

Abstract
The whole genome amplification (WGA) protocol evaluated during this study, GenomiPhi DNA amplification kit, is a novel method that is not based on polymerase chain reaction but rather relies on the highly processive and high fidelity Phi29 DNA polymerase to replicate linear genomic DNA by multiple strand displacement amplification. As little as 1 ng of genomic DNA template is sufficient to produce microgram quantities of high molecular weight DNA. The question explored during this study is whether such a WGA method is appropriate to reliably replenish and even recover depleted DNA samples that can be used for downstream genetic analysis. A series of human DNA samples was tested in our laboratory and validated using such analytical methods as gene-specific polymerase chain reaction, direct sequencing, microsatellite marker analysis, and single nucleotide polymorphism allelic discrimination using TaqMan and Pyrosequencing chemistries.
Although degraded genomic DNA is not a good template for Phi29 WGA, this method is a powerful tool to replenish depleted DNA stocks and to increase the amount of sample for which biological tissue availability is scarce. The testing performed during the validation phase of the study indicates no discernable difference between WGA samples and the original DNA templates. Thus, GenomiPhi WGA can be used to increase precious or depleted DNA stocks, thereby extending the life of a family-based linkage analysis project and increasing statistical power.
Keywords: Whole genome amplification, Phi29, strand displacement amplification
One of the challenges faced by many core facilities and research laboratories involved in genetic analysis is how to solve the problem of stock DNA limitation when additional tissue procurement is not possible. As the study progresses, these precious samples can be depleted faster than expected, resulting in missed data points and decreased statistical power. To resolve the problem of stock DNA limitation for one of our ongoing family-based linkage studies and extend the genetic analysis of precious pediatric samples, we have explored the method of whole genome amplification (WGA) developed by Amersham Biosciences (Piscataway, NJ) GenomiPhi DNA Amplification Kit). This WGA technology is based on the unique property of Phi29 DNA polymerase to replicate, under optimized buffer conditions, linear genomic DNAs by strand displacement amplification.1,2 The reaction uses random hexamers as primers for DNA synthesis and is performed routinely overnight at 30°C. Initial limited testing performed on high-quality genomic DNA isolated in our laboratory from human blood, surgical tissue, and embryonic rat tissues demonstrated that this powerful technology allowed up to 1000-fold amplification of starting material. The lowest amount of template tested (5 ng) routinely gave the best amplification yield. The quality of the genomic DNA (GP-DNA) amplified using this method was verified by microsatellite-(short tandem repeat; STR) and gene-specific amplification. The results suggested that this method produced high molecular weight GP-DNA that was representative of the original genomic DNA template.
In the present study, we evaluated the capability of this WGA technology to increase stocks of DNAs isolated from pediatric patients for which very small amounts of tissues were available. Direct sequencing and single-nucleotide polymorphisms (SNPs) analysis of the DNAs pre- and post-WGA generated identical profiles, and additional testing using pyrosequencing confirmed that this method of WGA could be used with confidence to increase DNA stocks.
To further explore the capability of this WGA method on samples of different quality, we tested a total of 64 human genomic DNA samples from our original heterogeneous and “aging” collection of DNAs for which we had depleted stocks (45 samples) or noticed poor performance (19 samples) during STR testing. Of the original 45 depleted stock samples, 36 were successfully amplified as indicated by the STR-polymerase chain reaction (PCR) profiles which were identical to that of the original template genomic DNA. The remaining 9 depleted DNA stocks failed our attempt at STR amplification even with the smallest of microsatellite markers. The successfully amplified WGA samples were used to replenish the stocks of DNAs belonging to a collection of samples used in one of our family-based linkage studies, and complete a whole-genome scan using ABI Prism Linkage Mapping Set v2.5. No Mendelian errors were detected for the successfully amplified samples, thus confirming that the Phi29 DNA polymerase demonstrate a high degree of fidelity during DNA replication.3 Additional testing of the replenished DNA cohort was performed by SNP analysis using both TaqMan chemistry and pyrosequencing, two platforms used in our laboratory for association studies. No Mendelian errors were detected in either platform. We also tested the 19 samples from this collection that consistently failed genotyping or gave low peaks for the smallest of the STR tested (thus suggesting degradation problems). None of the 19 degraded DNAs were recoverable by WGA.
METHODS
DNA Processing
Using the PureGene DNA Isolation Kit (Gentra Systems, Minneapolis, MN) genomic DNA was extracted from blood directly, or from lymphoblast cultures established from patients’ lymphocytes by Epstein Barr virus transformation. Whole genome amplification was performed using GenomiPhi DNA Amplification Kit from Amersham Biosciences following the manufacturer’s instruction. In addition, a no-template control was prepared, in which water instead of human genomic DNA was incubated overnight with Phi 29 and random primers. In some cases, the amplified GP-DNA was subjected to five successive rounds of whole genome amplification by Phi29 (GP1 to GP5). The overnight amplification process generated samples of high viscosity, which were diluted in water to facilitate downstream applications. All GP-DNAs were column purified, as recommended in the manufacturer’s protocol prior to downstream analysis, using MicroBio-Spin P-30 Tris Chromatography columns (BioRad, Hercules, CA). Briefly, the P-30 columns were centrifuged at 1000 × g for 2 min to settle the matrix and remove the flowthrough. The columns were washed once with 500 μL 1xTE and centrifuged at 1000 × g for 2 min. Sample volume was increased to 50 μL with 1xTE prior to loading onto the P-30 spin columns, to facilitate DNA purification. The DNA samples were eluted at 1000 g for 4 min.
Gene-Specific PCR
Genomic DNA, GP-DNAs, and the no-template water control were tested by PCR using primers specific for genes FGFR4 (accession number Y13901) and WDR8 (accession number NT_004321). PCR was performed with 100 ng genomic DNA in a 50-μL reaction in 1X Taq Buffer (5X Taq Buffer is 83 mM Tris-HCl pH 8.8, 83 mM (NH4)2SO4, 33.5 mM MgCl2, 850 μg/mL bovine serum album, 34 mM EDTA, 50 mM β-mercaptoethanol), 5% DMSO, 500 μM each dNTP, 1 μM of each forward and reverse primers, and 1.25 U Taq polymerase (Qiagen, Valencia, CA). PCR reactions were run on an ABI 9700 thermocycler for 30 cycles (30 sec at 94°C, 30 sec at 60°C and 1 min at 72°C). Products were detected via 2% agarose gel electrophoresis and ethidium bromide staining. All primers were purchased from Integrated DNA Technologies (Coralville, IA). The sequences of primers are as follows: WDR8 forward primer 5′Bio/CAAGGACCAGACGGTTATC, WDR8 reverse primer GATCCACTCCAGCAGTCAG; Actin forward primer TGTATGCCTCTGGTCGTACCA, Actin reverse primer ACAGAGTATTGCGCTAGGAG; FGFR4 forward primer GGTCTGCAAGAGGAAAAACAAG, FGFR4 reverse primer CGTTGATGACGATGTGCTTAGC. Molecular weight markers were purchased from Invitrogen (Carlsbad, CA).
Microsatellite Analysis
Using multicolor fluorescent dye technology, chromosome-specific microsatellite markers were amplified by single and multiplex PCR using ABI Prism Linkage Mapping Set v2.5 and optimized protocols developed in our laboratory (Picerno, Stabley, and Sol-Church, unpublished observations). The reactions were pooled using a Biomek 2000, combined with the size standard Genescan 400HD ROX (Applied Biosystem, Foster City, CA), heat denatured, and then loaded on an ABI Prism 377 and separated by gel electrophoresis. GeneScan 3.1 software was used to size the specific PCR products. Genotyper 2.1 was used for allele calling prior to statistical analysis. Mendelian errors were detected by means of the Mapmaker/Pedmanager software available at www.genome.wi.mit.edu/ftp/distribution/software/pedmanager for each STR and SNP genotype analysis.
SNP Analysis
Two methods were used with adult and pediatric sample to test GP-DNA performance under SNPs conditions.
Pyrosequencing: WDR8 (bp 3,298,379 to bp 3,351,145) was screened using 7 SNPs. Specific primer pairs were used to amplify the SNP-containing regions of gene WDR8. For each primer pair, one of the primers was biotinylated to allow subsequent immobilization of PCR products onto Streptavidine Sepharose HP (Amersham Pharmacia Biotech, Uppsala, Sweden). Specific sequencing primers, located adjacent to the SNP of interest, were then annealed to the single-stranded biotinylated template and samples subjected to pyrosequencing using the PSQ96 SNP Reagent Kit (Pyrosequencing AB, Uppsala, Sweden), following the manufacturer’s recommendation.
Assays-On-Demand: Genes ICMT, Gnb1, and CamTA1, which are located on 1p36, were screened for known SNP markers using TaqMan chemistry. Evenly spaced SNPs were selected to screen approximately 140 Kb of GNB1 locus (bp 1,655,032–1,794,701) at an average density of 1 SNP/15 Kb of DNA. Four SNPs were used to screen approximately 43 Kb of DNA containing ICMT (bp 5,372,339–5,415,006). CAMTA1 genomic region was scanned from bp 6,265,899 to bp 6,578,014 using 10 SNPs. In addition, the DNA family cohort was genotyped using SNP rs2294714 located on WDR8. Optimized Assays-on-Demand and TaqMan Universal PCR Master Mix (with UNG) were purchased from Applied Biosystems. Assays were arrayed on a 384-well optical plate using 20 ng of DNA in a 5-μL reaction volume. PCRs were performed and analyzed on an ABI 7900HT according to the manufacturer’s specification. Data were analyzed using SDS 2.1 software.
DNA Sequencing
A discrete region of the FGFR4 gene containing SNP rs393923 was amplified using as template either the GenomiPhi amplified DNA or the original genomic sample. PCR products were purified via the Quick Step PCR Purification Kit (Edge Biosystem, Gaithersburg, MD). Sequencing was performed in both directions using the ABI BigDye Terminator Cycle Sequencing Ready Reaction Kit v3.1 as recommended by the manufacture and analyzed on an ABI 377 DNA sequencer. Sequence analysis was performed using MacVector7.1.1 (Accelrys, San Diego, CA).
RESULTS
Analysis of GP-DNA
Samples ranging in DNA concentration as well as in DNA quality were subjected to whole genome amplification using Amersham Bioscience GenomiPhi DNA Amplification Kit. After overnight incubation with Phi29, the viscous samples (GP-DNA) were routinely diluted in water and column purified prior to gel electrophoresis and downstream analysis. Figure 1 shows a typical profile of GP-DNA visualized after ethidium bromide staining (lanes 1 to 10). Interestingly, the no-template water control (lane 11), which went through the same processing as GP-DNAs, shows an electrophoretic profile similar to that of other genomic DNA. Spectrophotometric analysis of the water control indicates high DNA yield which is consistent with the pattern observed by gel electrophoresis analysis; however, this control sample failed to amplify using gene-specific or microsatellite-specific primers. In the absence of genomic DNA template, it is the random hexamers which become template for Phi29 rolling circle mode of amplification. Thus, gel electrophoresis or spectrophotometric analysis of GP-DNA in itself is not sufficient to ascertain that the amplification was successful. Further downstream verification steps (such as gene-specific PCR) are needed to determine successful amplification and validate the GP products. Yields obtained from validated GP-DNAs are displayed in Table 1. Up to a 1000-fold increase in GP-DNA is obtainable when starting with 5 ng of genomic DNA. Interestingly, increasing template concentration to 25 ng did not increase the yield of amplified DNA as seen for samples 2-9, 6-2, and 7-32, indicating that the maximum amount of product that accumulates during the overnight incubation is limited by the manufacturer’s reagent formulation. Sample 6-2 is a typical example of DNA showing signs of degradation pre-WGA as depicted by a very high fail rate during STR analysis. Although gel electrophoresis and spectrophotometric analysis suggested that the amplification process was successful, STR analysis of the GP-DNA demonstrated that the WGA had failed (Table 1). As in the water control, Phi29 in the presence of degraded genomic DNAs will use the random hexamer primers as a template for amplification.
FIGURE 1.

Agarose (0.8%) gel electrophoresis of genomic DNAs amplified using the GenomiPhi Kit. M: 1-Kb molecular weight markers. Lanes 1–10 show typical profiles of human genomic DNA post amplification. Lane 11: Water control depicting random hexamer-mediated amplification after overnight incubation with Phi29.
TABLE 1.
Whole Genome Amplification Study of DNAs Used in Linkage and Association Analysis
| Sample | Starting Amount (ng) | Fold | STR D7S661 | STR D8S284 | SNP rs3765689 | SNP rs3818330 | AoD RPL 22 |
| 2-9 | 25 | 320.7 / 326.6 | |||||
| 2-9 GP | 192X | 320.7 / 326.4 | T / T | C / A | 1 / 2 | ||
| 26-3 | 5 | 322.7 / 322.7 | |||||
| 26-3 GP | 780X | 322.6 / 322.7 | T / C | C / C | 2 / 2 | ||
| 6-2 | 25 | Fail | Fail | ||||
| 6-2 GP | 212X | Fail | Fail | ||||
| 7-20 | 5 | 318.79 / 322.59 | 287.97 / 293.72 | ||||
| 7-20 GP | 860X | 318.79 / 322.74 | 287.97 / 293.83 | T / T | C / A | 2 / 2 | |
| 7-31 | 5 | 318.8 / 320.7 | 293.7 / 305.2 | ||||
| 7-31 GP | 960X | 318.8 / 320.7 | 293.7 / 305.3 | T / T | C / C | 2 / 2 | |
| 7-32 | 25 | Low | |||||
| 7-32 GP | 232X | 312.8 / 312.8 | T / T | C / C | 2 / 2 |
The samples tested in this group had been stored for several years and were starting to show signs of depletion and degradation (as for 6-2). This table displays characteristic samples from our set of GP-DNAs. GP-DNAs were validated as indicated by microsatellite (STR) and SNP analysis. GP samples that failed STR amplification were not tested further. AoD, Assays-on-Demand; SNP, single-nucleotide polymorphisms; STR, short tandem repeat.
Genotyping of Human GP-DNAs Using STR and SNP Markers
All GP-DNAs were validated using microsatellite markers included in panel 12 of ABI linkage mapping set v2.5. The PCR profiles were compared with those obtained for the original genomic DNA templates. There was 100% concordance between the genotyped markers pre- and post-WGA. Figure 2 displays an example of side-by-side comparison STR profiles at loci D7S661 and D8S284 for individual 7-20. In the case of GP-DNA 7-20, a differential allelic amplification was noted at locus D7S661, for the 322 bp allele, when compared with the original genomic DNA profile (top two electropherograms). In this study we have not observed any allelic dropout due to the WGA but on occasion have seen differential allelic amplification. However, this did not seem to be WGA-specific but rather a PCR-specific artifact.
FIGURE 2.

Typical genotyping profiles of human DNA pre- and post-GenomiPhi amplification using two microsatellites—D7S661 (blue) and D8S284 (green)—included in panel 12 of the ABI Prism Linkage Mapping Set v2.5. Depleted stock of sample 7-20 (see Table 1) was successfully replenished by WGA as indicated by identical genotypes for pre- and post-genomic amplification. The black arrow points to the 322-bp differentially amplified allele of D7S661.
All validated GP-DNAs were used to complete a screen of the chromosome X-specific microsatellite markers included in panel 28 (ABI linkage mapping set v2.5). Genotyping of the entire cohort of DNA collected for this family-based linkage study allowed us to further validate the genotype of the GP-DNA samples by checking for Mendelian inheritance. No Mendelian errors were noted during the STR study of GP-DNAs.
The entire DNA cohort was then genotyped with SNP markers using Assays-on-Demand (Fig. 3) and pyrosequencing (Fig. 4). Figure 3 is a typical example of the discrimination plot obtained for the entire DNA collection (including genomic and GP-DNA) for SNP rs2294714 of WDR8 intron 3. As mentioned above, the DNA collection is of heterogeneous quality and, as such, the data clusters are not as tight as those obtained with homogeneous quality DNA. However, one can easily discern between the three clusters of genotypes—the homozygous CC and TT, and the heterozygous CT. Three of the GP-DNAs included in Table 1 are indicated by red circles on the plot. In addition to TaqMan technology, pyrosequencing was also used for association studies. Figure 4 shows a typical example of pyrosequencing analysis of WDR8 gene SNP rs3818330. The sequencing primer (5′-AGACGGTCTCTGATGATG-3′) was used to interrogate this SNP. The pyrograms display the genotypes for two related individuals 7-20 (aunt) and 7-31 (nephew). A total of 31 SNPs located on four genes (Gnb1 WDR8, ICMT, and CamTA1) were used to genotype the replenished DNA cohort. No Mendelian inheritance discrepancies were noted in related individuals for whom DNA was replenished by WGA.
FIGURE 3.
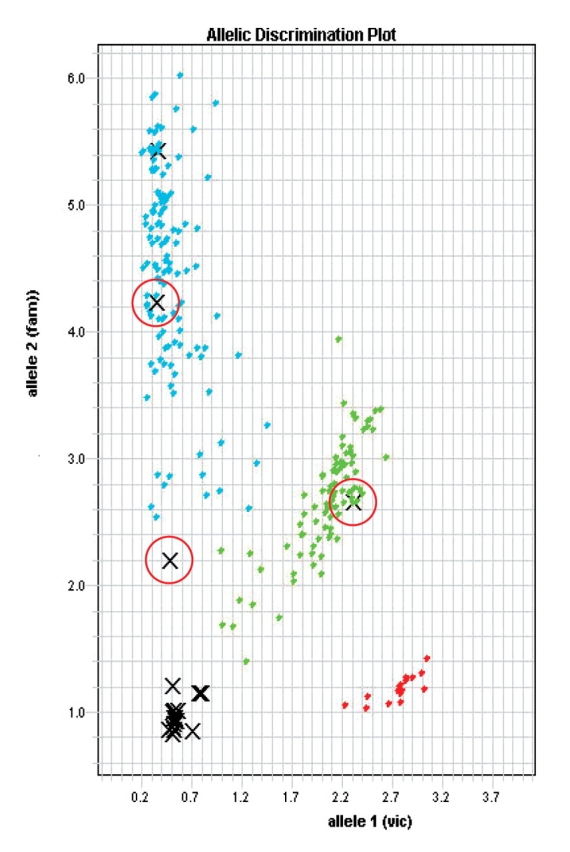
Allelic discrimination plot obtained for rs2294714 after screening adult human DNA using ABI Assays-On-Demand. Red circles indicate GP-DNAs 7-20 (aunt), 7-30 (niece) and 7-31 (nephew) (see Table 1). The individuals in the study cluster in three groups (blue and red are homozygous samples, green are heterozygous C/T). Negative (water and no template) controls and failed samples all cluster at the bottom left area of the plot.
FIGURE 4.

Screening of SNPs in genomic and GP samples from related individuals using pyrosequencing. A: Theoretical profiles of the WDR8 gene SNP rs3818330 for the homozygous C/C and heterozygous C/A genotypes. B: Experimental results of an individual with genotypes C/C (7-31, nephew) and C/A (7-20, aunt) using GP-DNA. The SNP site is highlighted in yellow. Additional sequence information obtained on the sample pyrograms (TAGAG) show 100% concordance with the known sequence adjacent to the SNP site.
Validation of WGA by Direct DNA Sequencing
Direct sequencing was performed, as described in Methods, on both pre- and post-WGA pediatric DNA samples. A region of the FGFR4 gene containing SNP rs393923 was first amplified by PCR. The 489-bp product was then sequenced in both forward and reverse directions. Figure 5 shows the sequencing electropherogram of a 70-bp region containing SNP rs393923 on pediatric samples 22, 26, and 35. The SNP is indicated by a red arrow. We also found an additional heterozygous site in sample 35 as indicated by the blue arrow. DNA variants were confirmed by sequencing both DNA strands. No mutations were detected in the GP-DNA samples when compared with the original genomic DNA template (see Table 1) for the FGFR4 region studied.
FIGURE 5.

Sequencing profiles of a region of FGFR4 in pediatric samples. A 489-bp PCR product was generated using genomic DNA before and after the genome amplification procedure (GP) and sequenced. SNP rs393923 is indicated by a red arrow. A blue arrow indicates an additional heterozygous site in 35. No mutation was detected in the GP-DNA samples when compared with the original genomic DNA templates (Table 2).
In order to determine if one could generate a large quantity of WGA DNA from a depleted sample without loss of genomic information, samples 29 and 35 were subjected to five successive rounds of WGA by Phi 29. The genotypes of the reamplified GP-DNAs were verified using WDR8 SNP rs3765689 and by sequencing of the FGFR4 region (Table 2). There was 100% concordance between the original genomic DNA and the WGA products generated after five rounds of reamplifications (GP5).
TABLE 2.
Reamplification of WGA Products
| Sample (10ng) | Amplification Fold | WDR8 rs3765689 | Sequencing FGFR4 |
| 29 | T / T | 486 bp | |
| 29 GP | 530X | T / T | No change |
| 29 GP2 | 530X | ||
| 29 GP5 | 610X | T / T | No change |
| 35 | T / T | 489 bp | |
| 35 GP | 660X | T / T | No change |
| 35 GP2 | 560X | ||
| 35 GP5 | 495X | T / T | No change |
Two pediatric sample DNA’s were subjected to five successive rounds of amplification as indicated in methods (GP1 to GP5). GP2 was obtained by amplification of 10 ng of GP1 DNA as template for both samples 29 and 35. Amplification of GP2 resulted in GP3 etc., through 5 cycles (GP5). Genomic and GP-DNAs were validated as indicated with PCR, direct sequencing, and pyrosequencing.
DISCUSSION
In early 2003, the Biomolecular Core Laboratory became the repository of a collection of DNA samples that was used in a family-based linkage study to map a complex disease trait. This collection of precious DNA was very heterogeneous in quality and quantity. Older samples often showed marked signs of degradation as depicted by problems amplifying even the smallest microsatellite repeats locus. Because additional tests were required, some of the DNA stocks were becoming rapidly exhausted. As the microsatellite study progressed, it became apparent that the sample depletion would result in data loss that might lead to a decrease in statistical power. Thus we started the present study to explore the potential of WGA to recover enough DNA to complete the family-based linkage study and prepare reagents for a candidate gene association study.
Several PCR-based methods are available for WGA such as PEP4 and DOP-PCR.5 Although these two WGA approaches allow for successful amplification of genomic DNA, we were concerned by the fact that PCR-based DNA polymerase is error prone.6 Thus we searched for a non–PCR-based method which would preserve the information of the template DNA while allowing us to replenish sample stocks for reliable downstream genetic analysis. At the time this study began, a new non–PCR-based WGA product was being launched by Amersham Biosciences, GenomiPhi DNA Amplification Kit. GenomiPhi WGA is based on multiple strand displacement amplification of linear genomic DNA performed by Phi29 DNA polymerase. Phi29 is a highly processive enzyme that has been shown to have 100-fold lower error rate than Taq polymerase.3 To evaluate this novel method of WGA, Phi29 reagent kit was chosen to amplify 45 “problem” human genomic DNA samples selected from our original heterogeneous collection of DNAs. Since gel electrophoresis and spectrophotometric analysis of DNA generated with this method are not indicative of the quality of the amplified products, validation was performed using microsatellite markers from chromosomes 7 and 8. Although we observed one instance of reduced amplification for one of the alleles (see Fig. 2), no allelic dropout was observed during this study. Of the original 45 samples, 36 were successfully amplified and stocks replenished, but 9 DNAs failed our attempts at amplification. GenomiPhi DNA Amplification Kit was also unable to recover DNA samples that showed characteristic hallmarks of degradation preamplification. Validated GP-DNAs were used to replenish our family-based cohort, which allowed us to complete a whole genome scan linkage study (manuscript in preparation). These samples were tested along with the rest of the family-based samples with the 18 chromosome X-specific microsatellite markers included in panel 28 of the ABI linkage mapping set. All genotypes were consistent with family allelic distribution as determined by PedManager. After microsatellite genotyping, sequence integrity of the amplified DNA was further confirmed by single nucleotide polymorphism analysis using TaqMan Assays-on-Demand as well as pyrosequencing. SNP selection was based on position of the SNP on the gene of interest as well as information contained in the Celera and NCBI databases regarding its validation status and allelic frequency (> 5%). Whenever possible, the distance between selected adjacent SNPs was kept constant (e.g., 1 SNP every 15 Kb of DNA on Gnb1). However, the distance between the SNPs used to scan the 300-Kb region of CamTA1, varied from 7.9 Kb to 88.1 Kb. A total of 24 SNP loci in genes Gnb1, WDR8, ICMT, and CamTA1 were interrogated for association study using TaqMan assays. The real time profile of WGA samples was similar to that of nonamplified genomic DNAs, thus suggesting no bias in the amplification of these specific loci by Phi29. Seven additional SNPs were used to screen WDR8, one of our candidate genes, using pyrosequencing. As with microsatellite genotyping, no Mendelian error was noted during pyrosequencing or TaqMan screening of the SNPs.
A second series of DNAs obtained from pediatric patients helped us test the GenomiPhi kit further by direct sequencing. Sequence alignments of a region of the FGFR4 gene (genomic and GP-DNA samples), revealed no new DNA variant introduced by the Phi29 amplification step. We also investigated whether additional rounds of amplification with Phi29 would introduce de novo mutations leading to genotyping errors. Samples were subjected to five cycles of successive reamplification. The WGA products generated after the fifth successive round of amplification with Phi29 were identical in sequence to the original template DNA, indicating that a large quantity of WGA DNA can be generate from a depleted sample by reamplification and can be used with confidence for genotyping studies. GenomiPhi routinely yielded a maximum of 6 μg of validated genomic DNA after overnight amplification in 20 μL reaction volume. We also demonstrated that it was possible to reduce the enzyme mix input by at least one quarter without any detectable loss in DNA quality or yield (data not shown). Thus, the limiting factors controlling the maximum yield for this reaction are the random hexamers or dNTPs found in the reaction mix.
We conclude from this study that GenomiPhi DNA Amplification Kit is a valuable tool suited for the recovery of precious DNA, and that the quality of the amplified material shows no detectable difference from the original starting material. Although we were able to validate this WGA in a family-based study in which genotyping errors can easily be ascertained, we are confident that this technology can be used for other types of studies (e.g., case/control). It is the high fidelity of the Phi29 enzyme at replicating high molecular weight genomic DNA which makes this technology an asset in core facilities and research labs alike.
Acknowledgments
The authors thank Drs. Marcella Devoto and Karen Gripp for support with the use of biological materials. This work was supported by funds to KSC from the Nemours Foundation and by NIH Grant Number 1 P20 RR020173-01 from the National Center for Research Resources.
REFERENCES
- 1.Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 2001;11(6):1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dean FB, Hosono S, Fang L, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA 2002;99(8): 5261–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esteban JA, Salas M, Blanco L. Fidelity of phi 29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. J Biol Chem 1993; 268(4):2719–2726. [PubMed] [Google Scholar]
- 4.Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Whole genome amplification from a single cell: Implications for genetic analysis. Proc Natl Acad Sci USA 1992;89(13):5847–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide-primed PCR: General amplification of target DNA by a single degenerate primer. Genomics 1992;13(3):718–725. [DOI] [PubMed] [Google Scholar]
- 6.Dunning AM, Talmud P, Humphries SE. Errors in the polymerase chain reaction. Nucleic Acids Res 1988; 6(21):10393. [DOI] [PMC free article] [PubMed] [Google Scholar]