

Abstract
We previously reported a protein expression profiling experiment conducted on human pancreatic tissues using 2D gel electrophoresis and mass spectrometry. Here, 18 spots that were identified in the gel at molecular weights more than 10 kDa lower than database values are characterized. The matrix-assisted laser desorption/ionization mass spectrometry coverage is sufficient to identify the protein region present in each spot. Most of the fragments correspond to processed chains and known structural or functional domains, which may result from limited proteolysis.
Keywords: Pancreatic tissue, protein fragments, 2D gel electrophoresis, MALDI, limited proteolysis
Two-dimensional (2D) gel electrophoresis for separation of complex protein samples coupled with mass spectrometry for protein identification has been used to analyze protein expression patterns for many sample types. Inherent in the use of this technique is information not only on full-length protein expression, but expression of modified, splice variant, cleavage product, and processed proteins. Any protein modification that leads to a change in overall protein charge and/or molecular weight (MW) will generate a different spot on the 2D gel. Modification specific staining can identify whether a specific post-translational modification is responsible for the shift, and mass spectrometry can potentially identify the source of isoelectric point (pI) and/or MW differences.1,2 Due to the lack of complete coverage for a protein’s amino acid sequence using either matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) or high-performance liquid chromatography (HPLC) tandem mass spectrometry (LC-MS/MS), there has been limited success in using MS to identify isoforms and post-translational modifications. While the theoretical MW is often slightly higher than the MW of the fully processed protein due to cleavage of signal and pro-peptides, there can also be post-translational modifications that increase the protein’s gel MW. Thus an exploration into the causes of the difference in the theoretical MW and the MW as seen in the gel can yield information about the state of the protein. When the gel MW of a given protein is significantly lower than the calculated weight, the gel spot represents a protein fragment.
The extent to which proteins are present as fragments or variants in tissues and fluids has not been determined, but the combination of 2D gel electrophoresis, Western blotting, and mass spectrometry–based protein identification makes such analyses possible. Two-dimensional gel electrophoresis of human mammary tissue, followed by immunoblotting, resulted in multiple spots at significantly differing molecular weights.3,4 Mattow et al. used 2D gel electrophoresis and MALDI-MS to examine the culture supernatant proteome of Mycobacterium tuberculosis, where 58% of the proteins were detected in more than one spot, including multiple truncations of elongation factor EF-Tu.5,6
The function of protein fragments is dependent on activation processes and localization properties. Proteins that are activated by limited proteolysis can be identified as either precursor protein or fully processed product based on this information. In other cases, there are transcriptional variants of different lengths, such as 54-kDa heat shock cognate protein (HSC54), which serves as a competitive inhibitor of the full-length transcript, and 71-kDa heat shock cognate protein (HSC71), with differing localization properties.7,8 Fragments are secreted into the serum, where they have been identified as antigens or markers for specific diseases.9,10 Several angiogenesis inhibitors are fragments of larger proteins that are themselves not active as angiogenesis inhibitors.11 Vasostatin, the N-terminal domain of calreticulin, is an angiogenesis inhibitor that exerts antitumor effects in vivo.12 In a surface-enhanced laser desorption/ionization (SELDI) identification of three serum biomarkers for the detection of early stage ovarian cancer,13 two of the biomarkers were identified as lower-MW protein fragments. Thus, fragments may be of greater importance as secreted proteins than in the tissue of origin.
Our previously published study used 2D gel electrophoresis and image analysis to identify proteins that were differentially expressed between normal pancreatic tissue, pancreatitis tissue, and pancreatic adenocarcinoma tissue.14 Sixty-eight differentially expressed gel spots were analyzed by MALDI-MS and database searching, and 40 different proteins were identified from these spots. In the course of that analysis, 10 proteins were identified in 18 gel spots at MW values significantly lower than their theoretical MW values. In this study, the protein regions observed in these fragments are identified by detailed analysis of the mass spectral results generated for the previous study. In vitro experiments are described that address the possibility of proteolytic activity as the source of protein fragments.
METHODS
Pancreatic tissue samples.
Twenty-one total pancreatic tissue samples were obtained from NCI Human Tissue Network as described in Supplemental Table 1 of the previous report.14 Normal tissues included autopsy samples (n = 5) collected within 8 h of death from patients with non-pancreatic diseases. The remaining tissues were surgical samples, including normal tissues (n = 2) adjacent to tumors, normal tissue (n = 1), tissues from pancreatitis patients (n = 7), and pancreatic ductal adenocarcinomas (n = 6). All samples were frozen within 30 min of resection. Selected samples were pooled by tissue type into three sets—normal, pancreatitis, and tumor.
TABLE 1.
Protein Regions Observed and Level of Coverage for Protein Fragments
| Residue Endpoints | ||||||||||
| Protein Name | Swiss-Prot Accession Number | Spot | DB MW (kDa) | Gel MW (kDa) | N | C | Protein Region | Protein N-Terminala | Coverageb (%) | Weighted Coveragec (%) |
| Cathepsin D | P07339 | 8 | 45 | 27 | 177 | 411 | NA | C-term. | 30 | 51 |
| 9 | 45 | 27 | 177 | 411 | NA | C-term. | 17 | 29 | ||
| Chymotrypsinogen B | P17538 | 16 | 28 | 28 | 80 | 125 | ND | full | 14 | |
| 17 | 28 | 17 | 19 | 125 | -signal | N-term. | 20 | 33 | ||
| 18 | 28 | 17 | 19 | 125 | -signal | N-term. | 20 | 33 | ||
| 19 | 28 | 15 | 80 | 125 | NA | central | 14 | 26 | ||
| 20 | 28 | 15 | 80 | 125 | NA | central | 14 | 26 | ||
| GRp78 d | P11021 | 21 | 72 | 72 | 50 | 633 | ND | full | 37 | |
| 22 | 72 | 48 | 50 | 386 | ND | N-term. | 28 | 41 | ||
| 23 | 72 | 48 | 50 | 386 | ND | N-term. | 28 | 41 | ||
| 24 | 72 | 25 | 447 | 633 | NA | C-term | 12 | 35 | ||
| α-Amylase 2A | P04746 | 27 | 58 | 16 | 16 | 155 | -signal | N-term. | 12 | 43 |
| 28 | 58 | 10 | 177 | 210 | NA | central | 6 | 36 | ||
| Pancreatic lipase related protein 2 | P54317 | 30 | 52 | 52 | 44 | 334 | -signal | full | 15 | |
| 31 | 52 | 31 | 44 | 251 | -signal | N-term. | 9 | 16 | ||
| Prolyl 4-hydroxylase β | P07237 | 34 | 57 | 57 | 18 | 461 | -signal | full | 42 | |
| 35 | 57 | 37 | 18 | 338 | -signal | N-term. | 20 | 31 | ||
| 36 | 57 | 25 | 18 | 230 | -signal | N-term. | 14 | 32 | ||
| Heat shock cognate protein 71 kDad | P11142 | 40 | 71 | 37 | 26 | 342 | ND | N-term. | 17 | 33 |
| Cytokeratin 8 | P05787 | 43 | 54 | 17 | 264 | 391 | NA | central | 19 | 60 |
| Actin, β and γe | P60709, P63261 | 56 | 42 | 40 | 19 | 372 | ND | full | 31 | |
| 59 | 42 | 26 | 197 | 372 | NA | C-term. | 10 | 17 | ||
| 14-3-3 ζ | P63104 | 66 | 28 | 17 | 28 | 41 | NA | N-term. | 6 | 9 |
a Detection of the protein N-terminal peptide for each gel spot. –signal indicates that the N-terminal peptide was detected with the signal sequence removed; NA:not applicable for protein fragments that do not cover the N-terminal of the protein or protein lacking a signal sequence; ND: not detected in the MS.
b (# of amino acid residues identified in MS/# of amino acids in database protein sequence) × 100.
c coverage for fragments is weighted by the MW of the gel spot, calculated as (% coverage) × (database MW of full-length protein / observed gel MW of protein fragment).
d Two proteins were previously reported under different names14: GRP78 was listed under the gene name HSPA5 and heat shock cognate protein 71 kDa under heat shock cognate protein 54 kDa. The names have been changed to be consistent with the Swiss-Prot names listed for these accession numbers.
e β and γ isoforms not distinguishable with our MS data.
Two-dimensional gel electrophoresis as performed in reference 14.
For each individual or pooled sample, 150 μg of protein was precipitated from the radioimmuno-precipitation assay (RIPA) buffer. Two-dimensional gel electrophoresis was performed for protein separation with isoelectric focusing over pI ranges of 3–10, 4–7, or 5–8 used for the first dimension and SDS-PAGE for the second dimension, as previously described.14,15 Gels were stained with SYPRO-Ruby and images captured on a Kodak Image Station 440CF. Integrated signal intensities were analyzed quantitatively by using Kodak 1D or Bio-Rad’s PDQuest 2-D gel image analysis software. Differential expression was then confirmed visually by two independent observers.
In-gel digestion and MALDI target preparation as performed in reference 14.
The selected spots were manually excised and subjected to in-gel tryptic digestion based on the procedure described by Rosenfeld et al.16 The samples were desalted using a Ziptip μ-C18 pipette tip (Millipore, Billerica, MA) according to the manufacturor’s protocol, using 1.45 μL of matrix to elute the bound peptides directly onto the MALDI target. Ten milligrams of α-cyano-4-hydroxycinnamic acid (Applied Biosystems, Framingham, CA) was dissolved in 1 mL of 50% acetonitrile/0.1% tri-fluoroacetic acid to produce a saturated solution. The supernatant was then diluted 1:1 with solvent and used as the matrix. Alternating rows on the target were spotted with 0.45 μL of Calibration Mixture 1 (Applied Biosystems) for external calibration.
Protein identification by peptide mass fingerprinting (PMF), originally performed in reference 14.
MALDI-TOF spectra were acquired for reference 14 on an Applied Biosystems Voyager DE-PRO in reflectron mode over the m/z range 700–2500. Automated data acquisition was performed by the Proteomics Solutions 1 Utility V1.0.0, with close external calibration performed for each sample spot. Database searching was repeated for this paper. Spectral processing utilized Data Explorer 3.5.0.0 to do baseline correction and de-isotoping of the mass spectra. The peak list was filtered to remove porcine trypsin and human keratin masses seen in blank gel digests or contaminated samples. The remaining 15 or 20 most intense peaks in the 900–2500 m/z range were entered into Auto-MS-Fit (v. 3.2.1)17 for PMF matching between the experimentally measured digest peptide masses and those generated by theoretical digest of a mammalian subset of the Swiss-Prot database (v. May 9, 2004)18 containing proteins up to 100 kDa (about 25,000 entries). The PMF searches were carried out assuming peptides were the result of tryptic digestion, with no more than two missed cleavages, and carbamidomethylation of the cysteine residues. Variable modifications considered were oxidation of methionines, protein N-terminal acetylation, and cyclization of peptide N-terminal Gln to form pyrrolidone carboxylic acid. The first-pass search in Auto MS-Fit required the experimentally measured masses to be within 80 ppm of the theoretical values, and generated a list of proteins that matched at least 5 out of the 20 masses submitted. A second pass utilized Intellical to recalibrate the spectrum based on the first-pass results as a pseudo-internal calibration curve, and required experimentally measured masses to be within 30 ppm of the calculated values. A list of proteins that matched at least 4 out of the 20 peak masses submitted was generated. False-positive hits typically had a maximum of 4 or 5 peptides matched out of 20 masses submitted under the search conditions described.
Protein identification by MALDI-PSD peptide fragmentation as performed in the previous study.14
Proteins with less than 6 peptide masses matched by PMF were subjected to post-source decay (PSD) fragmentation spectra on the most intense peptide ion(s) to identify the protein by peptide sequencing. For the PSD, an average of 10 segments were collected at different mirror ratios. In the <180 m/z range, ~3 × 10−6 torr collision gas (air) was used for CID. A list of fragment ions was entered into the MS-Tag search engine manually, using the same parameters as above, and queried against a database of expected peptide fragment ions based on tryptic digest of database proteins. The parent ion mass tolerance was 30 ppm, while the fragment ion mass tolerance was 1500 ppm. The search was performed first using the Swiss-Prot database. For this paper, the search was repeated with a shuffled database to estimate the rate of false positives.19 False-positive hits occurred when 76 ± 11% of ions were matched for peptides with fewer than 20 ions submitted, and false-postive hits matched at most 60 ± 6% of the ions for the peptide with more than 20 ions submitted.
Protease Screening Kit assay.
Tissue lysates were analyzed to detect any residual protease activity with the Protease Screening Kit (Genotech, St. Louis, MO) following the manufacturer’s protocol. Twenty tissue lysate samples (one sample was used up prior to this analysis) were analyzed. Five micrograms of protein extract in RIPA buffer from each sample was incubated with protease substrate at 37°C for 3 h. Following precipitation and centrifugation steps, assay buffer was added to the clear supernatant. A pink color developed in the presence of protease activity. The intensity of the color, which is proportional to the protease activity, was measured at λ = 574 nm using a Cary 50 Bio UV-Visible Spectrophotometer (Varian Instruments, Walnut Creek, CA).
RESULTS AND DISCUSSION
Protein Fragments Identified from the MALDI-MS Coverage Pattern
Ten proteins identified during the original study appeared in the gel at MW more than 10 kDa lower than the theoretical MW, and thus represent protein fragments. These ten proteins were seen at lower MW in 18 distinct gel spots, and five of the proteins also were detected at the expected MW for the full-length protein. On average, each protein was identified from two separate gels. Figure 1 is a representative 2D gel image from reference 14 showing the location of the 18 protein fragment and 5 full-length protein spots. The proteins were identified either by PMF of the MALDI-MS or peptide fragmentation of individual peptides using MALDI-PSD.14 For this paper, the same spectra were subjected to a new search using the Swiss-Prot database. The protein identification data for the 23 spots of interest from reference 14 is detailed in Supplementary Table 1. The full peak lists, peptide sequences, and residue numbers for the MALDI-matched masses for the new searches are given in Supplementary Table 2.
FIGURE 1.

Representative 2D gel electrophoresis image of normal human pancreatic tissue, covering pi range of 4 to 7. The locations of the spots are marked on the gel, but not every spot is detectable in this particular gel.
TABLE 2.
Summary of Domains Identified from Protein Fragments
| Protein | Domain | In vitroa | In vivoa |
| Chains generated by known processing pathways for enzyme activation | |||
| Cathepsin D | 8–9: heavy chain | ||
| Chymotrypsinogen B | 17–18: neo-chymotrypsinogen A-B chain 19–20: chymotrypsin B chain |
20–22 | |
| Protein fragments corresponding to structural domains | |||
| GRP78 | 22–23: N-terminal ATPase domain 24: C-terminal oligomerization domain |
24, 25, 39 | 23 |
| Pancreatic lipase related protein 2 | 31: N-terminal catalytic domain | 31, 32 | 30, 33 |
| Prolyl 4-hydroxylase β | 35: a (thioredoxin domain)-b-b′ 36: a-b |
26–28 | 29 |
| Heat shock cognate protein 71 kDa | 20: N-terminal ATPase domain with NLS, lacks NES and NLRS sequences, possible inhibitory variant like 54 kDa variant which is not able to move into nucleus but remains in cytoplasm even under stress. | 40 | 7, 8 |
| Cytokeratin 8 | 43: linker and coil 2 CK18 binding domain. Similar to cleavage produced by limited proteolysis studies where 20 kDa coil 2 generated with trypsin and chymotrypsin | 34 | 9, 34, 41–42 |
| Actin, β and γ | 59: C-terminal subdomains 3 and 4 | 43 | |
| Protein fragments not associated with domains | |||
| α-amylase 2A | 36–38 | ||
| 14-3-3-ζ | |||
aReferences are listed for previous observation of protein fragment domains for the proteins listed, either in vitro or in vivo.
For 78-kDa glucose-regulated protein GRP78 (previously reported via its gene name HSPA5), the full-length 72-kDa protein was identified in spot 21. The peptides detected in the MALDI-MS are shown in Figure 2A and are labeled in the spectrum on the upper panel of Figure 2B. Thirty-seven percent of the amino acids from the database sequence for GRP78 precursor protein were covered by the MALDI-MS. Of the MALDI-detected peptides for spot 21, the residue closest to the N-terminal was V50, while the residue closest to the C-terminal was K633. GRP78 was also identified in three lower-MW gel spots, spots 22–24. For spots 22–23, all the peptides detected were confined to the protein’s N-terminal, with V50 the residue closest to the N-terminal, and R386 the C-terminal extreme residue detected (Figure 2A and middle panel of 2B). In contrast, the peptides detected in spot 24 were confined to the protein C-terminal region, with peptides confined between residues K447 and K663 (Figure 2A and lower panel of 2B). Thus, spots 22–23 represent N-terminal protein fragments of GRP78, while spot 24 represents a C-terminal protein fragment of GRP78. The sequence coverage of spots 22 and 23 vs. spot 24 are mutually exclusive, as can be seen by the lack of overlapping peptides in the spectra from spots 23 and 24 in Figure 2B. Cleavage between residues 386 and 447 could have produced the N- and C-terminal fragments. Significantly, even in the spectrum of the full-length protein, no peptides are seen in the 386–447 region, due to a dearth of cleavage sites in that stretch. Similarly, the MALDI-MS for the other protein fragment spots contained peptides that were confined to a particular protein region, either N-terminal, C-terminal, or central. They are listed in Table 1, along with the MALDI-MS N- and C-terminal extreme residues detected.
FIGURE 2.
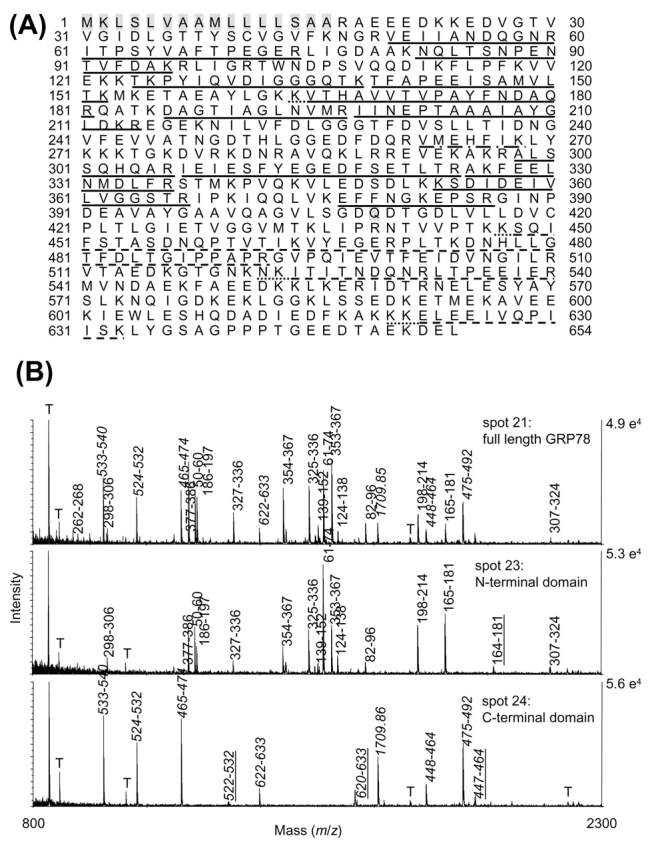
A: Sequence for human GRP78 precursor, with peptides identified by MALDI-MS in spots 21–24 labeled as follows: solid line, peptides seen in spots 21–23; dashed line, peptides seen in 21 and 24; dash-dot, peptide seen in 21 only; dotted lines, missed cleavages seen in 22–24. Shaded sequence is signal peptide at N-terminal, and shaded Q409 is boundary between N- and C-terminal domains. B: MALDI spectra of GRP78 spots 21 (full-length), 23 (N-terminal domain), and 24 (C-terminal domain). Peaks are labeled with residue numbers, with those from the C-terminal domain labeled in italics, and tryptic missed cleavage peptides in spectra 23 and 24 underlined. T denotes trypsin autolysis peaks.
Coverage values are also listed in Table 1, as calculated in the conventional manner, using the number of amino acid residues detected in each MALDI-MS divided by the number of amino acids in the database sequence. In addition, a weighted percent coverage is calculated for the protein fragment spots. Here the ratio of the database MW for the full-length protein over the gel MW of the protein fragment is used to reflect the fact that the spot contains only a fragment of the protein. When this is done, the percentage coverage for protein fragments is on average similar to that of full-length proteins. Using the percentage coverage values for the 18 protein fragments contained in Table 1, 33% coverage is obtained on average. From the data in our previous publication,14 an average of 24% coverage was observed for the 50 protein spots that appeared at expected MW values. In order to improve protein coverage, the MALDI-MS were examined manually to determine whether unmatched peaks could be identified manually, possibly as non-tryptic cleavage products. In general, the unmatched masses listed in the MS-Fit output represented noise. However, in several cases, an unmatched peptide peak was found to be the protein N-terminal peptide after removal of the signal sequence. Since the Swiss-Prot database lists the precursor protein, the signal sequence is not removed for the theoretical digest. However, removal of the signal sequence generates a new N-terminal tryptic peptide for the protein. This peptide was observed in eight MALDI-MS in this study (see Table 1), and confirmed by peptide fragmentation of the N-terminal peptide by MALDI-PSD in three cases. Incorporation of signal peptide information would help to improve database searching for PMF and MS/MS searches. Several other unmatched ions were results of sample oxidation during 2D gel electrophoresis: oxidized tryptophan, oxidized carbamidomethylated cysteines, and fragmentation products of oxidized residues. As seen by the GRP78 spectra in Figure 2B, most of the peptide peaks are identified as tryptic peptides.
Combining 2D gel electrophoresis and protein identification by mass spectrometry is an effective method for establishing the presence of protein fragments in biological samples. The peptides detected for protein fragments are confined to a particular region of the protein sequence, and listing the endpoints of the MS detected peptides helps identify the approximate boundaries, even with the 20–30% coverage typical of MALDI-MS. While region-specific antibodies can also be used for such analyses, the possibility of nonspecific binding necessitates confirmation by another method for identification of any protein not seen at the expected molecular weight. If the antibody is raised for only one region of the protein, it will not detect those fragments that do not contain the epitope sequence, while the MS analysis is largely sequence independent. Reporting of the region covered by the MS protein identification will be a useful tool in establishing the prevalence of fragments and more accurately reflect the information contained by the data.
Processed Protein Chains, Functional and Structural Domains Observed
A number of mechanisms are possible for the generation of the protein fragments. They may be generated at the RNA level as sequence variants and truncations, or post-translational modifications used to activate proteins or fragments generated for alternative purposes, such as inhibitors or for secretion. Some proteins are activated by cleavage into polypeptide chains held together by disulfide bridges. In the cell, they remain intact. However, during reduction and alkylation of the cysteines prior to isoelectric focusing, the disulfide bonds are disrupted and the individual chains are separated by 2D gel electrophoresis. This phenomenon is observed for two of the proteins in this study, cathepsin D and chymotrypsinogen. The active form of cathepsin D consists of a light and heavy chain, linked by disulfide bonds. The heavy chain includes residues 169–412, with a calculated MW of 27 kDa. The experimental MALDI data for spots 8 and 9 includes peptides in the region extending from 195–411, with a gel MW of 27 kDa, and can thus be assigned as the heavy chain of a processed cathepsin D (Table 1).
Five spots were identified as chymotrypsinogen (Table 1). Processing of the inactive zymogen chymotrypsinogen A to the fully active protease chymotrypsin proceeds via two pathways.20,21 In the fast activation pathway, trypsin cleaves after Arg-33, producing an active π-chymotrypsin. Chymotrypsin subsequently excises two dipeptides, residues 32–33 and 165–166, to form the fully activated α-chymotrypsin. The three chains are held together by disulfide bridges. An alternate, slow activation pathway involves chymotryptic cleavage of one or more of three chymotryptic cleavage sites to form inactive neo-chymotrypsinogens, as indicated in Figure 3A.22 Neo-chymotrypsinogens can be activated by tryptic cleavage after Arg-15, again progressing to a three-chain enzyme. The location of the dipeptides and final chains is shown in Figure 3A, highlighting the location of peptides detected by MALDI-MS and MALDI-PSD. Spot 16 migrated at the pI and MW expected for the 28-kDa inactive precursor protein chymotrypsinogen, similar to its position in 2D gel separations of pancreatic juice. Spots 17 and 18 (gel MW 17 kDa) were shown to contain peptides from both the A and B chains of chymotrypsin. The N-terminal peptide containing the A chain and first dipeptide (residues 19–33) is detected only in the spectra of spots 17 and 18. This peptide is generated by tryptic digestion only if the precursor signal peptide has been removed prior to 2D gel electrophoresis. The MALDI-PSD of this peptide at m/z 1562 is shown in Figure 3B. The A-B chain is produced via the neo-chymotrypsinogen pathway of activation. The MS of spots 19–20 combined with gel mobility suggest that these spots contain the B chain of the fully activated chymotrypsin enzyme.
FIGURE 3.
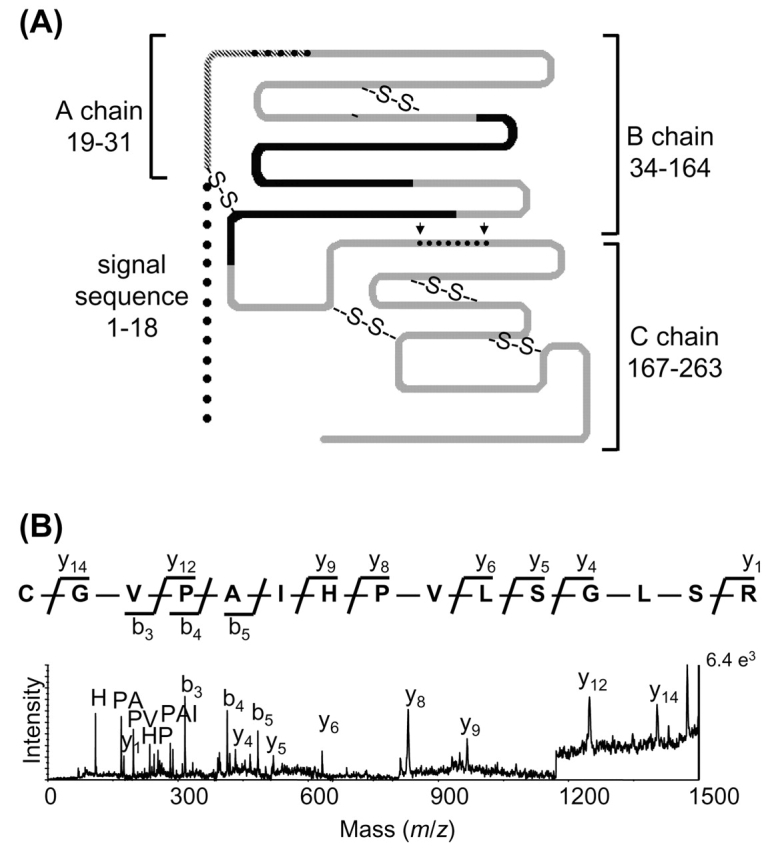
A: Schematic diagram of chymotrypsinogen precursor and chymotrypsin chains. Cleavage sites for neo-chymotrypsinogen activation pathway shown with arrows, with excised dipeptides as dots; adapted from reference 19. Peptides seen in spots 16–20 shown in black, peptide identified in spots 17 and 18 with crosshatching. See text for details. B: MALDI-PSD fragmentation spectra of peptide present only in spots 17 and 18, with parent ion mass m/z 1561. The peptide consists of residues 19–88, containing the A chain plus the dipeptide linker between the A and B chains. Immonium, di-, tripeptide, b, and y ions are labeled for the sequence CGVPAIHPVLSGLSR, assuming the cysteine is carbamidomethylated.
The other eight proteins observed as protein fragments do not represent chains resulting from activation pathways required to produce a functional protein. However, most have been observed previously and represent structural or functional domains. A summary of the domains detected is given in Table 2, along with citations for the characterization of the domains in vitro, or previous observations of fragments from in vivo samples. GRP78 fragments have been observed previously in a global proteomics analysis of pancreatic tissue.23 In addition, in vitro studies have identified two domains, an N-terminal ATPase domain and a C-terminal oligomerization domain.24–26 The boundary between these in vitro domains is Q409, and that is consistent with our observed N-terminal domain spots 22 and 23 and the C-terminal domain in spot 24 (Figure 2).
Prolyl 4-hydroxylase β (gene P4HB), also known as protein disulfide isomerase, is a well-characterized protein containing a signal sequence and five structural domains, as shown in Figure 4A.27–29 It was identified in three gel spots in the original study, spots 34–36. Spot 34 is the full-length protein, and spots 35 and 36 are N-terminal protein fragments, with different cleavage sites producing the 37-kDa and 25-kDa protein fragments (Table 1). The MALDI spectra are shown in Figure 4B. Spot 34 contains peptides from a, b, b′, and a′ domains (upper pane), while spot 35 contains peptides from a, b, and b′ (middle pane) and spot 36 contains peptides only from the a and b domains (lower pane). The cleavage point that generated spot 36 is most likely localized between residues 230 and 247, as the peptide covering residues 231–247 is observed at m/z 1965 (peak labeled b/b′ in Figure 4B) in the spectra of spots 34 and 35, but not spot 36. Additionally, the peptide covering residues 223–230 is observed in spot 36 at m/z 991. The 231–247 region overlaps the boundary between the b and b′ domains. The signal peptide has again been removed, exposing a new N-terminal tryptic peptide that is observed in all spectra at m/z 1521 (peak with bold a label in Figure 4B). The same cleavage site that generated spot 36 was implicated in a 2D gel electrophoresis/mass spectrometry protein profiling experiment on barley seed development that detected both N- and C-terminal domain fragments.30
FIGURE 4.
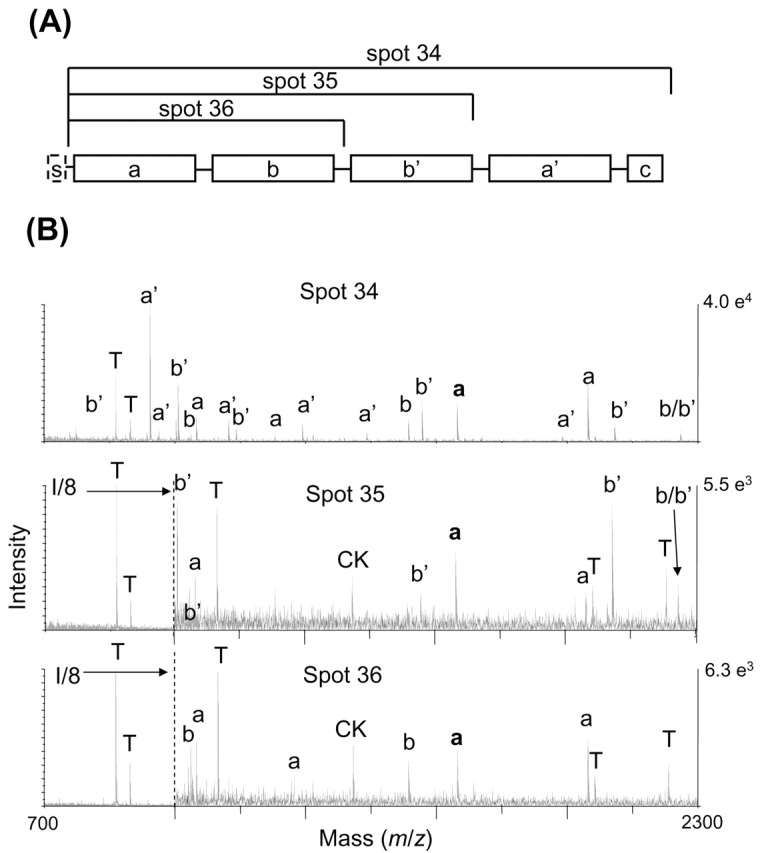
A: Schematic of domains of prolyl 4-hydroxylase β, s-signal; a and a′ are thioredoxin domains, b and b′ are inactive domains; adapted from reference 36. B: MALDI spectra for spots 34–36 with peak labeled according to protein domain. The N-terminal peptide after removal of the precursor protein signal peptide is shown in bold. It is present in all spectra. Peptide labeled b/b′ spans the b and b′ domains. T denotes trypsin autolysis peak, and CK is cytokeratin peak also seen in blank spectrum.
In several studies, limited proteolysis has identified susceptible cleavage sites that are in agreement with our data. In horse pancreatic lipase–related protein 2 (PLRP2), it has been noted that the Ser262Thr263 location is very susceptible to proteolytic cleavage.31 Cleavage at this site is consistent with our data for the protein fragment of PLRP2 in spot 31, with MALDI-MS amino acid residues detected between 44 and 251. In vitro studies have noted that this N-terminal region is the enzyme’s catalytic domain.32,33 Similarly, the linker and coil 2 regions of cytokeratin 8 are contained in the 17-kDa fragment seen in spot 43, which has been observed as 20-kDa fragments from limited proteolysis of cytokeratin 8 and 18 protofilaments.34 Cytokeratin 8 fragments are found in the serum and in tumors as a component of tissue poly-peptide specific antigen (TS1), as reviewed by Sundstrom and Stigbrand.35 Interestingly, the immunodominant region of the cytokeratin 8 fragments in serum was determined to be the conserved residues 340–365, a region present in the 17-kDa fragment at spot 43.
Heat shock cognate protein 71 kDa (HSC71) has a 54-kDa variant (HSC54) containing the N-terminal portion of the protein, with deletions in the C-terminal domain, which has been identified at the mRNA level in various types of human cells and tissues.7 There is evidence that it functions as an endogenous inhibitory regulator of HSC71 by competing for the cochaperones. In addition, the 54-kDa variant is not able to translocate into the nucleus during stress conditions because it lacks a nuclear localization–related sequence.8 The 37-kDa ATPase domain in spot 40 contains N-terminal peptides common to both HSC71 and HSC54, and may represent another variant, or a fragment of either of the known variants.
For two of the proteins detected as protein fragments, the fragments were not associated with known domains. α-Amylase was identified in spots 27 and 28, which correspond to N-terminal and central domain fragments. However, these fragments do not correlate with the known structural domains of the protein.36–38 For 14-3-3 ζ, a single peptide was detected near the protein’s N-terminal, but the coverage is too low to make any connection to protein domains. In summary, 18 protein fragment spots were identified, with 6 spots corresponding to post-translational processing for activation by limited proteolysis, 9 spots representing structural domains, and 3 that could not be correlated with protein domains.
Residual Protease Activity Is Detected in the Pancreatic tissue Protein extracts during extended Incubation times
The cleavages identified suggest that limited proteolysis was the mechanism of protein-fragment production. This would be facilitated by activation of endogenous proteases, as high levels of protease precursors are found normally in pancreatic tissue. Activation could occur as part of normal cellular processes, during sample collection, and/or during processing of the lysates prior to 2D gel electrophoresis. After collection, the tissue samples used in this study were frozen at −80°C prior to analysis and then lysed in protease inhibitor containing RIPA buffer. After lysis, they were kept in 8 M urea briefly on ice prior to 2D gel electrophoresis. Thus, it is unlikely that significant proteolytic activity occurred during sample processing prior to 2D gel electrophoresis.
In order to determine whether any active proteases are present in the sample, extended incubation at 37°C was conducted on 20 of the original tissue samples, using a commercial protease screening kit. A color change indicates proteolytic activity, and absorbance values are given in Table 3. A control sample, without tissue, lacked proteolytic activity. Normal pancreatic tissue (n = 8) derived from both autopsy and surgical samples, as well as the pancreatitis patient tissue (n = 6), showed higher levels of proteolytic activity than the tumor tissue on average. The method of sample collection does not explain the difference in proteolytic activity, as surgical and autopsy normal samples gave a similar range of values. Tumor tissue has lower levels of the precursor proteases,14 and it is therefore not surprising that a relatively lower level of proteolytic activity was observed. When protease inhibitor levels were doubled, a slight reduction in activity was noted in the normal and pancreatitis samples (Table 3). This result indicates that some proteases in these samples are not affected by the protease inhibitors used in standard preparations.
TABLE 3.
Protease Activity Detection with Screening Kit after Extended Incubationa
| Absorbance for 1X PIb | Absorbance for 2X PIb | |
| Normal (n = 8) | 0.072 ± 0.023 | 0.056 ± 0.026 |
| Pancreatitis (n = 6) | 0.062 ± 0.028 | 0.051 ± 0.025 |
| Tumor (n = 6) | 0.036 ± 0.034 | 0.041 ± 0.045 |
a Tissue samples were incubated at 37°C for 3 h in RIPA buffer containing protease inhibitors, then assayed with the Protease Screening Kit for determination of protease activity. Absorbance was measured at λmax 574 nm. Higher levels of absorbance indicate higher levels of protease activity in the sample.
b1X: Incubation with normal 1X concentration of protease inhibitors; 2X: incubation with 2X concentration of protease inhibitors.
While these experiments indicate that high levels of endogenous proteases may increase the abundance of protein fragments, protein fragments have been observed in other sample types. Other investigators using 2D gel electrophoresis combined with Western blotting or mass spectrometry have identified protein fragments in multiple gel spots when analyzing tissue, plant material, and microorganisms.3–5,23,30 Increased use of 2D Western blotting and 2D gel electrophoresis/mass spectrometry identification in a variety of sample types and species will enable determination of the prevalence and variety of protein fragments.
CONCLUSION
Two-dimensional gel electrophoresis and MALDI-MS are an effective strategy for determining the protein domains present in those gel spots that are observed at significantly lower MW values than are given in the database. While average sequence coverage is only 30%, the peptides detected are confined to a specific region of the protein, such as the protein N- or C-terminal. This information could easily be incorporated into protein identification tables. Regional coverage information is not readily available from either LC-MS/MS analysis of digests of cellular lysates or from epitope-specific antibodies. Some of the protein fragments correspond to chains produced by known cellular processing and activation pathways. Others have been detected as functional and structural domains during in vitro experiments or noted in other in vivo studies, indicating they function intra- or extra-cellularly (see Table2). By using tools that allow both protein identification and measurement of MW, we can assess the abundance and distribution of protein fragments. Correlation of these results with targeted functional studies on specific proteins will elucidate the biological function of protein fragments.
Acknowledgments
We thank Lisa Schroeder for her excellent technical support, Rong Wang for providing the shuffled database, and Asma Sharif for assistance with the figures. This study was supported by National Institutes of Health (NIH) grants RO1 CA098380, P20 CA101936, P30 ES07784, NIH Cancer Center Core grant CA16672, and a research grant from the Topfer Research Funds.
REFERENCES
- 1.Hart C, Schulenberg B, Steinberg TH, Leung WY, Patton WF. Detection of glycoproteins in polyacrylamide gels and on electroblots using Pro-Q Emerald 488 dye, a fluorescent periodate Schiff-base stain. Electrophoresis 2003;24:588–598. [DOI] [PubMed] [Google Scholar]
- 2.Schulenberg B, Goodman TN, Aggeler R, Capaldi RA, Patton WF. Characterization of dynamic and steady-state protein phosphorylation using a fluorescent phosphoprotein gel stain and mass spectrometry. Electrophoresis 2004;25:2526–2532. [DOI] [PubMed] [Google Scholar]
- 3.Roberts K, Bhatia K, Stanton P, Lord R. Proteomic analysis of selected prognostic factors of breast cancer. Proteomics 2004;4:784–792. [DOI] [PubMed] [Google Scholar]
- 4.Somiari RI, Sullivan A, Russell S, Somiari S, Hu H, Jordan R, et al. High-throughput proteomic analysis of human infiltrating ductal carcinoma of the breast. Proteomics 2003;3:1863–1873. [DOI] [PubMed] [Google Scholar]
- 5.Mattow J, Schmidt F, Hohenwarter W, Siejak F, Schaible UE, Kaufmann SH. Protein identification and tracking in two-dimensional electrophoretic gels by minimal protein identifiers. Proteomics 2004;4:2927–2941. [DOI] [PubMed] [Google Scholar]
- 6.Mattow J, Schaible UE, Schmidt F, Hagens K, Siejak F, Brestrich G, et al. Comparative proteome analysis of culture supernatant proteins from virulent Mycobacterium tuberculosis H37Rv and attenuated M. bovis BCG Copenhagen. Electrophoresis 2003;24:3405–3420. [DOI] [PubMed] [Google Scholar]
- 7.Tsukahara F, Yoshioka T, Muraki T. Molecular and functional characterization of HSC54, a novel variant of human heat-shock cognate protein 70. Mol Pharmacol 2000;58:1257–1263. [DOI] [PubMed] [Google Scholar]
- 8.Tsukahara F, Maru Y. Identification of novel nuclear export and nuclear localization-related signals in human heat shock cognate protein 70. J Biol Chem 2004;279:8867–8872. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Quintela A, Mella C, Abdulkader I, Pérez LF, Campos J, Otero E, et al. Serum levels of tissue polypeptide specific antigen are correlated with hepatocyte cytokeratin expression in alcoholic liver disease. Alcohol Clin Exp Res 2004;28:1413–1418. [DOI] [PubMed] [Google Scholar]
- 10.Nomura F, Tomonaga T, Sogawa K, Ohashi T, Nezu M, Sunaga M, et al. Identification of novel and downregulated biomarkers for alcoholism by surface enhanced laser desorption/ionization-mass spectrometry. Proteomics 2004;4:1187–1194. [DOI] [PubMed] [Google Scholar]
- 11.Pike SE, Yao L, Setsuda J. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood 1999;94:2461–2468. [PubMed] [Google Scholar]
- 12.Xiao F, Wei Y, Yang L, Zhao X, Tian L, Ding Z, et al. A gene therapy for cancer based on the angiogenesis inhibitor, vasostatin. Gene Ther 2002;9:1207–1213. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Bast RC, Jr., Yu Y, Li J, Sokoll LJ, Rai AJ, et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res 2004;64:5882–5890. [DOI] [PubMed] [Google Scholar]
- 14.Shen J, Person MD, Zhu J, Abbruzzese JL, Li D. Protein expression profiles in pancreatic adenocarcinoma compared with normal pancreatic tissue and tissue affected by pancreatitis as detected by two-dimensional gel electrophoresis and mass spectrometry. Cancer Res 2004;64:9018–9026. [DOI] [PubMed] [Google Scholar]
- 15.Liu J-W, Shen J-J, Tanzillo-Swarts A, Bhatia B, Maldonado CM, Person MD, et al. Annexin II expression is reduced or lost in prostate cancer cells and its re-expression inhibits prostate cancer cell migration. Oncogene 2003;22:1475–1485. [DOI] [PubMed] [Google Scholar]
- 16.Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal Biochem 1992;203:173–179. [DOI] [PubMed] [Google Scholar]
- 17.Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (±10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 1999;71:2871–2882. [DOI] [PubMed] [Google Scholar]
- 18.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 2004;32 (Database issue):D115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang R, Prince JT, Marcotte EM. Mass spectrometry of the M. smegmatis proteome: protein expression levels correlate with function, operons, and codon bias. Genome Res 2005;15:1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraut J. Chymotrypsinogen: X-ray structure. In Hydrolysis: Peptide Bonds Vol. 3, 3rd ed. NY: Academic Press, 1971, pp. 165–169.
- 21.Guy O, Rovery M, Desnuelle P. Formation and activation of neo-chymotrypsinogen B. Biochim Biophys Acta 1966;124:402–405. [DOI] [PubMed] [Google Scholar]
- 22.Rovery M, Poilroux M, Yoshida A, Desnuelle P. Degradation of chymotrypsinogen by chymotrypsin. Biochim Biophys Acta 1957;23:608–620. [DOI] [PubMed] [Google Scholar]
- 23.Hu L, Evers S, Lu ZH, Shen Y, Chen J. Two-dimensional protein database of human pancreas. Electrophoresis 2004;25:512–518. [DOI] [PubMed] [Google Scholar]
- 24.Chevalier M, King L, Wang C, Gething MJ, Elguindi E, Blond SY. Substrate binding induces depolymerization of the C-terminal peptide binding domain of murine GRP78/BiP. J Biol Chem 1998;273:26,827–26,835. [DOI] [PubMed] [Google Scholar]
- 25.Chevalier M, King L, Blond S. Purification and properties of BiP. Methods Enzymol 1998;290:384–409. [DOI] [PubMed] [Google Scholar]
- 26.King L, Chevalier M, Blond SY. Specificity of peptide-induced depolymerization of the recombinant carboxy-terminal fragment of BiP/GRP78. Biochem Biophys Res Commun 1999;263:181–186. [DOI] [PubMed] [Google Scholar]
- 27.Darby NJ, Penka E, Vincentelli R. The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency. J Mol Biol 1998;276:239–247. [DOI] [PubMed] [Google Scholar]
- 28.Darby NJ, van Straaten M, Penka E, Vincentelli R, Kemmink J. Identifying and characterizing a second structural domain of protein disulfide isomerase. FEBS Lett 1999;448:167–172. [DOI] [PubMed] [Google Scholar]
- 29.Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta 2004;1699:35–44. [DOI] [PubMed] [Google Scholar]
- 30.Finnie C, Melchior S, Roepstorff P, Svensson B. Proteome analysis of grain filling and seed maturation in barley. Plant Physiol 2002;129:1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jayne S, Kerfelec B, Foglizzo E, Chapus C, Crenon I. High expression in adult horse of PLRP2 displaying a low phospholipase activity. Biochim Biophys Acta 2002;1594:255–265. [DOI] [PubMed] [Google Scholar]
- 32.Lowe ME. Properties and function of pancreatic lipase related protein 2. Biochimie 2000;82:997–1004. [DOI] [PubMed] [Google Scholar]
- 33.Roussel A, Yang Y, Ferrato F, Verger R, Cambillau C, Lowe M. Structure and activity of rat pancreatic lipase-related protein 2. J Biol Chem 1998;273:32,121–32,128. [DOI] [PubMed] [Google Scholar]
- 34.Hatzfeld M, Maier G, Franke WW. Cytokeratin domains involved in heterotypic complex formation determined by in-vitro binding assays. J Mol Biol 1987;197:237–255. [DOI] [PubMed] [Google Scholar]
- 35.Sundstrom BE, Stigbrand TI. Cytokeratins and tissue polypeptide antigen. Int J Biol Markers 1994;9:102–108. [DOI] [PubMed] [Google Scholar]
- 36.Buisson G, Duee E, Haser R, Payan F. Three dimensional structure of porcine pancreatic alpha-amylase at 2.9 A resolution. Role of calcium in structure and activity. EMBO J 1987;6:3909–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larson SB, Greenwood A, Cascio D, Day J, McPherson A. Refined molecular structure of pig pancreatic alpha-amylase at 2.1 Å resolution. J Mol Biol 1994;235:1560–1584. [DOI] [PubMed] [Google Scholar]
- 38.Qian M, Haser R, Payan F. Structure and molecular model refinement of pig pancreatic alpha-amylase at 2.1 Å resolution. J Mol Biol 1993;231:785–799. [DOI] [PubMed] [Google Scholar]
- 39.Chevalier M, Rhee H, Elguindi EC, Blond SY. Interaction of murine BiP/GRP78 with the DnaJ homologue MTJ1. J Biol Chem 2000;275:19,620–19,627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flaherty KM, McKay DB, Kabsch W, Holmes KC. Similarity of the three-dimensional structures of actin and the ATPase fragment of a 70-kDa heat shock cognate protein. Proc Natl Acad Sci USA 1991;88:5041–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ditzel HJ, Garrigues U, Andersen CB, Larsen MK, Garrigues HJ, Svejgaard A, et al. Modified cytokeratins expressed on the surface of carcinoma cells undergo endocytosis upon binding of human monoclonal antibody and its recombinant Fab fragment. Proc Natl Acad Sci USA 1997;94:8110–8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishibori H, Matsuno Y, Iwaya M, Osada T, Kubomura N, Iwamatsu A, et al. Human colorectal carcinomas specifically accumulate Mr 42,000 ubiquitin-conjugated cytokeratin 8 fragments. Cancer Res 1996;56:2752–2757. [PubMed] [Google Scholar]
- 43.Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin:DNase I complex. Nature 1990;347:37–44. [DOI] [PubMed] [Google Scholar]