

Abstract
Shotgun proteomics, where a tryptic digest of a complex proteome sample is directly analyzed by either single dimensional or multidimensional liquid chromatography tandem mass spectrometry, has gained acceptance in the proteomics community at large and is widely used in core facilities. Here we review the development in our laboratory of an alternative first-dimension separation technique for shotgun proteomics, immobilized pH gradient isoelectric focusing (IPG-IEF). The key advantages of the technology over other multidimensional separation formats (simplicity, high resolution, and high sensitivity) are discussed. The concept of using peptide pI to filter large shotgun proteomics datasets generated by the IPG-IEF technique to minimize false positives and negatives is also introduced. Finally, an account of the comparison of the technique with the established gold standard for multidimensional separation of peptides, strong cation exchange chromatography, is presented, along with the prospects for the use of peptide pI along with accurate mass measurement for the identification of peptides.
Keywords: Isoelectric focusing, immobilized pH gradient, mass spectrometry, protein identification, proteomics, two-dimensional chromatography
An area of extensive interest to the proteomics community at large is the development of alternative strategies to the conventional 2D electrophoresis (2DE) approach for the analysis of proteins in complex sample mixtures. Typically, such alternative approaches focus on the use of some form of liquid chromatography (LC) coupled to electrospray ionization tandem mass spectrometry (ESI-MS/MS) on three-dimensional or linear ion traps as well as quadrupole time of fight instruments, although the recent development of high-performance tandem time of flight mass spectrometers employing matrix-assisted laser desorption/ionization (MALDI) by several manufacturers has spurred interest in the development of LC-MALDI techniques to analyze such samples.1 This experimental format, where the peptides are directly analyzed by tandem mass spectrometry and then analyzed and recombined into protein identifications using bioinformatics tools has been termed “shotgun” proteomics, and has gained wide acceptance by both specialist research groups and core proteomics service facilities.
A single dimension separation by reversed-phase LC (RPLC) does not have sufficient peak capacity to handle the tens of thousands of peptide species present in a typical unfractionated tryptic digest of a complicated proteome sample derived from microbes, eukaryotic cells, or tissues. Consequently, multidimensional separation techniques must be employed to adequately resolve these mixtures. The most common of these is typically strong cation exchange (SCX) chromatography, either performed on-line (the so-called MudPIT approach)2 or offline,3 although recently the use of 1D sodium dodecylsulfate-polyacrylamide gel electrophoresis separation of the initial protein mixture followed by LC-MS/MS (“GeLC-MS”) has gained favor with some investigators.4 These methods have enabled the successful separation and identification of large numbers of proteins in complex samples, but typically suffer from less than optimal resolution due to overloading, particularly when the multidimensional separation is performed online in biphasic capillary columns, which have relatively low sample loading capacity. This phenomenon is reflected in a reduction in unique peptide identifications observed in these techniques after the first few SCX fractions are analyzed. Often, high-abundance peptides can be observed in every elution step of the SCX separation.
The concept of using isoelectric focusing (IEF), where molecules are separated on the basis of their isoelectric points, for the separation of protein mixtures has been widely employed not only in the first dimension of separation in 2-dimensional electrophoresis experiments but as well for preparative purifications of proteins in both liquid5 and gel6 formats. Preparative IEF of protein mixtures has also been employed for a prefractionation technique as a prelude to 2DE analysis7,8 as well as for LC-MS/MS based separation regimens.9
In contrast, few investigators have examined the concept of using IEF for separating complex mixtures of peptides. C. S. Lee and colleagues have developed and demonstrated a multidimensional system for the separation of peptides based on capillary IEF, coupled to nano–-reverse-phase high-performance liquid chromatography.10,11 Although this work demonstrated a successful integration of peptide IEF with RPLC, a significant disadvantage of this approach is the complex system of switching valves and capillary traps necessary to couple the two modes of separation, making it challenging to implement on a routine basis in a nonspecialist laboratory setting. In addition, since capillary IEF technology is used in this system, it also suffers from a relatively limited sample loading capacity, which could ultimately limit the amount of information (peptide protein identifications) obtainable. This may not be the case, however, since the authors report to identify greater than 1000 (1132) proteins from a 9.6-μg sample load of Saccharomyces cerevisiae lysate.10 In evaluating the quality of the data in this report, it is important to note that no statistical tests were employed to evaluate the degree of confidence in this relatively large number of identifications. This implies that a significant number of the reported data may represent false-positive results.12 Ultimately, the shortcomings discussed above could limit the utility of this approach for widespread adaptation.
Several recent reports have employed preparative liquid-phase IEF platforms, similar to what has been employed in the past for protein mixtures, for analyzing tryptic peptides. Xiao and co-workers examined peptides derived from a digest of human serum13 which employed a Rotofor device. Simpson’s group14 in contrast used a continuous free flow electrophoresis device to focus tryptic peptides derived from cultured human colon cells. Baczek used a home-constructed solution IEF platform to separate a simple mixture of peptides derived from tryptic digest of five proteins,15 as well as for analysis of the S. cerevisiae proteome.16
One major disadvantage of using liquid-based IEF for such a separation is the modest resolving power obtainable with such technology. For example, in Baczek’s report on shotgun analysis of the yeast pro-teome,16 38% of peptides were found in more than one IEF fraction, and 15% were present in four or more fractions. This is due in part to the number of fractions that can be collected from such devices, which is typically limited by the instrumental design. Another potential problem with a liquid-phase IEF approach is that a relatively large concentration of carrier ampholytes must be employed to generate the pH gradient. These compounds can then in turn interfere with LC-MS/MS analysis. In the instance of the colon cancer study, the presence of the liquid ampholytes may account for the relatively modest number of identifications obtained (254 peptides from 77 proteins) from a sample that should theoretically contain thousands of proteins. Xaio et al. demonstrated that such a separation could be performed in an ampholyte-free system, since the peptides themselves can form a pH gradient. These investigators were more successful in obtaining a larger number of peptides/proteins identifications (844/437) from their experiment, using relatively simple technology.
Previously, some investigators have employed the well-known immobilized pH gradient isoelectric focusing (IPG-IEF) gel strips typically used for the first-dimension separation in 2D electrophoresis work17,18 for direct MALDI mass spectrometric analysis of intact proteins (“virtual 2D gel electrophoresis”)19 as well as for the first dimension of a shotgun type experimental strategy, following digestion of the IEF separated peptides.20 Expanding upon this work, our group recently has explored the use of IPG-IEF of peptides as the first-dimension technique for multidimensional peptide separations.21–23 This review will provide a brief introduction to the use of the IPG-IEF method for peptide separations and provide an account of the work done to date in developing and evaluating this method as a first-dimension separation methodology for shotgun proteomics experiments.
USING IPG-IEF IN SHOTGUN PROTEOMICS
The integration of this technique within a shotgun proteomics workflow is outlined graphically in Figure 1. A sample for analysis is initially loaded onto an IPG strip via the rehydration method. The presence of a filter paper wick at the anode allows peptides outside the isoelectric point (pI) range of the strip to exit. This significantly improves the quality of the data obtained at the basic end of the strip, especially in narrow-range (one pI unit) separations. The peptides are then focused using a commercially available IEF unit, using programs similar to what are employed for protein separations. An important advantage of using IPG technology over other IEF formats such as liquid IEF or slab gels21 is that the pH gradient in the manufactured strips is known and highly reproducible, making prediction of the pI range of a particular fraction relatively straightforward.22 Following focusing, the strip is cut into a number of fractions which are collected into Eppendorf tubes (currently, this is done manually with scissors, although we are in the process of designing an automated strip cutting device).
FIGURE 1.

The overall strategy for the integration of immobilized pH gradient isoelectric focusing (IPG-IEF) in shotgun proteomics. IEF, isoelectric focusing; K, lysine; R, arginine.
Several facts worth mentioning here are that producing IEF strip “cuts” that are square and similar in size is important, so that theoretical pI ranges can be established for each fraction. If the strip is not cut squarely, a given peptide band can be dispersed across multiple fractions, which complicates the use of pI for data filtering. Another issue that we initially considered in the fractionation process was that over the time necessary to cut the strip into fractions, significant peptide diffusion may occur, leading to decreased resolution. We have recently obtained some data using fluorescently labeled peptides in cooperation with collaborators at Amersham Biosciences that showed, over a time frame of 2 h, peptide bands increased in width approximately twofold. This implies that, on the time scale of the manual strip cutting process (10–15 min), diffusion should not be a significant issue, which is corroborated by our experience in the laboratory. It is also important to note that the IPG sample buffer contains 8 M urea, which significantly increases the solution viscosity, which also limits the rate of diffusion in the strip.
Following the fractionation process, peptides are extracted from the strip using a series of washes in acidic solvents of progressively higher organic content, similar to what is done for in-gel digests of intact proteins. Although we have not empirically determined the peptide extraction efficiency from the IEF gel, it is expected to be high since one is dealing with preformed peptides rather than the products of a relatively inefficient in-gel digest of a protein. In addition the IEF gel has a relatively low (ca. 4%) crosslinker content, so diffusion of the peptides into the extraction solvent should not be significantly hindered. These resultant extracts are concentrated via centrifugal vacuum evaporation on a Speedvac. It is important to note that the extracts need to be subjected to further cleanup using C18 solid phase extraction before LC-MS/MS, to remove residual overlay oil from the IEF process and other interferents. Several other investigators using the technique have noted a problem with excessive background noise from direct injection of the extracts without this preparation step. For an 18-cm IEF strip, we typically process 45 fractions of 4 mm in length. This can be tedious to do using Eppendorf tubes/C18 spin columns, so we have recently begun to perform the extraction and cleanup steps in a 96-well format, using microwell plates and 96-well SPE devices. Following sample preparation, the peptide extracts are analyzed by nano–RPLC-MS/MS, followed by database searching and filtering, exploiting the use of pI as a method to filter data in order to reduce spurious matches.12
An in silico tryptic digest of the Escherichia coli proteome, plotted against pI for no missed cleavage sites and one missed cleavage, is depicted in Figure 2.21 It can be seen from this plot that peptides are well spread out over the pI range for a given mass, implying that separation based on pI should provide good resolution for isobaric peptide species. As expected, when the number of missed cleavages considered increases, the regions of maximum peptide density also shift to the basic region, since such peptides contain an increased number of lysine and arginine residues. An increase in density is also observed in the basic regions, due to the increased probability of a peptide having multiple acidic (aspartic acid or glutamic acid) residues. When a sample of E. coli cellular lysate was analyzed by IEF-IPG on a 3–10 range strip, the distribution of results plotted as a function of IEF fraction number and pI (Figure 3) agreed quite well with theory, as a small number of peptides were identified from the range corresponding to pI 7–8, and the largest number of peptides were identified in the 3–4 pI range. The steep slope of the peaks in this plot also illustrates the high resolution of the IEF separation format. Typically, the observed standard deviation of the pIs of the peptides identified in the fractions was on the order of ± 0.2 pI units.22 The experimental and theoretical data also suggest that a narrow-range pI strip covering the 3.5–4.5 pI range may be most effective in resolving the greatest number of tryptic peptides in a proteome, as approximately 80% of the E. coli proteome is represented by at least one tryptic peptide within this pI range. Similar trends are observed when proteome data from other organisms is examined in this manner.
FIGURE 2.
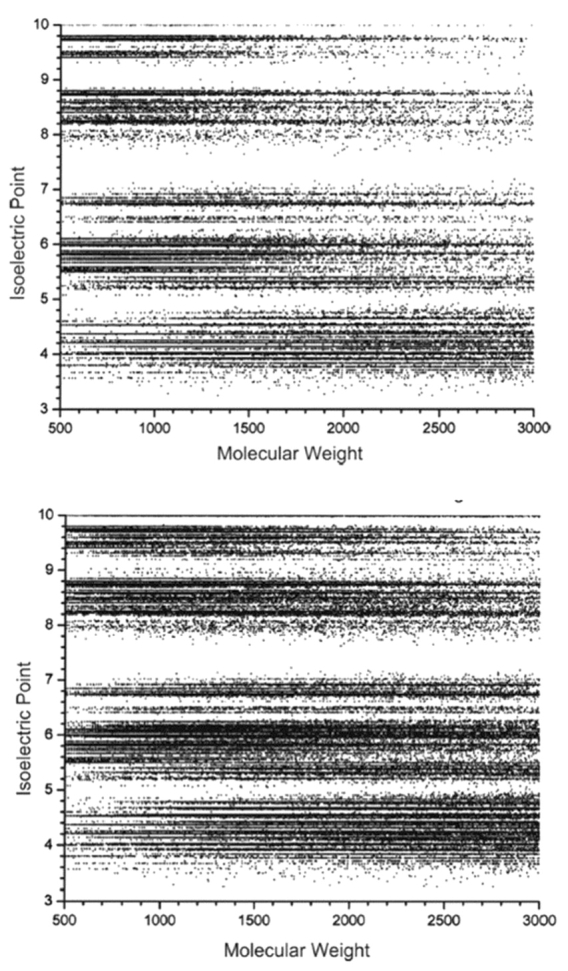
Theoretical pI of peptides from the E. coli proteome as a function of mass for no missed cleavages of trypsin (upper panel) and one missed cleavage of trypsin (lower panel). (Reproduced with permission from J Proteome Res, 2004;3:112–119, Copyright © 2004 American Chemical Society.)
FIGURE 3.

Three-dimensional plot of data obtained from a wide range (pH 3–10) IPG-IEF separation of the soluble E. coli proteome as a function of number of peptides identified, IPG fraction number, and mean peptide pI. (Reproduced with permission from Electrophoresis 2004;25:936–945. Copyright © 2004, John Wiley and Sons.)
Another advantage of IPG-IEF is the enhanced sensitivity of the technique. In our hands, the observed sensitivity of the technique has been on the order of 50–100 times greater than a similar SCX fraction. In our initial experiments with the soluble proteome of E. coli, it was only necessary to inject ~1% of each individual IEF fraction for satisfactory LC-MS/MS analysis. Larger injections produced chromatograms typical of sample overloading. This observation of increased sensitivity is similar to what has previously been reported for capillary IEF-RPLC.10 A potential explanation of this observation is that all of the peptides in a given IPG-IEF fraction are of a similar pI and therefore a similar net charge at a given pH. This should result in these species having a similar response in ESI-MS. This assertion was tested by mixing fractions from the acidic and basic ends of a wide-range IPG strip and subjecting them to LC-MS/MS analysis, using a longer gradient than before. Both fractions produced approximately the same number of identifications when originally analyzed alone. In all cases, there was a strong bias (85:15) towards peptide identifications from the acidic end, where one would expect this ratio to be nearly 50:50. This represents the results from a single experiment; obviously this effect needs to be explored in further detail to provide a sound basis for explanation.22
USING PEPTIDE pI AS A DATA FILTERING CRITERION
Since in IPG-IEF, peptides are separated on the basis of pI, this can be exploited as a useful tool to filter protein and peptide database search results. One of the major issues in MS-based peptide and protein identifications is validation of database matches produced by such searching programs as Mascot24 and SEQUEST.25 Several strategies typically used, especially with SEQUEST data, are search score cutoffs26 and/or “manual validation” of individual tandem mass spectra based on arbitrary rules,27 which are typically empirically determined and lack any firm mathematical basis for implementation. A recent report from our laboratory12—where the use of relatively stringent filtering criteria resulted in the match of greater than 1300 protein loci identified by a single peptide match to a protein “database” of random amino acid sequences—illustrates the need for development of more effective data filtering tools. Since all of the peptides in a given IEF fraction should be within a predicable pI range, pI should be able to be employed to filter spurious peptide identifications from database search results. Lee’s group has also calculated the average pI of the proteins identified in their IEF/RPLC experiments,10,28 but it is not clear if they have implemented it as a means of filtering data. A program we have recently developed in-house for analysis of data generated from SEQUEST searches, IDSieve, includes a feature that allows one to filter data in a given SEQUEST output file by predicted peptide pI range.
As shown for our Rattus norvigicus data in Figure 4A–C, the use of the pI constraints allows one to mine a significantly larger number of identifications, while maintaining an acceptably low (< 1%) false-positive identification rate. Figure 4A shows the distribution of SEQUEST cross correlation scores (Xcorr) for all peptide charge states as a function of peptide pI for a sample of R. norvigicus testis analyzed by IEF-IPG. In this case, the bulk of high Xcorr identifications cluster in the pI range of 3.5–4.5, which was the pI range of the strip used. When the reverse database search hits are also plotted, as shown in Figure 4B, one can see that the number of reverse hits (false-positive identifications) greatly increases in the area outside of the IEF strip’s pI range. Then, one can determine the Xcorr cutoff scores that produce a peptide false-positive identification rate of 1%. This “cutoff line” is shown in composite for all charge states for simplicity in Figure 4B. However, if one uses the pI range of the strip (3.5–4.5) as a data-filtering criterion, the Xcorr score cutoffs may be lowered, as shown graphically in Figure 4C, allowing one to mine more identifications from a dataset while maintaining an acceptably low false-positive identification rate. In the case of the E. coli proteome, for fully tryptic peptides, this enabled the identification of 23% more peptides than the use of Xcorr alone. One advantage of using pI as a data filter over other data validation methods is that it is an invariant physiochemical property of the peptide, and can be incorporated into a data analysis regimen regardless of the database searching scheme being employed.
FIGURE 4.
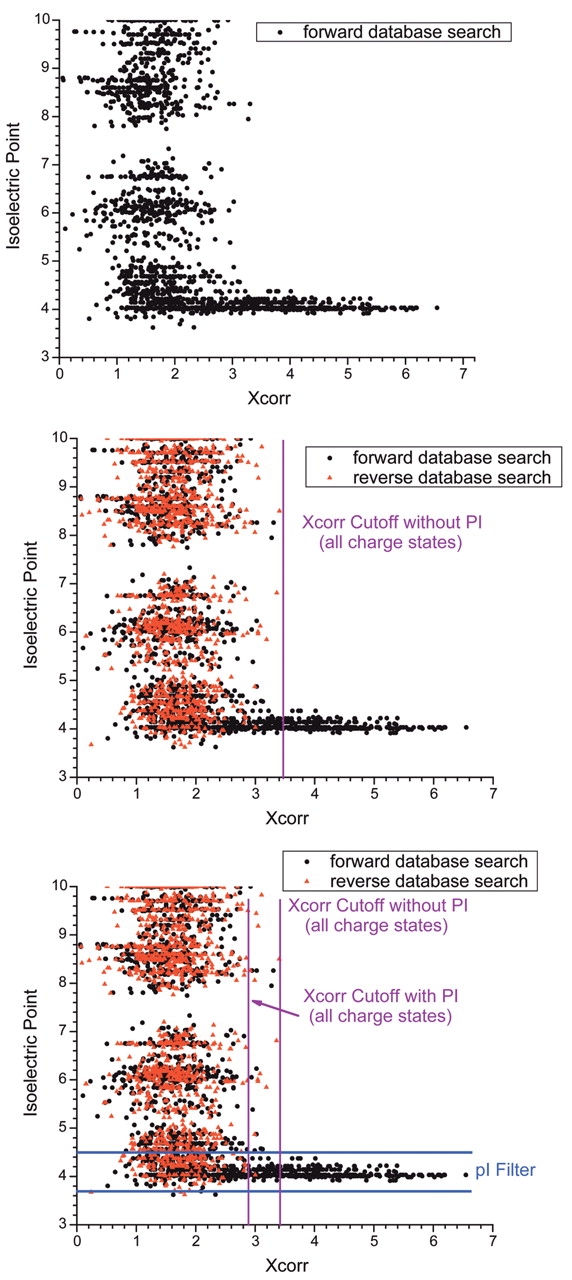
The use of pI as a filtering criterion. The graphs depict all protein hits from SEQUEST searches of an nrIPG-IEF shotgun proteomics analysis of the R. norvigicus testicular proteome, plotted as a function of SEQUEST cross-correlation score (Xcorr) and pI. A: Hits obtained from searching the forward orientation of the database only. B: Hits obtained when the reversed database approach was used to determine Xcorr cutoffs for a peptide false positive identification rate of ~1%. Reversed database hits are shown in red. C: Application of the pI 3.5–4.5 filter to the data, showing that the Xcorr cutoffs can be relaxed while maintaining a false positive peptide identification rate of ~1%. (Figure based on data originally published in reference 12.)
IPG-IEF VERSUS SCX FOR SHOTGUN PROTEOMICS
As mentioned before, the current “gold standard” for multidimensional separation of complex peptide mixtures is strong cation exchange chromatography either performed on-line or off-line from the LC-MS/MS analysis. We have recently completed23 a comparative study of the IPG-IEF technique with off-line SCX chromatography, using the R. norvigicus testis proteome as a model case of a complex eukaryotic system. The IPG-IEF separation in this case employed a narrow-range strip (pI 3.5–4.5). This was done because, as mentioned before, the bulk of fully tryptic peptides should lie within this range, so such a strip should be useful in maximizing the resolution of peptides and therefore the number of identifications from a sample of this complexity. In addition, since the pI spread in a given fraction should be relatively small, sensitivity should be maximized for reasons mentioned previously.
A multidimensional separation method that possesses high efficiency should result in a maximal number of unique identifications in a given fraction. Although the detection of a peptide across multiple fractions allows increased confidence in identification, a technique that exhibits a high identification redundancy per fraction is compromised ultimately in efficiency. Figure 5 shows a comparison between the numbers of unique protein loci unambiguously29 identified per fraction by IEF (circles) and SCX (triangles) in this comparative study. It should be noted here that in the case of the SCX experiment, in order to minimize comparative bias, we only analyzed every other of the 128 fractions to maximize the number of unique identifications obtained by SCX, and omitted the flowthrough (first 17 collected fractions) to minimize redundancy. One would expect the slope of such a curve to be representative of the separation efficiency of the technique. As can be seen, IEF clearly is favored here, as the slope of the curve remains relatively steep over the majority of the run. In contrast, the SCX curve, although exhibiting a similar slope to the IEF curve over the first half of the run, exhibits an abrupt leveling off. As the conditions used here were nearly optimal for SCX,23 one would expect the redundancy to be even greater in a classical MudPIT experimental setup.
FIGURE 5.

Graph of the cumulative number of unique proteins identified from a sample of R. norvigicus testis lysate as a function of fraction number for IPG-IEF (triangles) and SCX (circles). Note that in the SCX experiment, although 128 fractions were collected, every other fraction was analyzed to minimize peptide redundancy. (Figure based on data originally published in reference 23.)
A Venn diagram summarizing the number of proteins that were unambiguously identified by IEF and SCX respectively, after statistical validation and filtering via IDSieve, is shown in Figure 6. Although IEF clearly identified more peptides (7626 vs 6776), only 2418 were in common between the two techniques, implying that neither technique provided comprehensive coverage. However, 75% of the peptides exclusively identified in the SCX separation were of pI greater than 4.5, whereas the vast majority of the identified peptides from the IEF separation fell within a single pI unit. Based on these results, the IPG-IEF technique provides superior “depth of coverage” of the proteome as compared to SCX. It also is logical to conclude that if one was to run multiple narrow range strips spanning the entire pI range, many more peptides and proteins would be identified than with the SCX approach.
FIGURE 6.

Venn diagram depicting the number of unique proteins and peptides identified by nrIEF-IPG and SCX experiments on the R. norvigicus testicular proteome. Note that approximately 75% of the “unique” peptides found via SCX were greater than pI 4.6. SCX, strong cation exchange.
Beyond the many advantages outlined herein for the use of IEF as a first-dimension separation for shotgun proteomics, the use of accurate mass and pI alone shows promise as a rapid means to identify proteins in shotgun experiments.30 Previously, accurate mass alone in the form of the accurate mass tag (AMT) strategy has been developed by Smith and colleagues as a means of reducing the time spent on tandem mass spectrometric analysis of peptide mixtures. However, if one examines the theoretical false-positive rate of such an approach, it is revealed that even at a mass accuracy of 0.5 ppm, an unacceptable protein false-positive identification rate is observed when applied to organisms of genome size greater than E. coli, unless a large number of peptides per protein (5+) are required for a positive identification. In order to address this shortcoming of the AMT strategy we have recently proposed the integration of the accurate mass tag approach with the predicted peptide pI from samples separated by IPG-IEF.30 Using the pI as an additional constraint, one can relax the stringency of the mass accuracy, while maintaining an acceptable false-positive identification rate. As an example, using a combination of pI and accurate mass for the E. coli proteome, a false-positive protein identification rate can be achieved of 1% at 20 ppm mass accuracy and 0.1 unit pI prediction accuracy, with 2 peptides per protein used as a basis for identification. This implies that instruments other than expensive and complex Fourier transform mass spectrometry platforms could be used to implement this approach. In addition, the use of pI allows one to distinguish between isobaric peptides that could not otherwise be discriminated by accurate mass alone. Initial experiments are in progress with a linear ion trap/Fourier transform mass spectrometry hybrid instrument, which exhibits routine mass measurement accuracy at the 2–5 ppm level to test the efficacy of this strategy. The interested reader is referred to the report in reference 31 for a detailed theoretical discussion of the accurate mass/pI identification strategy.
FUTURE DIRECTIONS
As evidenced by the work reviewed here, IPG-IEF shows great promise as a high-performance separation for shotgun proteomics. In order to ultimately adapt the technology for routine use in proteomics laboratories, several areas need to be addressed. First, as mentioned before, the processing of the IPG strip would be greatly facilitated by the development of a device to automatically or semiautomatically cut the strip into a number of fractions of a predefined size. Besides having the advantage of producing reproducible fraction sizes, this device should aid in limiting any diffusion effects by decreasing the amount of time necessary to generate the IEF fractions. In addition, to date we have concentrated our investigations on the soluble protein complement. As many protein species of interest are present in biological membranes, the efficacy of this approach for analysis of membrane protein digests needs to be examined. The ultimate sensitivity and dynamic range of the technique also needs to be evaluated. To date, we have loaded samples up to 10 mg and as little as 100 μg (starting material) onto the IPG strips with successful LC-MS/MS results. It remains to be seen, though, if the IPG-IEF technique can be used successfully with the relatively modest amount of protein (~10 μg) obtained from such sampling strategies as laser capture microdissection.31
Recent technological developments have resulted in the commercialization of several new instrumental systems based on two-dimensional (linear) ion traps.32–34 These platforms offer increased ion storage capacity and faster scan speeds, leading to significant gains in sensitivity and dynamic range over conventional (3D) ion traps. In an initial set of experiments using this instrument (Bundy J. L. and Brown K. J., unpublished data), we identified approximately 38% more peptides than are reported here in an equivalent narrow-range IEF experiment with rat testicular tissue.23
Finally, improvements in the prediction of peptide pI (to ± 0.1 pI unit) will allow more identifications to be mined from the data, as well as aid in the implementation of the accurate mass/pI strategy for high-throughput protein identification. We have recently completed initial development of a new peptide pI prediction algorithm that has allowed us to realize this goal.35
NOTE
Since the submission of this report, there have been a number of publications in the use of isoelectric focusing for peptide separation in shotgun proteomics by several groups.1–6 The most significant of these reports has been by Griffin and colleagues, whom have reported the use of a free-flow electrophoresis apparatus for the analysis of a shotgun digest of the chromatin enriched fraction of Sacchromyces cerevisiae.4 Moreover, they employed peptide isoelectric point as a filtering criterion, in concert with probability-based scoring. Several questionable protein identifications from single peptide hits and/or partially tryptic peptides were cross-validated with immunoblotting.
Acknowledgments
The authors would like to acknowledge Bengt Bjellqvist (Amersham Biosciences) and Angelika Görg (Technical University of Munich) for helpful conversations on IEF-IPG technology and prediction of peptide pI. Financial support for research performed in our laboratories was provided by Internal Research and Development funds from RTI and an unrestricted grant from Merck and Co.
REFERENCES
- 1.Rejtar T, Hu P, Juhasz P, et al. Off-line coupling of high-resolution capillary electrophoresis to MALDI-TOF and TOF/TOF MS. J Proteome Res 2002;1:171–179. [DOI] [PubMed] [Google Scholar]
- 2.Washburn MP, Wolters D, Yates JR, 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 2001; 19:242–247. [DOI] [PubMed] [Google Scholar]
- 3.Peng J, Elias JE, Thoreen CC, et al. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome. J Proteome Res 2003; 2:43–50. [DOI] [PubMed] [Google Scholar]
- 4.Beausoleil SA, Jedrychowski M, Schwartz D, et al. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA 2004;101:12130–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egen NB, Bliss M, Mayersohn M, et al. Isolation of monoclonal antibodies to phencyclidine from ascites fluid by preparative isoelectric focusing in the Rotofor. Anal Biochem 1988;172:488–494. [DOI] [PubMed] [Google Scholar]
- 6.Righetti PG, Gianazza E. Isoelectric-focusing in immobilized pH gradients—Theory and newer methodology. Methods Biochem Analysis 1987;32:215–278. [DOI] [PubMed] [Google Scholar]
- 7.Zuo X, Speicher DW. A method for global analysis of complex proteomes using sample prefractionation by solution isoelectrofocusing prior to two-dimensional electrophoresis. Anal Biochem 2000;284:266–278. [DOI] [PubMed] [Google Scholar]
- 8.Gorg A, Boguth G, Kopf A, et al. Sample prefractionation with Sephadex isoelectric focusing prior to narrow pH range two-dimensional gels. Proteomics 2002;2: 1652–1657. [DOI] [PubMed] [Google Scholar]
- 9.Janini GM, Conrads TP, Veenstra TD, et al. Development of a two-dimensional protein-peptide separation protocol for comprehensive proteome measurements. J Chromatogr B: Anal Technol Biomed Life Sci 2003;787: 43–51. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Balgley BM, DeVoe DL, et al. Capillary isoelectric focusing-based multidimensional concentration/separation platform for proteome analysis. Anal Chem 2003;75:3145–3152. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Lee CS, Shen Y, et al. Integration of capillary isoelectric focusing with capillary reversed-phase liquid chromatography for two-dimensional proteomics separation. Electrophoresis 2002;23:3143–3148. [DOI] [PubMed] [Google Scholar]
- 12.Cargile BJ, Bundy JL, Stephenson JL, Jr. Potential for false positive identifications from large databases through tandem mass spectrometry. J Proteome Res 2004;3(5):1082–1085. [DOI] [PubMed] [Google Scholar]
- 13.Xiao Z, Conrads TP, Lucas DA, et al. Direct ampholyte-free liquid-phase isoelectric peptide focusing: Application to the human serum proteome. Electrophoresis 2004;25:128–133. [DOI] [PubMed] [Google Scholar]
- 14.Moritz RL, Ji H, Schutz F, et al. A proteome strategy for fractionating proteins and peptides using continuous free-flow electrophoresis coupled off-line to reversed-phase high-performance liquid chromatography. Anal Chem 2004;76:4811–4824. [DOI] [PubMed] [Google Scholar]
- 15.Baczek T. Fractionation of peptides in proteomics with the use of pI-based approach and ZipTip pipette tips. J Pharm Biomed Anal 2004;34:851–860. [DOI] [PubMed] [Google Scholar]
- 16.Baczek T. Fractionation of peptides and identification of proteins from Saccharomyces cerevisiae in proteomics with the use of reversed-phase capillary liquid chromatography and pI-based approach. J Pharm Biomed Anal 2004;35:895–904. [DOI] [PubMed] [Google Scholar]
- 17.Bjellqvist B, Ek K, Righetti PG, et al. Isoelectric-focusing in immobilized ph gradients—Principle, Methodology and Some Applications. J Biochem Biophys Methods 1982;6:317–339. [DOI] [PubMed] [Google Scholar]
- 18.Gorg A, Obermaier C, Boguth G, et al. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 2000;21:1037–1053. [DOI] [PubMed] [Google Scholar]
- 19.Loo RR, Cavalcoli JD, VanBogelen RA, et al. Virtual 2-D gel electrophoresis: Visualization and analysis of the E. coli proteome by mass spectrometry. Anal Chem 2001;73:4063–4070. [DOI] [PubMed] [Google Scholar]
- 20.Giorgianni F, Desiderio DM, Beranova-Giorgianni S. Proteome analysis using isoelectric focusing in immobilized pH gradient gels followed by mass spectrometry. Electrophoresis 2003;24:253–259. [DOI] [PubMed] [Google Scholar]
- 21.Cargile BJ, Bundy JL, Freeman TW, et al. Gel based isoelectric focusing of peptides and the utility of isoelectric point in protein identification. J Proteome Res 2004;3:112–119. [DOI] [PubMed] [Google Scholar]
- 22.Cargile BJ, Talley DL, Stephenson JL, Jr. Immobilized pH gradients as a first dimension in shotgun proteomics and analysis of the accuracy of pI predictability of peptides. Electrophoresis 2004;25:936–945. [DOI] [PubMed] [Google Scholar]
- 23.Essader AS, Cargile BJ, Bundy JL, et al. A comparison of immobilized pH gradient isoelectric focusing and strong cation exchange chromatography as a first dimension in shotgun proteomics. Proteomics 2005; 5(1):24–34. [DOI] [PubMed] [Google Scholar]
- 24.Perkins DN, Pappin DJ, Creasy DM, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999;20:3551–3567. [DOI] [PubMed] [Google Scholar]
- 25.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 1994;5:976–989. [DOI] [PubMed] [Google Scholar]
- 26.Tabb DL, McDonald WH, Yates JR, 3rd. DTASelect and Contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 2002;1:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Link AJ, Eng J, Schieltz DM, et al. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol 1999;17:676–82. [DOI] [PubMed] [Google Scholar]
- 28.Mohan D, Pasa-Tolic L, Masselon CD, et al. Integration of electrokinetic-based multidimensional separation/concentration platform with electrospray ionization-Fourier transform ion cyclotron resonance-mass spectrometry for proteome analysis of Shewanella oneidensis. Anal Chem 2003;75:4432–40. [DOI] [PubMed] [Google Scholar]
- 29.Rappsilber J, Mann M. What does it mean to identify a protein in proteomics? Trends Biochem Sci 2002;27: 74–78. [DOI] [PubMed] [Google Scholar]
- 30.Cargile BJ, Stephenson JL, Jr. An alternative to tandem mass spectrometry: Isoelectric point and accurate mass for the identification of peptides. Anal Chem 2004;76: 267–275. [DOI] [PubMed] [Google Scholar]
- 31.Wulfkuhle JD, Paweletz CP, Steeg PS, et al. Proteomic approaches to the diagnosis, treatment, and monitoring of cancer. Adv Exp Med Biol 2003;532:59–68. [DOI] [PubMed] [Google Scholar]
- 32.Douglas DJ, Frank AJ, Mao D. Linear ion traps in mass spectrometry. Mass Spectrom Rev 2005;24(1):1–29 [DOI] [PubMed] [Google Scholar]
- 33.Schwartz JC, Senko MW, Syka JE. A two-dimensional quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom 2002;13:659–669. [DOI] [PubMed] [Google Scholar]
- 34.Hager JW. A new linear ion trap mass spectrometer. Rapid Commun Mass Spectrom 2002;16:512–526. [Google Scholar]
- 35.Cargile BJ, Sevinsky JR, Essader AS,Stephenson, Jr., JL. Prediction of the isoelectric point of tryptic peptides based on adjacent amino acid effects. Electrophoresis, submitted for publication. [DOI] [PubMed]
References
- 1.Storms HF, van der Heijden R, Tjaden UR, et al. Optimization of protein identification from digests as analyzed by capillary isoelectric focusing-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2005;824:189–200. [DOI] [PubMed] [Google Scholar]
- 2.Storms HF, van der Heijden R, Tjaden UR, et al. Capillary isoelectric focusing-mass spectrometry for shotgun approach in proteomics. Electrophoresis 2004;25:3461–3467. [DOI] [PubMed] [Google Scholar]
- 3.Xie H, Rhodus NL, Griffin RJ, et al. A catalogue of human saliva proteins identified by free flow electrophoresis-based peptide separation and tandem mass spectrometry. Mol Cell Proteomics 2005, posted electronically August 15th. [DOI] [PubMed]
- 4.Xie H, Bandhakavi S, Griffin TJ. Evaluating preparative isoelectric focusing of complex peptide mixtures for tandem mass spectrometry-based proteomics: A case study in profiling chromatin-enriched subcellular fractions in Saccharomyces cerevisiae. Anal Chem 2005;77:3198–207. [DOI] [PubMed] [Google Scholar]
- 5.Tu CJ, Dai J, Li SJ, et al. High-sensitivity analysis, posted electronically August 15th of human plasma proteome by immobilized isoelectric focusing fractionation coupled to mass spectrometry identification. J Proteome Res 2005;4:1265–1273. [DOI] [PubMed] [Google Scholar]
- 6.Gan CS, Reardon KF, Wright PC. Comparison of protein and peptide prefractionation methods for the shotgun proteomic analysis of Synechocystis sp. PCC 6803. Proteomics 2005;5:2468–478. [DOI] [PubMed] [Google Scholar]