

Abstract
Polymerase chain reaction (PCR)-based assays can target either DNA (the genome) or RNA (the transcriptome). Targeting the genome generates robust data that are informative and, most importantly, generally applicable. This is because the information contained within the genome is context-independent; i.e., generally, every normal cell contains the same DNA sequence—the same mutations and polymorphisms. The transcriptome, on the other hand, is context-dependent; i.e., the mRNA complement and level varies with physiology, pathology, or development. This makes the information contained within the transcriptome intrinsically flexible and variable. If this variability is combined with the technical limitations inherent in any reverse-transcription (RT)-PCR assay, it can be difficult to achieve not just a technically accurate but a biologically relevant result. Template quality, operator variability, the RT step itself, and subjectivity in data analysis and reporting are just a few technical aspects that make real-time RT-PCR appear to be a fragile assay that makes accurate data interpretation difficult. There can be little doubt that in the future, transcriptome-based analysis will become a routine technique. However, for the time being it remains a research tool, and it is important to recognize the considerable pitfalls associated with transcriptome analysis, with the successful application of RTPCR depending on careful experimental design, application, and validation.
Keywords: Transcriptome, gene expression, RNA, reverse transcription, PCR
Functional genomics explores the role of the numerous ligand, receptor, and signaling networks that converge on transcriptional regulation and that are essential to the understanding of the molecular and mechanistic details of these complex events at the level of the individual tissue or cell. One consequence of this focus is the prominence afforded to techniques that permit transcriptome analysis; the aim is to ascribe functional significance to expression signature changes revealed between tissues, disease states, or following treatment. While high-throughput microarray analysis constitutes the sledgehammer that permits large-scale analysis of expression patterns, the reverse transcription (RT) followed by the polymerase chain reaction (PCR) represents the forceps that affords the sensitivity necessary to validate its findings for individual genes.
Reverse-transcription polymerase chain reaction remains the most sensitive technique for the detection of often-rare mRNA targets, and its application in a real-time setting has become the most popular method of quantitating steady-state mRNA levels.1 However, it has also become clear that while the use of real-time assays has addressed some of the problems associated with conventional, gel-based RT-PCR assays, it has also introduced new challenges that must be appreciated and dealt with, if data are to be reported in a biologically relevant way.2 Areas that require critical consideration are the standardization of quantitative RT-PCR (qRT-PCR) protocols3; attention to and consistency with regards to reagents used4,5; and the careful consideration of assay design, template preparation, and analytical methods.6 This latter point, which includes the analysis, reporting, and interpretation of real-time data, is of particular importance when the aim is the quantification of very low copy number targets—for example, when extracting mRNA from tiny biopsies such as those derived from colonoscopies, single cells, or laser-capture microdissected samples. Unfortunately, in these circumstances, qRT-PCR data may be used in an inappropriate manner to support conclusions that are not reliably related to the actual results obtained.
QUALITY OF RNA
Sample acquisition and purification of its RNA mark the initial step of every qRT-PCR assay, and the quality of the template is arguably the most important determinant of the reproducibility and biological relevance of subsequent qRT-PCR results. Any problems that affect reproducibility, and hence the relevance of results, are likely to have originated here.7 Many samples, especially biopsies of human tissue, are unique; hence, a wasted nucleic acid preparation means that the opportunity to record data from that sample is irretrievably lost. A separate consideration concerns the waste of money, as one of the distinguishing features of real-time PCR assays is their outrageous running cost. It is therefore prudent to expend extensive efforts on getting every stage of this process absolutely right, starting with consistency when collecting, transporting, and storing samples. This continues with rigorous adherence to protocols when extracting nucleic acids and with the appropriate storage of purified material; continued care must be exercised every time the sample is taken out of storage for analysis.
Unlike DNA, which is as tough as old boots, RNA is extremely delicate once removed from its cellular environment. Therefore, its purification is much trickier than that of DNA and a template suitable for inclusion in an RT-PCR assay must fulfill the following criteria:
It must be of the highest quality if quantitative results are to be relevant.
It should be free of DNA, especially if the target is an intronless gene.
There must be no copurification of inhibitors of the RT-step.
It must be free of nucleases for extended storage.
The most obvious problem concerns the degradation of the RNA and this is best addressed by insisting that every RNA preparation is rigorously assessed for quality. The assessment of RNA integrity by inspection of the 28S and 18S ribosomal RNA bands using gel electrophoresis is a cumbersome, low-throughput method and requires significant amounts of precious RNA. We recommend the use of a system such as Agilent’s (Palo Alto, CA) RNA LabChip and 2100 Bioanalyzer (Fig. 1). Certainly, in terms of routinely analyzing large numbers of RNA preparations, it is by far the most convenient and objective way of assessing the quality of RNA. Clearly, there is little point in observing significant differences between samples if these differences are simply due to one sample being degraded. The importance of using high-quality RNA is demonstrated by the results shown in Figure 2.
FIGURE 1.

RNA quality assessment. These plots and electropherogram show 12 RNA preparations extracted from fresh human colonic biopsies. The quality of these preparations, as judged by the absence of any bands other than the 28S, 18S, and 5S rRNA, is very high.
FIGURE 2.

Importance of assaying high-quality RNA. A: RNA was extracted from 19 fresh colonic biopsies using Qiagen RNeasy columns (Crawley, UK) and analyzed on the Agilent Bioanalyzer using the RNA LabChip. The intact RNA preparation on the left shows the 18S and 28S rRNA peaks as well as a small amount of 5S RNA. Degradation of the RNA sample on the right produces a shift in the RNA size distribution toward smaller fragments and a decrease in fluorescence signal as dye intercalation sites are destroyed. Where such analysis revealed degradation, RNA was re-extracted from the same sample. Real-time RT-PCR assays were carried out for seven target genes and for each sample the copy number obtained from intact RNA was divided by the copy number obtained from the degraded RNA. If RNA quality was irrelevant, the ratio of the two would be expected to be close to 1, since these are identical samples. If RNA quality was important, the ratio should be greater than 1, since the copy number calculated from the intact RNA should be higher than the copy number from the degraded RNA. B: The result shows clearly that RNA quality does matter, for some genes (e.g., IGF-I) more than for others (IGF-IR). The anomalous result obtained for GH may be due to the secondary structure of its mRNA, which is reduced by degradation, thus making it more accessible to priming. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; PCNA, proliferating cell nuclear antigen; GH, growth hormone; IGF-1, insulin-like growth factor-1; 1α-OH, 1α-hydroxylase; IGF-1R, insulin-like growth factor-1receptor.
A second question relates to the presence of inhibitors in template RNA preparations. There are numerous components within blood and tissue that can inhibit RT-PCR assays. Mammalian blood, especially the heme compound,8 is well known for containing inhibitors of the PCR assay,9,10 with as little as 1% v/v blood inhibiting Taq polymerase.11 Humic acid is an inhibitor of PCR reactions carried out on samples extracted from soil,12 and inhibitors are present in food,13 with calcium an important culprit.14 One important aspect of any inhibition of the PCR assay is that this may compromise PCR as a diagnostic tool. For example, chain-terminating drugs, such as acyclovir used in the treatment of retro viruses, inhibit Taq DNA polymerase, producing a false negative result in some patients.15 High levels of copurified RNA can also result in failure of the PCR assay.16 Culture media, components of nucleic extraction reagents,13 and even the use of wooden toothpicks to pick bacterial colonies have been reported as inhibiting the PCR reaction.17 Last but not least, inhibitors can be selective: Skeletal muscle has been reported to contain inhibitors that inhibit one polymerase—e.g., Taq, but not Thermus thermophilus polymerase.18 Figure 3 shows an example of inhibition of an RT-PCR assay by DNA.
FIGURE 3.

Assessment of inhibitors in RNA preparations. A test mastermix was prepared using the Brilliant 1-step qRT-PCR mastermix (Stratagene), to which a plant gene sense strand amplicon, primers (200 nM), and FAM-BHQ-labeled TaqMan probe (500 nM) (Applied Biosystems, Warrington, Cheshire, UK) were added. Total RNA was prepared from fresh colonic biopsies (5 mg) using Qiagen RNeasy columns and resuspended in 80 μL of Tris-Cl/EDTA buffer. RNA (100 ng) was added to the test mastermix and a one-tube RTPCR assay (final volume 25 μL, 10 hr RT at 50°C, 40 cycles of 30 min at 70°C) was carried out on a Stratagene MX-4000 real-time PCR instrument (white bars). Seven control amplifications containing water rather than RNA (black bars) were set up and run at the same time. Most of the RNA samples recorded the same Ct values as the water controls, and all but three were within 1 Ct of the control. However, one sample did record a significantly higher Ct, suggesting that this preparation contained a contaminant. Upon dilution, this sample recorded the same Ct as the control samples (not shown). Ct, threshold cycle.
Clearly, there is little point in recording spurious differences in mRNA levels that are based simply on the presence of inhibitors in the different templates affecting either the RT or the PCR assay. One way of avoiding this is to test each RNA preparation for inhibitors by amplifying an amplicon set that has no sequence identity with any known sequence within the target RNA. For example, if one is investigating human gene expression, a plant or artificial amplicons could be used to test each RNA preparation for inhibitors. Practically, this involves preparing a mastermix that includes the plant or artificial amplicon, both primers, and the specific probe set. A benchmark Ct (threshold cycle) that is characteristic for that assay in the absence of any inhibitor is recorded by adding water to that mastermix (the “no added template” control). This acts as a reference point for Ct values obtained when the water is substituted with RNA prepared from cells, biopsies, or body fluids. In the absence of inhibitor, the Ct remains the same; in the presence of inhibitor, the Ct increases. This is illustrated in Figure 3.
It might be thought that the use of a reference gene as an endogenous control can also identify the presence of inhibitors. This may well be possible for experiments involving RNA extracted from tissue culture cells, although one would have to show that the particular reference gene used is not affected by experimental conditions. However, for experiments involving biopsies, the problem with this approach is that the mRNA levels of reference genes vary significantly between different individuals and tissues. Without a priori knowledge of mRNA levels in a particular tissue, it is not possible to determine whether a particularly low Ct is caused by an inhibitor or by low levels of that particular mRNA in that sample. Therefore, we do not recommend the use of reference genes for this purpose.
Historically, so-called housekeeping genes, believed to be constitutively expressed and minimally regulated, have been used widely as internal RNA references for Northern blotting, RNAse protection, and qualitative RT-PCR analyses. They remain widely used as reference genes (endogenous controls) for quantitative analysis in real-time RT-PCR assays, usually without any real investigation as to how invariant their mRNA levels really are under the experimental conditions being investigated. A recent systematic analysis and comparison of their usefulness on in vivo tissue biopsies has concluded that a single housekeeping gene should not be used for normalization.19 It seems reasonable to assume that most genes are regulated and that this will cause significant unpredictable differences in their expression patterns between and even within the same individual. If housekeeping genes are to be used, they must be validated for the specific experimental setup and it is probably necessary to choose more than one—as was done, for example, for expression profiling of T helper cell differentiation.20 The problems associated with the selection of appropriate reference genes were described recently in a clear and authoritative manner, wherein the authors recommended using the geometric mean of multiple, carefully selected reference genes for normalization.21 These authors helpfully provide a program that aids in selecting the most suitable reference genes.
SPECIFIC VERSUS NONSPECIFIC CHEMISTRIES
There are two types of homogeneous fluorescent reporting chemistries: nonspecific detection and specific detection.
Nonspecific Detection
Nonspecific detection uses intercalating dyes such as SYBR Green that bind to any double-stranded DNA generated during the PCR reaction and emit enhanced fluorescence.22 These are simply added as a reagent to the PCR cocktail of standard reactions and, although intrinsically nonspecific, can yield quasitemplate specific data if DNA melt curves are used to identify specific amplification products.23 Assays using DNA-binding dyes have two advantages over probe-based ones: (1) they can be incorporated into optimized and long-established protocols that use legacy primers and experimental conditions, and (2) they are significantly cheaper, as there is no probe-associated cost. This makes them very useful for optimizing a PCR reaction; for example, when testing any interaction between the primers by melt curve analysis, and carrying out initial, exploratory screens of multiple amplicons before using a probe-based protocol. Indeed, despite the nonspecific nature of amplification detection, DNA-binding dye-based assays need not be less reliable than probe-based assays. Interestingly, there is at least one report that suggests that SYBR Green I detection is more precise and produces a more linear decay plot than TaqMan detection.24 Disadvantages include their indiscriminate binding to any double-stranded DNA, which can result in fluorescence readings in the “no template controls” (NTC) due to dye molecules binding to primer dimers. This can be minimized by using separate RT and PCR steps.
A second problem is that since this assay is no more specific than conventional PCR, the use of melt curves is obligatory, thus adding to the complexity of data analysis. A third drawback is that multiple dye molecules bind to a single amplified molecule and consequently the amount of signal generated following irradiation is dependent on the mass of double-stranded DNA produced in the reaction. Assuming the same amplification efficiencies, amplification of a longer product will generate more signal than a shorter one. If amplification efficiencies are different, quantification will be even more inaccurate.
Specific Detection
Template-specific analysis requires the design and synthesis of one or more custom-made fluorescent probes for each PCR assay. Most reporting systems utilize fluorescent resonance energy transfer (FRET) or similar interactions between donor and quencher molecules as the basis of detection. The types of reporters used for these probes include fluorescein, rhodamine, and cyanine dyes, and derivatives thereof; some also have either fluorescent or nonfluorescent acceptors on the same or on a complementary molecule. There is a huge selection of fluorescent dyes, mainly because the chemistries for label incorporation into nucleic acid probes are well developed since they are used in other molecular biology procedures such as DNA sequencing. All chemistries follow the same principle: A fluorescent signal is only generated if the amplicon-specific probe hybridizes to its complementary target. In addition, some probes may also be used in melt-point analyses to provide additional identification of amplified product. The main advantage of specific chemistries is that specificity no longer resides in the primers; instead, the use of a probe introduces an additional level of specificity. Nonspecific amplification due to mispriming or primer–dimer artifacts does not generate a signal and is ignored by the fluorescence detector. This obviates the need for post-PCR Southern blotting, sequence analysis, or melt curves to confirm the identity of the amplicon. Another advantage over intercalating dyes is that the probes can be labeled with different, distinguishable reporter dyes that allow the detection of amplification products from several distinct sequences in a single PCR reaction (multiplex). However, the absence of detection is not the same as the absence of artifacts, and nonspecific amplification can, and indeed does, affect amplification efficiency and any subsequent quantification. The major disadvantage is that because of its specificity, artifacts that interfere with amplification efficiency cannot be detected. Therefore, intercalating dyes should be used to optimize primers and reaction conditions prior to any quantification experiments to ensure the absence of amplification artifacts. Another disadvantage is the cost associated with these chemistries: Each target requires its own specific probe. This becomes particularly painful when quantifying multiple targets, as costs escalate very rapidly.
LINEARITY OF THE REVERSE-TRANSCRIPTION STEP
One of the key advantages of real-time PCR assays is their wide dynamic range, which allows the researcher to compare Ct values obtained from samples containing hugely different levels of DNA. The difference between a PCR assay and an RT-PCR assay is that the latter reaction can be initiated in three different ways, which of course has the potential to result in variable results. cDNA priming can be carried out using random primers, oligo-dT, or target-specific primers. Each of the three methods differ significantly with respect to cDNA yield and variety as well as specificity and, since the choice of primer can cause marked variation in calculated mRNA copy numbers,25 the implications of using any particular method should be considered carefully.26 It is worth pointing out that the melting temperature of both random primers and oligo-dT is well below the optimum temperature of thermostable RTs; hence, neither can be used with thermostable RT enzymes without some low-temperature preincubation step or primer modification (e.g., locked nucleic acid substitution of a nucleotide27).
Ambion have shown that unintended endogenous priming can occur regardless of which primers are used to prime the RT reaction. Using 32P-labeled avian myeloblastosis virus (AMV), Moloney murine leukemia virus (MMLV), and RNaseH−MMLV reverse transcriptases, they performed standard RT reactions with and without primers. They found that the resulting products were identical, and concluded that the cDNA generated in the RT reactions was the result of endogenous random priming (http://www.ambion.com/catalog/CatNum.php?1740). Such nonspecific priming can lead to lowered and/or variable signal in the subsequent PCR assay, although how much of a problem this is in real life remains unclear. Not surprisingly, Ambion’s EndoFree RT kit addresses this problem.
Random primers prime RT at multiple points along the transcript, hence producing more than one cDNA transcript per original target. Thus this method is by definition nonspecific, but yields the most cDNA and is most useful for transcripts with significant secondary structure. First-strand cDNA synthesis with random primers should be conducted at room temperature. However, the majority of cDNA synthesized from total RNA will be ribosomal RNA-derived. This could create real problems if the target of interest is present at low levels, as it may not be primed effectively by random primers and its amplification may not be quantitative.
Indeed, it has been demonstrated that random hexamers can overestimate mRNA copy numbers by up to 19-fold compared with a sequence-specific primer.25 It has been described as the least reliable method of priming cDNA26; nevertheless, as with any experimental protocol, this random priming of cDNA can yield reliable and reproducible results if it is carried out in a careful, competent manner. One added advantage of random priming is that it generates the least bias in the resulting cDNA.6
cDNA synthesis using oligo-dT is more specific to mRNA than random priming, as it will not transcribe rRNA. It can struggle to generate transcripts from mRNAs with significant secondary structure, and obviously it will not prime any RNAs that lack a polyA tail, e.g., those specifying histones or viral RNAs. However, since oligo-dT priming requires very high-quality RNA that is full length, it is not a good choice for transcribing RNA that is likely to be fragmented, such as that typically obtained from laser capture microdissected tissue or from archival material. Furthermore, the RT may fail to reach the primer probe binding site if secondary structures exist that impede its processivity or if the primer/probe binding site is at the extreme 5′-end of a long mRNA. This may be the case if the mRNA contains a very long untranslated 3′-region or if splice variants differ at the 5′-end of the mRNA (e.g., the MHC class II transactivator isoforms I, III, and IV).
Target-specific primers synthesize the most specific cDNA and, all things being equal are probably the most sensitive option for quantification.26 The main disadvantage of this method is that it requires separate priming reactions for each target; hence it is not possible to return to the same preparation and amplify other targets at a later stage. It is also wasteful if only limited amounts of RNA are available.
In our experience, the use of target-specific oligonucleotides to prime cDNA gives superior results to using random primers. In particular, we find that a reaction primed by target-specific primers is linear over a wider range than a similar reaction primed by random primers. This is illustrated in Figure 4. However, there does appear to be gene-specific variation and, as always, it is important to validate individual assays using standard curve dilutions before coming to conclusions about results obtained from actual samples. As always, dogma is the enemy of progress, and a properly validated, executed, analyzed, and interpreted real-time RT-PCR assay carried out using random primers is infinitely preferable to a poorly designed, hastily executed, inappropriately analyzed and gene-specific primed assay.
FIGURE 4.
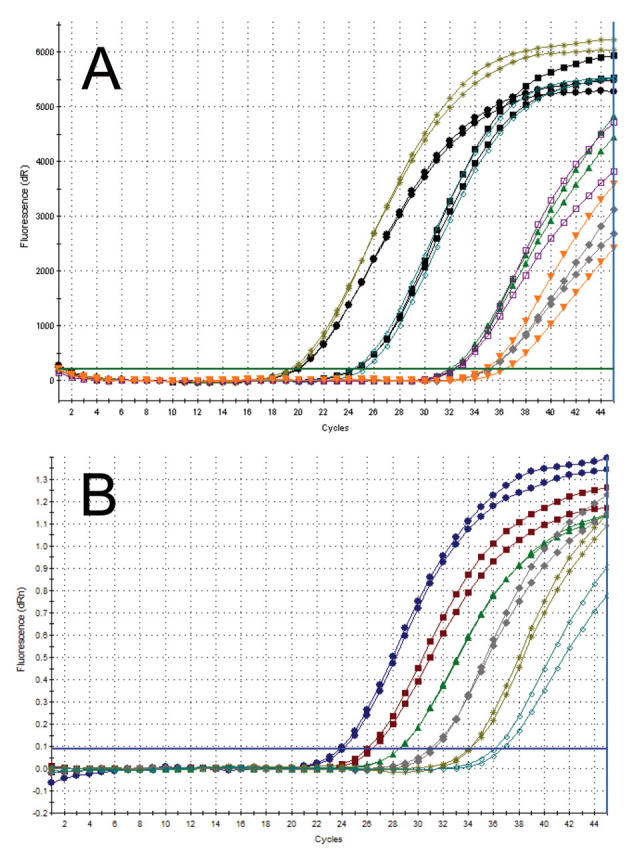
Comparison of RT-PCR assays primed using random (A) or target-specific (B) primers. Both reactions were carried out using the Brilliant 2-step RT-PCR kit. The only difference was that assay A was primed using random nonamers primers (Applied Biosystems), whereas assay B was primed using a target-specific primer (Proligo, Paris, France). In each case, 100 ng of total RNA was subjected to 10-fold (A) or 5-fold (B) serial dilutions and reverse transcribed using standard RT conditions as specified by the manufacturer. One tenth of each cDNA preparation was then included in a PCR assay. dR, baseline-corrected raw fluorescence; dRn, baseline-corrected normalized fluorescence.
The resolving power of RT-PCR is also limited by the efficiency of RNA-to-cDNA conversion, which depends on the enzyme used. However, the conversion efficiency is significantly (greater than 3-fold) lower when target templates are rare and it is negatively affected by nonspecific or background RNA present in the RT reaction.28 Of course, considerations of linearity of the RT step are just one side of the equation. Another consideration concerns the “Monte Carlo” effect, an inherent limitation of PCR amplification from small amounts of any complex template due to differences in amplification efficiency between individual templates in an amplifying cDNA population.29 Every template has a certain probability of being amplified or being lost and, once diluted past a certain threshold, copy number will display large variations in amplification. The Monte Carlo effect is dependent upon template concentration: The lower the abundance of any template, the less likely its true abundance will be reflected in the amplified product. One model for this phenomenon considers primer annealing to any individual template molecule during each PCR cycle as a random event. Under conditions of primer excess, the probability of primer annealing is dependent upon annealing temperature, annealing time, and the number of available templates. If the number of molecules of a particular template is limiting, then that template within a complex mixture will have slight and random differences in amplification efficiencies depending upon whether the primers were able to anneal. If these differences occur early in the PCR assay, large variations in final product concentration can be produced during the exponential phase of the amplification reaction. cDNAs of lower abundance will be more likely to experience the Monte Carlo effect, since their probability of primer annealing is lower.
Unfortunately, this situation is difficult to resolve, since many experiments are designed to identify very low target mRNAs. One solution is to use mRNA, rather than total RNA preparations. This may improve primer-binding efficiency, as it would reduce significantly the complexity and quantity of unrelated template present during primer/target annealing. However, preparation of mRNA involves additional steps, may lead to the loss of some mRNA, and it is more difficult to assess the quality of the final product. Nevertheless, if ultimate sensitivity is the main consideration, the use of mRNA may be advisable. In addition, all assays quantitating very low target copy numbers should be run in triplicate and be repeated at least once, so that any problems with reproducibility become immediately apparent. Nevertheless, it is worth emphasising that real-time RT-PCR, like any other assay, will not generate quantitative results at the limits of its sensitivity. One of the major advantages of including a standard curve with every run is that its highest dilutions provide an immediate benchmark for the assessment of the quality of the results obtained from the unknown samples. The highest dilution of the standard curve to report consistently concordant Ct values delineates the lowest copy number that can be quantitated with confidence. If the Ct values recorded by any unknowns translate into copy numbers lower than that benchmark, they should be recorded as qualitative (yes/no) results.
DATA ANALYSIS
The Ct has become the parameter most conveniently and most frequently quoted when reporting qRT-PCR results. However, it is important to consider carefully what the Ct actually reveals and to ask whether quoting a Ct is sufficiently informative to allow a confident assessment of any conclusion drawn from a real-time RT-PCR experiment.
The threshold cycle (Ct) is defined as the cycle when sample fluorescence exceeds a chosen threshold above calculated background fluorescence. The critical word is “chosen,” since background fluorescence is not a constant or absolute value but is influenced by changing reaction conditions. Hence, if background fluorescence varies, the value of a Ct recorded for any particular sample is also going to be variable. Since the Ct is central to an appropriate understanding of the real-time assay, and at the same time is frequently misunderstood, it is important to spell out the parameters governing its value. The Ct is at the heart of the qRT-PCR assay, as it is used to determine copy numbers, which is of course the whole point of carrying out a quantitative assay. A positive Ct (defined as a fluorescence reading of less than the final cycle number) can arise due to genuine amplification, but some Ct values are not due to genuine amplification and some genuine amplification does not record a Ct.
One important reason for a real amplification not recording a Ct is the wandering (drifting) baseline caused by an incorrectly set background cycle range. This range specifies the cycles that will be used to calculate the threshold fluorescence levels. Typically, it encompasses only early PCR cycles prior to the accumulation of significant amplification products, e.g., 3 through 15 on the Applied Biosystems PRISM 7700 or 5 through 9 on the Stratagene instruments. The background signal in all wells is used to determine the “baseline fluorescence” across the entire reaction plate. However, sometimes this does not generate an accurate background reading for that individual well. A comparison of two amplification plots shows that they have very similar ΔRn values (baseline-corrected normalized fluorescence) (0.023 vs. 0.02), but whereas one evidently crosses the default threshold, the other one remains well below it (see Fig. 5). This is because the fluorescence levels in the green well remain fairly constant throughout the early stages of the PCR assay, and start rising from approximately the same level recorded at the end of the baseline cycle. The fluorescence represented by the red line, on the other hand, drifts downwards significantly by 0.015 units, and the rise recorded following probe hydrolysis is not sufficient to allow the amplification plot to cross the default threshold.
FIGURE 5.

The wandering (drifting) baseline. A illustrates the problem of two amplification plots recording approximately the same ΔRn values, yet only one crossing the threshold established by the default baseline setting of 3–15 cycles on a PRISM 7700. B shows how an upwards adjustment of the baseline corrects for the downward drift and establishes a level playing field, allowing the sample analyzed in the red well to be recorded as a positive. ΔRn, baseline-corrected normalized fluorescence.
It is useful to use an analogy here: If two individuals, A and B, jump up in the air, it is important to ascertain that both started their jump from the same level before declaring that Jumper A can jump higher than Jumper B, even though the former has crossed the bar and the latter has not (see Fig. 5A). Similarly, it is clear from the amplification plots that it is not correct to report one well as recording a positive Ct and the other well a negative one. This is where appropriate baseline correction comes in: An adjustment of the baseline cycles to include the lowest point of the amplification plot corrects for this fluorescence drift and allows this well to record a correct Ct that is very similar to the one recorded by the green well. Using the above analogy, raising the platform of Jumper B (Fig. 5B) shows that he actually jumps higher, but, most importantly, that both jumpers have now crossed the bar.
The most valuable application of such baseline corrections is when analyzing negative controls and detecting clear evidence of amplification, which is too low to cross the default threshold level. Indeed, if these corrections result in a negative control becoming positive, this becomes the critical component of the analysis. Many instruments now provide the choice of a default adaptive baseline enhancement, which automatically calculates the best baseline for each plot individually, thereby providing the most accurate Ct.
THE THRESHOLD
The threshold calculated by the real-time instrument depends on the baseline, and the default settings are usually not altered in standard runs. However, they may need to be changed if specific conditions arise, usually linked to the high Ct and low ΔRn values associated with very low target copy numbers.
Two points are worth noting:
There need not be a single threshold for each run. For example, Applied Biosystems acknowledge that data from a single run can be analyzed with multiple threshold values and they refer to a “window or range of values within which a threshold setting will fit.”30 Indeed, the thresholds calculated by other instruments (e.g., those from Stratagene and the latest Applied Biosystems instruments) vary depending on what well or combination of wells are being analyzed.
-
The threshold must intersect the exponential phase of individual amplification plots. If the Ct values are very high and the ΔRn values are very low, there may well not be a clearly defined exponential phase of the amplification plot. In such cases it will be necessary to make threshold adjustments that generate a (qualitative) positive sample, with actual quantification quite irrelevant, as it would only generate an inaccurate copy number.
However, it is also clear from experience that multiple thresholds are the exception rather than the rule for the vast majority of runs that target medium-level mRNAs. Nevertheless, sometimes multiple thresholds are the only way that the data can be analyzed fairly; this is of particular importance when negative controls are involved. One example of when to use multiple thresholds is when there are clear signs of amplification in a negative control, and application of the default baseline and/or threshold would result in a negative Ct. Altering the threshold, or the baseline if a wandering baseline is the problem, usually corrects this technical inconsistency and allows the operator to record a positive Ct. The threshold problem is illustrated in Figure 6 using an assay that targets a low-abundance mRNA and generates very low ΔRn values.
FIGURE 6.
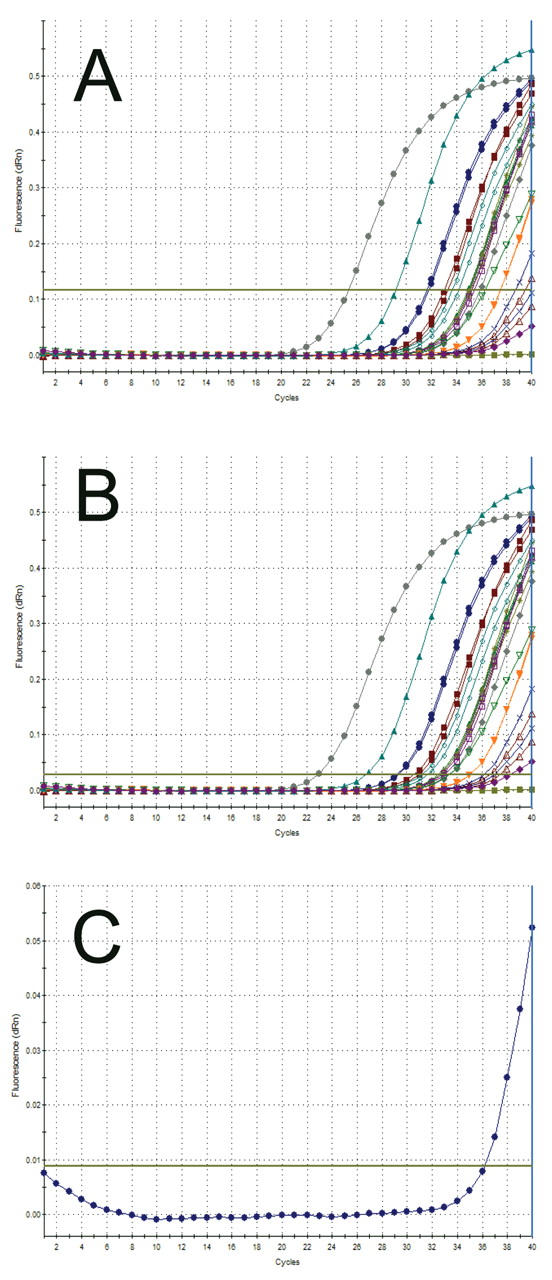
Altered thresholds alter results. A shows amplification plots from an assay that aims to detect a low copy number target and at the same time records very low ΔRn values. An analysis of all wells places the threshold at 0.12, and leaves several amplification plots, that are clearly positive below that default threshold. One of these is a “no template control” (NTC). B shows how manual reduction of the threshold allows the recording of positive threshold cycles (Ct) for all samples, including the negative control. C shows how the use of an adaptive baseline also allows the recording of a positive Ct for the NTC.
Of course, the whole question of how to interpret a positive NTC is the subject of many a heated debate. Interestingly, no guidelines have been published on this matter. Therefore, the proposals below are based on our views and are not meant to be definitive. We acknowledge that the cut-off points are arbitrary, but they represent a common-sense approach and a starting point for a discussion of this subject that needs to be carried out. In some instances, the situation is quite clear: If all unknown samples record Ct values of around, say 18–25, and the NTC records a Ct of 39, then it is legitimate to ignore the very high Ct values recorded for the NTC and use the data. However, we recommend that the fact that the NTC did record a positive Ct be noted in the results section of any publication reporting those data. The only exception to this rule would be an NTC recording a Ct less than 30, for this suggests the presence of high levels of contamination somewhere in the laboratory and assay set-up and requires urgent attention.
The situation is quite different if the values for unknown sample and NTC Ct are more comparable. Again, this is usually an issue only when quantitating RNA from single cells or from laser capture microdissected tissue, but it is a crucial one since “Caesar’s wife must be above suspicion.” We suggest that any Ct that differs by more than 5 from the NTC be regarded as probably not caused by any contaminant, especially when the replicate wells also record positive, similar Ct values. Of course, if one replicate records a positive Ct, and the other(s) is negative, then that sample must be treated with the utmost suspicion and certainly can never be called a positive. At the very least, the sample must be rerun, ideally using more template, and generate its positive Ct in the absence of any NTC contamination.
If the ΔCt separating the unknown sample from the NTC is greater than 5 Ct, that sample is as likely to have become contaminated as not and must be rerun, again using more template RNA. The rationale behind using more RNA is that for every doubling of input template, the Ct should increase by about 1. For example, if the unknown recorded a Ct of 37.5 and the NTC a Ct of 39.0, then the only acceptable result, in our opinion, would be a new Ct of around 36.5, if the amount of template RNA had been doubled. Of course, the NTC would have to record a Ct of 45. The NTC is such a crucial part of a good experimental setup that the requirement for an absolutely negative result cannot ever be compromised. Therefore, we propose that if the Ct values recorded by unknowns are above 33–35, then the NTC must always be negative for any results to be valid. Finally, if the unknowns record Ct values in the region of 37–39, it is important to run the reaction for 45 cycles, to be certain that the NTC comes up negative. Clearly, if an unknown records a Ct of 39.5, and the run ends after 40 cycles, any NTC that would have recorded a Ct of 40.01 would come up as a negative. Additional advice would be to try and redesign the assay to make it as efficient as possible, thus lowering the cycle number when the instrument first detects amplification product. The whole question of amplification efficiency is very well discussed elsewhere.31,32
Incidentally, the question of where to place the NTC and how many NTCs are required per well is also worth a brief mention. In our opinion, in a 96- or 384-well assay, NTCs should always be run in the row below and next to the lowest dilution of the standards. There should be at least two NTC controls with triplicate replicates. One NTC should be sealed prior to the addition of any unknowns, positive controls, or standard templates. The second NTC should be sealed after the addition of any unknowns, positive controls, or standard templates.
CONCLUSION
Real-time RT-PCR is extremely powerful and can generate reliable, reproducible, and biologically meaningful results. However, this brief review of some of the underlying problems should also have made it clear that great care must be taken in planning and analyzing real-time RT-PCR assays. We have barely touched on the problems of normalization and reference genes (previously known as housekeeping genes), and have not mentioned “absolute” versus relative quantification or the need for standard curves and how they should be generated. Because the reporting of Ct values alone can conceal as much as it reports, we believe it is necessary to begin a concerted effort to introduce more standard analysis and reporting procedures, as has been done for microarray technology in the establishment of the MIAME guidelines (www.mged.org/miame). Certainly, in the absence of such standards for real-time RT-PCR, it falls to the editors of journals to ensure that papers that include this technology are appropriately reviewed, and that any conclusions are rigorously supported by the actual data.
For the researcher, it is vital to consider each stage of the experimental protocol, starting with the laboratory setup and proceeding through sample acquisition, template preparation, RT, and finally the PCR step. Only if every one of these stages is properly validated is it possible to obtain reliable quantitative data. Of course, choice of chemistries, primers and probes, and instruments must be appropriate to whatever is being quantitated. Finally, data must be interpreted, and this remains a real problem. Clearly, real-time qPCR is a valuable, versatile, and powerful technique. But, like anything powerful, it needs to be treated with respect.
Acknowledgments
SAB is grateful to Bowel and Cancer Research for supporting some of the research described here.
REFERENCES
- 1.Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 2000;25:169. [DOI] [PubMed] [Google Scholar]
- 2.Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J Mol Endocrinol 2002;29:23–39. [DOI] [PubMed] [Google Scholar]
- 3.Gabert J, Beillard E, van der Velden VH, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia 2003;17:2318–2357. [DOI] [PubMed] [Google Scholar]
- 4.Wolffs P, Grage H, Hagberg O, Radstrom P. Impact of DNA polymerases and their buffer systems on quantitative real-time PCR. J Clin Microbiol 2004;42:408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeung AT, Holloway BP, Adams PS, Shipley GL. Evaluation of dual-labeled fluorescent DNA probe purity versus performance in real-time PCR. Biotechniques 2004;36:266–270, 272, 274–265. [DOI] [PubMed] [Google Scholar]
- 6.Ginzinger DG. Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp Hematol 2002;30:503–512. [DOI] [PubMed] [Google Scholar]
- 7.Bomjen G, Raina A, Sulaiman IM, Hasnain SE, Dogra TD. Effect of storage of blood samples on DNA yield, quality and fingerprinting: A forensic approach. Indian J Exp. Biol 1996;34:384–386. [PubMed] [Google Scholar]
- 8.Akane A, Matsubara K, Nakamura H, Takahashi S, Kimura K. Identification of the heme compound copurified with deoxyribonucleic acid (DNA) from bloodstains, a major inhibitor of polymerase chain reaction (PCR) amplification. J Forensic Sci 1994;39:362–372. [PubMed] [Google Scholar]
- 9.Al Soud WA, Jonsson LJ, Radstrom P. Identification and characterization of immunoglobulin G in blood as a major inhibitor of diagnostic PCR. J Clin Microbiol 2000;38:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al Soud WA, Radstrom P. Purification and characterization of PCR-inhibitory components in blood cells. J Clin Microbiol 2001;39:485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panaccio M, Lew A. PCR based diagnosis in the presence of 8% (v/v) blood. Nucleic Acids Res 1991;19:1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Int J Food Microbiol 1996;62: 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossen L, Norskov P, Holmstrom K, Rasmussen OF. Inhibition of PCR by components of food samples, microbial diagnostic assays and DNA-extraction solutions. Int J Food Microbiol 1992;17:37–45. [DOI] [PubMed] [Google Scholar]
- 14.Bickley J, Short JK, McDowell DG, Parkes HC. Polymerase chain reaction (PCR) detection of Listeria monocytogenes in diluted milk and reversal of PCR inhibition caused by calcium ions. Lett Appl Microbiol 1996;22:153–158. [DOI] [PubMed] [Google Scholar]
- 15.Yedidag EN, Koffron AJ, Mueller KH, et al. Acyclovir triphosphate inhibits the diagnostic polymerase chain reaction for cytomegalovirus. Transplantation 1996;62: 238–242. [DOI] [PubMed] [Google Scholar]
- 16.Pikaart MJ, Villeponteau B. Suppression of PCR amplification by high levels of RNA. Biotechniques 1993; 14:24–25. [PubMed] [Google Scholar]
- 17.Lee AB, Cooper TA. Improved direct PCR screen for bacterial colonies: Wooden toothpicks inhibit PCR amplification. Biotechniques 1995;18:225–226. [PubMed] [Google Scholar]
- 18.Belec L, Authier J, Eliezer-Vanerot MC, et al. Myoglobin as a polymerase chain reaction (PCR) inhibitor: A limitation for PCR from skeletal muscle tissue avoided by the use of Thermus thermophilus polymerase. Muscle Nerve 1998;21:1064–1067. [DOI] [PubMed] [Google Scholar]
- 19.Tricarico C, Pinzani P, Bianchi S, et al. Quantitative real-time reverse transcription polymerase chain reaction: Normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies 2002;309:293–300. [DOI] [PubMed] [Google Scholar]
- 20.Hamalainen HK, Tubman JC, Vikman S, et al. Identification and validation of endogenous reference genes for expression profiling of T helper cell differentiation by quantitative real-time RT-PCR. Anal Biochem 2001;299:63–70. [DOI] [PubMed] [Google Scholar]
- 21.Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time geometric averaging of multiple internal control genes. Genome Biol 2002:3: research0034.1–0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishiguro T, Saitoh J, Yawata H, et al. Homogeneous quantitative assay of hepatitis C virus RNA by polymerase chain reaction in the presence of a fluorescent intercalater. Anal. Biochem 1995;229:207–213. [DOI] [PubMed] [Google Scholar]
- 23.Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 1997;245: 154–160. [DOI] [PubMed] [Google Scholar]
- 24.Schmittgen TD, Zakrajsek BA, Mills AG, et al. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: Comparison of endpoint and real-time methods. Anal Biochem 2000;285:194–204. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Byrne CD. Differential priming of RNA templates during cDNA synthesis markedly affects both accuracy and reproducibility of quantitative competitive reverse-transcriptase PCR. Biochem J 1999;337:231–241. [PMC free article] [PubMed] [Google Scholar]
- 26.Lekanne Deprez RH, Fijnvandraat AC, Ruijter JM, Moorman AF. Sensitivity and accuracy of quantitative real-time polymerase chain reaction using SYBR green I depends on cDNA synthesis conditions. Anal Biochem 2002;307:63. [DOI] [PubMed] [Google Scholar]
- 27.Latorra D, Arar K, Hurley JM. Design considerations and effects of LNA in PCR primers. Mol Cell Probes 2003;17:253–259. [DOI] [PubMed] [Google Scholar]
- 28.Curry J, McHale C, Smith MT. Low efficiency of the Moloney murineleukemia virus reverse transcriptase during reverse transcription of rare t(8;21) fusion gene transcripts. Biotechniques 2002;32:768–770. [DOI] [PubMed] [Google Scholar]
- 29.Karrer EE, Lincoln JE, Hogenhout S, et al. In situ isolation of mRNA from individual plant cells: Creation of cell-specific cDNA libraries. Proc Natl Acad Sci USA 1995;92:3814–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Applied Biosystems: Data analysis on the ABI PRISM 7700 Sequence Detection System: Setting baselines and thresholds (http://www.appliedbiosystems.com/support/tutorials/pdf/data_analysis_7700.pdf).
- 31.Pfaffl MW, Georgieva TM, Georgiev IP, et al. Real-time RT-PCR quantification of insulin-like growth factor (IGF)-1, IGF-1 receptor, IGF-2, IGF-2 receptor, insulin receptor, growth hormone receptor, IGF-binding proteins 1, 2 and 3 in the bovine species. Domest Anim Endocrinol 2002;22:91–102. [DOI] [PubMed] [Google Scholar]
- 32.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001;29:E45. [DOI] [PMC free article] [PubMed] [Google Scholar]