

Abstract
Edman degradation sequencing relies on comparing high-performance liquid chromatography retention times of the sample phenylthiohydantoin amino acids with phenylthiohydantoin amino acid standards. The elution characteristics of the twenty common amino acids have been well characterized, which aids in making confident assignments. Modified amino acids may present more of a challenge since they are not part of the commonly used standards and because the protein sequencer analyst may not have experience with them. Laboratories requesting a sample were sent a tube containing approximately 775 pmoles of a 20-amino-acid synthetic peptide composed of several modified amino acids that may be found in proteins or are generated during sample preparation. In addition to filling in an assignment sheet, which included retention times and peak areas, participants were asked to provide specific details about the parameters used for the sequencing run. References for some of the modified amino acid elution characteristics were provided and the participants had the option of viewing a list of the modified amino acids present in the peptide at the Edman Sequencing Research Group website (ESRG). The goal of the study consisted of two parts: assessment of the ability to correctly assign all the amino acids in the peptide, including the modified amino acids; and the collection and compiling of elution time characteristics of modified amino acids for instruments used in the study. The resulting compilation of the modified amino acid elution times and running conditions will be accessible at the Association of Biomolecular Resource Facilities (ABRF) ESRG website for future reference. The ABRF ESRG 2004 sample is the 16th in a series of studies designed to aid laboratories in evaluating their abilities to obtain and interpret amino acid sequence data.
Keywords: Edman sequencing, chemical sequencing, modified amino acid, phenylthiohydantoin (PTH) amino acid, retention time
The Edman Sequencing Research Group (ESRG) is a research group of the Association of Bio-molecular Resource Facilities (ABRF), which from its inception has been dedicated to promoting excellence in the field of protein and peptide sequence analysis by Edman chemistry, a chemical procedure that analyzes a protein or peptide one amino acid at a time, starting from the N-terminus. Over the years, this group has directed numerous studies that have focused on various aspects of Edman sequencing, often allowing the participating facilities to directly evaluate their own performance compared with others.1–15 These studies have also strived to benefit the general scientific community by collecting and disseminating methods and instrument-specific data related to each study.
In this the 16th ESRG study, we chose to analyze the elution characteristics of several common modified amino acids on currently operating sequencer systems. The last attempt to compile elution conditions of modified amino acids was performed by Mark Crankshaw and Greg Grant in 1992.16 To investigate any changes resulting from sequencer or chromatography differences since then, a synthetic peptide containing seven modified amino acids was synthesized by the ESRG and distributed to interested ABRF members as well as to others from the general scientific community. Participants were asked to return their results electronically, filling in a spread sheet with the retention times and peak areas of the phenylthiohydantoin amino acid (PTH-AA) standards and the amino acids from the synthetic peptide. A list of the modified amino acids contained in the peptide was placed on the ESRG web site, available as an optional aid in calling the sequence, and participants were challenged to identify as many modified amino acids as possible. The ESRG tabulated retention times and peak areas for both the known standards and the called amino acids in the peptide. We also calculated repetitive yields and other data related to the specific instruments used in the analyses.
MATERIALS AND METHODS
The sequence of the synthetic peptide was chosen carefully, and is shown in Figure 1A. Note the repeats of Tyr (at positions 1, 10, and 17) and Ala (at positions 3, 8 and 18), which were used to calculate initial and repetitive yields. For each of the modified amino acids [except Met(O) and Cys-S-β-PAM] there was at least one unmodified version of the same amino acid to allow accurate comparison of each in a single sequence run. The inclusion of two Trp residues (4 and 15) enabled a reasonable level of confidence in the analysis of this more difficult amino acid. These residues, in addition to the multiple Tyr residues, also permitted quantitation by absorption at γ = 280 nm if this was deemed necessary. The C-terminal Arg residue imparted a strong basic residue to minimize sample washout from the reaction cartridge during sequence analysis and mimicked a typical tryptic peptide’s behavior in this regard. Overall, the length and composition of the peptide were also deemed reasonable for synthesis. Figure 1B shows the structures of the modified amino acids that were incorporated into the synthetic peptide for the 2004 study.
FIGURE 1.

Synthetic peptide that was sent to participating laboratories. A: Sequence of the synthetic polypeptide. B: Structures of modified amino acids in the peptide.
Peptide Synthesis and Preparation
The 2004 ESRG peptide was synthesized using Fmoc chemistry. With the exception of Fmoc-Lys(Me) and Fmoc-Arg(Me)2, which were coupled manually, all couplings were done on an Applied Biosystems (ABI, Foster City, CA) 433A peptide synthesizer using ABI reagents and standard protected Fmoc amino acids. All protected Fmoc amino acids were from ABI except for the following. Fmoc-Arg[Pbf]-Wang resin and Fmoc-Hyp(tBu)-OH were obtained from Nova-Biochem (San Diego, CA). Fmoc-Arg(Me)2-OH and Fmoc-Lys(Me)(Boc)-OH were obtained from Bachem Bioscience (King of Prussia, PA). Fmoc-Lys(Ac)-OH, Fmoc-Met(O)-OH and Fmoc-Ser[PO(OBzl)-OH]-OH were obtained from AnaSpec (San Jose, CA). After each coupling cycle, unreacted residues were capped with acetic anhydride. The completed peptide was cleaved and deprotected using ethanedithiol:thioanisole:trifluoroacetic acid (1:1:8) for 3 h, precipitated and washed with diethyl ether, purified on a 10 × 250-mm Vydac C18 reversed-phase column, and lyophilized. The Cys alkylation was performed with a one-milligram aliquot of lyophylized peptide, solubilized in 450 μL 50 mM Tris (pH 8.1)/50% acetonitrile plus 50 μL of 30% acrylamide/water. After sitting at ambient temperature for 50 min, the peptide was purified with multiple injections on a Phenomenex Jupiter C18 2.1 mm × 220-mm column, eluting with 0.1% trifluoroacetic acid/acetonitrile solvents. The peptide peaks from all runs were pooled and 25-μL aliquots were placed in polymerase chain reaction (PCR) tubes, dried on a Speed Vac spinning concentrator, and stored at −20°C until shipped. Amino acid analysis indicated approximately 775 pmol of peptide per tube. A total of 200 tubes were prepared.
Sample Distribution
The ESRG announced the 2004 study by e-mail to all ABRF members via the discussion board, as well as on the main ABRF page under “Open Research Studies” and on the ESRG page. A total of 42 requests for samples were received. Samples were sent out by regular mail to all who requested samples.
Sample Analysis
ESRG members performed initial analyses to evaluate the quality of the peptide and data. All eight members analyzed the samples as they would normally for similar synthetic peptides. They obtained very comparable results, which imparted confidence in the design and synthesis of the peptide for the study. Their results for retention times for the modified PTH-AA were included in the overall tabulation of retention times (see Table 3 below).
TABLE 3A.
Relative Retention Times of Amino Acids Normalized to Ala for the Procise Sequencers

TABLE 3B.
Relative Retention Times of Amino Acids Normalized to Ala for the ABI 477, HP G1005A, and the Porton 2090e

Participating laboratories were told that each sample contained approximately 775 pmol of peptide that would be readily soluble in 50–100 μL of 30% acetonitrile with 0.1% trifluoroacetic acid. Using this knowledge of sample concentration, the laboratories were asked to load enough to obtain a good quality read for 20 cycles using their normal analytical conditions for this run.
Data Reporting
Each laboratory was asked to return data to the ESRG using an electronically transmitted Excel table that was sent to all participating laboratories. The table had entry slots for the retention times, picomole values, and peak areas of all the standard PTH-AAs as well as each of the 20 cycles for the synthetic peptide. As several of the modified PTH-AAs result in more than one peak, the laboratories were asked to report the observed peaks in order of their elution from the column. Laboratories were asked to report known amino acids using the common three-letter amino acid code, to report “X” for unknown amino acids, and to report “–” for no observed amino acid peaks for each cycle. They were also asked to indicate their confidence level in the call by placing parentheses around tentative calls.
In order to help participating laboratories identify the modified amino acids, the ESRG posted a list of the modified amino acids found in the peptide, but not their relative position in chromatograms. In addition, a reference of elution characteristics for commonly found modified amino acids originally compiled by Crankshaw and Grant16 in 1992 was also available on the ESRG web site. Participants were asked to identify, as best they could, the unknown PTH-AAs using this reference or their own expertise and to report their assignments at each cycle.
The study also included an instrument survey to determine the analytical conditions for each instrument used, including gradient conditions, buffers and solvents, chemistry cycles, and other parameters that may have affected the results of the study. All of these data, compiled from a total of 28 participating laboratories (including those of the ESRG) were analyzed for this report.
RESULTS AND DISCUSSION
The most commonly used instruments for this year’s study were from Applied Biosystems, Inc. The reason for this lies in the fact that ABI is the only manufacturer still selling and supporting high-sensitivity chemical sequencers in North America. Other chemical sequencers used in prior years and to a lesser extent in this year’s study were from Porton, Hewlett Packard, Shimadzu, or Beckman, and are either not high-sensitivity instruments (< 10 pmol minimal load), are discontinued, obsolete, or are no longer supported by the manufacturers. Some of these sequencers, however, can still be found in operation in some core laboratories generating excellent data. Instruments such as these are often operated by highly skilled chemists who know how to maintain and tweak the instruments for maximum sensitivity and accuracy. The Hewlett Packard G1005A is an example. This instrument has consistently out performed the ABI instruments in repetitive yield over the years, but its use has declined and this year only one laboratory used it for analyzing the synthetic peptide.
IDENTIFICATION ACCURACY
The accuracy of the calls returned by the participating laboratories is found in Figure 2 and in Table 1 . Each entry shows the facility number, instrument type, and percent sample loaded on the instrument. As in previous studies, “PC” stands for a positive (high confidence) correct determination, “TC” for a tentative correct determination, “PW” for a positive (high confidence) wrong determination, “TW” for a tentative wrong determination, and “X” indicates no call (“–” or “X” reported) at any given cycle.
FIGURE 2.

Graphic representation of accuracy in identifying amino acids during sequencing. PC, positive correct (solid bars); TC, tentative correct (vertical hatched lines); PW, positive wrong (horizontal hatched lines); TW, tentative wrong (diagonal hatched lines); X, no identification given (open bars).
TABLE 1A.
Facility Number and Submitted Amino Acid Calls for Every Position in the Synthetic Peptide

TABLE 1B.
Analysis of data submitted by each facility, including % loaded, initial yield, repetitive yield, and correct and incorrect calls

The overall accuracy of the calls for the unmodified amino acids was excellent for all the participating laboratories. This is hardly surprising, as the sequence was repetitive and a large amount of sample was provided for each laboratory. Accuracy of the determinations of the modified amino acids was significantly lower, however, possibly reflecting the lack of experience of participating laboratories in this determination by Edman sequencing. Few references of retention times of modified amino acids have been published. We had posted the Crankshaw/Grant reference16 on the ESRG web site to give all participants an equal chance at identifying the modified residues.
The first modified amino acid in the peptide, N-N-dimethyl arginine (dmR) was at cycle 2, and was the most poorly identified residue in the entire study, with only 5 PC, 1 TC, 8 PW, and 3 TW calls out of 20. In one instance, dmR eluted with essentially the same retention time as unmodified Arg, and was identified as Arg. In most other cases of incorrect identification, this residue was called as N-ɛ-methyl lysine (also called monomethyl lysine, or mmK). The most probable reason for this low accuracy was the fact that the elution profile for dmR was not included in the Crankshaw/Grant reference,16 demonstrating the general lack of other resources to help identify rare elution profiles of modified amino acids. Since we did list on the ESRG web site the modified amino acids included in the peptide, those who did identify it might have done so by the process of elimination. But this is speculation since we did not ask how the individual laboratories figured out each call. The major peak observed eluted near Tyr, particularly on the Procise cLC. The reported peak areas (Relative Yields in Table 3 below) were near the full average peak areas expected from calculated repetitive yields (e.g., Fig. 3).
FIGURE 3.

Trend line for plot of log10 (peak area) vs. sequencing cycle used to calculate repetitive yields. The data were taken from areas of Tyr and Ala peaks reported by facility 19. The slope of the trend line is the log10 of the repetitive yield.
The second modified amino acid in the peptide was N-ɛ-methyl lysine (mmK) at position 5. This was the second most difficult to identify amino acid in the study, with seven PC, one TW, four PW and three TW calls out of 20. The most likely reason for this low score was that again this amino acid was not correctly located in the chromatogram from the Crankshaw/Grant reference16 that was placed on the ABRF web site. In that reference a single peak eluting near Tyr (and thus approximately where dmR eluted in this study) is reported for mmK. A peak from mmK eluting at this earlier position may be indicative of incomplete phenylthiocarbamylation of the methylated Lys side chain. In their discussion, Crankshaw/Grant16 mention that there was variability in the reported positions where the PTH derivatives of mono- and dimethyl lysine eluted. Therefore they tested samples of the methylated lysines on an ABI 477A sequencer and found that the major peak for mmK eluted just after Leu, with a smaller peak between DMPTU and Ala. Unfortunately the position of that major peak was omitted from their summary chromatogram. It is clear from the present ESRG study that the major peak indeed elutes after Leu, with only a minor peak eluting between Ala and Arg. In three instances, the raw data presented by study participants did not include any peak eluting after Leu at position 5, indicating that the most likely cause for missing this peak in those cases was that data collection and integration stopped prior to elution of the main mmK peak. Peak area observed was again comparable to the average area expected from calculated repetitive yields.
The third modified amino acid occurred at position 7. This residue was phosphorylated Ser (pS). There were 10 PC, 2 TC, 2 PW, 2 TW, and 4 NC reported. This residue is generally unstable to Edman degradation, and no significant peak is typically observed for this amino acid. More than half of the facilities, however, correctly identified phosphorylated Ser, either by observing a small peak at the normal position for Ser and a large peak at the position for Ser′(dehydroalanine adduct). They could also compare the results from this residue with the results obtained in cycle 14 for a normal, unmodified Ser residue. One laboratory derivatized the peptide by treatment with aminoethane thiol to the PTH-Ser-2-aminoethanethiol and they were able to identify the modified amino acid.17,18
The fourth modified amino acid was an oxidized Met (oxM). This modification was difficult to identify because the PTH-Met(O) is readily converted to PTH-Met in the presence of reducing agents commonly found in some of the sequencer chemicals, e.g. dithiothreitol (DTT) in ABI ethyl acetate (S2B) or normally present in R4. Thus, we counted both Met and Met(O) as correct for this position, giving a total of 18 PC calls, one TC, and one NC. Of the 19 correct calls, 14 identified the residue as Met. Of the 5 that called the modified amino acid correctly, one laboratory using a Procise-cLC reported a peak between Gln and Thr. Three facilities deduced the presence of Met(O) by mass spectrometry, although 2 of these still reported PTH-Met in their tabulations with a footnote stating that mass spectrometry indicated that the initial peptide contained Met(O). The list of modified amino acids posted on the web site contained an oxidized Met, which might have helped some groups identify it as Met(O), since there were no other Met residues present in the peptide. These observations demonstrate the problem with using Edman sequencing as a means for identifying oxidized methionine.
The fifth modified amino acid was a fairly common one, cysteine-S-β-propionamide (spC), formed by reaction of free Cys with acrylamide,19 a frequent side reaction of protein purification on SDS-PAGE gels. It was well identified, with 15 PC calls reported. A major peak eluting after Glu and before His was easily identified as this common Cys derivative.
The sixth modified amino acid in the peptide was N-ɛ-acetyl lysine (acK), which was also well identified, with 15 PC calls reported. The peak eluted in a similar position as was described by Crankshaw and Grant16 and by Ross et al.,20 and was detected eluting before Ala on the ABI sequencers in this study. The yield or recovery was slightly greater than for most of the amino acids, allowing for clear detection.
The final modified amino acid was another fairly common one, 4-hydroxyproline (hyP), which was reported with 14 PC calls. This amino acid gives a pair of peaks, one eluting just before His and the other after Ala with a ratio of roughly 3:5 as observed with the 49X HT sequencers. Some respondents reported only one peak, which is likely due to the chromatography conditions used by those laboratories, making it possible for one of the peaks to coelute with either His or Ala. This amino acid is commonly observed in collagen sequences and many experienced sequencers are familiar with its identification.
Repetitive and Initial Yields
All participating facilities were asked to return primary data to the ESRG to allow a systematic procedure to derive initial yields and repetitive yields for the study. For this purpose, peak areas of all PTH-AA standards and those of called amino acids at each cycle were tabulated to obtain the initial yield using the following formula:
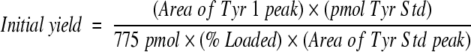 |
where “775 pmol” is the total supplied sample for the study, “% Loaded” is as reported by each facility, and the remaining values are as stated for each data set reported. The results for each facility are shown in Table 1.
Repetitive yields were calculated using the multiple Tyr and Ala residues designed specifically for this purpose in the synthetic peptide. Tyr was at positions 1, 10, and 17, while Ala was at positions 3, 8, and 18, giving 6 positions out of 20 from which to calculate the repetitive yield. Repetitive yields were calculated from the slope of the Excel trend line of a plot of log A as a function of sequencing cycle, where values of A are peak areas of Tyr or Ala residues in the sample sequence. The slope of the trend line is the log of the repetitive yield, as shown in Figure 3. The average repetitive yield for the study was 86.6 % for the 13 participating ABI 49x HT sequencers, 86.6% for the 6 ABI 49x cLC sequencers, 95.6% for 1 ABI 477 from which data were supplied, and 91.2% for 1 Hewlett Packard G1005A. Initial and repetitive yields obtained on sequencers operated by members of the ESRG were similar to those calculated for participating facilities. These repetitive yields were lower than expected. Although repetitive yields when sequencing peptides are sometimes low due to sample washout, this was not the case in this study. Analyzing the data showed that the yield held steady as sequencing progressed, rather than dropping as the peptide became shorter, which would have indicated sample washout. In fact, for several facilities, repetitive yields calculated using amino acids at positions 8, 10, 17, and 18 were higher than those that included Tyr 1 and Ala 3, suggesting that sequencer performance improved after the initial cycles. We do not have an explanation for this unexpected result. Table 1B shows the repetitive yields by facility.
Retention Times of Standard PTH-AAs
Retention times for the PTH-AA standards for the two major instruments used in this study were tabulated for further analysis. These included 16 ABI 49x-HT systems and 8 ABI 49x-cLC systems. Other results returned included those from 2 ABI 477As, one Hewlett Packard G1005A, and one Porton 2090e. Of the two ABI 477s, one facility did not return PTH-AA standard retention times and peak areas, eliminating this facility from further analysis.
The absolute retention times reported by the facilities varied considerably from each other due to column and gradient differences as well as other parameters influenced by the type of instrument used. The actual elution order for the amino acids on all the instruments was the same, with the exception of the Hewlett Packard G1005A. In order to be able to compare the facilities to each other, we first separated the results by instrument type and then normalized the retention time data within each set. Normalization of the retention times for the standard amino acids allowed us to directly compare the retention times of instruments even though they had significantly different total chromatographic start and end points. To do this normalization, we used the retention time of Ala as the common reference point, and applied the following formula:
 |
where RTnA is the retention time of any amino acid normalized to Ala; RTx is the raw retention time of amino acid x; RTA is the raw retention time of Ala; RTD is the raw retention time of Asp, the first amino acid to elute; and RTL is the raw retention time of Leu, the last amino acid to elute.
This formula divides the retention time difference between amino acid x and Ala by the total length of the chromatographic run from Asp to Leu. The results shown in Table 2 give RTnAs of around −0.3 min for Asp, RTnAs of 0 min for Ala, and RTnAs of around 0.7 min for Leu. With these normalized retention time values, standard deviations for the individual amino acids were calculated. The greatest standard deviation (0.05) was observed at the front end of the chromatographic separation for Asp and Glu, which can be explained by their higher than normal retention time variability. This is due to their charge and the interaction with the pH of the buffers used and the age of the column. At the other end of the chromatogram, there was a gradual increase in standard deviation towards Leu, again explained by greater than proportional differences in retention times due to total gradient length. As may be expected, Lys showed the greatest overall standard deviation (0.06), as its position relative to Ile and Leu is variable and can easily and frequently be adjusted in the ABI HPLC systems.
Retention Times of the PTH-AAs in the Sample
In a similar manner, we normalized the retention times reported for the called amino acids in the synthetic peptide. The raw retention times for each cycle were normalized using the data supplied for the standards from each participating facility. Again, Ala was used as the anchor point, and reported raw retention times were normalized to decimal values using the following formula:
 |
where SRTnA is the calculated retention time of a sample amino acid normalizd to Ala; SRTx is the raw retention time of amino acid x; RTA is the raw retention time of Ala; RTD is the raw retention time of Asp; and RTL is the raw retention time of Leu.
This formula allowed all reported retention times to be compared directly while minimizing differences caused by gradients or column variability. The range of values extended from −0.3 to 0.75. This was a wider range than seen for the standards since one major peak (N-ɛ-methyl lysine; mmK) eluted after Leu. Table 3 shows the results of these calculations sorted by instrument type, as well as the averages and standard deviations of these values.
Scaled Retention Times and Relative Yields of Modified PTH-AA Peaks
Once the averages of the normalized retention times (RT) of the reported peaks for the modified PTH-AAs were calculated, the following formula was used to return these decimal values to actual minutes (scaled retention times; SRT). This was done to allow a more normal description of the retention times of modified PTH-AAs compared with the standard unmodified PTH-AAs (Table 3 and Figure 4).
FIGURE 4.

Scaled elution times for standard and modified PTH amino acids for the ABI Procise HT and the ABI Procise cLC.
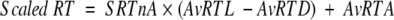 |
AvRTL, AvRTD, and AvRTA are the average retention times for Leu, Asp, and Ala, respectively, for each instrument type. (These have been drawn into a typical chromatographic separation for easy reference in Fig. 4.)
In an attempt to determine relative yields of the modified amino acids compared with the yields of the common 20 amino acids, we developed a percent yield value based on the repetitive yield and the theoretical yield of each amino acid. The TREND function in Excel used the log values of the Tyr and Ala peak areas to create a straight trend line (see Figure 3 for an example). The value of log A at each cycle number on this plot is the log of the theoretical peak area for the that cycle, and taking the antilog of that value thus gives a theoretical peak area for each cycle of sequencing. The relative yield column shown in Table 3 is the average of the reported observed peak area divided by the theoretical peak area of each sequencer group. The intention in determining these values was to show how the modified amino acid peak areas compared with the unmodified amino acids, and in the cases where there were multiple peaks, what the relative recovery was for each peak. Relative yields varied between 2% to 154% for the various instruments, showing the difficulty in seeing all of the peaks for some of the modified amino acids.
Instrument Survey
The instrument and methods survey is found in Tables 4 and 5. In brief, there were 16 ABI Procise 49x-HT systems, 8 ABI Procise 49x-cLC systems, 2 ABI 477As, one HP G1005A, and one Porton 2090e contributing to this year’s study. Nearly all of these instruments were used with standard manufacturer’s reagents and solvents, columns and chemistry cycles. This approach appears to work well, keeping the average instrument in reasonably good analytical condition, judging from the results submitted for this study. The lack of competing manufacturers means that little effort is devoted to improving the performance of current instruments or designing newer, more powerful instruments. Also, there are few supply alternatives.
TABLE 4.
Information About the Sequencers Used in This Study

TABLE 5.
Information About HPLC Systems Used in This Study

CONCLUSIONS
Five of the modified amino acids examined in this study, N,N-dimethylarginine, N-ɛ-methyl lysine, S-β-propionamidocysteine, N-ɛ-acetyl lysine, and 4-hydroxyproline yielded stable phenylthiohydantoins with distinctive retention times during Edman degradation, making them easy to identify. Phosphoserine, however, is unstable during sequencing, making its identification difficult. Nonetheless, experienced labs were able to identify it by observing increases in the areas of certain small peaks normally associated with unmodified serine. Alternatively, a more definitive identification of phosphoserine was obtained by derivatizing the peptide with aminoethane thiol. Methionine sulfoxide was reduced to Met during sequencing and thus was identified as Met. In a couple of cases, reduction was incomplete and a small peak due to PTH-Met(O) was observed between Gln and Thr.
The accuracy of the modified amino acid assignments illustrates that not all protein sequencing analysts are as familiar with the elution characteristics of the PTH-modified amino acids as they are with the 20 common PTH-amino acids. By using a carefully designed synthetic peptide containing the modified amino acids, N,N-dimethylarginine, N-ɛ-methyl lysine, phospho Ser, S-β-propionamidocysteine, methionine sulfoxide, N-ɛ-acetyl lysine, and 4-hydroxyproline, this study provided the means to compile a list of the elution characteristics of these amino acids from multiple samplings of a variety of the currently operating sequencers. Because many of these sequencers are run under nearly identical conditions, this list should provide a means to make confident assignments of these modified amino acids. The relative retention times for the modified PTH-AAs have been posted on the ABRF website at www.abrf/esrg.org. Additional modified amino acids will be added to this list with the 2005 study.
REFERENCES
- 1.Niece RL, Williams KR, Wadsworth CL, et al. A synthetic peptide for evaluating protein sequencer and amino acid analyzer performance in core facilities: Design and results. In Hugli TE (ed.): Techniques in Protein Chemistry. San Diego: Academic Press, 1989:89–101.
- 2.Speicher DW, Grant GA, Niece RL, Blacher RW, Fowler AV, Williams KR. Design, characterization and results of ABRF-89SEQ: A test sample for evaluating protein sequencer performance in protein microchemistry core facilities. In Hugli TE (ed.): Current Research in Protein Chemistry, San Diego: Academic Press, 1990:159–166.
- 3.Yuksel KU, Grant GA, Mende-Muller LM, Niece RL, Williams KR, Speicher DW. Protein sequencing from polyvinylidene difluoride membranes: Design and characterization of a test sample (ABRF-90SEQ) and evaluation of results. In Villafranca JJ (ed.): Techniques in Protein Chemistry II. San Diego: Academic Press, 1991:151–162.
- 4.Crimmins DL, Grant GA, Mende-Muller LM, et al. Evaluation of protein sequencing core facilities: Design, characterization, and results from a test sample (ABRF-91SEQ). In Angeletti RH (ed.): Techniques in Protein Chemistry III. San Diego: Academic Press, 1992:35–43.
- 5.Mische SM, Yuksel KU, Mende-Muller LM, Matsudaira P, Crimmins DL, Andrews PC. Protein sequencing of post-translationally modified peptides and proteins: Design, characterization and results of ABRF-92SEQ. In Angeletti RH (ed.): Techniques in Protein Chemistry IV. San Diego: Academic Press, 1993:453–461.
- 6.Rush J, Andrews PC, Crimmins DL, et al. Synthetic peptide for evaluating protein sequencing capabilities: Design of ABRF-93SEQ and results. In Crabb JW (ed.): Techniques in Protein Chemistry V. San Diego: Academic Press, 1994:133–141.
- 7.Gambee JE, Andrews PC, Grant GA, Merrill B, Mische SM, Rush J. Assignment of cysteine and tryptophan residues during protein sequencing: Results of ABRF-94SEQ. In Crabb JW (ed.): Techniques in Protein Chemistry VI. San Diego: Academic Press, 1995:209–217.
- 8.DeJongh KS, Fernandez J, Gambee JE, et al. Design and analysis of ABRF-95SEQ, a recombinant protein with sequence heterogeneity. In Marshak D (ed.): Techniques in Protein Chemistry VII. San Diego: Academic Press, 1996:347–358.
- 9.Fernandez J, Admon A, DeJongh K, et al. Evaluation of ABRF-96SEQ: A sequence assignment exercise. In Marshak DR (ed.): Techniques in Protein Chemistry VIII. San Diego: Academic Press, 1997:69–78.
- 10.Stone K, Fernandez J, Admon A, et al. ABRF-97SEQ: Sequencing results of a low-level sample. J Biomol Tech 1999;10:26–32. [Google Scholar]
- 11.Carr S, Crabb J, Davis G, et al. ABRF-99SEQ: Analysis of a peptide and protein. Presented at ABRF ’99: Bioinformatics and Biomolecular Technologies: Linking Genomes, Proteomes, and Biochemistry; March 1999; Durham, NC.
- 12.Henzel W, Admon A, Carr S, et al. ABRF-98SEQ: Evaluation of peptide sequencing at high sensitivity. J Biomol Tech 2000;11:92–99. [PMC free article] [PubMed] [Google Scholar]
- 13.Carr J, Crabb J, Davis G, et al. ABRF-00SEQ: Sequence analysis of a post-translationally modified peptide. Presented at ABRF ’00: From Singular to Global Analysis of Biological Systems, February 2000; Bellevue, WA.
- 14.Buckel SD, Cook RG, Crawford JM, et al. ABRF-2002ESRG, a Difficult Sequence: Analysis of a PVDF-Bound Known Protein with a Heterogenous Amino-Terminus. J Biomol Tech 2002;13:246–257. [PMC free article] [PubMed] [Google Scholar]
- 15.Buckel SB, Cook RG, Crawford JM, et al. ABRF-ESRG’03: Analysis of a PVDF-Bound Known Protein with a Homogeneous Amino-Terminus. J Biomol Tech 2003;14: 278–288. [PMC free article] [PubMed] [Google Scholar]
- 16.Grant GA, Crankshaw MW. Identification of PTH-amino acids by HPLC. In Smith BJ (ed.): Protein Sequencing Protocols. Totowa, NJ: Humana Press, 2003:247–268. [DOI] [PubMed]
- 17.Weckwerth W, Willmitzer L, Fiehn O. Comparative quantification and identification of phosphoproteins using stable isotope labeling and liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom 2000;14:1677–168. [DOI] [PubMed] [Google Scholar]
- 18.Morishima-Kawashima M, Hasegawa M, Takio K, et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem 1995;270:823–829. [DOI] [PubMed] [Google Scholar]
- 19.Brune D. Alkylation of cysteine with acrylamide for protein sequence analysis. Anal Biochem 1992;207:285–290. [DOI] [PubMed] [Google Scholar]
- 20.Ross FE, Zamborelli T, Herman AC, Yeh C, Tedeschi NI, Luedke ES. Detection of acetlylated lysine residues using sequencing by Edman degradation and mass spectrometry. Techniques in Protein Chemistry VII. San Diego: Academic Press, 1996:201–208.