-acon [220], multiple species of IRP1 with Cys residues in different oxidation states [224,225], or tyrosine nitrated IRP1 [219,220]. Examination of these issues is needed in order to establish the physiological relevance of ONOO−in influencing c-acon/IRP1 function.
Exposure of c-acon to oxidants including ONOO− can lead to loss of the Fe–S cluster and generation of an oxidized form of IRP1 that does not bind RNA unless a reductant is added [106,219,224]. However, in response to ONOO−, a significant fraction of IRP1 appears to be permanently inactivated leading to the conclusion that this oxidant disrupts the normal role of IRP1 in controlling iron metabolism [106,219,220,224]. In contrast, some studies suggest that exposure of c-acon to low levels of ONOO− might activate RNA binding with higher levels inactivating the RNA binding [216]. It is not clear how ONOO− alone leads to production of the reduced apoprotein form of IRP1 since it appears that no reductants were included in the assays [216]. Future studies need to determine which species of IRP1 protein are produced after exposure of purified c-acon to ONOO− as well as how, and if, they can be converted to the active RNA binding form. Furthermore, determining which of these species of IRP1 are formed in cells and can be converted to an RNA binding state or are degraded is another important goal.
Translating these in vitro studies with purified c-acon or cell lysates into an understanding of which RNS affect IRP1 in vivo and which species are the most effective is a challenging task. When considering the impact of ONOO− on cellular function one must contend with its ability to react with a broader range of compounds than NO, since NO is more restricted in the target proteins it can react with (e.g., proteins containing transition metals cofactors), the greater ability of NO relative to ONOO− to cross cell membranes, and the fact that ONOO− has an extremely short biological half-life [221]. Evidence suggesting a role of ONOO− in influencing IRP1 in vivo comes from studies demonstrating nitration of purified c-acon and the detection of nitrated IRP1 in activated macrophages, although these findings in cell culture could reflect the action of ONOO− on c-acon or the apoprotein form of IRP1 [219,226]. Since ONOO−, but not NO, promotes tyrosine nitration of proteins, these studies support the idea that like NO, there is a role for ONOO− in vivo [221]. However, the efficient generation of IRP1 RNA binding activity in cells by pharmacological and physiological agents that increase NO production, coupled with the short half-life and reduced specificity of ONOO-, suggest that NO may be the primary RNS controlling IRP1 function. Taken together, important future goals in this aspect of the IRP1 field includes determination of the species of IRP1 produced in cells in response to RNS and increased focus on the physiological or pathological circumstances under which these agents control iron metabolism.
3.3. Phosphorylation dictates the mechanism of iron regulation of IRP1
Both IRPs are regulated by phosphorylation [179,227] and recent advances have been made in this area regarding IRP1. The RNA binding and aconitase functions of IRP1 can be controlled through protein kinase C (PKC)-dependent phosphorylation [106,179,227–231]. Two PKC phosphorylation sites are present in IRP1, S138 and S711 [179,229,231]. Genetic approaches using S711 phosphomimetic mutants of IRP1 indicate that the aconitase function is targeted selectively in that the forward reaction (citrate to isocitrate) is severely inhibited while the reverse reaction is much less affected [229,231]. Aconitases operate in two modes and it appears that phosphorylation at S711 impairs the citrate but not the isocitrate mode. S711 phosphomimetic mutants can retain high affinity IRE binding activity although the protein appears to be unstable [229,231]. Further studies are needed to determine how S711 phosphorylation affects citrate and isocitrate metabolism as well as IRP1 RNA binding activity in cells.
Recent evidence indicates that IRP1 can be iron-regulated by Fe–S cluster-dependent or Fe–S cluster-independent means and that S138 phosphorylation favors the latter. The classical model for iron regulation of IRP1 involves control of RNA binding activity by insertion or loss of the [4Fe–4S] cluster with no change in total IRP1/c-acon protein level. However, it had been known for years that IRP1 protein is degraded in response to high levels of heme or genetic iron overload but the underlying mechanism for this response was not clear [232–234]. The recent finding that S138 phosphorylation provides a means to invoke the protein degradation response begins to describe such a mechanism [179,230] (Fig. 4). S138 phosphomimetic mutants of IRP1 display a so-called cluster instability phenotype in that they can assemble a [4Fe–4S] cluster, and exhibit robust aconitase activity, but the Fe–S cluster is markedly more sensitive to cluster perturbants [203,204]. A consequence of this phenotype is increased accumulation of IRP1 in the RNA binding form and the suggestion that the Fe–S cluster might not be required for regulation of RNA binding activity. Interestingly, the S138E phos-phomimetic mutant undergoes iron-dependent protein degradation [179,230] in a manner that does not require the [4Fe–4S] cluster [179]. The fact that S138 phosphorylated IRP1 also undergoes iron-dependent degradation argues that phosphorylation at this residue invokes a cluster-independent means for regulating IRP1 [179]. Furthermore, a mutant of IRP1 that cannot be phosphory-lated at S138, nor form the Fe–S cluster, was also subject to iron-dependent degradation. This indicates that S138 phosphorylation can promote, but is not required for, Fe–S cluster independent regulation of IRP1. Further support for the cluster-independent mechanism of IRP1 regulation came from studies of mice with genetic defects in Fe–S cluster assembly or disassembly where, unlike the situation in wildtype mice, liver IRP1 was primarily regulated by protein degradation [179]. These studies indicate that a second mechanism is available to control the accumulation of IRP1 RNA binding activity that can serve as a safeguard when the assembly or disassembly of Fe–S clusters is improperly regulated (Fig. 4). Furthermore, given the different RNA binding characteristics and kinetics of regulation of S138 phosphorylated IRP1 this alternative mechanism for iron-regulation can be invoked to meet the specialized iron needs of proliferating cells or during other situations where the metabolic fate of iron is diverted to specialized needs [179].
Taken together, progress in recent years has illustrated the critical role of the [4Fe–4S] cluster in the regulation of IRP1 function in response to oxidative or inflammatory stresses but has also revealed that IRP1 can be iron regulated in a manner not requiring the [4Fe–4S] cluster. Clearly, multiple agents capable of disrupting the Fe–S cluster in c-acon can promote accumulation of IRP1 RNA binding activity. It is clear that cluster perturbants act together with iron to control the distribution of IRP1 between its RNA binding an aconitase forms. However, the recent findings that high levels of IRP1 RNA binding activity can be deleterious to cell function [183] supports the concept that multiple means are needed to control accumulation of IRE binding activity by IRP1. Iron-dependent regulation of the stability of IRP1 protein provides a mechanism to control accumulation of IRE binding activity that does not require the [4Fe–4S] cluster and which results in different kinetics and/or magnitude of response of IRP1 to changes in iron status or to factors other than iron (e.g., growth factors). Differences in the capacity of cells and tissues to control the expression of IRP1 RNA binding activity using the cluster-dependent or cluster-independent mechanisms may be predictive of their response to various pathological insults.
3.4. Functions for aconitases in lower organisms
In plants and some microorganisms, c-acon is involved in the glyoxylate pathway, allowing these organisms to convert lipids into glucose and other essential compounds. Although c-acon in Caernorhabditis elegans shares striking resemblance to mammalian IRP1, it fails to bind RNA, which is consistent with the lack of IREs in the C. elegans genome [235]. Other IRP1-related proteins in organisms such as Arapidopsis thaliana, Nicotiana tabacum and Trypanosome brucei were reported to possess aconitase activity, but no RNA binding activity was reported for these proteins [236–238]. Finally, recent work demonstrates that of the two cytosolic aconitases in Drosophila, only one can bind RNA [239]. This interesting finding supports the concept that both the aconitase and RNA binding functions of IRP1 have important roles in cell function.
The bifunctional roles of aconitases are not limited to animal cells. Genetic and biochemical studies of bacterial aconitases suggest that they can post-transcriptionally control aspects of iron and energy metabolism arguing that the dual function of this unique family of proteins has been exploited for some time [84–86]. Perhaps most striking is the recent finding that mito-chondrial aconitase has a major role in the maintenance of mitochondrial DNA in yeast [240]. This function of yeast aconitase does not require formation of the Fe–S cluster and appears to involve association of the protein with mitochondrial DNA. Hence, aconitases continue to surprise us with their unexpected roles in cell function.
3.5. Mechanisms of IRP2 regulation
Although human IRP1 and IRP2 are 64% identical and 75% conserved, several differences between the proteins are apparent. The foremost structural variation is the presence of a unique 73 amino acid (73-aa) region at the N-terminus of IRP2. Even though IRP2 contains the conserved cysteines involved in [4Fe–4S] cluster assembly, it cannot assemble a [4Fe–4S] cluster [241] and fails to function as c-acon during iron-sufficient conditions [100]. Instead, the decrease in IRP2 RNA binding activity during iron-sufficient conditions is accompanied by a reduction in protein levels [98–100,242].
The primary regulators of IRP2 protein abundance are iron and oxygen. IRP2 protein levels are diminished during iron-sufficient conditions [98–100,242] by a post-translational mechanism, since mRNA stability and protein synthesis are unaffected by cellular iron status [98,194]. The iron-dependent reduction of IRP2 protein levels is mediated by proteasomal degradation [98,99] and requires the synthesis of an unidentified labile protein(s) [243]. Unlike iron, hypoxia stabilizes IRP2 [244–247]. As cellular oxygen tension decreases from 21% to 3%, IRP2 protein levels and RNA binding activity increase [195]. The hypoxic stabilization of IRP2 can be reversed by treatment with iron [195,244] showing that IRP2 stabilization is not due to a general decrease in proteasomal function during hypoxia. The regulation of IRP2 by oxygen deserves significant consideration due to the hypoxic nature of most tissues. At physiological oxygen tensions, IRP2 is the predominant RNA binding protein since IRP1 is mainly in its c-acon form [137,195], although this can depend on iron status [137]. IRP2 may have evolved to regulate IRE-containing mRNAs in vertebrates since it is conserved in mammals, frogs, and fish and is absent in flies, worms, and trypanosomes. The IRP2 binding preference for the H- and L-ferritin IREs [108,156] supports this concept, since translational regulation is the primary mechanism for ferritin regulation in vertebrates.
Three models for iron-mediated IRP2 degradation have been proposed (Fig. 5). For many years, the predominant model for iron-dependent IRP2 degradation has involved the unique 73-aa region of the protein. In this first model, three of the five cysteines in the 73-aa region (C168, C174 and C178) facilitate an iron- and oxygen-dependent oxidative modification that signals for protein ubiquitination and ultimately proteasomal degradation [99,248]. In vitro studies with a 63-aa peptide, which corresponds to a subset of the 73-aa region, indicate that these three cysteines coordinate ferrous iron and that one cysteine is oxidatively modified to dehydrocysteine and other products [249]. Removal of the 73-aa region from IRP2 abolishes its iron-dependent degradation in the human rhabdomyosarcoma RD4 cell line, while insertion of the 73-aa sequence into IRP1 confers iron-dependent degradation to IRP1 [99,250]. Taken together, this work suggests that the 73-aa region of IRP2 is necessary for iron-dependent degradation. More recent studies using human embryonic kidney 293 (HEK293) and human H1299 lung cancer cells show that an IRP2 mutant protein lacking the 73-aa region and an IRP2 (C168S/C174S/C178S) triple mutant protein are both degraded during iron-replete conditions [246,251,252]. The reason for these conflicting data is uncertain. However, these data show that the 73-aa region of IRP2 is not necessary for iron-mediated degradation at least in HEK293 or H1299 cells. This suggests that multiple cell type specific mechanisms may exist to regulate iron-mediated IRP2 degradation.
Fig. 5.
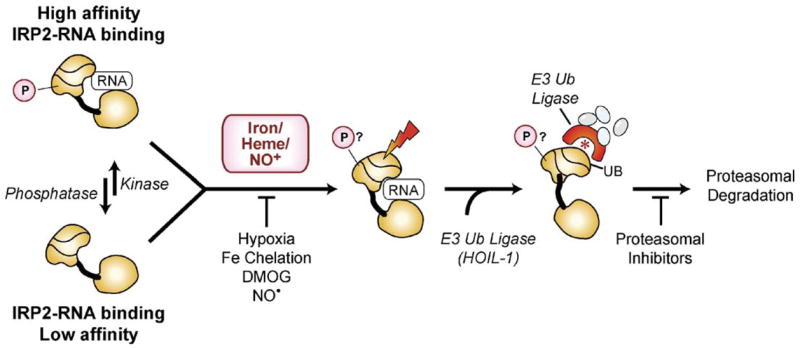
Proposed models for iron-dependent and iron-independent IRP2 regulation. Iron-dependent pathway: iron and/or heme transiently bind IRP2 modifying a specific amino acid(s), which is then recognized by a specific E3-ubiquitin ligase (HOIL-1) leading to IRP2 ubiquitination (UB) and proteasomal degradation. Alternatively, iron/oxygen activates a 2-OG-dioxygenase that hydroxylates IRP2, which provides a target site for a specific E3-ubiquitin ligase. NO+ stimulates IRP2 degradation while NO−stabilizes IRP2. Hypoxia, iron chelators and the 2-OG-dioxygenase inhibitor DMOG also stabilize IRP2. Proteasome inhibitors block IRP2 degradation. IRP2 iron-independent pathway: phosphorylation regulates IRP2 RNA binding activity independent of iron. IRP2 can switch between a high-affinity phosphorylated form and a low-affinity dephosphorylated form, which are regulated by specific protein kinases and phosphatases. Both forms are substrates for iron-mediated degradation. IRP2 phosphorylation may allow IRP2 to alter iron-homeostasis independent of cellular iron concentration. IRP2 is displayed based on m-acon structure with three domains (I–III) connected by a linker to domain IV.
A second model for IRP2 degradation involves heme-mediated protein oxidation. IRP2 degradation can be stimulated by the administration of exogenous heme [232,251,253], although it is not clear whether exogenous heme is directly affecting IRP2 protein levels or if it is providing a source of free iron following liberation from the porphyrin ring. Evidence for a direct heme effect includes oxygen-dependent in vitro binding of heme to C168 and the subsequent oxygen-dependent oxidation of C174 and C178 in the IRP2 63-aa peptide [250,254,255]. Other in vitro studies, however, show that heme binds to C201 and H204 located within the 73-aa region [250], but outside the region corresponding to the 63-aa peptide used by Jeong et al. [255]. These amino acids are found in the sequence C201PFH204, which resembles a canonical heme regulatory motif found in proteins regulated by heme [256,257]. It is proposed that redox exchange between these ligands generates an oxidative modification that targets IRP2 for degradation. In RD4 cells, mutation of C201 and H204 attenuates heme-mediated IRP2 degradation but does not completely abolish it, which suggests that other amino acids are required to coordinate heme [250]. Mutation of the five cysteines (C135, C168, C174, C178 and C201) in an IRP1 molecule containing the 73-aa region fails to completely block heme-mediated degradation in RD4 cells [99,250], suggesting that heme may mediate its effect on IRP2 degradation at a location outside the 73-aa region. Other studies show that an IRP2 protein lacking the 73-aa region or IRP2 proteins containing C168S/C174S/C178S or C137A/C168A/C174A/C178A/ C201A substitutions undergo proteasomal degradation in hemin-treated H1299 cells and HEK293 cells [251,252]. One possible explanation for these results is that like iron-mediated IRP2 degradation, heme-mediated degradation mechanisms may be cell type specific. The evidence for a direct heme effect on IRP2 protein levels is primarily based on heme binding to the 73-aa region of IRP2 in vitro since in vivo studies have yielded conflicting results. While this is still a plausible model, it is important to note the promiscuous nature of heme binding to proteins in vitro [258].
Other studies indicate that exogenous heme functions only as an iron source, since iron chelation abrogates heme-mediated IRP2 degradation [251]. Interestingly, inhibition of heme synthesis with the 5-aminolevulinate dehydratase inhibitor succinylacetone reduces both iron-mediated IRP2 degradation [232,250,253,254] and, to a lesser extent, heme-mediated IRP2 degradation [250,251]. This implies that iron mediates IRP2 degradation, at least partially, by stimulating heme synthesis, and that exogenous heme may be degraded intracellularly or processed differently from endogenous heme. Whether IRP2 senses non-heme or heme iron, or both forms of iron, is unresolved. Cellular heme content correlates with iron availability when iron stores are limited [250]. During iron-sufficient conditions, however, IRP2 degradation occurs without an observed enhancement of heme synthesis or cellular heme content [250]. Therefore, multiple mechanisms involving both heme and iron sensing may exist to regulate IRP2 during different cellular conditions.
Since the involvement of the 73-aa region of IRP2 in iron- and heme-mediated degradation is controversial, the role of the recently identified IRP2 E3 ubiquitin ligase HOIL-1 (heme-oxidized IRP2 ubiquitin ligase-1) as the primary IRP2 E3 ligase is unexpected. HOIL-1 was identified in a yeast two-hybrid screen that utilized the oxidized recombinant 73-aa region of IRP2 as bait [254]. HOIL-1 ubiquitinates heme- or iron-treated IRP2 in vitro and binds to the 73-aa region of IRP2 in RD4 cells treated with iron or heme, but not in cells treated with desferi-oxamine [254]. These data show that the binding of HOIL-1 to the 73-aa region is iron/heme dependent. HOIL-1 has been previously identified in other yeast two-hybrid screens where it interacted with the hepatitis B virus X-protein [259], the E2 ligase UbcM4/UbcH7 [260], the kinases PKCβI and PKCζ [261,262] and the suppressor of cytokine signaling protein SOCS6 [263]. The role of HOIL-1 in ubiquitination and degradation of these proteins has only been reported for SOCS6-associated proteins. In some studies HOIL-1/RBCK1 was shown to act as a transcriptional activator that shuttled in and out of the nucleus [261, 262]. The interaction of HOIL-1 with many partners and the dispensable nature of the 73-aa region of IRP2 for degradation in some cell types suggest that another IRP2 E3 ubiquitin ligase must exist.
A third model suggests that iron-mediated degradation of IRP2 may involve the activity of a 2-oxoglutarate (2-OG)-dependent dioxygenase, which requires iron, oxygen and 2-OG for substrate hydroxylation [264]. Since both IRP2 degradation and 2-OG-dependent dioxygenase activity are inhibited by desferioxamine, hypoxia and the hypoxia mimetic cobalt chloride, the involvement of 2-OG-dependent dioxygenases in iron-mediated IRP2 degradation was examined. DMOG, an inhibitor of 2-OG-dependent dioxygenases, prevents iron-mediated IRP2 degradation [246,252]. DMOG does not appear to function as an iron chelator since IRP1 RNA binding activity is not affected [246, 252]. This mechanism of IRP2 regulation may only be effective when previously iron deficient cells are stimulated with iron, since DMOG is less effective as an inhibitor of IRP2 degradation in cells that do not receive pretreatment with desferioxamine [252]. 2-OG-dependent dioxygenase activity may be required for direct IRP2 hydroxylation but the identification of a hydroxylated site is required for definitive proof. It is also possible that hydroxylation of an upstream component of the IRP2 degradation machinery is responsible for the stabilization of IRP2 by DMOG.
Additional regulation of IRP2 is facilitated by nitric oxide (NO). NO functions to stabilize or degrade IRP2 depending on the type of NO donor used. Kim et al. show that sodium nitro-prusside (SNP) treatment may induce S-nitrosylation at C178 in the 73-aa region, which targets IRP2 for degradation in COS-1 cells [265]. Recent studies show that SNP-mediated IRP2 degradation may be due to the release of iron, not NO+, from SNP. For example, IRP2 degradation can be blocked by simultaneous treatment with SNP and desferioxamine [251,266] and treatment of H1299 cells with photodegraded SNP, which cannot release NO+, stimulates IRP2 degradation [266]. These data show that SNP mediates its effect on IRP2 degradation by releasing the iron of nitroprusside leading to an increase in intracellular iron levels [266]. S-nitrosylation of IRP2 may occur at C178 but it is unlikely that this modification stimulates IRP2 degradation since an IRP2 protein lacking the 73-aa region or an IRP2 (C168S/C174S/C178S) mutant protein both degrade in SNP- treated H1299 cells [266]. In contrast to the stimulation of IRP2 degradation caused by the release of iron from SNP, NO generated by the pharmacological NO donor S-nitoso-N-acetyl-penicillamine (SNAP) or NO generated physiologically by B6.NOS cells stabilizes IRP2 [267]. IRP2 stabilization is apparently not due to a general decrease in proteasomal function in response to NO and does not require the 73-aa domain [267]. The SNAP-generated NO may indirectly regulate IRP2 by decreasing intracellular iron levels through the formation of iron–nitrosyl complexes [268]. The mechanisms of NO-mediated IRP2 regulation are not entirely elucidated. Pharmacologically or biologically generated NO appears to indirectly affect IRP2 stability by modulation of the intracellular iron pool. Seemingly contradictory data may be a result of technical or cell type specific differences.
IRP2 is also regulated by phosphorylation. Induction of HL-60 cell differentiation by phorbol 12-myristate 13-acetate (PMA) treatment stimulates IRP2 phosphorylation and increases RNA binding activity [227]. Phosphorylation activates a latent pool of IRP2, since the increase in RNA binding activity is not attributable to an increase in IRP2 protein synthesis [227]. IRP2 phos-phorylation provides a mechanism for converting IRP2 from a low-affinity RNA binding form to a high-affinity RNA binding form in an iron-independent manner. This mechanism may be important for regulating Tf R1 and ferritin during periods of cellular proliferation, differentiation or stress.
Multiple mechanisms of IRP2 regulation may exist to regulate iron homeostasis in different cell types or in response to different cellular conditions. These mechanisms include the iron- and/or heme-mediated regulation of IRP2 protein stability and the iron-independent regulation of IRP2 RNA binding activity by phosphorylation (Fig. 5). The exact manner by which these regulators affect IRP2 protein stability and/or RNA binding activity has yet to be determined. The 73-aa region of IRP2 does not appear to be necessary for iron- and/or heme-mediated degradation of IRP2 as previously thought. The use of pharmacological agents like DMOG, SNP and SNAP has yielded insight into the regulation of IRP2 by oxygen and nitric oxide. However, these results should be interpreted carefully as these agents may be indirectly modulating intracellular iron levels. Future work will focus on the identification of iron- and/ or heme-mediated IRP2 protein modifications, IRP2 structural changes or protein–protein interactions that modulate protein stability and RNA binding activity. Additionally, expansion of studies into animal models would provide a more physiological setting to examine the complex regulation of IRP2.
3.6. Pathological effects of IRP1 and IRP2 deficiencies in mice
Genetically engineered mouse models of IRP1 and IRP2 deficiency have been generated. Mice with a targeted disruption of the Irp1 gene display no overt phenotype [165,269] except for mild dysregulation of ferritin expression in kidney and brown fat [165]. The lack of an overt phenotype in Irp1−/− mice is surprising given the conservation of c-acon in diverse organisms. The physiological function of c-acon still remains elusive. These data suggest that the c-acon and RNA binding forms of IRP1 are dispensable, at least during normal physiology.
Mouse models of IRP2 deficiency have been generated by constitutive and conditional inactivation of the Irp2 gene. Mice homozygous for mutant Irp2 alleles were generated by the insertion of a PGK-neomycin gene into exon 3/4 of the Irp2 gene [29]. These mice developed mild microcytic anemia, erythro-poietic protoporphyria and a progressive neurodegenerative disease characterized by an unsteady wide-based gait, ataxia, vestibular dysfunction, bradykinesia, tremors and postural defects that develop after 6 months of age [29,163,270]. In these mice, iron accumulates in liver, where it is associated with elevated ferritin and reduced Tf R1 expression, and in duodenum where it is associated with elevated ferritin and DMT1 expression [29]. In bone marrow, low iron stores and increased protopor-phyrin IX and zinc protoporphyrin levels correlate with reduced Tf R1 expression and increased eALAS biosynthesis [163,164]. The microcytic anemia can be explained by translational derepression of eALAS-IRE mRNA and the destabilization of Tf R1-IRE mRNA due to IRP2 deficiency. While many tissues acquire iron by TF-dependent as well as TF-independent pathways, precursor erythroid cells are dependent mainly on TF-bound iron for heme synthesis [271,272], and consequently, IRP2 deficiency leads to reduced Tf R1 expression and reduced iron acquisition. Interestingly, a genetic approach in zebrafish indicates that over-expression of IRP1 RNA binding activity can also produce a microcytic anemia, due in this case to excessive repression of eALAS mRNA translation [183].
Inactivation of both Irp1 and Irp2 (Irp2−/ − Irp1−/ −) genes results in embryonic lethality at the blastocyst stage [164,165,273]. Mice that lack both copies of IRP2 and one copy of IRP1 (Irp2−/ −Irp1+/−) develop more severe neurode-generative disease at an earlier age than Irp2−/ − mice [274]. The neurodegeneration is characterized by widespread axonpathy and vacuolization in several regions of brain, notably the substantia nigra. Irp2−/ −Irp1+/− mice also show marked alterations in ferritin and Tf R1 expression in forebrain, develop microcytic anemia and display abnormal iron homeostasis in liver. These studies show that IRP2 functionally substitutes for IRP1, but that IRP1 only partially substitutes for IRP2.
The molecular basis for the neurodegenerative disease in Irp2−/ − and Irp2−/ −Irp1+/− mice is not clear. However, dysregulation of iron homeostasis is associated with neurodegenerative diseases including Alzheimer’s disease, Friedreich’s ataxia, Parkinson’s disease and Pantothenate Kinase deficiency [30,275]. In Irp2−/ − and Irp2−/ −Irp1+/− mice, ferritin and ferric iron are elevated in neurons located in multiple regions of the brain, including substantia nigra, hippocampus, caudate putamen and cerebellum, and this correlates with axonal degeneration [29,274]. Tf R1 expression decreases in cerebellum and forebrain extracts [29,165]. It is also possible that other IRE mRNAs, as well as other genes, are dysregulated in brains from Irp2−/ − and Irp2−/ −Irp1+/− mice, and that this dysregulation contributes to axonal degeneration. Electron tomography shows that ferritin in Irp2−/ −Irp1+/− mouse brains is located in doubled-wall vesicular compartments that are thought to be invaginations of oliogoden-drocyte membranes into axons, while ferritin content in axons is reduced [276]. The mechanism responsible for the reduced axonal ferritin and axonal degeneration is not clear. Reduced ferritin content in axons, however, may be associated with iron deficiency, which ultimately could affect neuronal integrity. The importance of ferritin in brain iron metabolism is made evident by other studies showing that patients carrying a mutation in the ferritin-L mRNA, which alters ferritin structure and function, develop dominant adult-onset basal ganglia disease [28], while Fth+/− mice display evidence of oxidative stress and iron deficiency in brain [31].
A second model of IRP2 deficiency was generated by the conditional inactivation of the Irp2 gene [164,269]. A β-gala-ctosidase-neomycin (β-Geo) cassette flanked by Frt sites was inserted into intron 2 of the Irp2 gene, disrupting the Irp2 open reading frame and generating a null allele. The β-Geo was inserted with LoxP sites flanking exon 3 to allow excision of exon 3 by Cre recombinase to generate complete null alleles lacking the β-Geo cassette. Like the Irp2−/ − mice generated by LaVaute et al. [29], Irp2−/− mice carrying the β-Geo cassette or mice lacking this cassette develop microcytosis, with reduced Tf R1 mRNA expression in bone marrow and accumulation of iron and ferritin in duodenum and liver [164]. Nonheme iron is reduced in spleen and correlates with reduced ferritin and FPN expression. Unlike LaVaute’s Irp2−/ − mice, FPN and DMT1 expression are not elevated in duodenum. Interestingly, 12-month-old mice did not display the severe neurodegenerative disease that was observed in the same age mice generated by LaVaute et. al. [29]. The reason for the strikingly different neuro-degenerative phenotypes between the two Irp2−/ − mouse strains is not clear. It is known, however, that different gene targeting approaches can lead to different phenotypes. This could be due to silencing of neighboring genes caused by the generation of truncated gene products with biological activity or the retention of selection cassettes [277]. A relevant example of the latter case is the Irp1−/ − mouse carrying the β-Geo cassette, in which the presence of the cassette causes partially penetrant embryonic lethality unrelated to IRP1 deficiency, while its removal results in Irp1−/ − mice born in Mendelian ratios [269]. Another relevant example is the iron-loaded phenotype of Usf2−/ − (Upstream Stimulatory Factor 2) null mice [278]. This phenotype was not due to Usf2 deficiency, but rather to the silencing of adjacent hepcidin genes, since a distinct Usf2−/ − mouse generated by a different targeting strategy was not iron-loaded [279,280]. The phenotype of mutant mice can also be influenced by their genetic background [281,282]. For example, natural variation in iron metabolism in inbred mouse strains has been reported [283,284], which has been shown to affect the severity of iron accumulation in Hfe−/ −mice [285]. Whether the different neurodegenerative phenotypes displayed by the two independent Irp2−/ − mouse strains are caused by different targeting strategies or are attributable to their genetic background remains to be determined.
Why is IRP1 unable to fully compensate for the loss of IRP2 function in these models? A clue comes from studies showing that the RNA binding activity of IRP1, but not IRP2, appears refractory to activation in tissues of some, but not all [179], strains of mice fed a low iron diet [165]. These studies suggest that IRP1 is less sensitive to iron depletion in vivo, suggesting that IRP2 could be the predominant iron sensor in mammalian cells. One possible explanation for the reduced sensitivity of IRP1 RNA binding activity to iron depletion is that in the hypoxic environment of most tissues, IRP1 exists predominately in its c-acon [4Fe–4S] cluster form [137,165,195,286]. The notion is that the [4Fe–4S] cluster of IRP1 is stable in a low oxygen environment, where free radical generation is expected to be low, rendering IRP1 less sensitive to changes in iron concentration. Other studies show that both IRP1 and IRP2 RNA binding activities are activated in liver lysates isolated from rats and mice fed a low iron diet, though RNA binding activity of IRP2 is somewhat more sensitive to iron depletion than that of IRP1 in rats [137,179,287,288]. IRP1 still has an important role in iron homeostasis, based on the more severe neurodegenerative phenotype in Irp1+/−/Irp2−/ − mice and the fact that Irp1−/ −/Irp2−/ −mice are not viable past the blastocyst stage [164,274]. As mentioned earlier, recent work in zebrafish also indicates that over-activation of IRP1 can have severe pathological consequences [183].
Different tissue-specific expression patterns of IRP1 and IRP2 could also contribute to the distinct phenotypes of Irp1−/ − and Irp2−/ − mice [122,243]. IRP2 shows the highest expression in brain, skeletal muscle and heart, while IRP1 predominates in kidney, brown fat and liver. IRP1 and IRP2 also show distinct expression patterns in mouse brain with respect to cell types when analyzed by immunohistochemistry [289]. The fact that IRP1 expression is highest in kidney and brown fat is consistent with dysregulation of ferritin synthesis in these tissues in Irp1−/ −mice [165].
Another factor that might contribute to the distinct phenotypes of Irp1−/ − and Irp2−/ − mice is the apparently difference in binding affinities of IRP1 and IRP2 to IRE-mRNAs. This is due, at least in part, to structural differences in the stem of the hairpin loop of the IRE, which contains either a C-bulge (m-aconitase, eALAS or Tf R1) or an IL/B (ferritin) [108,145,147,290–292]. While the ferritin-H IRE and the five Tf R1 IREs bind IRP1 and IRP2 with similar apparent affinities, m-acon, eALAS and single Tf R IREs may preferentially bind IRP1 [100,108,122]. Collectively, these studies suggest that different mechanisms of regulation, expression patterns and RNA binding affinities of IRP1 and IRP2 contribute to IRE-mRNA regulation.
3.7. Consequences of dysregulation of IRE-containing mRNAs
In addition to dysregulation of IRE-containing mRNAs due to IRP1 and/or IRP2 deficiencies in mice, mutations in IREs or in the coding or regulatory sequences of IRE genes can be deleterious. Targeted disruptions of the ferritin-H gene or of the Tf R1 gene in mice result in embryonic lethality [31,293,294]. In humans, mutations in the ferritin-L IRE cause hyperferritinemia cataract syndrome [158,295–297], while a mutation in the ferritin-H IRE, which increases IRP binding, is associated with high serum ferritin levels and iron overload [159]. In a poly-cythaemia (Pcm) mouse strain, a 58-bp deletion in the FPN promoter results in aberrant transcriptional initiation and the elimination of the 5′ IRE sequence in FPN mRNA, leading to the accumulation of FPN protein in liver and gut [298]. This suggests that the loss of the IRE may be responsible for altered FPN expression. Missense mutations in the Fpn gene in humans cause hemochromatosis type IV also known as autosomal dominant ferroportin disease [299]. In one hemochromatosis patient, a novel mutation in the Fpn gene was identified in the 5′ UTR downstream of the IRE. Whether this mutation alters IRP binding to the IRE has not been yet defined [161]. Finally, mutations in the eALAS gene cause X-linked sideroblastic anemia [174]. These studies show that improper expression of proteins encoded by IRE-containing mRNAs disrupt iron metabolism at the cellular and organismal levels, leading to neurodegenerative diseases, iron overload and other diseases in humans, and lethality in animal models.
4. Outlook
Future research in the IRP field will focus on elucidating the multiple mechanisms responsible for the regulation of IRP1 and IRP2, the consequences of loss or over-expression of each IRP on cellular and organismal iron homeostasis, determining how each IRP selectively regulates the use of specific target mRNAs and identifying novel IRE-containing mRNAs. Further analysis of the relationship between Fe–S assembly and disassembly and the control of iron metabolism via IRP1 is needed. This includes determination of the specific Fe-S assembly factors involved in converting IRP1 to c-acon. The pathways used and species produced during disassembly of the Fe-S cluster in c-acon in response to cluster perturbants also needs further attention in order to fully understand the mechanism of the Fe-S switch. Additional mouse models for determining how disruption of Fe–S cluster assembly or disassembly affects IRP1 and iron metabolism are needed. Furthermore, determining how IRP1 is controlled through phosphorylation and its role in cluster-independent regulation is needed. Finally, a better understanding of the tissue-specific mechanisms underlying the adaptive responses to iron deficiency and the role of IRPs in this process should shed light on the metabolic remodeling that likely occurs in response to variations in iron availability. For IRP2, it will be important to identify iron and/or heme induced protein modifications and to determine whether multiple mechanisms exist to regulate IRP2 protein stability in different cell types. The mechanisms by which other effectors such as NO, phosphorylation and DMOG affect IRP2 activity requires further study. Additional mouse models of IRP2 deficiency are needed to definitively determine if IRP2 deficiency causes neurodegenerative disease, and if so, how dysregulation of iron homeostasis in brain contributes to neurodegenerative disorders.
Acknowledgments
Support for this work was from the NIH DK47219 and DK66600 (RSE) and NIH GM45201 (EAL). RSE also acknowledges support from USDA grant #2006-35200-16604 and UW Madison Hatch project #4885. We thank Eric Hanson and Kate Deck for helpful comments.
References
- 1.Wood RJ, Ronnenberg AG. Iron. 10. Lippincott Williams and Wilkins; Philadelphia, PA: 2006. [Google Scholar]
- 2.Lynch SR. The impact of iron fortification on nutritional anaemia. Best Pract Res Clin Haematol. 2005;18:333–346. doi: 10.1016/j.beha.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Hunt JM. Reversing productivity losses from iron deficiency: the economic case. J Nutr. 2002;132:794S–801S. doi: 10.1093/jn/132.4.794S. [DOI] [PubMed] [Google Scholar]
- 4.WHO. The World Health Report 2002-Reducing Risks, Promoting Health Life Styles. World Health Organization; 2002. [Google Scholar]
- 5.Anonymous. Iron deficiency—United States, 1999–2000. MMWR. 2002;51:897–899. [PubMed] [Google Scholar]
- 6.Allen LH. Biological mechanisms that might underlie iron’s effects on fetal growth and preterm birth. J Nutr. 2001;131:581S–589S. doi: 10.1093/jn/131.2.581S. [DOI] [PubMed] [Google Scholar]
- 7.Beard J. Iron deficiency alters brain development and functioning. J Nutr. 2003;133:1468S–1472S. doi: 10.1093/jn/133.5.1468S. [DOI] [PubMed] [Google Scholar]
- 8.Beard JL. Iron biology in immune function, muscle metabolism and neuronal functioning. J Nutr. 2001;131:568S–579S. doi: 10.1093/jn/131.2.568S. [DOI] [PubMed] [Google Scholar]
- 9.Dallman PR. Biochemical basis for the manifestations of iron deficiency. Annu Rev Nutr. 1986;6:13–40. doi: 10.1146/annurev.nu.06.070186.000305. [DOI] [PubMed] [Google Scholar]
- 10.Eisenstein RS. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu Rev Nutr. 2000;20:627–662. doi: 10.1146/annurev.nutr.20.1.627. [DOI] [PubMed] [Google Scholar]
- 11.Haas TB JD., IV Iron deficiency and reduced work capacity: a critical review of the research to determine a causal relationship. J Nutr. 2001;131:676S–690S. doi: 10.1093/jn/131.2.676S. [DOI] [PubMed] [Google Scholar]
- 12.I.N.A.C.G. (INACG) XII INACG Meeting; Washington D.C. 1990. [Google Scholar]
- 13.Cook JD, Flowers CH, Skikne BS. The quantitative assessment of body iron. Blood. 2003;101:3359–3364. doi: 10.1182/blood-2002-10-3071. [DOI] [PubMed] [Google Scholar]
- 14.Ross KL, Eisenstein RS. Novel roles for iron regulatory protein in the adaptive response to iron deficiency. J Nutr. 2003;133:1510S–1516S. doi: 10.1093/jn/133.5.1510S. [DOI] [PubMed] [Google Scholar]
- 15.Ajioka RS, Kushner JP. Hereditary hemochromatosis. Semin Hematol. 2002;39:235–241. doi: 10.1053/shem.2002.35634. [DOI] [PubMed] [Google Scholar]
- 16.Beutler E. Hemochromatosis: genetics and pathophysiology. Annu Rev Med. 2006;57:331–347. doi: 10.1146/annurev.med.57.121304.131310. [DOI] [PubMed] [Google Scholar]
- 17.Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106:3710–3717. doi: 10.1182/blood-2005-05-1857. [DOI] [PubMed] [Google Scholar]
- 18.Heeney MM, Andrews NC. Iron homeostasis and inherited iron overload disorders: an overview. Hematol Oncol Clin North Am. 2004;18:1379–1403. doi: 10.1016/j.hoc.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 20.Aisen P, Cohen G, Kang JO. Iron toxicosis. Int Rev Exp Pathol. 1990;31:1–46. doi: 10.1016/b978-0-12-364931-7.50006-9. [DOI] [PubMed] [Google Scholar]
- 21.Bacon BR, Britton RS. The pathology of iron overload: a free radical-mediated process? Hepatology. 1990;11:127–136. doi: 10.1002/hep.1840110122. [DOI] [PubMed] [Google Scholar]
- 22.Gregory A, Hayflick SJ. Neurodegeneration with brain iron accumulation. Folia Neuropathol. 2005;43:286–296. [PMC free article] [PubMed] [Google Scholar]
- 23.Puccio H, Koenig M. Friedrich ataxia: a paradigm for mitochondrial diseases. Curr Opin Genet Dev. 2002;12:272–277. doi: 10.1016/s0959-437x(02)00298-8. [DOI] [PubMed] [Google Scholar]
- 24.Sipe JC, Lee P, Beutler E. Brain iron metabolism and neurodegenerative disorders. Dev Neurosci. 2002;24:188–196. doi: 10.1159/000065701. [DOI] [PubMed] [Google Scholar]
- 25.Wolozin B, Golts N. Iron and Parkinson’s disease. Neuroscientist. 2002;8:22–32. doi: 10.1177/107385840200800107. [DOI] [PubMed] [Google Scholar]
- 26.Xu X, Pin S, Gathinji M, Fuchs R, Harris ZL. Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Ann N Y Acad Sci. 2004;1012:299–305. doi: 10.1196/annals.1306.024. [DOI] [PubMed] [Google Scholar]
- 27.Brunt EM. Pathology of hepatic iron overload. Semin Liver Dis. 2005;25:392–401. doi: 10.1055/s-2005-923311. [DOI] [PubMed] [Google Scholar]
- 28.Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J. Mutation in the gene encoding ferritin light polypeptide causes adult dominant basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- 29.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos RS M, III, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 30.Rouault TA. Iron on the brain. Nat Genet. 2001;28:299–300. doi: 10.1038/91036. [DOI] [PubMed] [Google Scholar]
- 31.Thompson K, Menzies S, Muckenthaler M, Torti FM, Wood T, Torti SV, Hentze MW, Beard J, Connor J. Mouse brains deficient in H-ferritin have normal iron concentration but a profile of iron deficiency and increased evidence of oxidative stress. J Neurosci Res. 2003;71:46–63. doi: 10.1002/jnr.10463. [DOI] [PubMed] [Google Scholar]
- 32.Frazer DM, Anderson GJ. Iron imports: I. Intestinal iron absorption and its regulation. Am J Physiol. 2005;289:G631–G635. doi: 10.1152/ajpgi.00220.2005. [DOI] [PubMed] [Google Scholar]
- 33.Mackenzie B, Garrick MD. Iron imports: II. Iron uptake at the apical membrane in the intestine. Am J Physiol. 2005;289:G981–G986. doi: 10.1152/ajpgi.00363.2005. [DOI] [PubMed] [Google Scholar]
- 34.Wessling-Resnick M. Iron imports: III. Transfer of iron from the mucosa into circulation. Am J Physiol. 2006;290:G1–G6. doi: 10.1152/ajpgi.00415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT. Identification of an intestinal heme transporter. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 36.Andrews NC. Understanding heme transport. New Eng J Med. 2005;353:2508–2509. doi: 10.1056/NEJMcibr053987. [DOI] [PubMed] [Google Scholar]
- 37.Fleming CCT MD, III, Su MA, Foernzler D, Bier DR, Dietrich WF, Andrews NC. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 38.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metalion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 39.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 40.Gunshin H, Fujiwara Y, Custodio AO, DiRenzo C, Robine S, Andrews NC. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Invest. 2005;115:1258–1266. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lam-Yuk-Tseung S, Gros P. Distinct targeting and recycling properties of two isoforms of the iron transporter DMT1 (NRAMP2, SlcIIA2) Biochemistry. 2006;45:2294–2301. doi: 10.1021/bi052307m. [DOI] [PubMed] [Google Scholar]
- 42.Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. 1998;95:1148–1153. doi: 10.1073/pnas.95.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iolascon A, d’Apolito M, Servedio V, Cimmino F, Piga A, Camaschella C. Microcytic anemia and hepatic iron overload in a child with compound heterozygous mutations in DMT1 (SCL11A2) Blood. 2006;107:349–354. doi: 10.1182/blood-2005-06-2477. [DOI] [PubMed] [Google Scholar]
- 44.Mims MP, Guan Y, Pospisilova D, Priwitzerova M, Indrak K, Ponka P, Divoky V, Prchal JT. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood. 2005;105:1337–1342. doi: 10.1182/blood-2004-07-2966. [DOI] [PubMed] [Google Scholar]
- 45.Ma Y, Specian RD, Yeh KY, Yeh M, Rodriguez-Paris J, Glass J. The transcytosis of divalent metal transporter 1 and apo-transferrin during iron uptake in intestinal epithelium. Am J Physiol. 2002;283:G965–G974. doi: 10.1152/ajpgi.00005.2002. [DOI] [PubMed] [Google Scholar]
- 46.Gunshin H, Starr CN, Direnzo C, Fleming MD, Jin J, Greer EL, Sellers VM, Galica SM, Andrews NC. Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood. 2005;106:2879–2883. doi: 10.1182/blood-2005-02-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Y, Yeh M, Yeh KY, Glass J. Iron imports. V. Transport of iron through the intestinal epithelium. Am J Physiol. 2006;290:G417–G422. doi: 10.1152/ajpgi.00489.2005. [DOI] [PubMed] [Google Scholar]
- 48.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 49.Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 50.Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL. Unexpected role of ceruloplasmic in intestinal iron absorption. Cell Metab. 2005;2:309–319. doi: 10.1016/j.cmet.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Jeong SY, David S. Glycosylphosphatidylinositol-anchored cerulo-plasmin is required for iron efflux from cells in the central nervous system. J Biol Chem. 2003;278:27144–27148. doi: 10.1074/jbc.M301988200. [DOI] [PubMed] [Google Scholar]
- 52.Stearman R, Yuan DS, Yamaguchi-Iwai Y, Klausner RD, Dancis A. A permease–oxidase complex involved in high-affinity iron uptake by yeast. Science. 1996;271:1552–1557. doi: 10.1126/science.271.5255.1552. [DOI] [PubMed] [Google Scholar]
- 53.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 54.Knutson MD, Oukka M, Ross LM, Adyemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 over expression and down-regulated by hepcidin. Proc Natl Acad Sci U S A. 2005;102:1324–1328. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knutson M, Wessling-Resnick M. Iron metabolism in the reticuloendothelial system. Crit Rev Biochem Mol Biol. 2003;38:61–88. doi: 10.1080/713609210. [DOI] [PubMed] [Google Scholar]
- 56.Aisen P. Transferrin receptor 1. Int J Biochem Cell Biol. 2004;36:2137–2143. doi: 10.1016/j.biocel.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Enns CA. In: Molecular and Cellular Iron Transport. Templeton DM, editor. Marcel Dekker; New York: 2002. pp. 71–94. [Google Scholar]
- 58.Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005;37:1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Giannetti AM, Halbrooks PJ, Mason AB, Vogt TM, Enns CA, Bjorkman PJ. The molecular mechanism for receptor-stimulated iron release from the plasma iron transport protein transferrin. Structure. 2005;13:1613–1623. doi: 10.1016/j.str.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 60.Lill R, Muhlenhoff U. Iron–sulfur–protein biogenesis in eukaryotes. Trends Biochem Sci. 2005;30:133–141. doi: 10.1016/j.tibs.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 61.Napier I, Ponka P, Richardson D. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood. 2005;105:1867–1874. doi: 10.1182/blood-2004-10-3856. [DOI] [PubMed] [Google Scholar]
- 62.Rouault TA, Tong WH. Iron–sulphur cluster biogenesis and mito-chondrial iron homeostasis. Nat Rev, Mol Cell Biol. 2005;6:345–351. doi: 10.1038/nrm1620. [DOI] [PubMed] [Google Scholar]
- 63.Theil EC. Iron, ferritin, and nutrition. Annu Rev Nutr. 2004;24:327–343. doi: 10.1146/annurev.nutr.24.012003.132212. [DOI] [PubMed] [Google Scholar]
- 64.Laufberger V. Sur la cristallisation de la ferritine. Bull Soc Chim Biol. 1937;19:1575. [Google Scholar]
- 65.Fineberg RA, Greenberg DM. Ferritin biosynthesis II. Acceleration of synthesis by the administration of iron. J Biol Chem. 1955;214:97–106. [PubMed] [Google Scholar]
- 66.Granick S. Ferritin. IX. Increase of the protein apoferritin in the gastrointestinal mucosa as a direct response to iron feeding. The function of ferritin in the regulation of iron absorption. J Biol Chem. 1946;164:737–746. [PubMed] [Google Scholar]
- 67.Korey SR, Levine B. Effect of iron on the biosynthesis of hepatic apoferritin. J Clin Invest. 1955;34:756–760. doi: 10.1172/JCI103130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drysdale JW, Munro HN. Failure of actinomycin D to prevent induction of liver apoferritin after iron administration. Biochim Biophys Acta. 1965;103:185–188. doi: 10.1016/0005-2787(65)90554-x. [DOI] [PubMed] [Google Scholar]
- 69.Drysdale JW, Munro HN. Regulation of synthesis and turnover of ferritin in rat liver. J Biol Chem. 1966;241:3630–3637. [PubMed] [Google Scholar]
- 70.Saddi R, Decken AVD. Specific stimulation of amino acid incorporation into ferritin by rat-liver slices after injection of iron in vivo. Biochim Biophys Acta. 1964;90:196–198. doi: 10.1016/0304-4165(64)90141-2. [DOI] [PubMed] [Google Scholar]
- 71.Zahringer J, Baliga BS, Munro HN. Novel mechanism for translational control in regulation of ferritin synthesis by iron. Proc Natl Acad Sci U S A. 1976;73:857–861. doi: 10.1073/pnas.73.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aziz N, Munro HN. Iron regulates ferritin mRNA translation through a segment of its 5′ untranslated region. Proc Natl Acad Sci U S A. 1987;84:8478–8482. doi: 10.1073/pnas.84.23.8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hentze MW, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, Klausner RD. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. 1987;238:1570–1573. doi: 10.1126/science.3685996. [DOI] [PubMed] [Google Scholar]
- 74.Leibold EA, Munro HN. Characterization and evolution of the expressed rat ferritin light subunit gene and its pseudogene family. Conservation of sequences within noncoding regions of ferritin genes. J Biol Chem. 1987;262:7335–7341. [PubMed] [Google Scholar]
- 75.Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a conserved sequence in the 5′ untranslated region of ferritin H- and L-chain mRNAs. Proc Natl Acad Sci U S A. 1988;85:2171–2175. doi: 10.1073/pnas.85.7.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rouault TA, Hentze MW, Caughman SW, Harford JB, Klausner RD. Binding of a cytosolic protein to the iron-responsive element of human ferritin messenger RNA. Science. 1988;241:1207–1210. doi: 10.1126/science.3413484. [DOI] [PubMed] [Google Scholar]
- 77.Owen D, Kuhn LC. Noncoding 3′ sequences of the transferrin receptor gene are required for mRNA regulation by iron. EMBO J. 1987;6:1287–1293. doi: 10.1002/j.1460-2075.1987.tb02366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mullner EW, Kuhn LC. A stem–loop in the 3′ untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cell. 1988;53:815–825. doi: 10.1016/0092-8674(88)90098-0. [DOI] [PubMed] [Google Scholar]
- 79.Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA, Klausner RD, Harford JB. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science. 1988;240:924–928. doi: 10.1126/science.2452485. [DOI] [PubMed] [Google Scholar]
- 80.Mullner EW, Neupert B, Kuhn LC. A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cell. 1989;58:373–382. doi: 10.1016/0092-8674(89)90851-9. [DOI] [PubMed] [Google Scholar]
- 81.Kohler SA, Henderson BR, Kuhn LC. Succinate dehydrogenase B mRNA of Drosophilia melanogaster has a functional iron-responsive element in its 5′-untranslated region. J Biol Chem. 1995;270:30781–30786. doi: 10.1074/jbc.270.51.30781. [DOI] [PubMed] [Google Scholar]
- 82.Felice MR, DeDomenico I, Li L, Ward DM, Bartok B, Musci G, Kaplan J. Post-transcriptional regulation of the yeast high affinity iron transport system. J Biol Chem. 2005;280:22181–22190. doi: 10.1074/jbc.M414663200. [DOI] [PubMed] [Google Scholar]
- 83.Puig S, Askeland E, Theile DJ. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell. 2005;120:99–110. doi: 10.1016/j.cell.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 84.Alen C, Sonenshein AL. Bacillus subtilis aconitase is an RNA binding protein. Proc Natl Acad Sci U S A. 1999;96:10412–10417. doi: 10.1073/pnas.96.18.10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang Y, Guest JR. Direct evidence for mRNA binding and post-transcriptional regulation by Escherichia coli aconitases. Microbiology. 1999;145:3069–3079. doi: 10.1099/00221287-145-11-3069. [DOI] [PubMed] [Google Scholar]
- 86.Tang Y, Quail MA, Artymiuk PJ, Guest JR, Green J. Escherichia coli aconitases and oxidative stress: post-transcriptional regulation of sodA expression. Microbiology. 2002;148:1027–1037. doi: 10.1099/00221287-148-4-1027. [DOI] [PubMed] [Google Scholar]
- 87.Kohler SA, Menotti E, Kuhn LC. Molecular cloning of mouse glycolate oxidase. High evolutionary conservation and presence of an iron-responsive element-like sequence in the mRNA. J Biol Chem. 1999;274:2401–2407. doi: 10.1074/jbc.274.4.2401. [DOI] [PubMed] [Google Scholar]
- 88.Cmejla R, Petrak J, Cmejlova J. A novel iron responsive element in the 3′UTR of human MRCK alpha. Biochem Biophys Res Commun. 2006;341:158–166. doi: 10.1016/j.bbrc.2005.12.155. [DOI] [PubMed] [Google Scholar]
- 89.Frishman D, Hentze MW. Conservation of aconitase residues revealed by multiple sequence analysis: implications for structure/function relationships. Eur J Biochem. 1996;239:197–200. doi: 10.1111/j.1432-1033.1996.0197u.x. [DOI] [PubMed] [Google Scholar]
- 90.Gruer MJ, Artymiuk PJ, Guest JR. The aconitase family: three structural variations on a common theme. Trends Biochem Sci. 1997;22:3–6. doi: 10.1016/s0968-0004(96)10069-4. [DOI] [PubMed] [Google Scholar]
- 91.Haile DJ, Rouault TA, Harford JB, Kennedy MC, Blondin GA, Beinert H, Klausner RD. Cellular regulation of the iron-responsive element binding protein: disassembly of the cubane iron–sulfur cluster results in high-affinity RNA binding. Proc Natl Acad Sci U S A. 1992;89:11735–11739. doi: 10.1073/pnas.89.24.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haile DJ, Rouault TA, Tang CK, Chin J, Harford JB, Klausner RD. Reciprocal control of RNA-binding and aconitase activity in the regulation of the iron-responsive element binding protein: role of the iron–sulfur cluster. Proc Natl Acad Sci. 1992;89:7536–7540. doi: 10.1073/pnas.89.16.7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hentze MW, Argos P. Homology Between IRE-BP, a regulatory RNA-binding protein, aconitase, and isopropylmalate isomerase. Nucleic Acids Res. 1991;19:1739–1740. doi: 10.1093/nar/19.8.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kennedy MC, Mende-Mueller L, Blondin GA, Beinert H. Purification and characterization of cytosolic aconitase from beef liver and its relationship to the iron-responsive element binding protein. Proc Natl Acad Sci U S A. 1992;89:11730–11734. doi: 10.1073/pnas.89.24.11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rouault TA, Stout CD, Kaptain S, Harford JB, Klausner RD. Structural relationship between an iron-regulated RNA-binding protein (IRE-BP) and aconitase: functional implications. Cell. 1991;64:881–883. doi: 10.1016/0092-8674(91)90312-m. [DOI] [PubMed] [Google Scholar]
- 96.Beinert H, Kennedy MC, Stout CD. Aconitase as iron–sulfur protein, enzyme, and iron regulatory protein. Chem Rev. 1996;96:2335–2373. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 97.Walden W. From bacteria to mitochondria: aconitase yields surprises. Proc Natl Acad Sci U S A. 2002;99:4138–4140. doi: 10.1073/pnas.082108799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guo B, Phillips JD, Yu Y, Leibold EA. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J Biol Chem. 1995;270:21645–21651. doi: 10.1074/jbc.270.37.21645. [DOI] [PubMed] [Google Scholar]
- 99.Iwai K, Klausner RD, Rouault TA. Requirements for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J. 1995;21:5350–5357. doi: 10.1002/j.1460-2075.1995.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guo B, Yu Y, Leibold EA. Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J Biol Chem. 1994;269:24252–24260. [PubMed] [Google Scholar]
- 101.Basilion JP, Rouault TA, Massinople CM, Klausner RD, Burgess WH. The iron-responsive element-binding protein: Localization of the RNA-binding site to the aconitase active-site cleft. Proc Natl Acad Sci U S A. 1994;91:574–578. doi: 10.1073/pnas.91.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dupuy J, Volbeda A, Carpentier P, Darnault C, Moulis JM, Fontecilla-Camps JC. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure. 2006;14:129–139. doi: 10.1016/j.str.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 103.Klausner RD, Stout CD, Rouault TA. Dispensable iron–sulfur clusters: the interconversion of aconitase and the RNA-binding protein, IRE-BP. Chem Biol. 1994;1:viv–vxv. [Google Scholar]
- 104.Brazzolotto X, Timmins P, Dupont Y, Moulis JM. Structural changes associated with switching activities of human iron regulatory protein 1. J Biol Chem. 2002;277:11995–12000. doi: 10.1074/jbc.M110938200. [DOI] [PubMed] [Google Scholar]
- 105.Gegout V, Schlegl J, Schlager B, Hentze MW, Reinholt J, Ehresmann B, Ehresmann C, Romby P. Ligand-induced structural alterations in human iron-regulatory protein-1 revealed by protein footprinting. J Biol Chem. 1999;274:15052–15058. doi: 10.1074/jbc.274.21.15052. [DOI] [PubMed] [Google Scholar]
- 106.Schalinske KL, Anderson SA, Tuazon PT, Chen OS, Eisenstein RS. The iron–sulfur cluster of iron regulatory protein 1 modulates accessibility of RNA binding and phosphorylation sites. Biochemistry. 1997;36:3950–3958. doi: 10.1021/bi9624447. [DOI] [PubMed] [Google Scholar]
- 107.Theil EC, Eisenstein RS. Combinatorial mRNA regulation: iron regulatory proteins and Iso-iron responsive elements (iso-IREs) J Biol Chem. 2000;275:40659–40662. doi: 10.1074/jbc.R000019200. [DOI] [PubMed] [Google Scholar]
- 108.Ke Y, Wu J, Leibold EA, Walden WE, Theil EC. Loops and bulge/ loops in iron-responsive element isoforms influence iron regulatory protein binding. J Biol Chem. 1998;273:23637–23640. doi: 10.1074/jbc.273.37.23637. [DOI] [PubMed] [Google Scholar]
- 109.Truty J, Malpe R, Linder MC. Iron prevents ferritin turnover in hepatic cells. J Biol Chem. 2001;276:48775–48780. doi: 10.1074/jbc.M105392200. [DOI] [PubMed] [Google Scholar]
- 110.White K, Munro HN. Induction of ferritin subunit synthesis by iron is regulated at both the transcriptional and translational levels. J Biol Chem. 1988;263:8938–8942. [PubMed] [Google Scholar]
- 111.Walden WE, Patino MM, Gaffield L. Purification of a specific repressor of ferritin mRNA translation from rabbit liver. J Biol Chem. 1989;264:13765–13769. [PubMed] [Google Scholar]
- 112.Goossen B, Caughman SW, Harford JB, Klausner RD, Hentze MW. Translational repression by a complex between the iron-responsive element of ferritin mRNA and its specific cytoplasmic binding protein is position-dependent in vivo. EMBO J. 1990;9:4127–4133. doi: 10.1002/j.1460-2075.1990.tb07635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goossen B, Hentze MW. Position is the critical determinant for function of iron-responsive elements as translational regulators. Mol Cell Biol. 1992;12:1959–1966. doi: 10.1128/mcb.12.5.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Muckenthaler M, Gray NK, Hentze MW. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4f. Mol Cell. 1998;2:383–388. doi: 10.1016/s1097-2765(00)80282-8. [DOI] [PubMed] [Google Scholar]
- 115.Goessling LS, Mascotti DP, Bhattacharyya-Pakrasi M, Gang H, Thach RE. Irreversible steps in the ferritin synthesis induction pathway. J Biol Chem. 1994;269:4343–4348. [PubMed] [Google Scholar]
- 116.Casey JL, Jeso BD, Rao K, Klausner RD, Harford JB. Two genetic loci participate in the regulation by iron of the gene for the human transferrin receptor. Proc Natl Acad Sci U S A. 1988;85:1787–1791. doi: 10.1073/pnas.85.6.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang J, Pantopoulos K. Conditional derepression of ferritin synthesis in cells expressing a constitutive IRP1 mutant. Mol Cell Biol. 2002;22:4638–4651. doi: 10.1128/MCB.22.13.4638-4651.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Seiser C, Teixeira S, Kuhn LC. Interleukin-2-dependent transcriptional and post-transcriptional regulation of transferrin receptor mRNA. J Biol Chem. 1993;268:13074–13080. [PubMed] [Google Scholar]
- 119.Tong X, Kawabata H, Koeffler HP. Iron deficiency can upregulate expression of transferrin receptor at both the mRNA and protein level. Br J Haematol. 2002;116:458–464. [PubMed] [Google Scholar]
- 120.Binder RA, Horowitz JA, Basilion JP, Koeller DM, Klausner RD, Harford JB. Evidence that the pathway of transferrin receptor mRNA degradation involves an endonucleolytic cleavage within the 3′ UTR and does not involve poly(A) tail shortening. EMBO J. 1994;13:1969–1980. doi: 10.1002/j.1460-2075.1994.tb06466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Casey JL, Koeller DM, Ramin VC, Klausner RD, Harford JB. Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3′-untranslated region of the mRNA. EMBO J. 1989;8:3693–3699. doi: 10.1002/j.1460-2075.1989.tb08544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Erlitzki R, Long JC, Theil EC. Multiple, conserved iron-responsive elements in the 3′ untranslated region of transferrin receptor mRNA enhance binding of iron regulatory protein 2. J Biol Chem. 2002;277:42579–42587. doi: 10.1074/jbc.M207918200. [DOI] [PubMed] [Google Scholar]
- 123.Johnson MB, Enns CA. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood. 2004;104:4287–4293. doi: 10.1182/blood-2004-06-2477. [DOI] [PubMed] [Google Scholar]
- 124.Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP. Molecular cloning of transferrin receptor 2. J Biol Chem. 1999;274:20826–20832. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 125.Robb A, Wessling-Resnick M. Regulation of transferrin receptor 2 protein levels by transferrin. Blood. 2004;104:4294–4299. doi: 10.1182/blood-2004-06-2481. [DOI] [PubMed] [Google Scholar]
- 126.Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze MW, Rouault TA, Andrews NC, Hediger MA. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509:309–316. doi: 10.1016/s0014-5793(01)03189-1. [DOI] [PubMed] [Google Scholar]
- 127.Hubert N, Hentze MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci U S A. 2002;99:12345–12350. doi: 10.1073/pnas.192423399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ke Y, Chen YY, Chang YZ, Duan XL, Ho KP, de-Jiang H, Wang K, Qian ZM. Post-transcriptional expression of DMT1 in the heart of rat. J Cell Physiol. 2003;196:124–130. doi: 10.1002/jcp.10284. [DOI] [PubMed] [Google Scholar]
- 129.Tchernitchko D, Bourgeois M, Martin ME, Beaumont C. Expression of the two mRNA isoforms of the iron transporter Nramp2/DMT1 in mice and function of the iron-responsive element. Biochem J. 2002;363:449–455. doi: 10.1042/0264-6021:3630449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wardrop SL, Richardson DR. The effect of intracellular iron concentration and nitrogen monooxide on nRAMP2 expression and non-transferrin bound iron uptake. Eur J Biochem. 1999;263:41–49. doi: 10.1046/j.1432-1327.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- 131.Bhasker CR, Burgiel G, Neupert B, Emery-Goodman A, Kuhn LC, May BK. The putative iron-responsive element in the human erythroid 5-aminolevulinate synthase mRNA mediates translational control. J Biol Chem. 1993;268:12699–12705. [PubMed] [Google Scholar]
- 132.Cox TC, Bawden MJ, Martin A, May BK. Human erythroid 5-ami-nolevulinate synthase: promoter analysis and identification of an IRE in the mRNA. EMBO J. 1991;10:1891–1902. doi: 10.1002/j.1460-2075.1991.tb07715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gray NK, Pantopoulos K, Dandekar T, Ackrell B, Hentze MW. Translational regulation of mammalian and drosophila citric acid cycle enzymes via iron-responsive elements. Proc Natl Acad Sci U S A. 1996;93:4925–4930. doi: 10.1073/pnas.93.10.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim HY, LaVaute T, Iwai K, Klausner RD, Rouault TA. Identification of a conserved and functional iron-responsive element in the 5′-untranslated region of mammalian mitochondrial aconitase. J Biol Chem. 1996;271:24226–24230. doi: 10.1074/jbc.271.39.24226. [DOI] [PubMed] [Google Scholar]
- 135.Melefors O, Goossen B, Johansson HE, Stripecke R, Gray NK, Hentze MW. Translational control of 5-aminolevulinate synthase mRNA by iron-responsive elements in erythroid cells. J Biol Chem. 1993;268:5974–5978. [PubMed] [Google Scholar]
- 136.Mikulits W, Schranzhofer M, Bauer A, Dolznig H, Lobmayr L, Infante AA, Beug H, Mullner EW. Impaired ferritin mRNA translation in erythroid progenitors: shift to iron-dependent regulation by the v-ErbA oncoprotein. Blood. 1999;94:4321–4332. [PubMed] [Google Scholar]
- 137.Chen OS, Schalinske KL, Eisenstein RS. Dietary iron intake modulates the activity of iron regulatory proteins (IRPs) and the abundance of ferritin and mitochondrial aconitase in rat liver. J Nutr. 1997;127:238–248. doi: 10.1093/jn/127.2.238. [DOI] [PubMed] [Google Scholar]
- 138.Schalinske KL, Chen OS, Eisenstein RS. Iron differentially stimulates translation of mitochondrial aconitase and ferritin mRNAs in mammalian cells. J Biol Chem. 1998;273:3740–3746. doi: 10.1074/jbc.273.6.3740. [DOI] [PubMed] [Google Scholar]
- 139.Schranzhofer M, Schifrer M, Cabrera JA, Kopp S, Chiba P, Beug H, Mullner EW. Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood. 2006;107:4159–4167. doi: 10.1182/blood-2005-05-1809. [DOI] [PubMed] [Google Scholar]
- 140.Butt J, Kim HY, Basilion JP, Cohen S, Iwai K, Philpott CC, Altschul S, Klausner RD, Rouault TA. Differences in the RNA binding sites of iron regulatory proteins and potential target diversity. Proc Natl Acad Sci U S A. 1996;93:4345–4349. doi: 10.1073/pnas.93.9.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Henderson BR, Menotti E, Kuhn LC. Iron regulatory proteins 1 and 2 bind distinct sets of RNA target sequences. J Biol Chem. 1996;271:4900–4908. doi: 10.1074/jbc.271.9.4900. [DOI] [PubMed] [Google Scholar]
- 142.Meehan HA, Connell GJ. The hairpin loop but not the bulged C of the iron responsive element is essential for high affinity binding to the iron regulatory protein-1. J Biol Chem. 2001;276:14791–14796. doi: 10.1074/jbc.M010295200. [DOI] [PubMed] [Google Scholar]
- 143.Bettany AJE, Eisenstein RS, Munro HN. Mutagenesis of the iron regulatory element binding protein further defines a role for RNA secondary structure in the regulation of ferritin and transferrin receptor expression. J Biol Chem. 1992;267:16531–16537. [PubMed] [Google Scholar]
- 144.Leibold EA, Laudano A, Yu Y. Structural requirements of iron-responsive elements for binding of the protein involved in both Tf R and ferritin mRNA post-transcriptional regulation. Nucleic Acids Res. 1990;18:1819–1824. doi: 10.1093/nar/18.7.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Addess KJ, Basilion JP, Klausner RD, Rouault TA, Pardi A. Structure and dynamics of the iron responsive element RNA: implications for binding of the RNA by iron regulatory binding proteins. J Mol Biol. 1997;274:72–83. doi: 10.1006/jmbi.1997.1377. [DOI] [PubMed] [Google Scholar]
- 146.Liang LG, Hall KB. A model of the iron responsive element RNA hairpin loop structure determined from NMR and thermodynamic data. Biochemistry. 1996;35:13586–13596. doi: 10.1021/bi961310q. [DOI] [PubMed] [Google Scholar]
- 147.Sierzputowska-Gracz H, McKenzie RA, Theil EC. The importance of a single G in the hairpin loop of the iron responsive element (IRE) in ferritin mRNA for structure: an NMR spectroscopy study. Nucleic Acids Res. 1995;23:146–153. doi: 10.1093/nar/23.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Theil EC. Targeting mRNA to regulate iron and oxygen metabolism. Biochem Pharmacol. 2000;59:87–93. doi: 10.1016/s0006-2952(99)00300-7. [DOI] [PubMed] [Google Scholar]
- 149.Ke Y, Theil EC. An mRNA loop/bulge in the ferritin iron-responsive element forms in vivo and was detected by radical probing with Cu-1, 10-phenanthroline and iron regulatory protein footprinting. J Biol Chem. 2002;277:2373–2376. doi: 10.1074/jbc.C100614200. [DOI] [PubMed] [Google Scholar]
- 150.Harrell CM, McKenzie AR, Patino MM, Walden WE, Theil EC. Ferritin mRNA: interactions of iron regulatory element with translational regulatory protein P-90 and the effect on base-paired flanking regions. Proc Natl Acad Sci U S A. 1991;88:4166–4170. doi: 10.1073/pnas.88.10.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bhattacharyya A, Murchie AIH, Lilley DMJ. RNA bulges and the helical periodicity of double-stranded RNA. Nature. 1990;343:484–487. doi: 10.1038/343484a0. [DOI] [PubMed] [Google Scholar]
- 152.Lilley DMJ. Linking of DNA and RNA by base bulges. Proc Natl Acad Sci U S A. 1995;92:7140–7142. doi: 10.1073/pnas.92.16.7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Moras D, Poterszman A. Protein–RNA interactions: getting into the major groove. Curr Biol. 1996;6:530–532. doi: 10.1016/s0960-9822(02)00534-1. [DOI] [PubMed] [Google Scholar]
- 154.Riordan FA, Bhattacharyya A, McAteer S, Lilley AMJ. Linking of RNA helices by bulged bases, and the structure of the HIV transactivator response element. J Mol Biol. 1992;226:305–310. doi: 10.1016/0022-2836(92)90947-i. [DOI] [PubMed] [Google Scholar]
- 155.Weeks KM, Crothers DM. RNA recognition by TAT-derived peptides: interaction in the major groove. Cell. 1991;66:577–588. doi: 10.1016/0092-8674(81)90020-9. [DOI] [PubMed] [Google Scholar]
- 156.Ke YH, Sierzputowska-Gracz H, Gdaniec Z, Theil EC. Internal loop/ bulge and hairpin loop of the iron-responsive element of ferritin mRNA contribute to maximal iron regulatory protein 2 binding and translational regulation in the iso-iron-responsive element/iso-iron regulatory protein family. Biochemistry. 2000;39:6235–6242. doi: 10.1021/bi9924765. [DOI] [PubMed] [Google Scholar]
- 157.Dix DJ, Lin PN, McKenzie AR, Walden WE, Theil EC. The influence of the base-paired flanking region on structure and function of the ferritin mRNA iron regulatory element. J Mol Biol. 1993;231:230–240. doi: 10.1006/jmbi.1993.1278. [DOI] [PubMed] [Google Scholar]
- 158.Allerson CR, Cazzola M, Rouault TA. Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia–cataract syndrome. J Biol Chem. 1999;274:26439–26447. doi: 10.1074/jbc.274.37.26439. [DOI] [PubMed] [Google Scholar]
- 159.Kato J, Fujikawa K, Kanda M, Fukuda N, Sasaki K, Takayama T, Kobune M, Takada K, Takimoto R, Hamada H, Ikeda T, Niitsu Y. A mutation, in the iron responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet. 2001;69:191–197. doi: 10.1086/321261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Levi S, Girelli D, Perrone F, Pasti M, Beaumont C, Corrocher R, Albertini A, Arosio P. Analysis of ferritins in lymphoblastoid cell lines and in the lens of subjects with hereditary hyperferritinemia–cataract syndrome. Blood. 1998;91:4180–4187. [PubMed] [Google Scholar]
- 161.Liu W, Shimomura S, Imanishi H, Iwamoto Y, Ikeda N, Saito M, Ohno M, Hara N, Yamamoto T, Nakamura H, Hada T. Hemochromatosis with mutation of the ferroportin 1 (IREG1) gene. Intern Med. 2005;44:285–289. doi: 10.2169/internalmedicine.44.285. [DOI] [PubMed] [Google Scholar]
- 162.Menotti E, Henderson BR, Kühn LC. Translational regulation of mRNAs with distinct IRE sequences by iron regulatory proteins 1 and 2. J Biol Chem. 1998;273:1821–1824. doi: 10.1074/jbc.273.3.1821. [DOI] [PubMed] [Google Scholar]
- 163.Cooperman SS, Meyron-Holtz EG, Olivierre-Wilson H, Ghosh MC, McConnell JP, Rouault TA. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood. 2005;106:1084–1091. doi: 10.1182/blood-2004-12-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, Schumann K, Hentze MW. Altered body iron distribution and microcytosis in mice deficient for iron regulatory protein 2 (IRP2) Blood. 2005;106:2580–2589. doi: 10.1182/blood-2005-04-1365. [DOI] [PubMed] [Google Scholar]
- 165.Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Olivarerre-Wilson H, Grinberg A, Love P, Rouault TA. Genetic ablations of iron regulatory protein 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004;23:386–395. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim HY, Klausner RD, Rouault TA. Translational repressor activity is equivalent and is quantitatively predicted by in vitro RNA binding for two iron-responsive element-binding proteins, IRP1 and IRP2. J Biol Chem. 1995;279:4983–4986. doi: 10.1074/jbc.270.10.4983. [DOI] [PubMed] [Google Scholar]
- 167.Festa M, Colonna A, Pietropaolo C, Ruffo A. Oxalomalate, a competitive inhibitor of aconitase, modulates the RNA-binding activity of iron-regulatory proteins. Biochem J. 2000;348:315–320. [PMC free article] [PubMed] [Google Scholar]
- 168.Santamaria R, Irace C, Festa M, Maffettone C, Colonna A. Induction of ferritin synthesis by oxalomalate. Biochim Biophys Acta. 2004;1691:151–159. doi: 10.1016/j.bbamcr.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 169.Chen O, Crisp RJ, Valachovic M, Bard M, Winge DR, Kaplan J. Transcription of the yeast iron regulon does not directly respond to iron but rather to iron–sulfur cluster biosynthesis. J Biol Chem. 2004;279:29513–29518. doi: 10.1074/jbc.M403209200. [DOI] [PubMed] [Google Scholar]
- 170.Li J, Kogan M, Knight SAB, Pain D, Dancis A. Yeast mitochondrial protein, Nfs1p, coordinately regulates iron–sulfur cluster proteins, cellular iron uptake, and iron distribution. J Biol Chem. 1999;274:33025–33034. doi: 10.1074/jbc.274.46.33025. [DOI] [PubMed] [Google Scholar]
- 171.Rutherford JC, Ojeda L, Balk J, Muhlenhoff U, Lill R, Winge DR. Activation of the iron regulon by yeast Aft1/Aft2 transcription factors depends on mitochondria but not cytosolic iron–sulfur protein biogenesis. J Biol Chem. 2005;280:10135–10140. doi: 10.1074/jbc.M413731200. [DOI] [PubMed] [Google Scholar]
- 172.Gakh O, Park S, Liu G, Macomber L, Imlay JA, Ferreira GC, Isaya G. Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum Mol Genet. 2006;15:467–479. doi: 10.1093/hmg/ddi461. [DOI] [PubMed] [Google Scholar]
- 173.Pandolfo M. Friedreich ataxia. Semin Pediatr Neurol. 2003;10:163–172. doi: 10.1016/s1071-9091(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 174.Fleming MD. The genetics of inherited sideroblastic anemias. Semin Hematol. 2002;39:270–281. doi: 10.1053/shem.2002.35637. [DOI] [PubMed] [Google Scholar]
- 175.Roy A, Solodovnikova A, Nicholson T, Antholine W, Walden WE. A novel eukaryotic factor for cytosolic Fe–S assembly. EMBO J. 2003;22:4835–4836. doi: 10.1093/emboj/cdg455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Li K, Tong W-H, Hughes RM, Rouault TA. Roles of the mammalian cytosolic cystein desulfurase, ISCS, and scaffold protein, ISCU, in the iron–sulfur cluster assembly. J Biol Chem. 2006;281:12344–12351. doi: 10.1074/jbc.M600582200. [DOI] [PubMed] [Google Scholar]
- 177.Tong W-H, Rouault TA. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron–sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 178.Bouton C, Chauveau MJ, Lazereg S, Drapier JC. Recycling of RNA binding iron regulatory protein 1 into an aconitase after nitric oxide removal depends on mitochondrial ATP. J Biol Chem. 2002;277:31220–31227. doi: 10.1074/jbc.M203276200. [DOI] [PubMed] [Google Scholar]
- 179.Clarke SL, Vasanthakumar A, Anderson SA, Pondarre C, Koh CM, Deck KM, Pitula JS, Epstein CJ, Fleming MD, Eisenstein RS. Iron-responsive degradation of iron regulatory protein 1 does not require the Fe–S cluster. EMBO J. 2006;25:544–553. doi: 10.1038/sj.emboj.7600954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pondarre C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han AP, Medlock A, Kutok J, Anderson SA, Eisenstein RS, Fleming MD. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic Fe–S cluster biogenesis. Hum Mol Genet. 2006;15:953–964. doi: 10.1093/hmg/ddl012. [DOI] [PubMed] [Google Scholar]
- 181.Seznec H, Simon D, Bouton C, Reutenauer L, Hertzog A, Golik P, Procaccio V, Patel M, Drapier JC, Koenig M, Puccio H. Friedreich ataxia: the oxidative stress paradox. Hum Mol Genet. 2005;14:463–474. doi: 10.1093/hmg/ddi042. [DOI] [PubMed] [Google Scholar]
- 182.Stehling O, Elasser HP, Bruckel B, Muhlenhoff U, Lill R. Iron–sulfur protein maturation in human cells: evidence for a function of frataxin. Hum Mol Genet. 2004;13:3007–3015. doi: 10.1093/hmg/ddh324. [DOI] [PubMed] [Google Scholar]
- 183.Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, Weber GJ, Dooley K, Davidson AJ, Schmidt B, Paw BH, Shaw GC, Kingsley P, Palis J, Schubert H, Chen O, Kaplan J, Consortium TTS, Zon LI. Deficiency of glutaredoxin 5 reveals Fe–S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 184.Bekri S, Kispal G, Lange H, Fitzsimmons E, Tolmie J, Lill R, Bishop DF. Human ABC7 transporter:gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron–sulfur protein maturation. Blood. 2000;96:3256–3264. [PubMed] [Google Scholar]
- 185.Cairo G, Recalcati S, Pietrangelo A, Minotti G. The iron regulatory proteins: targets and modulators of free radical reactions and oxidative damage. Free Radic Biol Med. 2002;32:1237–1243. doi: 10.1016/s0891-5849(02)00825-0. [DOI] [PubMed] [Google Scholar]
- 186.Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- 187.Mueller S, Pantopoulos K, Huebner CA, Stremmel W, Stremmel MW. IRP1 activation by extracellular oxidative stress in the perfused rat liver. J Biol Chem. 2001;276:23192–23196. doi: 10.1074/jbc.M100654200. [DOI] [PubMed] [Google Scholar]
- 188.Pantopoulos K, Hentze MW. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc Natl Acad Sci. 1998;95:10559–10563. doi: 10.1073/pnas.95.18.10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Minotti G, Recalcati S, Menna P, Salvatorelli E, Corna G, Cairo G. Doxorubicin cardiotoxicity and the control of iron metabolism: quinone-dependent and independent mechanisms. Methods Enzymol. 2004;378:340–361. doi: 10.1016/S0076-6879(04)78025-8. [DOI] [PubMed] [Google Scholar]
- 190.Minotti G, Ronchi R, Salvatorelli E, Menna P, Cairo G. Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes: evidence for distinct metabolic pathways and implications for iron-mediated cardiotoxicity of antitumor therapy. Cancer Res. 2001;61:8422–8428. [PubMed] [Google Scholar]
- 191.Ayaki H, Lee MJ, Sumino K, Nishio H. Different cytoprotective effect of antioxidants and change in the iron regulatory system in rodent cells exposed to paraquat and formaldehyde. Toxicology. 2005;208:73–79. doi: 10.1016/j.tox.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 192.Pantopoulos K, Mueller S, Atzberger A, Ansorge W, Stremmel W, Hentze MW. Differences in the regulation of iron regulatory protein-1 (IRP-1) by extra- and intracellular oxidative stress. J Biol Chem. 1997;272:9802–9808. doi: 10.1074/jbc.272.15.9802. [DOI] [PubMed] [Google Scholar]
- 193.Cairo G, Castrusini E, Minotti G, Bernelli-Zazzera A. Superoxide and hydrogen peroxide-dependent inhibition of iron regulatory protein activity: a protective stratagem against oxidative injury. FASEB J. 1996;10:1326–1335. doi: 10.1096/fasebj.10.11.8836047. [DOI] [PubMed] [Google Scholar]
- 194.Hanson ES, Leibold EA. Regulation of iron regulatory protein 1 during hypoxia and hypoxia/reoxygenation. J Biol Chem. 1998;273:7588–7593. doi: 10.1074/jbc.273.13.7588. [DOI] [PubMed] [Google Scholar]
- 195.Meyron-Holtz EG, Ghosh MC, Rouault TA. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science. 2004;306:2087–2090. doi: 10.1126/science.1103786. [DOI] [PubMed] [Google Scholar]
- 196.Pantopoulos K, Hentze MW. Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 1995;14:2917–2924. doi: 10.1002/j.1460-2075.1995.tb07291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Starzynski RR, Lipinski P, Drapier JC, Diet A, Smuda E, Barthlomiejczyk T, Gralak MA, Kruszweski M. Down-regulation of iron regulatory protein 1 activities and expression in superoxide 1 knockout mice is not associated with alterations in iron metabolism. J Biol Chem. 2005;280:4207–4212. doi: 10.1074/jbc.M411055200. [DOI] [PubMed] [Google Scholar]
- 198.Irace C, Scorziello A, Maffettone C, Pignataro G, Matrone C, Adornetto A, Santamaria R, Annunziato L, Colonna A. Divergent modulation of iron regulatory proteins and ferritin biosynthesis by hypoxia/reoxygenation in neurons and glial cells. J Neurochem. 2005;95:1321–1331. doi: 10.1111/j.1471-4159.2005.03449.x. [DOI] [PubMed] [Google Scholar]
- 199.Oliveira L, Drapier JC. Down-regulation of iron regulatory protein 1 gene expression by nitric oxide. Proc Natl Acad Sci U S A. 2000;97:6550–6555. doi: 10.1073/pnas.120571797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Brazzolotto X, Gaillard J, Pantopoulos K, Hentze MW, Moulis JM. Human cytoplasmic aconitase (Iron regulatory protein 1) is converted into its [3Fe–4S] form by hydrogen peroxide in vitro but is not activated for iron-responsive element binding. J Biol Chem. 1999;274:21625–21630. doi: 10.1074/jbc.274.31.21625. [DOI] [PubMed] [Google Scholar]
- 201.Mutze S, Hebling U, Stremmel W, Wang J, Arnhold J, Pantopoulos K, Mueller S. Myeloperoxidase-derived hypochlorous acid antagonizes the oxidative stress-mediated activation of iron regulatory protein 1. J Biol Chem. 2003;278:40542–40549. doi: 10.1074/jbc.M307159200. [DOI] [PubMed] [Google Scholar]
- 202.Varghese S, Tang Y, Imlay JA. Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J Bacteriol. 2003;185:221–230. doi: 10.1128/JB.185.1.221-230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Brown NM, Anderson SA, Steffen DW, Carpenter TB, Kennedy MC, Walden WE, Eisenstein RS. Novel role of phosphorylation in Fe–S cluster stability revealed by phosphomimetic mutations at Ser-138 of iron regulatory protein 1. Proc Natl Acad Sci U S A. 1998;95:15235–15240. doi: 10.1073/pnas.95.26.15235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Brown NM, Kennedy MC, Antholine WE, Eisenstein RS, Walden WE. Detection of a [3Fe–4S] cluster intermediate of cytosolic aconitase in yeast expressing iron regulatory protein 1. Insights into the mechanism of Fe–S cluster cycling. J Biol Chem. 2002;277:7246–7254. doi: 10.1074/jbc.M110282200. [DOI] [PubMed] [Google Scholar]
- 205.Drapier JC, Hibbs JB., Jr Murine cytotoxic activated macrophages inhibit aconitase in tumor cells. J Clin Invest. 1986;78:790–797. doi: 10.1172/JCI112642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bouton C, Drapier JC. Iron regulatory proteins as NO signal transducers. Sci STKE. 2003;2003:pe17. doi: 10.1126/stke.2003.182.pe17. [DOI] [PubMed] [Google Scholar]
- 207.Kim S, Ponka P. Role of nitric oxide in cellular iron metabolism. BioMetals. 2003;16:125–135. doi: 10.1023/a:1020788603046. [DOI] [PubMed] [Google Scholar]
- 208.Castro LA, Robalinho RL, Cayota A, Meneghini R, Radi R. Nitric oxide and peroxynitritie-dependent aconitase inactivation and iron-regulatory protein-1 activation in mammalian fibroblast. Arch Biochem Biophys. 1998;359:215–224. doi: 10.1006/abbi.1998.0898. [DOI] [PubMed] [Google Scholar]
- 209.Drapier JC, Hirling H, Wietzerbin J, Kaldy P, Kuhn LC. Biosynthesis of nitric oxide activates iron regulatory factor in macrophages. EMBO J. 1993;12:3643–3649. doi: 10.1002/j.1460-2075.1993.tb06038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kennedy MC, Antholine WE, Beinert H. An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J Biol Chem. 1997;272:20340–20347. doi: 10.1074/jbc.272.33.20340. [DOI] [PubMed] [Google Scholar]
- 211.Pantopoulos K, Hentze MW. Nitric oxide signaling to iron-regulatory protein: direct control of ferritin mRNA translation and transferrin receptor RNA stability in transfected fibroblasts. Proc Natl Acad Sci U S A. 1995;92:1267–1271. doi: 10.1073/pnas.92.5.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pantopoulos K, Weiss G, Hentze M. Nitric oxide and oxidative stress (H2O2) control mammalian iron metabolism by different pathways. Mol Cell Biol. 1996;16:3781–3788. doi: 10.1128/mcb.16.7.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Phillips JD, Kinikini DV, Yu Y, Guo B, Leibold EA. Differential regulation of IRP1 and IRP2 by nitric oxide in rat hepatoma cells. Blood. 1996;87:2983–2992. [PubMed] [Google Scholar]
- 214.Wardrop SL, Watts RN, Richardson DR. Nitrogen monoxide activates Iron Regulatory Protein 1 RNA-binding activity by two possible mechanisms: effect on the [4Fe–4S] cluster and iron mobilization from cells. Biochemistry. 2000;39:2748–2758. doi: 10.1021/bi991099t. [DOI] [PubMed] [Google Scholar]
- 215.Weiss G, Goossen B, Doppler W, Fuchs D, Pantopoulos K, Werner-Felmayer G, Wachter H, Hentze MW. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J. 1993;12:3651–3657. doi: 10.1002/j.1460-2075.1993.tb06039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cairo G, Ronchi R, Recalcati S, Campanella A, Minotti G. Nitric oxide and peroxynitrite activate the iron regulatory protein-1 of J774A.1 macrophages by direct disassembly of the Fe–S cluster of cytoplasmic aconitase. Biochemistry. 2002;41:7435–7442. doi: 10.1021/bi025756k. [DOI] [PubMed] [Google Scholar]
- 217.Bouton C, Raveau M, Drapier JC. Modulation of iron regulatory protein functions: further insight into the role of nitrogen- and oxygen-derived reactive species. J Biol Chem. 1996;271:2300–2306. doi: 10.1074/jbc.271.4.2300. [DOI] [PubMed] [Google Scholar]
- 218.Gardner PR, Costantino G, Szabo C, Salzman AL. Nitric oxide sensitivity of the aconitases. J Biol Chem. 1997;272:25071–25076. doi: 10.1074/jbc.272.40.25071. [DOI] [PubMed] [Google Scholar]
- 219.Soum E, Drapier JC. Nitric oxide and peroxynitrate promote the complete disruption of the [4Fe–4S] cluster of recombinant human iron regulatory protein 1. J Biol Inorg Chem. 2003;8:226–232. doi: 10.1007/s00775-002-0412-9. [DOI] [PubMed] [Google Scholar]
- 220.Soum E, Brozzolotto X, Goussias X, Bouton C, Moulis JM, Mattioli TA, Drapier JC. Peroxynitrite and nitric oxide differentially target the iron–sulfur cluster and amino acid residues of human iron regulatory protein-1. Biochemistry. 2003;42:7648–7654. doi: 10.1021/bi030041i. [DOI] [PubMed] [Google Scholar]
- 221.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 222.Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrate, but not by its precursor, nitric oxide. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- 223.Hausladen A. Superoxide and peroxynitrate inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994:29405–29408. [PubMed] [Google Scholar]
- 224.Bouton C, Hirling H, Drapier JC. Redox modulation of iron regulatory proteins by peroxynitrite. J Biol Chem. 1997;272:19969–19975. doi: 10.1074/jbc.272.32.19969. [DOI] [PubMed] [Google Scholar]
- 225.Han D, Canali R, Garcia J, Aguilera R, Gallaher T, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44:11986–11996. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 226.Gonzalez D, Drapier JC, Bouton C. Endogenous nitration of iron regulatory protein-1 (IRP-1) in nitric-oxide-producing murine macrophages: further insight into the mechanism of nitration in vivo an its impact on IRP1 functions. J Biol Chem. 2004;279:43345–43351. doi: 10.1074/jbc.M401889200. [DOI] [PubMed] [Google Scholar]
- 227.Schalinske KL, Eisenstein RS. Phosphorylation and activation of both iron regulatory protein 1 (IRP1) and IRP2 in HL60 cells. J Biol Chem. 1996;271:7168–7176. doi: 10.1074/jbc.271.12.7168. [DOI] [PubMed] [Google Scholar]
- 228.Eisenstein RS, Tuazon PT, Schalinske KL, Anderson SA, Traugh JA. Iron-responsive element binding protein: phosphorylation by protein kinase C. J Biol Chem. 1993;268:27363–27370. [PubMed] [Google Scholar]
- 229.Fillebeen C, Caltagirone A, Martelli A, Moulis JM, Pantopoulos K. IRP1 Ser-711 is a phosphorylation site, critical for regulation of RNA-binding and aconitase activities. Biochem J. 2005;388:143–150. doi: 10.1042/BJ20041623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Fillebeen C, Chahine D, Caltagirone A, Segel P, Pantopoulos K. A phosphomimetic mutation at Ser-138 renders iron regulatory protein 1 sensitive to iron-dependent degradation. Mol Cell Biol. 2003;23:6973–6981. doi: 10.1128/MCB.23.19.6973-6981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pitula JS, Deck KM, Clarke SL, Anderson SA, Vasanthakumar A, Eisenstein RS. Selective inhibition of the citrate-to-isocitrate reaction of cytosolic aconitase by phosphomimetic mutation of serine-711. Proc Natl Acad Sci U S A. 2004;101:10907–10912. doi: 10.1073/pnas.0404308101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Goessling LS, Daniels-McQueen S, Bhattacharyya-Pakrasi M, Lin J-J, Thach RE. Enhanced degradation of the ferritin repressor protein during induction of ferritin messenger RNA translation. Science. 1992;256:670–673. doi: 10.1126/science.1316633. [DOI] [PubMed] [Google Scholar]
- 233.Mascotti DP, Rup D, Thach RE. Regulation of iron metabolism: translational effects mediated by iron, heme, and cytokines. Annu Rev Nutr. 1995;15:239–261. doi: 10.1146/annurev.nu.15.070195.001323. [DOI] [PubMed] [Google Scholar]
- 234.Neonaki M, Graham DC, White KN, Bomford A. Down-regulation of liver iron-regulatory protein 1 in haemochromatosis. Biochem Soc Trans. 2001;30:726–728. doi: 10.1042/bst0300726. [DOI] [PubMed] [Google Scholar]
- 235.Gourley BL, Parker SB, Jones BJ, Zumbrennen KB, Leibold EA. Cytosolic aconitase and ferritin are regulated by iron in Caenorhabditis elegans. J Biol Chem. 2003;278:3227–3334. doi: 10.1074/jbc.M210333200. [DOI] [PubMed] [Google Scholar]
- 236.Navarre DA, Wendehenne D, Durner J, Noad R, Klessig DF. Nitric oxide modulates the activity of tobacco aconitase. Plant Physiol. 2000;122:573–582. doi: 10.1104/pp.122.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Peyret P, Perez P, Alric M. Structure, genomic organization, and expression of the Arabidopsis thaliana aconitase gene. Plant aconitase show significant homology with mammalian iron-responsive element-binding protein. J Biol Chem. 1995;270:8131–8137. doi: 10.1074/jbc.270.14.8131. [DOI] [PubMed] [Google Scholar]
- 238.Saas J, Ziegelbauer K, von-Haeseler A, Fast B, Boshart M. A developmentally regulated aconitase related to iron-regulatory protein-1 is localized in the cytoplasm and in the mitochondrion of Trypanosoma brucei. J Biol Chem. 2000;275:2745–2755. doi: 10.1074/jbc.275.4.2745. [DOI] [PubMed] [Google Scholar]
- 239.Lind MI, Missirlis F, Melefors O, Uhrigshardt H, Kirdy K, Phillips JP, Soderhall K, Rouault TA. Of two cytosolic aconitase expressed in Drosophila, only one functions as an iron regulatory protein. J Biol Chem. 281 doi: 10.1074/jbc.M603354200. in press. [DOI] [PubMed] [Google Scholar]
- 240.Chen XJ, Wang X, Kaufman BA, Butow RA. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307:714–717. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- 241.Phillips JD, Guo B, Yu Y, Brown FM, Leibold EA. Expression and biochemical characterization of iron regulatory proteins 1 and 2 in Sac-charomyces cerevisiae. Biochemistry. 1996;35:15704–15714. doi: 10.1021/bi960653l. [DOI] [PubMed] [Google Scholar]
- 242.Samaniego F, Chin J, Uwai K, Rouault TA, Klausner RD. Molecular characterization of a second iron responsive element binding protein, iron regulatory protein 2. J Biol Chem. 1994;269:30904–30910. [PubMed] [Google Scholar]
- 243.Guo B, Brown FM, Phillips JD, Yu Y, Leibold EA. Characterization and expression of iron regulatory protein 2 (IRP2) J Biol Chem. 1995;268:16529–116535. doi: 10.1074/jbc.270.28.16529. [DOI] [PubMed] [Google Scholar]
- 244.Hanson ES, Foot LM, Leibold EA. Hypoxia post-translationally activates iron-regulatory protein 2. J Biol Chem. 1999;274:5047–5052. doi: 10.1074/jbc.274.8.5047. [DOI] [PubMed] [Google Scholar]
- 245.Hanson ES, Leibold EA. Marcel Dekker; New York: 2002. [Google Scholar]
- 246.Hanson ES, Rawlins ML, Leibold EA. Oxygen and iron regulation of iron regulatory protein 2. J Biol Chem. 2003;278:40337–40342. doi: 10.1074/jbc.M302798200. [DOI] [PubMed] [Google Scholar]
- 247.Schneider BD, Leibold EA. Effects of iron regulatory protein regulation on iron homeostasis during hypoxia. Blood. 2003;102:3404–3411. doi: 10.1182/blood-2003-02-0433. [DOI] [PubMed] [Google Scholar]
- 248.Iwai K, Drake SK, Wehr NB, Weissman AM, LaVaute T, Minato N, Klausner RD, Levine RL, Rouault TA. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: implications for degradation of oxidized proteins. Proc Natl Acad Sci U S A. 1998;95:4924–4928. doi: 10.1073/pnas.95.9.4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kang DK, Jeong J, Drake SK, Wehr NB, Rouault TA, Levine RL. Iron regulatory protein 2 as iron sensor. Iron-dependent oxidative modification of cysteine. J Biol Chem. 2003;278:14857–14864. doi: 10.1074/jbc.M300616200. [DOI] [PubMed] [Google Scholar]
- 250.Ishikawa H, Kato M, Hori H, Ishimori K, Kirisako T, Tokunaga F, Iwai K. Involvement of the heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol Cell. 2005;19:171–181. doi: 10.1016/j.molcel.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 251.Bourdon E, Kang DK, Ghosh MC, Drake SK, Wey J, Levine RL, Rouault TA. The role of endogenous heme synthesis and degradation domain cysteins in cellular iron-dependent degradation of IRP2. Blood Cells Mol Diseases. 2003;31:247–255. doi: 10.1016/s1079-9796(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 252.Wang J, Chen G, Muckenthaler M, Galy B, Hentze MW, Pantopoulos K. Iron-mediated degradation of IRP2, an unexpected pathway involving a 2-oxoglutarate-dependent oxygenase activity. Mol Cell Biol. 2004;24:954–965. doi: 10.1128/MCB.24.3.954-965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Goessling LS, Mascotti DP, Thach RE. Involvement of heme in the degradation of iron regulatory protein 2. J Biol Chem. 1998;273:12555–12557. doi: 10.1074/jbc.273.20.12555. [DOI] [PubMed] [Google Scholar]
- 254.Yamanaka K, Ishikawa H, Megumi Y, Tokunaga F, Kanie M, Rouault TA, Morishima I, Minato N, Ishimori K, Iwai K. Identification of the ubiquitin-protein ligase that recognizes IRP2. Nat Cell Biol. 2003;5:336–340. doi: 10.1038/ncb952. [DOI] [PubMed] [Google Scholar]
- 255.Jeong J, Rouault TA, Levine RL. Identification of a hemesensing domain in iron regulatory protein 2. J Biol Chem. 2004;279:45450–45454. doi: 10.1074/jbc.M407562200. [DOI] [PubMed] [Google Scholar]
- 256.Lathrop JT, Timko MP. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science. 1993;259:522–555. doi: 10.1126/science.8424176. [DOI] [PubMed] [Google Scholar]
- 257.Zhang L, Guarente L. Heme binds to a short sequence that serves a regulatory function in diverse proteins. EMBO J. 1995;14:313–320. doi: 10.1002/j.1460-2075.1995.tb07005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Haile DJ, Rouault TA, Harford JB, Klausner RD. The inhibition of the iron responsive element RNA–protein interaction by heme does not mimic in vivo iron regulation. J Biol Chem. 1990;265:12786–12789. [PubMed] [Google Scholar]
- 259.Cong YS, Yao YL, Yang WM, Kuzhandaivelu N, Seto E. The hepatitis B virus X-associate protein, XAP3, is a protein kinase C-binding protein. J Biol Chem. 1997;272:16482–16489. doi: 10.1074/jbc.272.26.16482. [DOI] [PubMed] [Google Scholar]
- 260.Martinez-Noel G, Niedenthal R, Tamura T, Harbers K. A family of structurally related RING finger proteins interacts specifically with the ubiquitin-conjugating enzyme UbcM4. FEBS Lett. 1999;454:257–261. doi: 10.1016/s0014-5793(99)00823-6. [DOI] [PubMed] [Google Scholar]
- 261.Tatematsu K, Yoshimoto N, Koyanagi T, Tokunaga C, Tachibana T, Yoneda Y, Yoshida M, Okajima T, Tanizawa K, Kuroda S. Nuclear-cytoplasmic shuttling of a RING-IBR protein RBCK1 and its functional interaction with nuclear body proteins. J Biol Chem. 2005;280:22937–22944. doi: 10.1074/jbc.M413476200. [DOI] [PubMed] [Google Scholar]
- 262.Tokunaga C, Kuroda S, Tatematsu K, Nakagawa N, Ono Y, Kikkawa U. Molecular cloning and characterization of a novel protein kinase C-interacting protein with structural motifs related to RBCC family proteins. Biochem Biophys Res Commun. 1998;244:353–359. doi: 10.1006/bbrc.1998.8270. [DOI] [PubMed] [Google Scholar]
- 263.Bayle J, Lopez S, Iwai K, Dubreuil P, De-Sepulveda P. The E3 ubiquitin ligase HOIL-1 induces the polyubiquitination and degradation of SOCS6 associated proteins. FEBS Lett. 2006;580:2609–2614. doi: 10.1016/j.febslet.2006.03.093. [DOI] [PubMed] [Google Scholar]
- 264.Metzen E, Ratcliffe PJ. HIF hydroxylation and cellular oxygen sensing. Biol Chem. 2004;385:223–230. doi: 10.1515/BC.2004.016. [DOI] [PubMed] [Google Scholar]
- 265.Kim S, Wing SS, Ponka P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin–proteasome pathway. Mol Cell Biol. 2004;24:330–337. doi: 10.1128/MCB.24.1.330-337.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Wang J, Fillebeen C, Chen G, Andriopoulos B, Pantopoulos K. Sodium nitroprusside promotes IRP2 degradation via an increase in intracellular iron and in the absence of S-nitrosylation at C178. Mol Cell Biol. 2006;26:1948–1954. doi: 10.1128/MCB.26.5.1948-1954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Wang J, Chen G, Pantopoulos K. Nitric oxide inhibits the degradation of IRP2. Mol Cell Biol. 2005;25:1347–1353. doi: 10.1128/MCB.25.4.1347-1353.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Drapier JC, Pellat C, Henry Y. Generation of EPR-detectable nitrosyl–iron complexes in tumor target cells cocultured with activated macrophages. J Biol Chem. 1991;266:10162–10167. [PubMed] [Google Scholar]
- 269.Galy B, Ferring D, Hentze MW. Generation of conditional alleles of the murine iron regulatory protein (Irp)-1 and -2 genes. Genesis. 2005;43:181–188. doi: 10.1002/gene.20169. [DOI] [PubMed] [Google Scholar]
- 270.Salvatore MF, Fisher B, Surgener SP, Gerhardt GA, Rouault T. Neurochemical investigations of dopamine systems in iron-regulatory protein 2 (IRP-2) knockout mice. Mol Brain Res. 2005;139:341–347. doi: 10.1016/j.molbrainres.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 271.Andrews NC. Iron metabolism: iron deficiency and iron overload. Annu Rev Genomics Hum Genet. 2000;1:75–98. doi: 10.1146/annurev.genom.1.1.75. [DOI] [PubMed] [Google Scholar]
- 272.Ponka P, Schulman HM. Regulation of heme biosynthesis: distinct regulatory features in erythroid cells. Stem Cells. 1993;1:24–35. doi: 10.1002/stem.5530110607. [DOI] [PubMed] [Google Scholar]
- 273.Smith SR, Ghosh MC, Ollivierre-Wilson H, Tong WH, Rouault TA. Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyts stage of embryonic development. Blood Cells Mol Diseases. 2006;36:283–287. doi: 10.1016/j.bcmd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 274.Smith SR, Cooperman S, LaVaute T, Tresser N, Ghosh M, Meyron-Holz E, Land W, Oliverre H, Jortner B, Swizer R, Messing A, Rouault TA. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann N Y Acad Sci. 2003;1012:65–83. doi: 10.1196/annals.1306.006. [DOI] [PubMed] [Google Scholar]
- 275.Bush AI. Metals and neuroscience. Curr Opin Chem Biol. 2000;4:184–191. doi: 10.1016/s1367-5931(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 276.Zhang P, Land W, Lee S, Juliana J, Lefman J, Smith SR, Germain D, Kessel M, Leapman R, Rouault TA, Subramaniam S. Electron tomography of degenerating neurons in mice with abnormal regulation of iron metabolism. J Struct Biol. 2005;150:144–153. doi: 10.1016/j.jsb.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Pham CT, MacIvor DM, Hug BA, Heusel JW, Ley TJ. Long-range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci U S A. 1996;93:13090–13095. doi: 10.1073/pnas.93.23.13090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci U S A. 2002;99:4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sirito M, Lin Q, Deng JM, Behringer RR, Sawadongo M. Overlapping roles and asymmetrical cross-regulation of the USF proteins in mice. Proc Natl Acad Sci U S A. 1998;95:3758–3763. doi: 10.1073/pnas.95.7.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 282.Yoshiki A, Moriwaki K. Mouse phenome research: implications of genetic background. ILAR J. 2006;47:94–102. doi: 10.1093/ilar.47.2.94. [DOI] [PubMed] [Google Scholar]
- 283.Dupic F, Fruchon S, Bensaid M, Loreal O, Brissot P, Borot N, Roth MR, Coppin H. Duodenal mRNA expression of iron related genes in response to iron loading and iron deficiency in four strains of mice. Gut. 2002;51:648–653. doi: 10.1136/gut.51.5.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.LeBoeuf R, Tolson D, Heinecke JW. Dissociation between tissue iron concentrations and transferrin saturation among inbred mouse strains. J Lab Clin Med. 1995;126:128–136. [PubMed] [Google Scholar]
- 285.Fleming RE, Holden CC, Tomatsu S, Waheed A, Brunt EM, Britton RS, Bacon BR, Roopenian DC, Sly WS. Mouse strain differences determine severity of iron accumulation in Hfe knockout model of hereditary model of hemochromatosis. Proc Natl Acad Sci U S A. 2001;98:2707–2711. doi: 10.1073/pnas.051630898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Guyton AC, Hall JE. Textbook of Medical Physiology. 10. Saunders; Philadelphia, PA: 2000. [Google Scholar]
- 287.Chen OS, Blemings KP, Schalinske KL, Eisenstein RS. Dietary iron intake rapidly influences iron regulatory proteins, ferritin subunits and mitochondrial aconitase in rat liver. J Nutr. 1998;128:525–535. doi: 10.1093/jn/128.3.525. [DOI] [PubMed] [Google Scholar]
- 288.Ross KL, Eisenstein RS. Iron deficiency decreases mitochondrial aconitase abundance and citrate concentration without affecting tricarboxylic acid cycle capacity in rat liver. J Nutr. 2002;132:643–651. doi: 10.1093/jn/132.4.643. [DOI] [PubMed] [Google Scholar]
- 289.Leibold EA, Gahring LC, Rogers SW. Immunolocalization of iron regulatory protein expression in the murine central nervous system. Histochem Cell Biol. 2001;115:195–203. doi: 10.1007/s004180000246. [DOI] [PubMed] [Google Scholar]
- 290.Gdaniec Z, Sierzputowska-Gracz H, Theil EC. Iron regulatory element and internal loop/bulge structure for ferritin mRNA studied by cobalt(III) hexammine binding, molecular modeling, and NMR spectroscopy. Biochemistry. 1998;37:1505–1512. doi: 10.1021/bi9719814. [DOI] [PubMed] [Google Scholar]
- 291.Hall KB, Tang C. 13C relaxation and dynamics of the purine bases in the iron responsive element RNA hairpin. Biochemistry. 1998;37:9323–9332. doi: 10.1021/bi9805285. [DOI] [PubMed] [Google Scholar]
- 292.Hall KB, Williams DJ. Dynamics of the IRE RNA hairpin loop probed by 2-aminopurine fluorescence and stochastic dynamics simulations. RNA. 2004;10:34–47. doi: 10.1261/rna.5133404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ferreira C, Bucchini D, Martin ME, Levi S, Arosio P, Grandchamp B, Beaumont C. Early embryonic lethality of H-ferritin gene deletion in mice. J Biol Chem. 2000;275:3021–3024. doi: 10.1074/jbc.275.5.3021. [DOI] [PubMed] [Google Scholar]
- 294.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet. 1999;21:396–399. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- 295.Beaumont C, Leneuve P, Devaux I, Scoazec JY, Berthier M, Loiseau MN, Grandchamp B, Bonneau D. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyper-ferritinaemia and cataract. Nat Genet. 1995;11:444–446. doi: 10.1038/ng1295-444. [DOI] [PubMed] [Google Scholar]
- 296.Cazzola M, Bergamaschi G, Tonon L, Arbustini E, Grasso M, Vercesi E, Barosi G, Bianchi PE, Cairo G, Arosio P. Hereditary hyperferritinemia–cataract syndrome: relationship between phenotypes and specific mutations in the iron-responsive element of ferritin light-chain mRNA. Blood. 1997;90:814–821. [PubMed] [Google Scholar]
- 297.Girelli D, Corrocher R, Bisceglia L, Olivieri O, Franceschi LD, Zelante L, Gasparini P. Molecular basis for the recently described hereditary hyperferritinemia–cataract syndrome: a mutation in the iron-responsive element of ferritin L-subunit gene (the “Verona mutation”) Blood. 1995;86:4050–4053. [PubMed] [Google Scholar]
- 298.Mok H, Jelinek J, Pai S, Cattanach BM, Prchal JT, Youssoufian H, Schumacher A. Disruption of ferroportin 1 regulation causes dynamic alterations in iron homeostasis and erythropoiesis in polycythaemia mice. Development. 2004;131:1859–1868. doi: 10.1242/dev.01081. [DOI] [PubMed] [Google Scholar]
- 299.Pietrangelo A. The ferroportin disease. Blood Cells Mol Diseases. 2004;32:131–138. doi: 10.1016/j.bcmd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 300.Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. doi: 10.1182/blood-2006-02-003681. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Sanchez M, Galy B, Dandekar T, Bengert P, Vainshtein Y, Stollte J, Muckenthaler MU, Hentze MW. Iron regulation and the cell cycle: Identification of an iron-responsive element in the 3′ untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy. J Biol Chem. doi: 10.1074/jbc.M603876200. in press. [DOI] [PubMed] [Google Scholar]