

Abstract
Mechanisms that initiate lupus nephritis and cause progression to end-stage renal disease remain poorly understood. In this study, we show that lupus-prone New Zealand Mixed 2410 mice that develop a severe glomerulosclerosis and rapidly progressive renal disease overexpress IL-4 in vivo. In these mice, STAT6 deficiency or anti-IL-4 Ab treatment decreases type 2 cytokine responses and ameliorates kidney disease, particularly glomerulosclerosis, despite the presence of high levels of IgG anti-dsDNA Abs. STAT4 deficiency, however, decreases type 1 and increases type 2 cytokine responses, and accelerates nephritis, in the absence of high levels of IgG anti-dsDNA Abs. Thus, STAT6 and IL-4 may selectively contribute to the development of glomerulosclerosis, whereas STAT4 may play a role in autoantibody production.
Systemic lupus erythematosus (SLE),4 a heterogeneous systemic autoimmune disease, arises as a consequence of the failure of multiple immunological mechanisms (1, 2). Although some patients with SLE have severe inflammation in their kidneys, others develop rapidly progressive chronic renal disease that leads to glomerulosclerosis and renal failure. Mechanisms that initiate lupus nephritis and cause progression to end-stage renal disease remain poorly defined. Although the development of lupus nephritis is generally attributed to pathogenic autoantibodies such as anti-DNA Ab, some patients with SLE develop renal disease in the absence of anti-DNA Ab.
Pathogenic autoantibodies in lupus mice generally belong to the IgG2a and IgG3 subclasses (3, 4), which are predominantly dependent on the type 1 cytokine, IFN-γ, and are suppressed by the type 2 cytokine, IL-4 (5). IL-4, however, also promotes autoimmunity by inhibiting apoptosis of autoreactive B cells (6) and inhibits autoimmunity by inducing the production of the regulatory cytokine, TGF-β (7, 8). Type 1 and 2 cytokines can also directly participate in end organ damage (9). For example, the type 1 cytokine IFN-γ can exacerbate organ inflammation, whereas type 2 cytokines can exacerbate tissue fibrosis (10). It remains largely unresolved, however, which cytokines play the most vital roles in regulation of autoantibody production and in end organ damage during the development and progression of SLE.
Several laboratories have attempted to analyze the abnormalities of cytokine production in humans and mouse models with SLE (11–15). The data, however, are conflicting. Furthermore, difficulties in cytokine measurements may have contributed to the conflicting data obtained. For example, many studies have used in vitro methods for cytokine measurement following stimulation of cells with either anti-CD3, IL-2, or Con A under different culture conditions. In addition, a confusing picture for cytokine involvement in lupus has emerged using IL-4 and IFN-γ transgenic and knockout mice. For example, transgenic overexpression of IL-4 is associated with both protection from and development of a lupus-like disorder (16, 17). Also, in knockout mice, while germline deletion of IL-4 has an ameliorating effect on disease in MRL-lpr/lpr (MRL-lpr) mice (18), it has no effect on autoantibody levels and development of nephritis in chemical (HgCl2)-induced and BXSB mouse models (19, 20). These apparently conflicting results may be due to redundancy of cytokine functions, different backgrounds of the gene-targeted mice, and/or the fact that cytokines may play opposing roles, amplifying or inhibitory, during the development of lupus.
In this study, using an in vivo cytokine capture assay as well as other parameters that reflect type 1 or 2 cytokine production, we show that a lupus-prone strain, New Zealand Mixed (NZM.2410; see Ref. 21), overexpresses IL-4 and develops severe glomerulosclerosis, whereas another lupus-prone strain, MRL-lpr, overexpresses in vivo IFN-γ and develops severe kidney inflammation. The in vivo cytokine capture assay does not disturb the cytokine microenvironment within the cell and accurately measures in vivo cytokine secretion (22). We have rendered the NZM.2410 mice deficient in STAT4 or STAT6 function, or have treated them with an anti-IL-4 mAb to investigate the regulatory functions of type 1 and 2 cytokines on autoantibody production and renal disease. The STAT molecules are cytoplasmic proteins that play pivotal roles in development of type 1 and 2 cytokine responses and mediate many cytokine-induced responses. These proteins are activated following phosphorylation via the Janus kinase family of tyrosine kinases, which in turn are activated by interaction of a cytokine and its receptor (23). For example, STAT6 protein is activated following the interaction between IL-4 and the cell surface IL-4R, and is critical for the development of cells that secrete type 2 cytokines such as IL-4 and IL-13. STAT6-deficient mice have reduced type 2 cytokine production, reduced IgE levels, and IL-4-induced B cell proliferation (24–26). In contrast, STAT4 protein, which drives type 1 immune responses, is activated after the interaction between IL-12 and the IL-12R. This in turn induces the transcription of IFN-γ (23). STAT4-deficient mice lack IL-12-induced IFN-γ production and display a predominant type 2 cytokine phenotype (27).
In this work, we demonstrate that STAT6 deficiency or anti-IL-4 mAb treatment markedly inhibits the progression of lupus nephritis, despite the unaffected or even increased IgG anti-dsDNA Ab levels in NZM.2410 mice. STAT4 deficiency in these mice, however, increases renal disease, despite a decrease in IgG anti-dsDNA Ab levels.
Materials and Methods
Mice
Four strains of lupus-prone mice, NZM.2410, (New Zealand Black (NZB) × New Zealand White (NZW))F1 (BWF1), MRL-lpr, and NZB, and five nonautoimmune strains, NZW, BALB/c, (BALB/c × NZW)F1 (CWF1), B10.Pl, and (NZB × B10.Pl)F1 (BPF1), were used in this study. Breeding pairs of NZM.2410, NZB, NZW, MRL-lpr, BALB/c, and B10.Pl were purchased from The Jackson Laboratory (Bar Harbor, ME) and were bred locally. Nonautoimmune NZW and B10.Pl mice express MHC class II haplotype that is identical with that in NZM.2410 (H-2z = u); nonautoimmune CWF1 and BPF1 mice express MHC class II haplotype identical with BWF1 (H-2d/u); and BALB/c express MHC class II haplotype identical with that in NZB mice. All experiments were performed in accordance with the institutional animal research committee guidelines.
Generation of STAT4 and STAT6 null NZM.2410 mice
STAT4 and STAT6 null mutations (24, 27) on a 129, C57BL/6 mixed background were crossed onto the NZM.2410 background, using a marker-assisted selection strategy to accelerate the development of desired congenic mice. Each backcross generation was screened with microsatellite DNA markers at a density of ~25 cM. At least 25 males were screened at each generation. At each backcross generation, we specifically selected progeny that carried the target gene and had the lowest content of donor (129, C57BL/6) genes throughout the genome. To follow the STAT4 and STAT6 null mutations, we used PCR-based assays: one assay amplified the linked neomycin-resistance (neo) gene, and the other amplified the segment of the respective STAT gene spanning the null-generating deletion.
Abs and cytokines
The anti-IL-4 and anti-IFN-γ mAbs used for our studies have been previously described (22). Neutralizing anti-IL-4 mAb (11B11) was kindly provided by C. Reynolds (National Cancer Institute, Frederick, MD). Purified mouse rIL-4 and rIFN-γ were purchased from BD PharMingen (San Diego, CA).
Treatment protocol
Animals were injected i.p. with 250 μg of an anti-IL-4 mAb (11B11) or an isotype-matched rat IgG1 mAb (GL113) three times per week for 12 wk.
In vivo cytokine capture assay
In vivo IL-4 and IFN-γ levels were measured by an in vivo capture assay (22) with some modifications. In brief, mice were injected i.v. with 10 μg of biotinylated anti-mouse IL-4 (BVD4-1D11) or anti-mouse IFN-γ (R4-6A2) mAb in PBS containing 1% normal mouse serum, and were bled within 2–4 h (for IL-4) or 12–24 h (for IFN-γ). Sera (containing cytokine/anticytokine complexes) were collected. Flat-bottom white high-binding ELISA plates (ISC Bioexpress, Kaysville, UT) were coated with 100 μl of anti-IL-4 (24G2.3) or anti-IFN-γ (AN-18) mAbs at a concentration of 10 μg/ml in 0.1 M Tris-HCl, pH 8.3, for 2 h at room temperature (RT). Plates were then filled with 150 μl super block solution (Pierce, Rockford, IL) and emptied immediately by inversion. This process was repeated three times. Plates were then dried and washed once with distilled water using a Microwash II microtiter plate washer (Skatron, Sterling, VA). Positive controls were prepared by mixing 10 μl of 10 μg/ml cytokine with the appropriate biotinylated anti-cytokine mAb (5 μl of 2 mg/ml, BVD4-1D11.2 for IL-4 and R4-6A2 for IFN-γ) for 3 min at RT, and adding washing buffer to obtain a final IL-4 or IFN-γ concentration of 100 ng/ml. A total of 25 μl of positive control or serum samples diluted 2× (or 10× if the cytokine concentration was predicted to be high) was added to the plate for 1 h at 37°C. Plates were then rinsed twice with washing buffer (blocking buffer contained 10% Tween 20 solution and TBS), and then 10 times with distilled water. Streptavidin-HRP (1:20,000 in washing buffer; Pierce) was then added for 1 h at RT. After rinsing with washing buffer 5 times, and with water 20 times, plates were filled with BupH Tris saline (Pierce) and emptied by inversion. A total of 150 μl of freshly prepared Femto maximum sensitivity substrate (1 part enhancer, 1 part peroxidase, 18 part of BupH Tris saline; Pierce) was added, and plates were read immediately at 425 nm using a luminometer (Labsystems, Helsinki, Finland).
In vitro culture
Splenocytes were activated with 1 μg/ml plate-bound anti-CD3. After 1 wk in culture, cells were washed and restimulated with plate-bound anti-CD3 for 24 h. In some experiments, splenocytes were stimulated with Con A (2 μg/ml) for 72 h. Supernatants were harvested, and IL-4 and IFN-γ levels were quantified by ELISA, as described previously (28).
Measurement of serum IgE levels
Serum IgE levels were measured using flat-bottom white high-binding ELISA plates (ISC Bioexpress), which were coated with 50 μl of anti-mouse IgE mAb (EM-95; 10 μg/ml in 0.1 M Tris-HCl, pH 8.3) overnight at 4°C. Plates were blocked three times with 150 μl superblocking solution (Pierce). A total of 25 μl of a standard or test sera (diluted 2× to 10× in 10% blocking buffer in Tris saline/0.01% Tween) was added to the plates for 30 min at RT, followed by washing and incubation with 25 μl biotin-ylated anti-IgE (RIE4; 1/10,000 dilution) for 30 min at RT. Plates were then washed six times, and 25 μl immunopure streptavidin-HRP (1/20,000 in dilution buffer; Pierce) was added for 30 min at RT. Afterward, the plates were washed 10 times and filled with Buph Tris saline (Pierce) for 30 min at RT. After emptying, 150 μl of Femto substrate (Pierce) was added onto the plates, and the OD was read immediately at 425 nm using a luminometer (Labsystems).
Measurement of IgG1 and IgG2a in serum and kidney extracts
High-binding ELISA plates (Costar, Cambridge, MA) were first coated with 10 μg/ml goat anti-mouse IgG1 or IgG2a. After blocking with 10% FCS in PBS, standard, sera, or kidney extracts were added onto the plates for 1 h at RT. After washing, alkaline phosphatase-conjugated anti-mouse IgG1 or IgG2a (1/1000 dilution) was added for 1 h at RT. The plates were then developed with pNPP and read at 405 nm on a Multiskan MS ELISA reader. Total protein in kidney extracts was determined using bicinchoninic acid protein assay kit (Pierce). Renal Ig levels are represented as picograms Ig per milligram of kidney protein.
Detection of anti-dsDNA Ab
Anti-DNA Ab were detected, as described previously (29), using purified calf thymus DNA (Sigma-Aldrich, St. Louis, MO) to coat enhanced protein-binding ELISA plates (Costar, Corning, NY) overnight at 4°C. The next day, plates were washed once with PBS, and blocked with 200 μl 10% FCS for 1 h at RT. Standard and samples were added (50 μl/well) for 1 h at 37°C, followed by washing twice with PBS/0.05% Tween 20 (Sigma-Aldrich). Alkaline phosphatase-conjugated goat anti-mouse IgG, IgG1, or IgG2a (1:1000 in 1% FCS in PBS) was added for 1 h at RT. Plates were then developed with pNPP substrate, and read at 405 nm on a Labsystems ELISA reader.
Assessment of nephritis
Proteinuria was estimated utilizing Albustix assay strips (Bayer, Elkhart, IN), using a scale of 0–4+. Severe proteinuria was defined as ≥300 mg/dl (3+ or more) on two consecutive examinations, as described previously (30). For the assessment of renal histology, one-half of each kidney was fixed in 4% paraformaldehyde. Paraffin sections were subsequently stained with H&E, Jones silver stain, periodic acid Schiff, and Masson’s trichrome, and scored in a blind fashion for the following features (using a 0–3 scale): glomerular hypercellularity, necrotizing lesions, karyorrhexis, cellular crescents, and hyaline deposits (these features indicate glomerular activity score); interstitial inflammation, tubular cell necrosis, and epithelial cells or macrophages in tubular lumens (tubulointerstitial activity score); glomerulosclerosis, glomerular scars, fibrous crescents, tubular atrophy, and interstitial fibrosis (chronic lesion score). The raw scores assigned by various readers were averaged to obtain a mean score for each of the individual features. The mean scores for individual features were summed to obtain the three main scores (glomerular activity score, tubulointerstitial activity score, and chronic lesion score), and then the three main scores were summed to obtain a composite kidney biopsy score (KBS).
Statistical analysis
Mann-Whitney U and Student’s t tests were used to compare cytokine, Ig, and anti-DNA Ab levels in different groups of mice. The cumulative prevalence of proteinuria was compared between the test and control groups of mice using a log-rank test.
Results
NZM.2410 mice develop severe glomerulosclerosis
Glomerulonephritis in patients with SLE is a heterogeneous disorder: some patients experience severe glomerular and interstitial inflammation, while others develop rapidly progressive glomerulosclerosis and end-stage renal disease. Different lupus-prone mouse strains develop different forms of renal disease, which may be analogous to different subsets of human lupus nephritis. To test this idea, we compared components of renal histology in NZM.2410 and MRL-lpr mice at 3–4, 5–7, and 7–8 mo of age. Results from 5- to 7-mo-old mice are shown in Fig. 1. At each age tested, glomerulosclerosis was markedly increased in NZM.2410 mice as compared with MRL-lpr mice (p < 0.01; Mann-Whitney U test; Fig. 1), whereas inflammatory lesions, such as interstitial and perivascular inflammation (p < 0.01; Fig. 1) and glomerular infiltration (p = borderline (<0.1>0.05); data not shown), were more severe in MRL-lpr mice. Thus, different lupus-prone mouse strains exhibit different patterns of nephritis: NZM.2410 mice have more severe glomerulosclerosis, whereas MRL-lpr mice develop more severe glomerular and interstitial inflammation.
FIGURE 1.
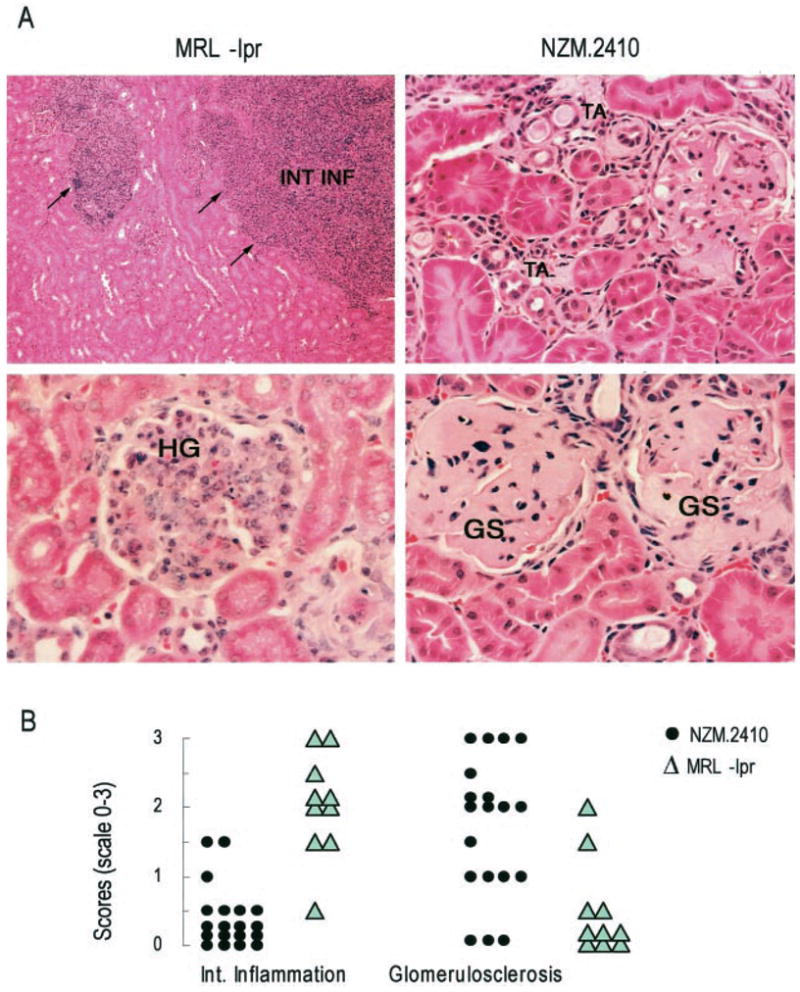
NZM.2410 mice develop more severe glomerulosclerosis than MRL-lpr mice. A, H&E-stained kidney sections show increased inflammatory cell infiltrates in the interstitium (arrows, INT INF) and glomeruli (HG (hypercellular glomerulus)) of MRL-lpr mice, and increased glomerulosclerosis (GS) and tubular atrophy (TA) in NZM.2410 mice. B, Nephritis scores in kidneys harvested from 5- to 7-mo-old mice (n = 15 NZM.2410 and 10 MRL-lpr). Interstitial inflammatory infiltrate (p < 0.01); glomerulosclerosis (p < 0.01).
Spontaneous IL-4 production is increased in the NZM.2410 mice
Because type 2 cytokines have been implicated in the development of tissue fibrosis (10, 31, 32), we examined whether expression of type 2 cytokines differs between lupus-prone strains that develop severe glomerulosclerosis vs those that develop predominantly inflammatory disease. Using a sensitive in vivo cytokine capture assay, we found that in vivo IL-4 production was significantly increased in the NZM.2410 strain when compared with other lupus-prone strains, MRL-lpr, BWF1, and NZB mice, and several nonautoimmune strains including BALB/c, B10.Pl, CWF1, and BPF1 mice (p = 0.04–0.00005, Student’s t test). Results from lupus-prone mice at the early disease stage are shown in Fig. 2A. Similar results were obtained at a very young age (5–9 wk old), prenephritic age (12–16 wk old), and advanced nephritic age (>25 wk old).
FIGURE 2.
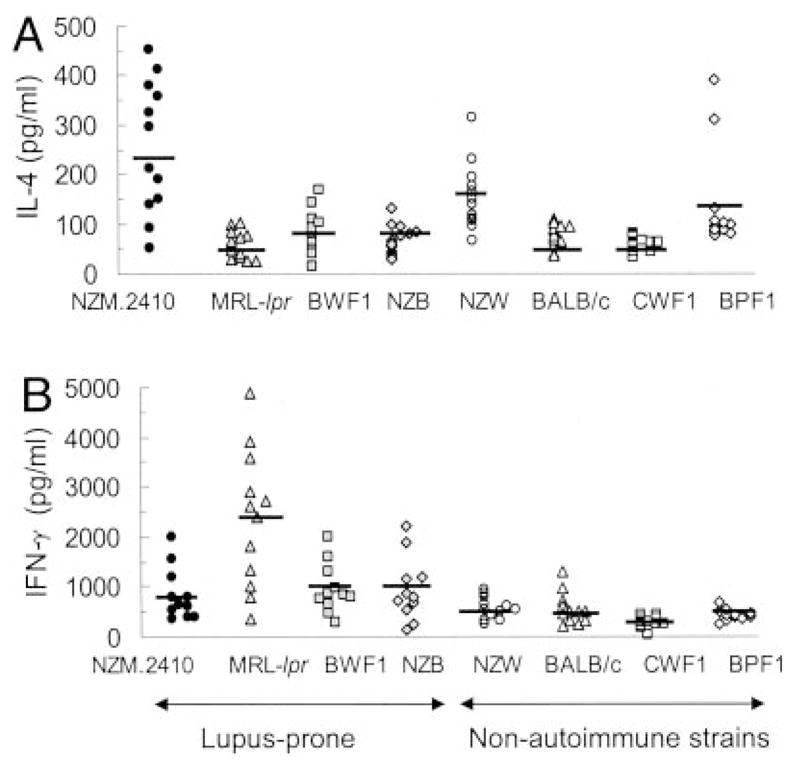
Intrinsic in vivo levels of IL-4 and IFN-γ in lupus-prone and nonautoimmune mice. Lupus-prone mice, assayed at an age when they begin to develop disease (16- to 20-wk-old NZM.2410 and BWF1; 12- to 16-wk-old MRL-lpr; and 24- to 32-wk-old NZB mice), and 16- to 20-wk-old nonautoimmune control mice were injected with 10 μg of biotinylated anti-IL-4 or anti-IFN-γ; their sera were obtained and tested for levels of Ab-captured IL-4 (A) or IFN-γ (B), as described in Materials and Methods. Values in pg/ml from individual mice are shown (n = 10–12 mice per group). Horizontal bars represent the arithmetic mean for each group.
IFN-γ levels, as determined by the in vivo capture assay, were not elevated in NZM.2410 mice as compared with other lupus-prone and most nonautoimmune strains tested, except when compared with CWF1 and BPF1 mice (p < 0.05). In contrast, MRL-lpr mice had higher IFN-γ levels than any other strain tested (p < 0.01; Fig. 2B).
Consistent with the increased IL-4 levels observed in NZM.2410 mice, serum IgE and IgG1 levels that are dependent on IL-4 (33) increased selectively in diseased NZM.2410 mice (p < 0.05–0.0001; data not shown). In contrast, serum IgG2a levels, which usually are dependent on IFN-γ (5), were not elevated in NZM.2410 mice as compared with other strains.
Neutralization of endogenous IL-4 in NZM.2410 mice reduces renal disease, but not serum IgG anti-dsDNA Ab level
To assess the contribution of IL-4 to disease expression in lupus-prone mice, we treated 20-wk-old NZM.2410 mice that already had circulating autoantibodies and early nephritic changes with a neutralizing anti-IL-4 mAb (11B11), an isotype-matched rat IgG1 mAb (GL113), or saline (PBS) for 12 wk. As expected, anti-IL-4 mAb treatment decreased IL-4 and increased IFN-γ production from cultured spleen cells (p < 0.05–0.01; Fig. 3A). The efficacy of this treatment was further shown by decreased serum total IgG1/IgG2a and IgE levels in anti-IL-4 mAb-treated mice (p < 0.05; Fig. 3, B and C).
FIGURE 3.
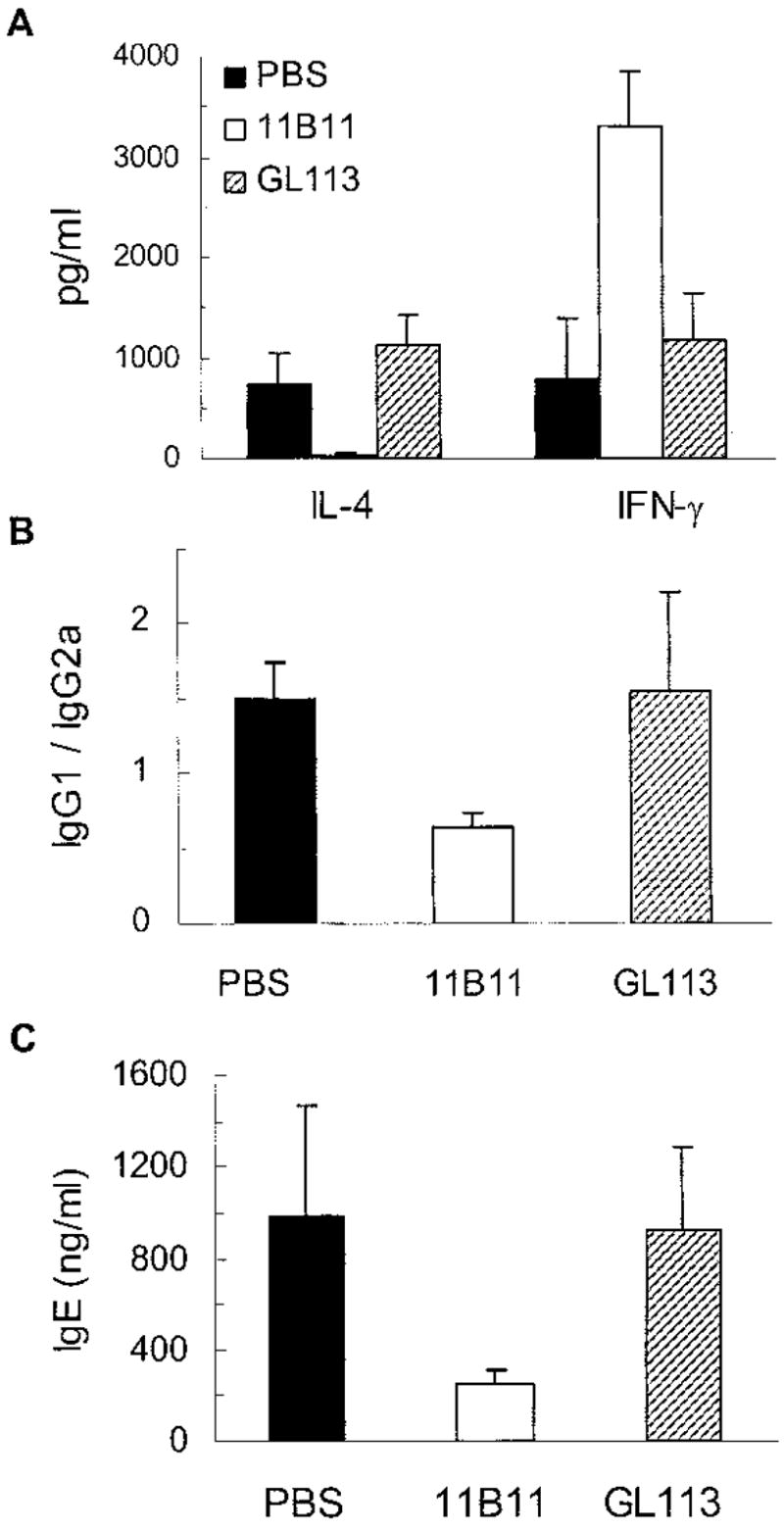
Treatment of NZM.2410 mice with anti-IL-4 mAb (11B11) inhibits IL-4 production, and reduces IgG1 and IgE levels. Twenty-week-old female NZM.2410 mice were treated with 11B11, GL113 (control Ig), or PBS. A, IL-4 and IFN-γ levels in supernatants of spleen cells cultured with Con A for 48–72 h. B and C, Total serum IgG1, IgG2a, and IgE levels were determined in 28- to 30-wk-old mice (p < 0.05–0.01 in 11B11-treated vs control groups; n = 5–8 mice per group). Results are expressed as the mean ± SE values from a representative of two independent experiments.
The proportion of mice developing severe proteinuria (≥300 mg/dl) was reduced in 11B11-treated vs GL113- or PBS-treated mice (p < 0.01, log-rank test; Fig. 4A). Consistent with the decreased proteinuria, a composite KBS was decreased in the 11B11-treated mice as compared with PBS- or GL113-treated mice (p < 0.01) (Fig. 4B).
FIGURE 4.

Treatment of NZM.2410 mice with an anti-IL-4 mAb inhibits progression of renal disease, but does not diminish IgG anti-dsDNA Ab production. Twenty-week-old NZM.2410 mice were treated with either 11B11, GL113, or PBS, as described in Results (n = 11–12 mice per group). A, Effect of 11B11 treatment on proteinuria. Data are represented as percentage of cumulative prevalence of severe (≥300 mg/dl) proteinuria. B, Kidneys harvested from treated and control mice were sectioned and scored. Glomerular and tubulointerstitial activity and chronic lesion scores were summed to obtain a total KBS, which is shown as individual sample scores. C, Comparison of individual mouse KBS representing the components of glomerular activity (glomerular cellularity score) and chronic glomerular lesions (glomerulosclerosis score) in treated and control mice. D, Representative H&E (upper panels)- and Jones silver-stained (lower panels) kidney sections are shown. GH, glomerular hypercellularity; GSc, glomerulosclerosis; Lo, glomerular lobulation; SL, segmental lesion; DT, dilated tubules. E, Serum IgG anti-dsDNA Ab levels (p < 0.05, 11B11-treated vs GL113 or PBS controls at 5 mo of age). F, Protein extracts from the kidneys of 11B11- or GL113-treated mice were tested for total IgG, IgG1, and IgG2a by ELISA. Results are expressed as the mean ± SE of Ig in pg/mg of protein in the kidney extracts (p = NS; n = 4–6 per group). Similar results were obtained in another experiment in which 6- to 8-wk-old NZM.2410 were treated with 11B11 or GL113 for 16 wk.
Kidney lesions in lupus glomerulonephritis include acute glomerular infiltration and chronic scarring (glomerulosclerosis). To assess the effect of 11B11 treatment on these lesions, we scored H&E-, Jones silver-, periodic acid Schiff-, and Masson’s trichrome-stained renal sections, as described in Materials and Methods. Although glomerular cellularity was not significantly different between the treated and control mice, glomerulosclerosis, both global and segmental, was decreased in the 11B11-treated mice (p < 0.01; Fig. 4, C and D).
Surprisingly, 11B11 treatment did not decrease IgG anti-dsDNA Ab production (Fig. 4E). In fact, serum IgG anti-DNA Ab levels were increased in 11B11-treated mice as compared with GL113-or PBS-treated controls (p < 0.05 at 5 mo of age). Anti-DNA Ab levels of both IgG1 and IgG2a isotypes were slightly higher in 11B11-treated mice than in control animals (p = NS; data not shown). The beneficial effects of 11B11 treatment on kidney disease did not appear to be due to changes in the degree of IgG deposits in kidneys (Fig. 4F). In kidney extracts, IgG1 levels were lower and IgG2a levels were higher in 11B11-treated than in GL113-injected control animals, although the differences were not statistically significant.
These observations suggest that elevated IL-4 levels in NZM.2410 mice may contribute to the development of glomerulosclerosis and chronic renal lesions, but may not be required for, or may even inhibit, autoantibody production.
Characterization of STAT4 and STAT6 knockout NZM.2410 mice
To further address the role of type 1/2 cytokines, we transferred the STAT4 and STAT6 null mutations onto the NZM.2410 background using a marker-assisted selection protocol, which accelerates the introgression of genes by using genetic markers distributed over the whole mouse genome. The final N6 backcross mice that were used in our experiments carried 79 of 79 (100%) NZM.2410 markers. Presence of the STAT4 and STAT6 null mutations was followed by PCR-based assays, and the absence of the respective STAT protein products was confirmed by Western blot analysis (not shown). To verify the effects of STAT4 or STAT6 null mutations on the production of type 1 and 2 cytokines, spleen cells from wild-type and mutant NZM.2410 mice were isolated and activated in vitro with plate-bound anti-CD3 Ab in the absence of exogenous cytokines. This allowed determination of the absence of STAT4 or STAT6 function on the developmental potential of spleen cells into Th1 or Th2 cytokine-secreting cells. After 1 wk in culture, the activated spleen cells were restimulated with anti-CD3 for 24 h, and IL-4 and IFN-γ production was quantified by ELISA. As shown in Fig. 5A, under neutral activation conditions, splenocytes from NZM.2410 secreted both IL-4 and IFN-γ. The STAT4−/− NZM.2410 mice secreted less IFN-γ (p < 0.05), but more IL-4, whereas STAT6−/− NZM.2410 mice secreted less IL-4 (p < 0.05), but more IFN-γ (p < 0.05).
FIGURE 5.
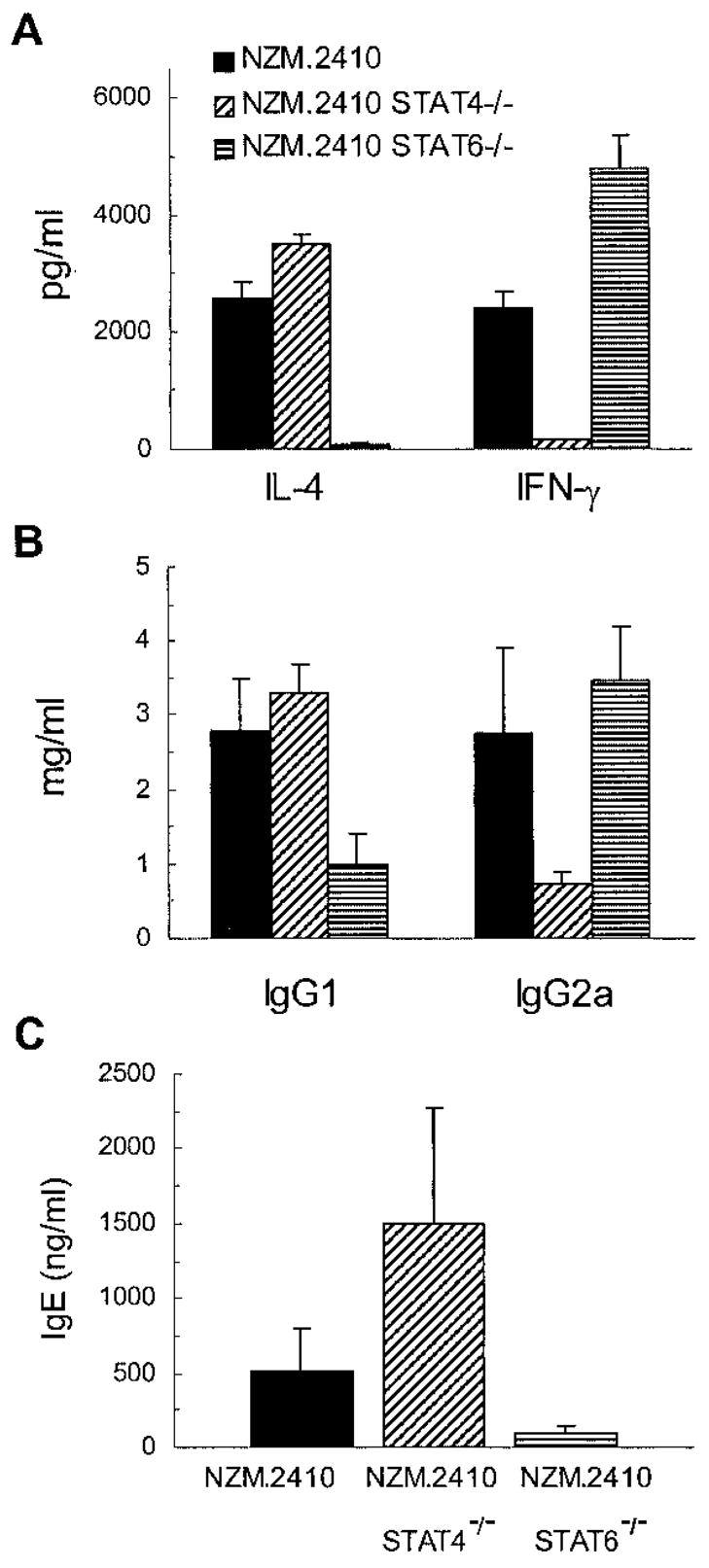
Cytokine and Ig levels in STAT4- and STAT6-deficient NZM.2410 mice. A, Splenic lymphocytes from 9- to 12-wk-old NZM.2410 mice with the indicated genotype (wild type, STAT4−/−, and STAT6−/−) were tested for IL-4 and IFN-γ production, as described in Results. B and C, Serum samples from these mice were tested for IgG1 and IgG2a isotypes and IgE levels. Results are expressed as the mean ± SE values of 12–15 mice per group.
Because IgE and IgG1 production is dependent on IL-4 (33), while IgG2a production is usually dependent on IFN-γ (5), IgG isotypes and IgE were evaluated in serial serum samples from the mutant and wild-type NZM.2410 mice. As shown in Fig. 5B, there was a significant isotype switch toward IgG1 in STAT4−/− NZM.2410 mice (p = 0.05), while STAT6−/− NZM.2410 mice showed a clear IgG2a predominance (p < 0.05). In addition, serum IgE levels were increased in STAT4−/− mice (p < 0.05), while IgE levels were decreased in STAT6−/− NZM.2410 mice compared with wild-type littermates (p = 0.02; Fig. 5C).
Targeted disruption of STAT6 reduces kidney disease in NZM.2410 mice
Long-term follow-up of STAT6−/− NZM.2410 mice showed a significant reduction in the development of kidney disease. Thus, the proportion of mice developing severe proteinuria (≥300 mg/dl) was markedly reduced in STAT6 null mice as compared with control mice (p = 0.005; Fig. 6A). Consistent with the decreased proteinuria, a composite KBS was decreased in these STAT6 null mice (p = 0.005; Fig. 6B). Furthermore, as with anti-IL-4 mAb treatment (Fig. 4, C and D), the decrease in glomerulosclerosis was the most consistent effect of STAT6 null mutation on kidney histology (Fig. 6C). Intriguingly, while we observed a significant effect on the development of kidney disease in STAT6−/− NZM.2410 mice, there was no decrease in IgG anti-dsDNA Ab levels in these mice when compared with their wild-type littermates (Fig. 6D).
FIGURE 6.
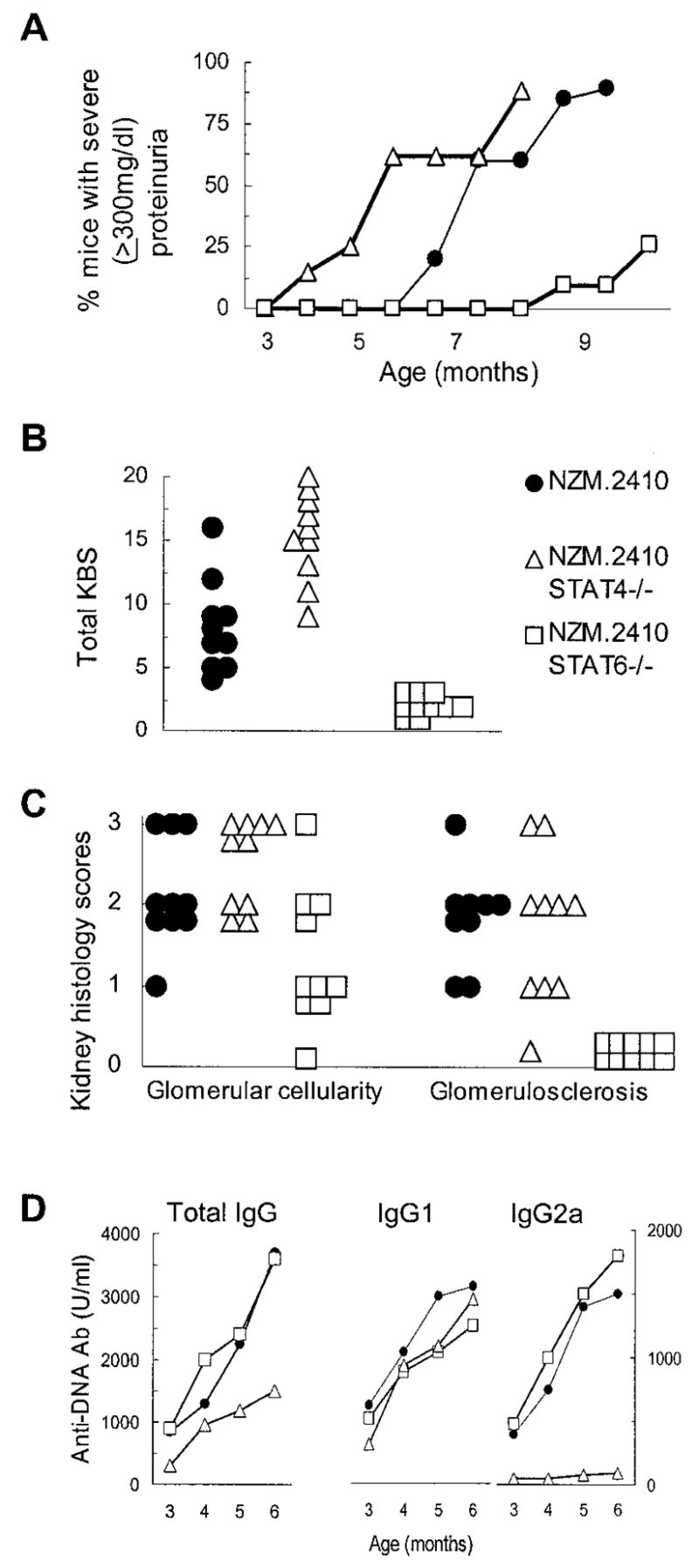
Characterization of kidney disease development in STAT4-and STAT6-deficient NZM.2410 mice. A, Proteinuria in NZM.2410 mice with the indicated genotypes. Data are presented as the percentage of cumulative prevalence of severe (≥300 mg/dl) proteinuria at various ages of the mice (n = 12–15 mice for each experimental group). B, Composite KBS of NZM.2410 mice with the indicated genotypes. Kidneys were harvested from 10 mice in each group at the age that the mice became severely proteinuric (≥300 mg/dl) or at the age of 11 mo if no severe proteinuria occurred at an earlier age. C, Comparison of individual mouse kidney scores representing the components of glomerular activity and chronic glomerular sclerosis in the three groups of NZM.2410 mice, as in B above. D, Serum IgG, IgG1, and IgG2a anti-dsDNA Ab levels in NZM.2410 mice with the indicated genotypes. The mean Ab level of 12–15 mice per group at various ages is shown.
Targeted disruption of STAT4 exacerbates kidney disease without increasing IgG anti-dsDNA Ab production in NZM.2410 mice
STAT4−/− NZM.2410 mice showed increased development of kidney disease, as evident by the accelerated development of severe proteinuria (≥300 mg/dl) compared with wild-type littermates (p = 0.02; Fig. 6A), and by increase in the composite KBS (p = 0.05; Fig. 6B). There was no significant difference in the effect of STAT4 null mutation on glomerular infiltration vs glomerulosclerosis (Fig. 6C). Other renal lesions, including tubular atrophy, crescent formation, and interstitial fibrosis, were increased in STAT4−/− NZM.2410, although the differences were not statistically significant (data not shown). Most interestingly, the exacerbated kidney disease in the STAT4 null mice was not associated with an increase in IgG anti-dsDNA autoantibodies. In fact, as shown in Fig. 6D, STAT4−/− NZM.2410 mice have significantly lower levels of IgG anti-dsDNA Abs compared with the STAT6−/− and control NZM.2410 mice (p = 0.05). Most auto-antibodies were of IgG1 isotype in STAT4−/− mice, whereas STAT6−/− mice showed a slight increase in IgG2a compared with wild-type mice, but this difference was not statistically significant.
Discussion
In this study, we explore the abnormalities of cytokine production in lupus, and examine their role in the development and progression of lupus nephritis. Previous attempts to identify cytokine abnormalities in lupus and other diseases have generally required stimulation of immune cells, which may not reflect the intrinsic abnormality of a cytokine. We therefore used an in vivo cytokine capture assay, which does not require any exogenous stimulation, and allows detection of physiological and quite small amounts of cytokines. Using this assay, we identified two strains of lupus-prone mice, one (NZM.2410) that overexpresses IL-4 and the other (MRL-lpr) that overexpresses IFN-γ (Fig. 2). The use of these mice should help to define the roles of these cytokines in the development and progression of lupus.
IL-4 can rescue B cells from apoptosis and enhance their survival (6, 34). The increased expression of IL-4, therefore, may result in the expansion and activation of autoreactive B cells, and thus may contribute to the development or aggravation of autoantibody-mediated disease. Indeed, IL-4 transgenic C3H mice develop an autoimmune-type disorder that resembles lupus (17). Thus, IL-4 may exacerbate B cell-mediated autoimmunity. IL-4 can also inhibit T cell activation in some in vivo systems (35). Consistent with this role, NZM.2410 mice that overexpress IL-4 have less renal inflammation compared with MRL-lpr mice that overexpress IFN-γ (Fig. 1). NZM.2410 mice, however, have more glomerulosclerosis than MRL-lpr mice (Fig. 1). Strikingly, the in vivo depletion of IL-4 or STAT6 gene deletion markedly inhibits chronic renal lesions and glomerulosclerosis (Figs. 4C, 4D, and 6C). These observations suggest a correlation between increased IL-4 levels and the development of glomerulosclerosis, and elevated IFN-γ levels with inflammatory cell infiltration. Thus, two different subsets or stages of disease may characterize lupus nephritis; one subset or stage of nephritis may be exemplified by MRL-lpr-type nephritis with marked renal inflammation, and the other subset or stage may be exemplified by the NZM.2410-type nephritis, which presents with glomerulosclerosis and less marked renal inflammation (Fig. 1).
Type 1 cytokines have been implicated in the development of anti-DNA Ab and lupus in MRL-lpr, BWF1, and BXSB mouse models of lupus (4, 18, 20, 36). Treatment of young, prenephritic BWF1 mice with an anti-IL-12 mAb decreases IgG anti-dsDNA Ab levels (36). Our results in STAT4-deficient NZM.2410 mice substantiate the importance of type 1 cytokines in autoantibody production (Fig. 6D). The STAT4-deficient mice, however, experienced accelerated nephritis in our study. Studies are underway to examine the following possibilities to explain this finding: 1) increased type 2 cytokine production in STAT4-deficient mice aggravates glomerulosclerosis in lupus-prone mice; 2) autoantibodies are not critical for the development of lupus nephritis; and 3) autoantibodies other than anti-dsDNA Ab may cause nephritis in NZM.2410 mice.
Our results show that STAT6 deficiency or anti-IL-4 mAb treatment markedly inhibits the progression of lupus nephritis in NZM.2410 mice (Figs. 4B, 4C, 6A, and 6B), even though IgG anti-dsDNA Ab levels are unchanged or slightly increased (Figs. 4E and 6D). STAT4-deficient NZM.2410 mice, by contrast, exhibited increased renal disease, despite a decrease in IgG anti-dsDNA Ab levels. These results appear to contradict a direct cause-effect relationship between the presence of autoantibodies and nephritis in NZM.2410 mice. One possibility is that some genes control the production and renal deposition of autoantibodies, while others contribute to the development and progression of renal disease (37). Consistent with this idea, mouse chromosome 11, which harbors the genes for IL-4, IL-5, and IL-13, also contains two loci, D11 Mit23 and D11 Mit164, which are linked to glomerulonephritis, but not to IgG anti-dsDNA Ab production in (NZM.2410 × C57BL/6)F2 progeny (38). Another possibility is that renal deposition of autoantibodies in SLE may represent an early event, which later triggers renal cells to secrete extra cytokines and growth factors, such as IL-4 and TGF-β, which may perpetuate glomerulosclerosis and chronic renal fibrosis. Finally, it is also possible that observed effects of the STAT4 or STAT6 knockout reflect the removal of lupus susceptibility or resistance genes during backcross of the mutated locus from the Sv129 onto the NZM.2410 genetic background. STAT4 and STAT6 genes, however, are on mouse chromosome 1 (25.9 cM) and mouse chromosome 10 (70.0 cM), respectively; thus, both genes are clearly outside any known lupus susceptibility or resistance region in NZM2410 mice. Therefore, it is unlikely that the phenotypes presented by these mice are due to any interference of the mutated locus with potential lupus susceptibility genes.
Our results raise the possibility that IL-4 and STAT6 may be directly involved in the development of lupus nephritis, particularly glomerulosclerosis and chronic renal fibrosis. IL-4 is known to promote fibroblast proliferation, collagen gene expression, and collagen synthesis in mouse models of pulmonary fibrosis (39–41). Another type 2 cytokine, IL-13, which is also significantly increased in NZM.2410 mice as compared with BALB/c mice (our unpublished data), can increase type 1 procollagen synthesis in vitro (42). Furthermore, IL-4 serves as a growth factor for cells that secrete TGF-β (7), a cytokine known to cause tissue fibrosis (43). IL-4 transgenic mice exhibit increased renal TGF-β expression and develop glomerulosclerosis, which is independent of Ig deposition (44). Studies are underway to examine these possibilities.
Finally, the cause of dysregulated IL-4 expression in NZM.2410 mice is not known. NZM.2410 mice have significantly increased numbers of IL-4-secreting CD4+ T cells (our unpublished data), which may be committed to produce IL-4 through a genetic regulation (45). Also, increased IL-4 expression may be regulated at the level of CD1d-restricted T cells (46), as CD1d null NZM.2410 mice have decreased IL-4 production (our unpublished data). Thus, CD1d-regulated events may contribute, at least in part, to the elevated IL-4 levels in NZM.2410 mice.
In summary, there may be several distinct immune pathways that can lead to the development of renal disease in mice (or probably humans) who have SLE. In the MRL-lpr mice, autoantibody and immune complex deposition probably have an important, although not exclusive role in the development of glomerulonephritis, proteinuria, and loss of renal function. In the NZM.2410, it is not at all clear that autoantibody and immune complex deposition is important; IL-4 and perhaps other cytokines induced by IL-4, such as IL-13, may be acting directly on glomerular cells to induce glomerulosclerosis. Thus, the immune system would be important in both strains for the induction of autoantibody production and cytokine production, but the autoantibodies would contribute importantly to disease in the MRL-lpr, while the cytokines would contribute more directly to disease in the NZM.2410. If these differences can be observed in mouse models of SLE, they may also subset human SLE patients, with the implication that different subsets of patients might have different prognoses and benefit from different therapies.
Acknowledgments
We thank Michael Grusby for providing STAT4−/− and STAT6−/− 129/C57BL mice; David Adams, Gurjit Khurana-Hershey, and Marsha Wills-Karp for critical reading of the manuscript; Suzanne Morris for helpful suggestions; and Pam Groen, Lisa McMillin, Tatyana Orekhova, Kathy Saalfeld, Neeraj Singh, and Ying Zhao for technical help.
Footnotes
This work was supported in part by grants from the National Institutes of Health (AR47322 and AR47363P&F to R.R.S.) and the Arthritis Foundation (to C.O.J.).
Abbreviations used in this paper: SLE, systemic lupus erythematosus; KBS, kidney biopsy score; NZB, New Zealand Black; NZM, New Zealand Mixed; NZW, New Zealand White; RT, room temperature.
References
- 1.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 2.Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345:340. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- 3.Ohnishi K, Ebling FM, Mitchell B, Singh RR, Hahn BH, Tsao BP. Comparison of pathogenic and nonpathogenic murine antibodies to DNA: antigen binding and structural characteristics. Int Immunol. 1994;6:817. doi: 10.1093/intimm/6.6.817. [DOI] [PubMed] [Google Scholar]
- 4.Theofilopoulos AN, Lawson BR. Tumor necrosis factor and other cytokines in lupus. Ann Rheum Dis. 1999;58:149. doi: 10.1136/ard.58.2008.i49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coffman RL, Seymour BW, Lebman DA, Hiraki DD, Christiansen JA, Shrader B, Cherwinski HM, Savelkoul HFJ, Finkelman FD, Bond MW, Mosmann TR. The role of helper T cell products in mouse B cell differentiation and isotype regulation. Immunol Rev. 1988;102:5. doi: 10.1111/j.1600-065x.1988.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 6.Illera VA, Perandones CE, Stunz LL, Mower DJ, Ashman RF. Apoptosis in splenic B-lymphocytes: regulation by protein kinase C and IL-4. J Immunol. 1993;151:2965. [PubMed] [Google Scholar]
- 7.Seder RA, Marth T, Sieve MC, Strober W, Letterio JJ, Roberts AB, Kelsall B. Factors involved in the differentiation of TGF-β-producing cells from näive CD4+ T cells: IL-4 and IFN-γ have opposing effects, while TGF-β positively regulates its own production. J Immunol. 1998;160:5719. [PubMed] [Google Scholar]
- 8.Prud’homme GJ, Piccirillo CA. The inhibitory effects of transforming growth factor-β1 (TGF-β1) in autoimmune diseases. J Autoimmun. 2000;14:23. doi: 10.1006/jaut.1999.0339. [DOI] [PubMed] [Google Scholar]
- 9.Kelley VR, Wuthrich RP. Cytokines in the pathogenesis of systemic lupus erythematosus. Semin Nephrol. 1999;19:57. [PubMed] [Google Scholar]
- 10.Sims PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 11.Lin LC, Chiang Y, Chou C, Hsieh K, Chiang BL. Dysregulation of T helper cell cytokines in autoimmune-prone NZB × NZW F1 mice. Scand J Immunol. 1995;42:466. doi: 10.1111/j.1365-3083.1995.tb03681.x. [DOI] [PubMed] [Google Scholar]
- 12.Shirai A, Conover J, Klinman DM. Increased activation and altered ratio of interferon-γ:interleukin-4 secreting cells in MRL-lpr/lpr mice. Autoimmunity. 1995;21:107. doi: 10.3109/08916939508993357. [DOI] [PubMed] [Google Scholar]
- 13.McMurray RW, Hoffman RW, Nelson W, Walker SE. Cytokine mRNA expression in the B/W mouse model of systematic lupus erythematosus: analyses of strain, gender and age effects. Clin Immunol Immunopathol. 1997;84:260. doi: 10.1006/clin.1997.4390. [DOI] [PubMed] [Google Scholar]
- 14.Funauchi M, Ikoma S, Enomoto H, Horiuchi A. Decreased Th1-like and increased Th2-like cells in systemic lupus erythematosus. Scand J Rheumatol. 1998;27:219. doi: 10.1080/030097498440859. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto S, Tokano Y, Kaneko H, Nozawa K, Amano H, Hashimoto H. The increased interleukin-13 in patients with systemic lupus erythematosus: relations to other Th1-, Th2-related cytokines and clinical findings. Autoimmunity. 2001;34:19. doi: 10.3109/08916930108994122. [DOI] [PubMed] [Google Scholar]
- 16.Santiago ML, Fossati L, Jacquet C, Muller W, Izui S, Reininger L. Interleukin-4 protects against a genetically linked lupus like syndrome. J Exp Med. 1997;185:65. doi: 10.1084/jem.185.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erb KJ, Beate R, Brevern V, Ryffel B, Schimpl A, Rivett K. Constitutive expression of interleukin-4 (IL-4) in vivo causes autoimmune-type disorders in mice. J Exp Med. 1997;185:329. doi: 10.1084/jem.185.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng SL, Moslehi V, Craft J. Role of interferon-γ and interleukin-4 in murine lupus. J Clin Invest. 1997;99:1936. doi: 10.1172/JCI119361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kono DH, Balomenos D, Pearson DL, Park MS, Hilderbrand B, Hultman BT, Pollard KM. The prototypic Th2 autoimmunity induced by mercury is dependent on IFN-γ and not Th1/Th2 imbalance. J Immunol. 1998;16:234. [PubMed] [Google Scholar]
- 20.Kono DH, Balomenos DM, Park S, Theofilopoulos AN. Development of lupus in BXSB mice is independent of IL-4. J Immunol. 2000;164:38. doi: 10.4049/jimmunol.164.1.38. [DOI] [PubMed] [Google Scholar]
- 21.Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice: origins and initial characterization. Lab Invest. 1993;68:419. [PubMed] [Google Scholar]
- 22.Finkelman FD, Morris SC. Development of an assay to measure in vivo cytokine production in the mouse. Int Immunol. 1999;11:1811. doi: 10.1093/intimm/11.11.1811. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Murphy KM. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat) 3 and Stat4. J Exp Med. 1995;181:1755. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 25.Shimoda K, Van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali BA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 26.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signaling. Nature. 1996;380:627. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 28.Singh RR, Hahn BH, Sercarz EE. Neonatal peptide exposure can prime T cells, and upon subsequent immunization induce their immune deviation: implications for antibody vs. T cell-mediated autoimmunity. J Exp Med. 1996;183:1613. doi: 10.1084/jem.183.4.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh RR, Kumar V, Ebling FM, Southwood S, Sette A, Sercarz EE, Hahn BH. T-cell determinants from autoantibodies to DNA can upregulate systemic autoimmunity in murine SLE. J Exp Med. 1995;181:2017. doi: 10.1084/jem.181.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan G, Singh RR. Vaccination with minigenes encoding V(H)-derived major histocompatibility complex class I-binding epitopes activates cytotoxic T cells that ablate autoantibody-producing B cells and inhibit lupus. J Exp Med. 2002;196:731. doi: 10.1084/jem.20020223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor β1. J Exp Med. 2001;17:809. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGaha T, Saito S, Phelps RG, Gorden R, Noben-Trauth N, Paul W, Bona C. Lack of skin fibrosis in tight skin (TSK) mice with targeted mutation in the interleukin-4Rα and transforming growth factor-β genes. J Invest Dermatol. 2001;116:136. doi: 10.1046/j.1523-1747.2001.00217.x. [DOI] [PubMed] [Google Scholar]
- 33.Snapper CM, Finkelman FD, Paul WE. Differential regulation of IgG1 and IgE synthesis by interleukin 4. J Exp Med. 1988;167:183. doi: 10.1084/jem.167.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mori M, Morris SC, Orekhova T, Marinaro M, Giannini E, Finkelman FD. IL-4 promotes the migration of circulating B cells to the spleen and increases splenic B cell survival. J Immunol. 2000;164:5704. doi: 10.4049/jimmunol.164.11.5704. [DOI] [PubMed] [Google Scholar]
- 35.Morris SC, Gause WC, Finkelman FD. IL-4 suppression of in vivo T cell activation and antibody production. J Immunol. 2000;164:1734. doi: 10.4049/jimmunol.164.4.1734. [DOI] [PubMed] [Google Scholar]
- 36.Nakajima A, Hirose S, Yagita H, Okumara K. Roles of IL-4 and IL-12 in the development of lupus in NZB/W F1 mice. J Immunol. 1997;158:1466. [PubMed] [Google Scholar]
- 37.Wakeland EK, Liu K, Graham RR, Behrens TW. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397. doi: 10.1016/s1074-7613(01)00201-1. [DOI] [PubMed] [Google Scholar]
- 38.Morel L, Mohan C, Yu Y, Schiffenbaur J, Rudofsky UH, Tian N, Longmate JA, Wakeland EK. Multiplex inheritance of component phenotypes in a murine model of lupus. Mamm Genome. 1999;10:176. doi: 10.1007/s003359900964. [DOI] [PubMed] [Google Scholar]
- 39.Gillery P, Fertin C, Nicolas JF, Chasang F, Kalis B, Banchereau J, Maquart FX. Interleukin-4 stimulates collagen gene expression in human fibroblast monolayer cultures: potential role in fibrosis. FEBS Lett. 1992;302:231. doi: 10.1016/0014-5793(92)80448-p. [DOI] [PubMed] [Google Scholar]
- 40.Postlethwaite AE, Holness MA, Katai H, Raghow H. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin-4. J Clin Invest. 1992;90:1479. doi: 10.1172/JCI116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors: role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J Immunol. 1994;152:3606. [PubMed] [Google Scholar]
- 42.Richter A, Puddicombe SM, Lordan JL, Bucchieri F, Wilson SJ, Djukanovic R, Dent G, Holgate ST, Daveis DE. The contribution of IL-4 and IL-13 to the epithelial-mesenchymal trophic unit in asthma. Am J Respir Cell Mol Biol. 2001;25:385. doi: 10.1165/ajrcmb.25.3.4437. [DOI] [PubMed] [Google Scholar]
- 43.Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thargeirsson SS. Hepatic expression of mature transforming growth factor β1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci USA. 1995;92:2572. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruger BM, Erb KJ, He Y, Lane JM, Davis PF, Hasan Q. Interleukin-4 transgenic mice develop glomerulosclerosis independent of immunoglobulin deposition. Eur J Immunol. 2000;30:2698. doi: 10.1002/1521-4141(200009)30:9<2698::AID-IMMU2698>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 45.Bix M, Wang ZE, Thiel B, Schork NJ, Locksley RM. Genetic regulation of commitment to interleukin 4 production by a CD4+ T cell-intrinsic mechanism. J Exp Med. 1998;188:2289. doi: 10.1084/jem.188.12.2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh N, Hong S, Scherer DC, Scherer I, Burdin N, Kronenberg M, Koezuka Y, Van Kaer L. Activation of NKT cells by CDld and α-galactosylceramide directs conventional T cells to the acquisition of Th2 phenotype. J Immunol. 1999;163:2373. [PubMed] [Google Scholar]