

Abstract
In the simplest view, aptamers can be thought of as nucleic acid analogs to antibodies. They are able to bind specifically to proteins, and, in many cases, that binding leads to a modulation of protein activity. New aptamers are rapidly generated through the SELEX (Systematic Evolution of Ligands by Exponential enrichment) process and have a very high target affinity and specificity (picomoles to nanomoles). Furthermore, aptamers composed of modified nucleotides have a long in vivo half-life (hours to days), are nontoxic and nonimmunogenic, and are easily produced using standard nucleic acid synthesis methods. These properties make aptamers ideal for target validation and as a new class of therapeutics. As a target validation tool, aptamers provide important information that complements that provided by other methods. For example, siRNA is widely used to demonstrate that protein knock-out in a cellular assay can lead to a biological effect. Aptamers extend that information by showing that the dose-dependent modulation of protein activity can be used to derive a therapeutic benefit. That is, aptamers can be used to demonstrate that the protein is a good target for drug development. As a new class of therapeutics, aptamers bridge the gap between small molecules and biologics. Like biologics, biologically active aptamers are rapidly discovered, have no class-specific toxicity, and are adept at disrupting protein-protein interaction. Like small molecules, aptamers can be rationally engineered and optimized, are nonimmunogenic, and are produced by scalable chemical procedures at moderate cost. As such, aptamers are emerging as an important source of new therapeutic molecules.
Keywords: Aptamer, target validation, therapeutic, cardiovascular disease, cancer
APTAMER DISCOVERY AND DEVELOPMENT
Properties of Aptamers
Aptamers are nucleic acid macromolecules that bind to molecular targets, including proteins, with high affinity and specificity. Aptamers are typically from 15 to 40 nucleotides in length and can be composed of DNA, RNA, or nucleotides with a chemically modified sugar backbone (i.e., 2′-fluoro, 2′-O-methyl, phosphorothioate). Complementary base pairing defines aptamer secondary structure, consisting primarily of short helical arms and single-stranded loops. Stable tertiary structure, resulting from combinations of these secondary structures, allows aptamers to bind to targets via van der Waals, hydrogen bonding, and electrostatic interactions. The huge number of possible tertiary structures allows aptamers to bind with high affinity via van der Waals, hydrogen bonding, and electrostatic interactions, to most small-molecule, peptide, or protein targets, with KD values ranging from 10 pM to 10 nM for proteins (Table 1). Aptamers can recognize their targets with great specificity. For instance, an aptamer to bFGF (FGF-2) binds with up to 20,000-fold greater affinity to bFGF than it does to its closely related fibroblast growth factor (FGF) -1, -4, -5, -6, and -7 homologues.1 Other aptamers distinguish between closely related members of a protein family, or between different functional or conformational states of the same protein.2
TABLE 1.
Protein Targets Against Which High-Affinity Aptamers Have Been Generated7
| Target | KD (nM) |
| Keratinocyte growth factor (KGF) | 0.0003 |
| HIV-1 reverse transcriptase (RT) | 0.02 |
| Transforming growth factor-beta 1 (TGF-β1) | 0.03 |
| P-selectin | 0.04 |
| Vascular endothelial growth factor receptor (VEGFr) | 0.07 |
| Platelet-derived growth factor (PDGF) | 0.09 |
| Immunoglobulin E (IgE) | 0.1 |
| Extracellular signal-regulated kinase (ERK) | 0.2 |
| CD4 antigen | 0.5 |
| HIV-1 RNAse H | 0.5 |
| Angiogenin | 0.7 |
| Complement factor C5 | 1.0 |
| Transforming growth factor-beta 2 (TGF-β2) | 1.0 |
| Interferon-gamma (IFN-γ) | 2.0 |
| Secretory phospholipase A2 (sPLA2) | 2.0 |
| Thrombin | 2.0 |
| L-selectin | 3.0 |
| Human neutrophil elastase | 5.0 |
| Integrin (αvβ3) | 8.0 |
| HCV NS3 protease | 10.0 |
| Yersinia pestis tyrosine phosphatase | 18.0 |
Aptamer History
The conceptual framework and process of aptamer generation emerged from pioneering experiments by independent groups, both of whom published their work in 1990. Tuerk and Gold described a process of in vitro selection, dubbed “SELEX” (Systematic Evolution of Ligands by Exponential enrichment), to determine the sequence requirements of T4 DNA polymerase.3 An eight-base region of an RNA that interacts with T4 DNA polymerase was chosen and randomized. Two different sequences were selected from a calculated pool of 65,536 species. SELEX identified the natural bacteriophage recognition sequence of the polymerase as the primary high-affinity ligand, along with a single major sequence variant that showed similar affinity for the enzyme.3 Shortly thereafter, Ellington and Szostak published further groundbreaking experiments utilizing in vitro selection to isolate nucleic acids with specific ligand binding properties.4 Starting nucleic acid libraries were comprised of molecules containing 100 nucleotides of randomized sequence. Affinity chromatography was used to isolate RNAs that demonstrated highly specific binding to several organic dye molecules, none of which had been known previously to bind nucleic acid. Approximately 1 in 1010 random sequence RNAs was found to fold in a manner that created specific dye binding sites. Ellington and Szostack originated the term “aptamer” to describe these nucleic acid-based ligands.4 Since 1990, aptamers have been generated against hundreds of molecular targets, ranging from small molecules and peptides to numerous proteins of therapeutic interest (e.g., growth factors, enzymes, immunoglobulins, and receptors).
In Vitro Selection of Aptamers
Aptamers are isolated by various SELEX methods that involve an iterative process of binding, partitioning, and amplifying novel nucleic acids from a combinatorial pool of up to 1016 variants (Fig. 1). SELEX can be performed manually at the bench top or by automated systems.5,6 Individual high-affinity aptamers are isolated after final refinement and optimization steps.7 Cloned aptamers are then screened for functional activity by, for example, their ability to modulate enzyme activity or their ability to neutralize their targets in cell-based assays. After the initial screen, it is usually desirable to determine the minimum sequence that still allows specific aptamer-target binding and efficacy. This process, minimization, is performed by mapping the critical parts of an aptamer via site-directed mutagenesis or chemical protection assays and eliminating superfluous regions. Minimization can often lead to increasing the affinity of the aptamer for the target, possibly due to a decrease in competing, nonbinding conformations. Other optimization steps include increasing the in vivo stability of an aptamer by substituting 2′-OHs with 2′-fluoro or 2′-O-methyl groups and chemically “capping” the 5′ and 3′ termini.
FIGURE 1.

Diagram outlining the SELEX process.
APTAMERS FOR TARGET VALIDATION
Target validation is the determination that a drug target is involved in disease pathology and is potentially a point of intervention for new drugs. The conventional approach for determining the relevance of a given protein to a particular disease is to inhibit the expression of the gene and observe the effects on an appropriate disease model. Target validation methods therefore have been devised that attack gene expression at the DNA, RNA, and protein levels (Fig. 2). One method that attacks gene expression at the DNA or genetic level is to construct a knock-out mouse by homologous recombination. The advantages of the gene knockout approach are that gene expression is completely abolished and that the function of a given gene can be examined in every system of the organism simultaneously. Two major disadvantages of gene knockouts are that the approach is extremely laborious and that results observed in mice may not translate to humans. Also, it is disquieting that knockout mice often show a phenotype that is inconsistent with extensive in vitro research.8 These conflicting phenotypes often manifest as either a very mild phenotype, possibly as a result of an additional redundant gene, or as an animal that has developmental defects resulting in lethality. Validating a target that falls into either the mild or lethal categories is extremely difficult.
FIGURE 2.

The levels of gene expression and corresponding target validation techniques.
Target validation approaches also include techniques that knockout gene expression at the level of the mRNA. RNA interference (RNAi)9 and antisense10 are two such approaches. RNAi uses double-stranded RNA to induce homology-dependent degradation of the cognate mRNA, and therefore block the expression of the desired protein. Initially, a major drawback of RNAi was that when applied to mammalian cells it triggered non-specific responses which obscured sequence-specific silencing. One of these nonspecific responses was the activation of the RNA-dependent protein kinase response pathway, which phosphorylates EIF-2a and nonspecifically arrests translation. Due to their small size, short inhibitory RNAs (siRNAs) can effectively block protein expression without inducing the protein kinase response.11 Although some nonspecific effects via the interferon response12,13 and possibly other pathways14 have been reported more recently. In the case of antisense, base pairing of an antisense strand of RNA (or DNA), with its corresponding mRNA, blocks translation of the mRNA by marking the RNA for degradation by RNases or by blocking the protein translation machinery. Specific antisense oligonucleotides can be generated rapidly in a cost-effective manner. Antisense technology does, however, have a number of disadvantages including toxicity, poor stability, and concerns about specificity.15 Recent advances in the chemical modification of antisense RNA backbones have alleviated some of these concerns.15
It is increasingly being recognized that in addition to validating the role of the protein in disease pathology, it is also important to validate the protein as a target for therapeutic intervention. Validation techniques that function at the gene level or mRNA level (siRNA, antisense, and knockouts) do not accurately mimic the effects of small molecule drugs which act at the protein level and exhibit a dose response. Also, since proteins sometimes have more than one function and are part of more than one multiprotein complex, deletion of the protein through altering mRNA levels can lead to the disruption of numerous pathways and regulatory cascades, some of which may not be relevant to the disease model in question.
Validating targets at the protein level is more direct and more closely mimics the action of highly specific small molecule drugs. Antibodies, which bind proteins with high affinity and specificity, are a powerful way to inactivate protein targets.16 However monoclonal antibodies are limited to extracellular targets as they do not function well within cells. Some progress has been made towards the use of single-chain antibody fragments for intracellular applications; however, even for these molecules the reducing conditions within the cytoplasm lead to misfolding and aggregation.16
The application of nucleic acid aptamers also inhibits gene expression at the protein level, and thus offers a compelling alternative to the above techniques. Aptamers have many attributes that make them suitable for target validation. They bind with high affinity and also exhibit a degree of specificity that allows them to distinguish between closely related molecules. Like monoclonal antibodies, aptamer binding to a target often, although not always, inhibits that targets activity. Like small molecule therapeutics, their dosage can be easily adjusted and aptamers exhibit a dose response. Importantly, an aptamer can inhibit target function in vivo by blocking or knocking out a single domain of a protein while leaving the remainder of the protein functional. Small-molecule drugs are well known to have this capability.17 Small molecules and aptamers share another characteristic in that they both, potentially, can act as agonists.
Another consequence of working at the protein level is that aptamers can provide information that is complementary to that obtained from gene-level validation approaches. Indeed, simultaneous application of siRNA and aptamer against the transcription factor NF-κB achieves 90% knock-down while either method alone can barely achieve 60%.18 Given that both aptamers and siRNA are RNA-based and can thus be simultaneously produced in mammalian cells from expression vectors, this combinatorial strategy may be useful against intractable targets.
Along with the above conceptual advantages, there are also practical advantages to working with aptamers. Being nucleic acids, aptamers are easy to produce, store, and modify. Large amounts of aptamers can be produced in vitro either enzymatically or synthetically. They can be easily delivered intracellularly by standard transfection techniques or produced in vivo with the appropriate expression vectors. Lyophilized aptamers can be stored for years without loss of activity and once they are reconstituted, aptamers can be boiled or subjected to numerous freeze-thaw cycles. Their stability both in vitro and in vivo can be further enhanced by various chemical modifications such as 2′-fluoro and 2′-O-methyl substitution.18
In general, the process of generating and stabilizing aptamers is straightforward, efficient, and relatively inexpensive. However, though aptamers have been generated against a broad range of ligands, not every protein target is equally amenable to aptamer selection. For example, highly acidic regions of proteins can be challenging aptamer targets, and aptamer access to particular sites on a protein may be subject to steric constraints. It is also worth noting that for target validation applications, delivery of aptamers into cells requires that the aptamer sequence be placed into an expression/stabilization/delivery vehicle or transcript. Encoding an aptamer into such a vehicle can lead to its inactivation due to improper folding of RNA expressed inside cells. In such instances, it may be necessary to design multiple aptamer expression constructs and validate aptamer activity in vitro prior to cellular delivery.
Validation of Intracellular Targets Using Aptamers
A number of studies have demonstrated the use of aptamers as intracellular target validation tools. Aptamers have been evolved against HIV-1 Rev and Tat, two RNA-binding proteins that are necessary for HIV-1 replication. Rev interacts with the Rev binding element and functions to transport the viral RNA from the nucleus to the cytoplasm. Tat is an RNA-binding transcription factor that is required for viral replication. When expressed intracellularly, anti-Rev or anti-Tat aptamers have inhibited HIV-1 production in cell culture.19,20
Transgenic aptamer-expressing Drosophila have been used to demonstrate the efficacy of aptamers as target validation tools in whole animals. Shi et al. showed that an anti-B52 aptamer could function in vivo to inhibit B52 function.21 B52 is a member of the Drosophila SR protein family, a group of nuclear proteins that are essential for pre-mRNA splicing. Shi et al. developed an RNA aptamer that specifically bound to B52 with high affinity and configured a multivalent aptamer for expression in cells and in flies.21 Previous work has shown that the level of B52 expression is critical for normal Drosophila development. B52 deletion results in death,22 and over-expression of B52 is associated with lethality and morphological defects including missing bristles and the absence of salivary glands.23 Shi et al. used anti-B52 aptamers to suppress the effects of B52 overexpression in Drosophila.21 They observed the reversal of abnormal bristle, wing, abdominal sternite, and salivary gland developmental associated with B52 overexpression. Moreover, an increase in the survival rate was observed for flies coexpressing B52 and the anti-B52 aptamer as compared with flies expressing B52 only. These results demonstrate that aptamers can be used to inhibit intracellular target function in a nonmammalian, whole animal system.
Further studies have demonstrated that aptamers can recognize proteins that mediate the mammalian signal transduction processes. For example, Blind et al. evolved aptamers that recognize the cytoplasmic domain of α1β2-integrin.24 β2-Integrins have been demonstrated to play a critical role in leukocyte cell adhesion. Anti-β2 aptamers were demonstrated to inhibit α1β2-integrin activity, as measured by a decrease in cell adhesion.24 Mayer et al. have extended these observations by knocking out cytohesin 1, a signaling protein downstream of α1β2-integrin.25 The anti-cytohesin 1 aptamer was demonstrated to inhibit the guanine nucleotide exchange factor activity of cytohesin 1. Moreover, cytohesin 1 inhibition translated into a biological effect as measured by cell adhesion and cytoskeletal rearrangement.
Validation of Extracellular Targets Using Aptamers
The ability to chemically stabilize aptamers against nuclease attack and to couple aptamers with high molecular weight polyethylene glycol (PEG) polymers (termed “PEGylation,” see below) allows them to persist and function in vivo, making them good tools for extracellular target validation. The best example of the use of aptamers for extracellular target validation is a series of experiments from Pietras et al. utilizing an anti-PDGF-B aptamer.26,27 Platelet-derived growth factor (PDGF) belongs to the cysteine-knot growth factor family and was originally isolated from platelets for promoting cellular mitogenic and migratory activity. The binding of PDGF isoforms to their cognate receptors induces the phosphorylation of specific residues in the intracellular tyrosine kinase domain of the receptors and activation of the signaling pathway. In general, PDGF isoforms are potent mitogens and thus are targeted for proliferative diseases such as cancer, diabetic retinopathy, glomerulonephritis, and restenosis.
PDGF-B has been implicated in the regulation of interstitial fluid pressure (IFP). Elevated IFP is a physiologically distinctive property of solid tumors that differs from healthy connective tissue and is considered to be a major obstacle limiting access of therapeutics to solid tumors.28 In some cases, IFP increases as a function of tumor size and malignancy, and high IFP in cancer patients can be associated with poor prognosis. Notably, many solid tumors which are currently being treated with chemotherapy regimens have high IFP and display paracrine signaling of PDGF in their stromal compartments. Current data indicate that tensile strength and mechanical stiffness of connective tissue are regulated by a complex interaction between cells such as fibroblasts with extracellular matrix components such as collagen and hyaluronan. PDGF is known to up-regulate synthesis of collagen and to mediate interactions of anchor proteins such as integrins with extracellular matrix components.
Pietras et al. wanted to validate the hypothesis that PDGF-B is involved in IFP and that blocking PDGF-B function could lead to increased uptake of chemotherapeutics into tumors.26,27 Using the KAT-4 thyroid carcinoma model, they used the tyrosine kinase inhibitor STI571 to block PDGF-B signaling in KAT-4 tumors and showed that this treatment significantly decreased tumor IFP in vivo (Fig. 3A), leading to increased uptake of Taxol. Most significantly, treatment also enhanced the anti-proliferative effect of Taxol (Fig. 4A). These data, along with previously published data, indicated that PDGF-B was indeed a good target for lowering IFP and increasing chemotherapeutic efficacy. However, since STI571, like all small-molecule kinase inhibitors, is known to affect other tyrosine kinases, it was impossible to know if the effect was due to PDGF-B blockage alone.
FIGURE 3.

Treatment with platelet-derived growth factor receptor (PDGFr) antagonists lowers interstitial fluid pressure in KAT-4 tumors. Tumor interstitial fluid pressure was measured 1–2 h after last administration of PDGF receptor inhibitor in KAT-4 tumors grown subcutaneously in severe combined immune deficiency (SCID) mice. PDGF receptor inhibitors were administered for a total of 4 days. A: Mice were treated with phosphate buffered saline (PBS) (n = 8) or STI571 (n = 9). B: Mice were treated with control aptamer (n = 8) or PDGF aptamer (n = 8). (Reproduced with permission from Pietras et al. Cancer Res 2002;62: 5476–5484.)
FIGURE 4.

Treatment with PDGF receptor antagonists enhances the effect of Taxol on KAT-4 tumors in vivo. Growth curves of KAT-4 tumors grown subcutaneously in SCID mice. A: Mice received no treatment (solid square, n = 8), STI571 (solid circle, n = 6), Taxol (solid triangle, n = 4), or STI571 and Taxol (cross bar, n = 8). B: Mice received polyethylene glycol (PEG; solid square, n = 8), PEG-conjugated PDGF aptamer (solid circle, n = 8), PEG and Taxol (solid triangle, n = 8), or PDGF aptamer and Taxol (cross bar, n = 8). *P < 0.05, PDGF-receptor antagonist and Taxol versus Taxol alone, Student’s t-test, **P < 0.01 PDGF-receptor antagonist and Taxol versus Taxol alone, Student’s t-test. (Reproduced with permission from Pietras et al. (Cancer Res 2002;62: 5476–5484.)
This dilemma was solved by using a highly specific aptamer to block PDGF-B in similar experiments. The PDGF-B aptamer was isolated through single stranded DNA SELEX.29 The aptamer has an affinity of 100 pM for PDGF-B and no appreciable affinity for the PDGF-A isoform. As with STI571, treatment of KAT-4 xenograft mice with PEG-conjugated PDGF-B aptamer lowered IFP (Fig. 3B) and dramatically increased tumor uptake of Taxol (Fig. 5). Most importantly, aptamer treatment strongly enhanced Taxol’s ability to inhibit tumor growth (Figure 4B). Given the outstanding specificity of aptamers, these experiments validate PDGF-B as a target for inhibition aimed towards enhancing the uptake and efficacy of chemotherapeutics. Furthermore, because aptamers in general and the PDGF-B directed aptamer in particular, already have many of the attributes required for a therapeutic, the anti-PDGF-B aptamer can directly enter into a therapeutic development program (see below).
FIGURE 5.
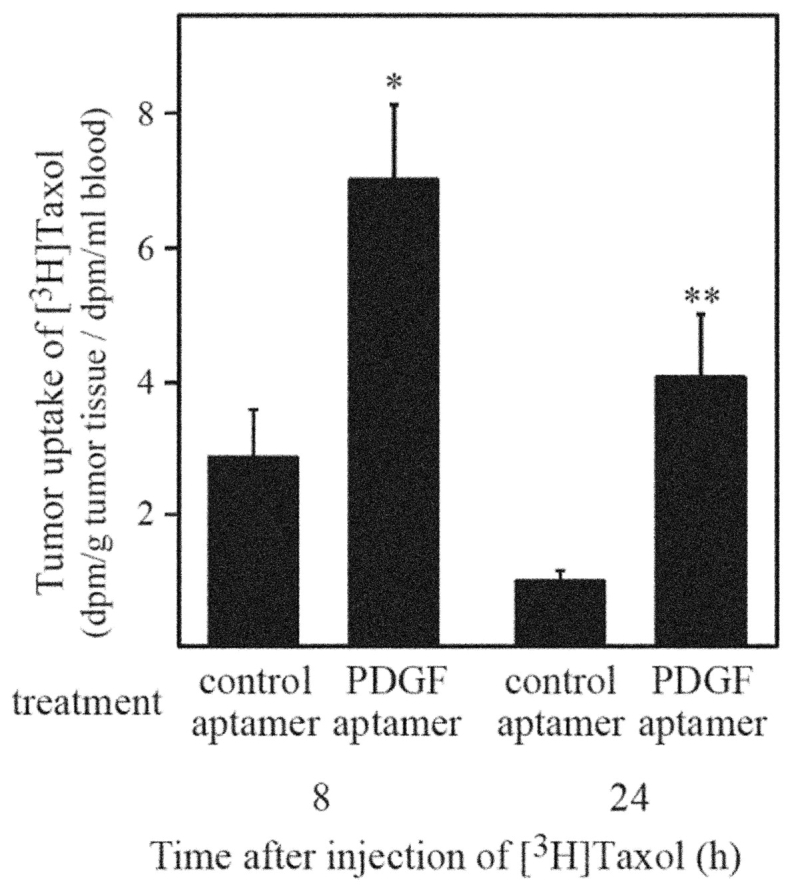
Treatment with PDGF aptamer antagonists increases uptake of [3H]Taxol in KAT-4 tumors. After treatment with PDGF-receptor antagonists or control, mice with KAT-4 tumors were injected subcutaneously with [3H] Taxol. Radioactivity was measured in homogenates of tumors and in blood samples 8 or 24 h after subcutaneous injection of radiolabeled compound. Mice were treated with control aptamer (8 h, n = 6; 24 h, n = 7) or PDGF aptamer (8 h, n = 6; 24h, n = 7); *P < 0.05, Student’s t test; **P < 0.01, Student’s t-test; bars, ± SE. (Reproduced with permission from Pietras et al. Cancer Res 2002;62:5476–5484.)
APTAMERS FOR THERAPEUTICS
In addition to target validation and research applications, aptamers are being developed as therapeutic agents. A number of aptamers have completed various stages of pre-clinical development, ranging from pharmacokinetic analysis, characterization of biological efficacy in cellular and animal disease models, and safety assessment. In particular, one aptamer, targeting vascular endothelial growth factor (VEGF), has completed phase III clinical trials for age-related macular degeneration (AMD), a leading cause of blindness.
General Properties of Therapeutic Aptamers
It is clear that aptamers have a number of desirable characteristics for use as therapeutics including high specificity and affinity, biological efficacy, and excellent pharmacokinetic properties. In addition, they offer specific competitive advantages over antibodies and other protein biologics.
Speed and Control
Aptamers are produced by an entirely in vitro process, allowing for the rapid generation of initial therapeutic leads. In vitro selection allows the specificity and affinity of the aptamer to be tightly controlled and allows the generation of leads against both toxic and nonimmunogenic targets.
Pharmacokinetics
Natural RNAs/DNAs have poor pharmacokinetics, primarily due to nuclease degradation and clearance via the kidneys. Both limitations can be addressed with appropriate chemical modifications as described below. Following optimization, aptamers typically exhibit half-lives on the order of 6 h in rats and 9–12 h in monkeys30–33 (Archemix Corp., unpublished data).
Nuclease Resistance
Nucleic acids are degraded in serum by a combination of endonucleases and 5′-3′ and 3′-5′ exonucleases. Appropriate chemical modifications block each activity.1,34–36 Notably, while the 2′-hydroxyl clearly reduces the plasma half-life of an aptamer, it often plays a key role in aptamer folding/structure, and global substitution generally leads to a loss of function. To identify aptamers modified at their 2′ position, the SELEX process can be carried out using random sequence pools containing pyrimidines modified at their 2′ positions. To further improve their stability, however, the purine nucleotides must be individually tested for the ability to accommodate stabilizing 2′-O-methyl modifications. While most purines can generally be substituted, a handful of positions are often required for function. For example, the lead clinical aptamer against VEGF (Macugen; Eyetech/Pfizer) contains two natural adenosines which are required to maintain binding affinity to the VEGF protein.
Exonucleases can be blocked by appropriate modifications to the 5′- and 3′-ends of an aptamer. Addition of a 3′-3′-linked thymidine cap prevents 3′-5′ exonuclease degradation from the 3′-terminus.35 Similarly, 5′-caps (such as PEG adducts described below) prevent exonuclease degradation from the 5′-terminus to increase aptamer residence times in the blood.
Clearance
Even with extensive modification to block nuclease degradation, stabilized molecules must also exhibit molecular weights of greater than 40 kD to remain in circulation for extended periods of time. A variety of studies have shown that complexation to form high molecular weight conjugates dramatically increases the serum half-life of aptamers. While several strategies have been enabled (including protein-aptamer complexation, tagging with lipids such as cholesterol, and attachment to liposomes), most efforts have been concentrated on PEGylation. High molecular weight PEGs can be covalently attached to aptamers without substantially altering their ability to tightly bind to targets. At the same time, these modifications have a profound effect on aptamer half-life in animals, extending aptamer half-life from minutes (no PEG) to several hours (40 kD PEG). Conjugation with 40 kD PEG at its 5′-terminus increases the in vivo half-life of the thrombin aptamer from 24 min to 6 h in rats, with little or no effect on thrombin binding affinity.34
Toxicity and Immunogenicity
Aptamers as a class have demonstrated little or no toxicity or immunogenicity. In chronic dosing of rats or woodchucks with high levels of aptamer (10 mg/kg daily for 90 days), no toxicity is observed by clinical, cellular, or biochemical measures.37,38 Whereas the efficacy of many monoclonal antibodies can be severely limited by immune responses against antibodies themselves, it is extremely difficult to elicit antibodies to aptamers (most likely because aptamers cannot be presented by T-cells via the major histocompatibility complex and the immune response is generally trained not to recognize nucleic acid fragments).
Administration
Whereas most antibody therapeutics are administered by intravenous infusion (typically over 2–4 h), aptamers can be administered by either intravenous or subcutaneous injection. This difference is primarily due to the comparatively low solubility and thus large volumes necessary for most therapeutic monoclonal antibodies. With good solubility (>150 mg/mL) and comparatively low molecular weight (aptamer: 10–50 kD; antibody: 150 kD), a weekly dose of aptamer may be delivered by injection in a volume of less than 0.5 mL. Aptamer bioavailability via subcutaneous administration is >80% in monkey studies.30 There is also evidence that nucleic acid-based therapeutics can be dosed topically and via pulmonary administration.
Scalability and Cost
Therapeutic aptamers are chemically synthesized and consequently can be readily scaled as needed to meet production demand. Whereas difficulties in scaling production have limited the availability of some biologics (e.g., Enbrel, Remicade) and the capital cost of a large-scale protein production plant is enormous, a single large-scale synthesizer can produce upwards of 100 kg of oligonucleotide per year and requires a relatively modest initial investment. The current cost of goods for aptamer synthesis at the kilogram scale is estimated at $500/g, comparable to that for highly optimized antibodies. Continuing improvements in process development are expected to lower the cost of goods to less than $100/g in 5 years.
Stability
Therapeutic aptamers are chemically robust. They are intrinsically adapted to regain activity following exposure to heat, denaturants, etc. and can be stored for extended periods (more than 1 yr) at room temperature as lyophilized powders.
Examples of Therapeutic Aptamers Now Under Development
Antithrombin Aptamer
The aptamer drug ARC183 (Archemix Corp) is a thrombin inhibitor in development for use as an anticoagulant during coronary artery bypass graft procedures. Currently, the only approved anticoagulant for coronary artery bypass graft is heparin. Significant advantages of heparin are low cost and the ability to rapidly reverse the action of heparin using protamine. However, heparin-protamine treatment has a number of serious side-effects including bleeding and thrombocytopenia (platelet count reduction) which is often asymptomatic but may be associated with life-threatening or fatal arterial or venous thrombosis. Consequently, a number of newer, higher-cost anticoagulants, such as low molecular weight heparins and Angiomax, are being developed for this market. However, these compounds have similar side-effects and their anticoagulation activity cannot be reversed rapidly. There is a significant unmet medical need for a safe, moderate-cost anticoagulant that does not require a separate reversing agent. An aptamer with a short in vivo half-life that specifically inhibits thrombin could fill this unmet need.
Anti-thrombin aptamer ARC183 is an all-DNA molecule, 15 nucleotides in length, and comprised entirely of unmodified G and T residues. The aptamer exhibits a KD of 2 nM for thrombin, 50 nM for prothrombin, and binding to other serum proteins or proteolytic enzymes is essentially undetectable.39 ARC183 is a strong anticoagulant in vitro, and inhibits thrombin-catalyzed activation of fibrinogen, and thrombin-induced platelet aggregation.42 ARC183 has key advantages in that it avoids heparin use and the risk of associated thrombocytopenia, is a specific inhibitor with rapid onset, is effective at inhibiting clot-bound thrombin, and has a short in vivo half-life of approximately 2 min which allows for rapid reversal of its effects and the avoidance of dose-adjusting complications of heparin and protamine. Neither significant toxicities nor excessive bleeding intra-operatively has been observed.
ARC183-dependent anti-coagulation occurs within minutes. It is a potent anticoagulant in dog and monkey models of cardiopulmonary bypass, yielding dose-dependent activated clotting time (ACT) values of 1500 seconds, at a 0.5 mg/kg/min dose.40,41 ARC183 exhibits a very short functional half-life in vivo of approximately 2 min, thus allowing for rapid reversal of the anticoagulant effects. No acute toxicities have been reported with ARC183, suggesting that the agent will continue to show an improved efficacy and safety profile over that of heparin (Archemix Corp., unpublished data). Clinical trials of ARC183 are now underway.
Anti-VEGF Aptamer
The furthest advanced aptamer in the clinic is an anti-VEGF aptamer, Macugen, being developed by Eye-tech Pharmaceuticals and Pfizer as an anti-angiogenesis factor for the potential treatment of AMD. AMD is a leading cause of blindness in the elderly.42,43 Choroidal neovascularization, associated with exudative AMD, is the mediator of this irreversible vision loss. Approximately 1.2 million people in the United States have exudative AMD with 200,000 new cases occurring yearly. A number of studies have suggested that anti-angiogenesis therapy may be useful in treating ocular neovascularization events.44–50 Much of this anti-angiogenesis work has focused on VEGF as a target. VEGF has also been implicated in the leakage of blood vessels. Anti-VEGF therapy, therefore, could have a dual effect, both inhibiting angiogenesis and blocking blood vessel leakage.
Macugen is a specific anti-VEGF-165 aptamer with a KD of approximately 50 pM that inhibits the binding of VEGF-165 to its receptors (fms-like tyrosine kinase-1 and to the kinase insert domain-containing region). Using experimental models, Macugen inhibited VEGF-mediated vascular leakage as measured by a corneal micropocket assay (Miles assay) in guinea pigs.51 Macugen has also shown efficacy as an anti-angiogenesis agent in a rat corneal angiogenesis model (65% decrease compared with controls) and a mouse retinopathy of prematurity model (80% inhibition compared with controls).51 Macugen was also effective in the inhibition of blood-retinal barrier breakdown in a diabetic rat model.52
The pharmacokinetic properties of Macugen have been studied. Dosed at 1 mg/kg by intravenous injection, Macugen exhibits a systemic half-life of around 9.3 h in monkeys. Macugen has a half-life of approximately 94 h when administered by intravitreal injection at a dose of 1 mg/eye in monkeys.53 No toxic effects were observed in either of these studies. Pre-clinical toxicity studies in rhesus monkeys further demonstrated no toxic effects and no immune response to Macugen.54
A Phase IA trial using 0.25–3.0 mg/eye intravitreal injections of Macugen in 15 patients with exudative AMD did not reach dose-limiting toxicity. Furthermore, 80% of these patients had improved or stabilized vision after 3 months, and 27% of these same patients had a three or more line improvements on the Early Treatment for Diabetic Retinopathy Study Chart (ETDRS).54 A Phase II study involving AMD patients undergoing repeated injections of Macugen revealed that 88% of patients had stabilized or improved vision after 3 months and 25% of eyes demonstrated a 3 lines or greater improvement in the ETDRS with Macugen. Sixty percent of patients who received photodynamic therapy in addition to Macugen had a 3-line improvement in the ETDRS at 3 months; 2.2% of patients treated with photodynamic therapy alone showed similar improvements in visual acuity.55 The Phase II/III trial enrolled 1186 patients. The patients were randomized to treatment with 0.3 or 3.0 mg/eye Macugen or sham injection and followed for 54 weeks. All patients in the Macugen treated group had significantly less disease progression than patients in the untreated group. Six percent of the Macugen-treated patients gained three or more ETDRS lines compared with 2% of the control group over the 54-week period. As in early Phase II studies, combining Macugen with photodynamic therapy could significantly increase the 6% success rate.
Macugen is the first aptamer to undergo clinical testing. Early results are promising. Macugen is well-tolerated in humans and has not demonstrated any toxicity. The results of these studies reveal that the inhibition of VEGF-mediated biological activity by an aptamer is a promising treatment strategy for the prevention of ocular neovascularization and macular edema.
Anti-PDGF Aptamer
Both in vitro and in vivo studies (see above) indicate that PDGF-B is a valid target for the relief of interstitial tumor pressure (IFP) as a method for increasing uptake of chemotherapeutics into tumors. Those same studies demonstrate that an aptamer can effectively inhibit PDGF-B–dependent IFP and increase the uptake of Taxol, leading to tumor growth inhibition.
An excellent example of a target validation tool being quickly converted to a therapeutic is ARC127, one of several PDGF-B binding aptamers that is currently being developed as a component of combination therapies for oncology indications by Archemix. ARC127 is 29 nucleotides in length. It binds specifically to PDGF-B and not PDGF-A, with 100 pM affinity. Modifications of certain residues in the aptamer with 2′-O-methyl moieties have been shown to dramatically increase in vitro stability without altering the aptamer affinity. Addition of an inverted thymidine residue at the 3′-end can provide further stability by increasing resistance of the aptamer to degradation by 3′ to 5′ exonucleases. A 40-kD PEG group covalently attached via an amine bond at the 5′-end has been shown to increase the in vivo half-life of the aptamer from minutes to 8 h when administered intravenously at a dosage of 10 mg/kg.56,57 ARC127 is also being considered for other indications where PDGF-B has been implicated, including glomerulonephritis, pulmonary hypertension, and intimal hyperplasia following percutaneous transluminal angioplasty.56,57
SUMMARY
Given their small size, ease of synthesis, and low cost, aptamers provide versatile tools for validation of intracellular and extracellular targets. In the case of extracellular targets, such as VEGF, thrombin, and PDGF discussed here, aptamer-based validation affords a direct path to therapeutic development. Therapeutic aptamer leads can be readily stabilized or shielded from renal filtration by chemical or compositional modification for evaluation in in vivo preclinical discovery programs. Results of clinical trials of Eyetech/Pfizer’s aptamer therapeutic for treatment of AMD are extremely encouraging and bode well for the future of aptamer therapeutics in general. Indeed, the FDA has granted fast-track status for Macugen, reflecting promise of this aptamer-based drug to offer treatment for prevention of ocular disease in patients who have few therapeutic options. With a number of additional aptamers expected to enter into clinical trials over the next year, aptamers appear poised to make a significant contribution to the treatment of acute and chronic diseases.
Acknowledgments
The authors thank Kristian Pietras for helpful comments concerning this manuscript and for permission to use his published figures.
REFERENCES
- 1.Jellinek D, et al. Potent 2′-amino-2′-deoxypyrimidine RNA inhibitors of basic fibroblast growth factor. Biochemistry 1995;34:11363–11372. [DOI] [PubMed] [Google Scholar]
- 2.Seiwert SD, Stines Nahreini T, Aigner S, Ahn NG, Uhlenbeck OC. RNA aptamers as pathway-specific MAP kinase inhibitors. Chem Biol 2000;7:833–843. [DOI] [PubMed] [Google Scholar]
- 3.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacterial T4 DNA polymerase. Science 1990;249:505–510. [DOI] [PubMed] [Google Scholar]
- 4.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 5.Cox JC, et al. Automated acquisition of aptamer sequences. Comb Chem High Throughput Screen 2002;5: 289–299. [DOI] [PubMed] [Google Scholar]
- 6.Cox JC, et al. Automated selection of aptamers against protein targets translated in vitro: From gene to aptamer. Nucleic Acids Res 2002;30:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marshall KA, Ellington AD. In vitro selection of RNA aptamers. Methods Enzymol 2000;318:193–214. [DOI] [PubMed] [Google Scholar]
- 8.Pich EM, Epping-Jordan MP. Transgenic mice in drug dependence research. Ann Med 1998;30:390–6. [DOI] [PubMed] [Google Scholar]
- 9.Hannon GJ. RNA interference. Nature 2002;418:244–251. [DOI] [PubMed] [Google Scholar]
- 10.Taylor MF, Wiederholt K, Sverdrup, F. Antisense oligonucleotides: A systematic high-throughput approach to target validation and gene function determination. Drug Discov Today 1999;4:562–567. [DOI] [PubMed] [Google Scholar]
- 11.Elbahir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494–498. [DOI] [PubMed] [Google Scholar]
- 12.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 2003;34:263–264. [DOI] [PubMed] [Google Scholar]
- 13.Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol 2002;5:834–839. [DOI] [PubMed] [Google Scholar]
- 14.Persengiev SP, Zhu X, Green MR. Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA 2004;10:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett CF. Efficiency of antisense oligonucleotide drug discovery. Antisense Nucleic Acid Drug Dev 2002; 12:215–24. [DOI] [PubMed] [Google Scholar]
- 16.Lichtlen P, Auf der Maur A, Barberis A. Target validation through protein-domain knockout-applications of intracellularly stable single-chain antibodies. Targets 2002;1:37–44. [Google Scholar]
- 17.Aramburu J, et al. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 1999;285:2129–2133. [DOI] [PubMed] [Google Scholar]
- 18.Chan R, Gilbert M, Thompson K, Marsh HN, Epstein D, Pendergrast PS. Co-expression of anti-NFKB RNA aptamers and siRNA leads to maximal suppression of NF-KB activity in mammalian cells. Nucleic Acids Res Methods (in press). [DOI] [PMC free article] [PubMed]
- 19.Lee SW, Gallardo HF, Gaspar O, Smith C, Gilboa E. Inhibition of HIV-1 in CEM cells by a potent TAR decoy. Gene Ther 1995;2:377–384. [PubMed] [Google Scholar]
- 20.Symensma TL, Giver L, Zapp M, Takle GB, Ellington AD. RNA aptamers selected to bind human immunodeficiency virus type 1 Rev in vitro are Rev responsive in vivo. J Virol 1996;70:179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi H, Hoffman BE, Lis JT. RNA aptamers as effective protein antagonists in a multicellular organism. Proc Natl Acad Sci USA 1999;96:10033–10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ring HZ, Lis JT. The SR protein B52/SRp55 is essential for Drosophila development. Mol Cell Biol 1994;14:7499–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraus ME, Lis JT. The concentration of B52, an essential splicing factor and regulator of splice site choice in vitro, is critical for Drosophila development. Mol Cell Biol 1994;14:5360–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blind M, Kolanus W, Famulok M. Cytoplasmic RNA modulators of an inside-out signal-transduction cascade. Proc Natl Acad Sci USA 1999;96:3606–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayer G, et al. Controlling small guanine-nucleotide-exchange factor function through cytoplasmic RNA intramers. Proc Natl Acad Sci USA 2001;98:4961–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pietras K, et al. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res 2001;61:2929–2934. [PubMed] [Google Scholar]
- 27.Pietras K, et al. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res 2002;62:5476–5484. [PubMed] [Google Scholar]
- 28.Heldin C, et al. Higher interstitial fluid pressure—An obstacle in cancer therapy. Nature 2004;4:806–813. [DOI] [PubMed] [Google Scholar]
- 29.Green LS, et al. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry 1996;35: 14413–14424. [DOI] [PubMed] [Google Scholar]
- 30.Tucker CE, et al. Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. 1999;732:203–12. [DOI] [PubMed]
- 31.Watson SR, et al. Anti-L-selectin aptamers: binding characteristics, pharmacokinetic parameters, and activity against an intravascular target in vivo. Antisense Nucleic Acid Drug Dev 2000;10:63–75. [DOI] [PubMed] [Google Scholar]
- 32.Reyderman L, Stavchansky S. Pharmacokinetics and biodistribution of a nucleotide-based thrombin inhibitor in rats. Pharm Res 1998;15:904–910. [DOI] [PubMed] [Google Scholar]
- 33.Reyderman L, Stavchansky S. Quantitative determination of short single-stranded oligonucleotides from blood plasma using capillary electrophoresis with laser-induced fluorescence. Anal Chem 1997;69:3218–3222. [DOI] [PubMed] [Google Scholar]
- 34.Pieken WA, et al. Structure-function relationship of hammerhead ribozymes as probed by 2′-modifications. Nucleic Acids Symp Ser 1991;51–53. [PubMed]
- 35.Dougan H, et al. Extending the lifetime of anticoagulant oligodeoxynucleotide aptamers in blood. Nucl Med Biol 2000;27:289–297. [DOI] [PubMed] [Google Scholar]
- 36.Cummins LL, et al. Characterization of fully 2′-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res 1995; 23:2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson FC, Kuchta RD, Mazurkiewicz A, Richardson KA. Polymerization of 2′-fluoro- and 2′-O-methyl-dNTPs by human DNA polymerase alpha, polymerase gamma, and primase. Biochem Pharmacol 2000;59: 1045–1052. [DOI] [PubMed] [Google Scholar]
- 38.Richardson FC, et al. An evaluation of the toxicities of 2′-fluorouridine and 2′-fluorocytidine-HCl in F344 rats and woodchucks (Marmota monax). Toxicol Pathol 1999;27:607–617. [DOI] [PubMed] [Google Scholar]
- 39.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992;355:564–566. [DOI] [PubMed] [Google Scholar]
- 40.Griffin LC, Tidmarsh GF, Bock LC, Toole JJ, Leung LL. In vivo anticoagulant properties of a novel nucleotide-based thrombin inhibitor and demonstration of regional anticoagulation in extracorporeal circuits. Blood 1993;81:3271–3276. [PubMed] [Google Scholar]
- 41.DeAnda A, Jr, et al. Pilot study of the efficacy of a thrombin inhibitor for use during cardiopulmonary bypass. Ann Thorac Surg 1994;58:344–350. [DOI] [PubMed] [Google Scholar]
- 42.Ghafour IM, Allan D, Foulds WS. Common causes of blindness and visual handicap in the west of Scotland. Br J Ophthalmol 1983;67:209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hyman L. Epidemiology of eye disease in the elderly. Eye 1987;1(Pt 2):330–341. [DOI] [PubMed] [Google Scholar]
- 44.Carmeliet P, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996;380:435–439. [DOI] [PubMed] [Google Scholar]
- 45.Carmeliet P, et al. Role of tissue factor in embryonic blood vessel development. Nature 1996;383:73–75. [DOI] [PubMed] [Google Scholar]
- 46.Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis. Recent Prog Horm Res 2000;55:15–35;discussion 35–36. [PubMed] [Google Scholar]
- 47.Ferrara N, Bunting S. Vascular endothelial growth factor, a specific regulator of angiogenesis. Curr Opin Nephrol Hypertens 1996;5:35–44. [DOI] [PubMed] [Google Scholar]
- 48.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989;246:1306–1309. [DOI] [PubMed] [Google Scholar]
- 49.Senger DR, et al. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev 1993; 12:303–324. [DOI] [PubMed] [Google Scholar]
- 50.Kim KJ, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993;362:841–844. [DOI] [PubMed] [Google Scholar]
- 51.The Eyetech Study Group. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina 2002;22:143–152. [DOI] [PubMed] [Google Scholar]
- 52.Ishida S, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med 2003;198:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drolet DW, et al. Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharmaceutical Research 2000; 17:1503–1510. [DOI] [PubMed] [Google Scholar]
- 54.Vinores SA. Technology evaluation: Pegaptanib, Eye-tech/Pfizer. Curr Opin Mol Ther 2003;5:673–679. [PubMed] [Google Scholar]
- 55.The Eyetech Study Group. Anti-vascular endothelial growth factor therapy for subfoveal choroidal neovascularization secondary to age-related macular degeneration: phase II study results. Ophthalmology 2003;110: 979–986. [DOI] [PubMed] [Google Scholar]
- 56.Floege J, et al. Novel approach to specific growth factor inhibition in vivo: Antagonism of platelet-derived growth factor in glomerulonephritis by aptamers. Am J Pathol 1999;154:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ostendorf T, et al. Specific antagonism of PDGF prevents renal scarring in experimental glomerulonephritis. J Am Soc Nephrol 2001;12:909–918. [DOI] [PubMed] [Google Scholar]