

Abstract
We have optimized a two-plasmid Tet-On system, the regulatory plasmid and the response plasmid, to produce tightly controlled inducible expression of the gene RAGE in cell-culture models. Two sets of plasmids were constructed: set 1 (universal; for broad range of cell types) and set 2 (neuron specific). For the response plasmid, the gene RAGE was cloned in pIRES2-EGFP plasmid (Clontech) and the CMV promoter replaced with TREtight (modified seven copies of Tet-operon fused with CMVm promoter). For the regulatory plasmid, rtTA (reverse tetracycline transactivator) was placed under either the CMV promoter or the cell-specific promoter neuronal specific enolase. Both plasmids have the mammalian selection marker neomycine; the EGFP reporter gene is only in the response plasmid and IRES is between the gene and EGFP. Following induction with doxycycline, cells expressing RAGE showed neomycine resistance and green fluorescence (EGFP). Our system has been tested in two different cell lines and showed negligible basal leakiness, high induction of the gene RAGE (142-fold), dose-dependent response to doxycycline, and strict cell-type specificity. This system is highly suitable for cell-specific expression of any gene of interest in primary cultures and mixed cell populations.
Keywords: Inducible system, Tet-On/Off, RAGE
The receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin superfamily of cell-surface molecules.1 RAGE has been implicated in the pathogenesis of several chronic disorders, including neurodegenerative diseases such as Alzheimer’s, Huntington’s, and Parkinson’s disease.1–3 Our main interest was to investigate the role of RAGE in neurodegeneration by over-expressing RAGE using cell-culture models. The salient feature of RAGE is that it binds to several classes of ligands; its repertoire of ligands includes advanced glycation end products (AGEs), amphoterin (highly expressed in neuronal development), β amyloid (a hallmark of Alzheimer’s disease), and S100/ calgranulins (a family of inflammatory mediators).1,4 The receptor (RAGE)-ligand interaction triggers cascades of cell signaling events including the release of TNF-α1 and caspase-8 (effector protein of TNF family cell-death pathway),5 both of which can be toxic to cells. It was therefore necessary to develop and optimize vectors enabling the tight regulation of this gene in terms of its induction in adequate amounts and at the desired time point.
A number of inducible gene expression systems have recently been described; the systems are named after their inducer drugs. Five systems that can be used for expressing genes in mammalian cell lines have been described: Tet-On/Off,6,7 Pip-On/Off,8 antiprogestin-dependent,9 ecdysone-dependent,10,11 and the rapamycin-dependent12,13 system. Among these, the Tet-On/Off system has been shown to exhibit the best features. The Tet system is well characterized and has been studied extensively in the context of both viral and non-viral vectors to regulate a variety of genes.14–17 The Tet-On/Off system originally comprised of two expression units: the response plasmid, where the gene of interest is placed under the Tet-inducible promoter/Tet responsive element (TRE), and the regulatory plasmid, which carries the tetracycline trans-activator (tTA) or reverse tetracycline trans-activator (rtTA), able to bind the inducer drug (dox/Tet) and mediate either repression or activation of the inducible gene.6,7,16,18 The response plasmid may carry a reporter gene to allow for assessment of the degree of success of the transfection and transfection efficiency. Both plasmids may carry selection markers, such as drug/antibiotic resistance genes for the selection of stably transfected cell lines. While many examples of successful applications of Tet-On/Off systems have been reported, certain limitations, such as basal leakiness and low induction of the genes, have necessitated further development to provide improved versions of this system. Thus, extensive development of the Tet-system has taken place since the time it was originally proposed by Gossen and Bujard (1992, Tet-Off system)6 and Gossen et al. (1995, Tet-on system).7 The most recent developments include incorporating both components of the Tet-system (rtTA and TRE) into one plasmid in both viral and non-viral vector.14,15,17
We wished to use the Tet-On system as an inducible expression system for the gene RAGE. Our early attempts to modify the pIRES2-EGFP plasmid (BD Biosciences, Clontech) by placing the TetCMV promoter (seven copies of Tet operon placed upstream of the CMV) upstream of the gene RAGE cloned at the multiple cloning site (MCS) of this plasmid and using the pCMV-rtTA (pTet-on, Clontech) plasmid as a regulatory plasmid, suffered from serious basal leakiness of the gene RAGE. Thus, the observed difference in gene expression between the non-induced and induced state was minimal. Therefore, our aim was to optimize the Tet-On system to achieve maximum induction of the gene RAGE with negligible basal leakiness of the gene. In addition, we also wanted to use enhanced green fluorescent protein (EGFP) as the reporter molecule, to make stable cell lines using a mammalian selection marker and have an internal ribososme entry site (IRES) between the MCS/gene of interest and EGFP, so that EGFP and the gene of interest could be translated from the same bicistronic mRNA. In a recent report by Lee et al.,16 it has been suggested that the individual components of the Tet system (rtTA and TRE) function optimally to control gene expression when they are incorporated into separate vectors, and that the ratio of rtTA to TRE is also very important to achieve efficient regulation of gene expression. We therefore optimized our dual-vector Tet-On system for the gene RAGE consisting of both a regulatory plasmid and a response plasmid.
MATERIALS AND METHODS
Plasmid Constructions
Two sets of plasmids, named as a universal set 1 (to transfect a broad range of cell lines) and a neuron-specific set 2 (for neuronal cell lines) were constructed. For set 1, we used pTet-On (BD Biosciences, Clontech; provided by a close colleague, Dr. Stephanie Hughes, Department of Pharmacology, Faculty of Medical and Health Sciences, University of Auckland, New Zealand) as a regulatory plasmid, where rtTA expression is driven by the CMV promoter; we named this plasmid pCMV-rtTA. To generate the response plasmid for set 1, pIRES2-EGFP (BD Biosciences, Clontech; provided by a close colleague, Dr. Debbie Young, Department of Molecular Medicine, Faculty of Medical and Health Sciences, University of Auckland) was modified. Full-length hRAGE cDNA in the plasmid pcDNA3 was provided by Dr. Shi-Fang Yan, Division of Surgical Science, Columbia University, New York. RAGE was excised from pcDNA3 using KpnI/XhoI restriction enzymes, blunted using T4 DNA polymerase (Epicentre, Canada) and cloned at the EcoRV site into the pBluescript II SK (+) plasmid (Stratagene). The gene RAGE was then cloned using SacII and SalI restriction sites at the MCS of pIRES2-EGFP. This plasmid was named pIRES-RAGE-EGFP. The CMV promoter of pIRES-RAGE-EGFP was replaced with TREtight, a modified Tet-responsive promoter that contains a Tet-response element with seven direct repeats of a 36-bp sequence; each repeat contains a 19-bp Tet operator sequence (Tet-O), and this is fused to a CMV minimal promoter region.19 For this replacement step, the TREtight fragment was amplified from the pTREtight plasmid (BD Biosciences, Clontech; provided by Dr. Stephanie Hughes). Cloning sites were added to the TREtight by PCR amplification using the forward primer with an AseI site, the reverse primer with a NheI site (primers, Table 1), and pfx Taq polymerase (proofreading polymerase), which gives blunt ended products. After running the PCR products on the agarose gel, the 362-bp fragment was purified using an Eppendorf kit. The blunt-ended TREtight fragment was first cloned into pZErO-2 (Invitrogen) at the EcoRV site (EcoRV restriction enzyme gives blunt-ended DNA fragments). This fragment was then excised using AseI and NheI restriction enzymes and cloned into the pIRES-RAGE-EGFP at these sites. This plasmid was named pTREtight-RAGE-EGFP.
TABLE 1.
Primer Specifications and PCR Conditions
| Target | Primer sequences (5′ → 3′) | PCR conditions | Product size | |
| NSE | Forward(AseI site at 5′) | attaatgagctcctcctctgctcgc | 94°C × 4 min | 1.8 Kb |
| Reverse(NheI site at 5′) | gctagcctcgaggactgcagactcag | 35 Cycles 94°C × 1 min 54°C × 1 min 72°C × 2 min Extension: 72°C ×10 min |
||
| rtTA | Forward (NheI site at 5′) | gctagcatgtctagattagataaaagtaaagtgatt | Same as above | 1.0 Kb |
| Reverse (EcoRI site at 5′) | gaattcctacccaccgtactcgtca | |||
| TREtight | Forward(AseI site at 5′) | attaatctcgagtttaccactccctatcag | 94°C × 4 min | 0.362 Kb |
| Reverse(NheI site at 5′) | gctagcaggatccccaggtaccgt | 35 Cycles 94°C × 30 sec 54°C × 30 sec 68°C × 1 min Extension: 68°C × 10 min |
To achieve neuronal cell specificity in set 2, pNSE-rtTA was made by again modifying the pIRES2-EGFP (BD Biosciences, Clontech). The CMV promoter was replaced with the neuronal specific enolase (NSE) promoter, and rtTA was cloned at the MCS after removing the EGFP fragment. For these modifications, the restriction sites AseI and NheI were added to the NSE promoter of pAAV/NSE-Luc (provided by Dr. Debbie Young) by PCR amplification (forward primer with AseI site and the reverse primer with NheI site; Table 1), and pfx Taq poly-merase was used for amplification. The amplified NSE promoter product, with the required restriction sites, was run on an agarose gel and purified using an Eppendorf kit. The product was then cloned into pZErO-2 vector (Invitrogen) at its EcoRV site, excised using AseI and NheI restriction enzymes, and cloned into the pIRES2-EGFP at these sites. The EGFP gene was removed from this plasmid using the restriction enzymes NotI and BstXI. The plasmid was then ligated by T4 DNA ligase (Roche) after blunt ending using T4 DNA polymerase (Epicentre, Canada). The rtTA fragment was then amplified from the pTet-On plasmid using primers with NheI and EcoRI sites in the forward and the reverse primers, respectively. After purifying the fragment, it was cloned into the pZErO-2 vector at the EcoRV site and then excised and cloned into the pNSE-IRES (without EGFP) plasmid between the NheI and EcoRI sites. These modifications gave a plasmid with rtTA under the NSE promoter without the reporter gene EGFP. This plasmid was named pNSE-rtTA. The response plasmid was the same for set 1 and set 2.
The cloning strategies are summarized in Figure 1. Two sets of plasmids were constructed: set 1, universal set/non-cell-type specific, consisting of the pCMV-rtTA (pTet-On) and pTREtight-RAGE-EGFP; and set 2, neuron-specific set consisting of pNSE-rtTA and pTRE-tight-RAGE-EGFP. The regulatory plasmids have neomycin resistance (Neor gene), which is used as a mammalian selection marker. This plasmid can therefore be used to make stable cell lines. The response plasmid also has the Neor and the EGFP reporter gene. Thus, after induction of the gene, only cells which are transfected with both the regulatory and the response plasmids show green fluorescence. Double-stable cells will show Neor resistance and green fluorescence.
FIGURE 1.


Cloning strategies used to construct the Tet-On system. A: Schematic representation of the cloning steps used to make the regulatory plasmids pCMV-rtTA and pNSE-rtTA. in PCMV-rtTA, expression of rtTA is driven by the CMV promoter, whereas in pNSE-rtTA, the CMV promoter of the pIRES2-EGFP plasmid was replaced with the NSE promoter and the rtTA fragment subcloned downstream of the NSE promoter after removing the EGFP fragment. B: Schematic illustration of the subcloning steps involved in the construction of the response plasmid. The CMV promoter was replaced with TREtight in the pIRES2-EGFP, and the gene RAGE subcloned downstream of the TREtight.
Transfections
Murine neuroblastoma Neuro2a (ATCC, CCL-131) and human cervical carcinoma HeLa (ATCC, CCL-2) cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). For routine cultures, cell lines were cultured in complete Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 1 mM sodium pyruvate. Cultures were maintained in 250 mL, 75 cm2 culture flasks and were passaged routinely on reaching confluency. For transfections, Neuro2a (2.4×105 cells) and HeLa (1.5×105 cells) were plated in 24-well plates. On reaching 90 95% confluency, cells were transiently co-transfected with either set 1 or set 2 plasmids by using a Lipofectamine 2000 transfection kit (Invitrogen). All plasmids were purified using a Qiagen kit. Opti-MEM (modified MEM medium, Invitrogen) was used to dilute DNA and the lipofectamine reagent according to the manufacturer’s instructions. The ratios of regulatory to response plasmids and the total amount of DNA (μg) to lipofectamine (μL) were determined from our preliminary experiments (data not shown). We found that a 1:10 nM ratio of regulatory to response plasmid and a 1:2.4 ratio of total DNA (μg) to lipofectamine (μL) were optimal; these ratios were used for all transient co-transfection experiments. The medium was replaced after 4 h of transfection with complete DMEM with 10% FBS, 2 mM glutamine, and 1 mM sodium pyruvate. As the response plasmid has the EGFP reporter gene, EGFP expression (green fluorescence) was used to monitor the transfection efficiency.
Induction of RAGE Expression, Dose Response to Doxycycline (dox, an Analogue of Tetracycline) and Basal Leakiness of the Gene
RAGE expression was induced 24 h after transfection by supplementing the medium with different doses of dox (0.1 2μg/mL). RAGE expression, shown as green fluorescence due to EGFP expression, was recorded using a Nikon inverted microscope; all images were taken under identical microscopic settings. Transfection efficiency was estimated by counting the number of EGFP-expressing cells/total number of cells. Cells were recovered after 24h of induction, washed twice with PBS, and lysed in whole cell lysis buffer (2% SDS, 2% glycerol, 62.5 mM Tris [pH 6.8], and 1 complete mini-tablet of proteases [Roche] per 10 mL of lysis buffer). Protein concentration was estimated by using a Bio-Rad protein assay. Western blot was performed using a monoclonal anti-human RAGE antibody (Chemicon) to detect the full length RAGE in the cell lysates obtained from these experiments. Briefly, 20 μg of protein was loaded into each well of a 10% PAGE gel and run at 200 V. A nitrocellulose membrane and Trisglycine transfer buffer (with 20% methanol) was used for transfer. Transfer was carried out at 120 V for 1 h at 4°C. The membrane was blocked with 5% nonfat milk in PBS-T (PBS with 0.1% Tween-20) for 16 18 h at 4°C. After blocking, the membrane was washed three times in PBS-T at room temperature (RT) followed by incubation for 2 h at RT with the monoclonal RAGE antibody (Chemicon), 1:1000 diluted in 1% milk in PBS-T. The membrane was then washed with PBS-T twice, followed by incubation with anti-mouse secondary antibody (Sigma, 1: 5000 diluted in 1% milk in PBS-T). β-actin was used as an internal loading control. ImageJ software was used to quantify the bands. The RAGE expression was normalized against the β-actin expression and the ratio of RAGE/actin optical density (OD) was used for analysis.
Cell Type Specificity
Neuro2a and HeLa cells were co-transfected either with the universal set 1 plasmids (pCMV-rtTA and pTREtight-RAGE-EGFP, 1:10 nM ratio) or with the neuron-specific set 2 plasmids (pNSE-rtTA and pTREtight-RAGE-EGFP, 1:10 nM ratio), as described above. Cell specificity of the promoter was investigated after supplementing the medium with or without dox (2 μg/mL) by microscopic detection of the green fluorescence using a Nikon inverted microscope and by detection of RAGE expression on Western blot.
RAGE Induction in the Presence of AGEs in both Neuro2a and HeLa Cells
Transfected cells were induced with 2 μg/mL of dox and supplemented with 50 μMμM AGEs (prepared in our laboratory as described below). Culture media and cell lysates were collected after 4, 6 and 24 h of RAGE induction. Cell lysate protein concentration was adjusted to 1 mg protein/ mL of lysate. TNF-α is the downstream effector of RAGE is the downstream effector of RAGE-AGE interaction, and caspase-8 the effector protein of the TNF-α-mediated cell death pathway.1,5 The presence of TNF-α and caspase-8 was therefore estimated in the culture supernatants and cell lysates. RAGE and caspase-8 were detected by Western blot using specific monoclonal antibodies (Chemicon and Cell Signaling, respectively). This anti-caspase-8 antibody (IC12) detects cleaved, un-cleaved, and intermediate caspase-8 fragments. TNF-α was measured in the culture supernatants and the corresponding cell lysates from Neuro2a and HeLa cells using both mouse and human ELISA kits (BD Biosciences), respectively. The minimum detectable limits of TNF-α for these kits were 5 pg/mL and 2 pg/mL, respectively. Samples and standards were run in duplicate according to the manufacturer’s instructions, and the mean absorbance value of each standard and the sample was recorded after subtracting the mean absorbance value of the zero standard.
Preparation of AGEs
AGEs were prepared by incubating 0.75 mM BSA with 0.75 M glucose and 0.01 M D-ribose in sterile PBS (pH 7.4) at 50°C for 6 wks. All solutions used were filter sterilized and all incubations done in sterile conditions. After incubation, the samples were dialyzed for 24 h to remove unbound sugars; the dialysis process was repeated three times. Dialyzed AGEs were filter sterilized and stored at 70°C until use. The AGEs preparation was analyzed for the presence of bacterial endotoxins by QCL-1000 endotoxin assay kit (BioWhittaker) according to the manufacturer’s instructions (endotoxins could not be detected in the AGE preparation). Protein concentrations were estimated by the Bradford protein assay (Bio-Rad) using BSA standards. AGE contents were determined by taking the optical measurements at 405 nm (OD at 405 nm; 0.588/mg protein) as described by Munch et al.20 and Kuhla et al.21 The presence of AGEs was also confirmed by running the samples on a 10% PAGE gel and performing Western blot using the monoclonal anti-AGE 6D12 antibody (TransGenic, Inc., Japan). This antibody is a Nɛ-carboxymethyllysine (CML) adduct; i.e., it recognizes the CML-complexed BSA/proteins, where the CML epitope of AGEs serves as the ligand for RAGE.4,22 The concentration of AGE-modified proteins was calculated using the molecular weight of non-glycated protein (BSA).
RESULTS
The Reduction of the Basal Leakiness of the Gene RAGE and Dose Response to dox
Following co-transfection of Neuro2a and HeLa cells with set 1 plasmids (pCMV-rtTA and pTREtight-RAGE-EGFP), in the absence of the inducer dox, expression of the EGFP reporter molecule and RAGE was undetectable (Figure 2). High inducibility (142-fold) of the full-length RAGE was seen in both Neuro2a and HeLa cell lines in the presence of dox (Figure 2, B and C). We observed two bands of RAGE, corresponding to 55 and 50 kd on the Western blot (Figure 2B). The level of expression of EGFP and RAGE correlated positively with the amount of dox used for induction. At 0.1 μg/mL of dox, Neuro2a cells showed 5.5-fold increase in the expression of RAGE; 1 μg/ mL dox showed 42.5-fold induction, while 2 μg/mL of dox showed 142-fold induction of RAGE compared to non-induced RAGE expression, which was negligible (Figure 2, A, B, and C). The presence of the reporter gene EGFP made it possible to monitor the efficiency of transfection (70–75%) and transfection homogeneity in cells. Furthermore, the use of a dual vector system, i.e., incorporating the TREtight promoter and transactivator elements in two separate vectors, allowed us to alter the ratio of regulatory plasmid to response/reporter plasmid. A ratio of 1:10 nM was the most efficient for transient co-transfections.
FIGURE 2.
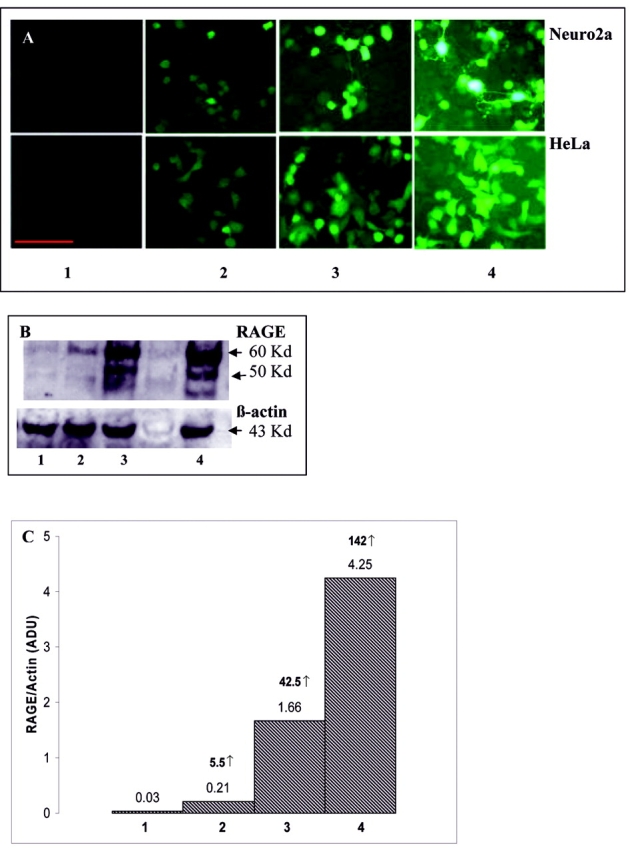
Induction of RAGE expression with different doses of dox. A: EGFP expression in Neuro2a and HeLa cell lines; scale bar: 50 μm. B: Representative Western blot for RAGE, using anti-RAGE monoclonal antibody (Chemicon) and Neuro2a cell lysates. Bands were quantified using imageJ. C: Histogram representing the fold increases in the induction of RAGE expression in response to the different doses of dox. Each bar represents the ratio of RAGE protein to β-actin. The arbitrary densitometric units (ADU) are depicted on each bar and the fold increase relative to non-induced RAGE shown in bold over the bars. 1 = no induction; 2 = induction with 0.1 μg/mL of dox; 3 = induction with 1 μg/mL of dox; 4 = Induction with 2 μg/mL of dox.
Cell-Type Specificity of Our System
The neuronal cell-type specificity of our system was investigated using both Neuro2a and HeLa cells, where cells were co-transfected with either universal set 1 (pCMV-rtTA and pTREtight-RAGE-EGFP, 1:10 nM ratio) or with neuronal-specific set 2 (pNSE-rtTA and pTREtight-RAGE-EGFP, 1:10 nM ratio) plasmids. After 24 h of transfection, RAGE expression was induced by supplementing the medium with 2 μg/mL dox. Induction of the gene was observed by microscopic detection of the EGFP expression (Figure 3) and by RAGE expression on Western blot (data not shown). Our system showed strict cell-type specificity. Our universal set 1 plasmid showed expression of EGFP and RAGE in both Neuro2a and HeLa cells, while neuron-specific set 2 showed EGFP and RAGE expression in Neuro2a cells only. In HeLa cells, gene expression with set 2 plasmids was undetectable (Figure 3).
FIGURE 3.

Neuro2a and HeLa cell lines transfected with our Tet-On-inducible system for the gene RAGE. To test the cell-type specificity of the neuronal-specific enolase (NSE) promoter, the cell lines were either co-transfected with set1 (pCMV-rtTA and pTREtight-RAGE-EGFP) or with set 2 (pNSE-rtTA and pTREtight-RAGE-EGFP) plasmids. After 24 h of transfection, RAGE expression was induced by supplementing the culture medium with 2 μg/mL of dox. EGFP expression was recorded using a nikon inverted microscope. 1 = no induction; 2 = cells co-transfected with set 1 plasmids and induced with 2 μg/mL of dox; 3 = cells co-transfected with set 2 plasmids and induced with 2 μg/mL of dox. Scale bar: 50μm.
Effects of the Induction of RAGE in the Presence of AGEs in Neuro2a and HeLa Cells
Up-regulation of TNF-α and caspase-8 protein was observed only in cells in which RAGE was induced with dox in the presence of its ligand AGE (Figure 4). Non-induced cells treated with AGEs or cells induced (for RAGE expression) but not supplemented with AGEs showed no detectable levels of TNF-α and caspase-8. TNF-α was secreted into cell supernatants by both Neuro2a and HeLa cells. After 24 h of induction for RAGE in the presence of AGEs, TNF-α secreted by HeLa cells was approximately twofold (1.77 times) higher than that secreted by Neuro2a cells (Figure 4B). Membrane-bound TNF-α, detected in the HeLa cell lysates, was 1.6-fold higher than that secreted into the corresponding supernatants by these cells (Figure 4B). We could not detect TNF-α in the cell lysates (membrane bound) of Neuro2a cells (data not shown).
FIGURE 4.

RAGE induction in the presence of AGEs. A: Representative Western blots showing expression of RAGE, caspase-8, and β-actin proteins in Neuro2a cell lysates detected using specific antibodies. Neuro2a cell lysates were collected following transfection (with our inducible expression system) and induction with dox in the presence and absence of AGEs. 1 = no induction; 2 = AGEs only (no induction but supplemented with 50 μm AGEs); 3 = RAGE only (induced with 2 μg/mL dox without AGEs supplements); 4 = RAGE-AGE (induced with 2 μg/mL dox and supplemented with 50 μm AGEs). B: Graph showing ELISA results representing the TNF-α levels in the culture supernatants collected from Neuro2a and HeLa cells and cell lysates from HeLa cells following transfection (with our Tet-inducible system) and induction to express RAGE in the presence and absence of 50 μm AGEs. In culture supernatants, TNF-α levels are expressed in pg/mL of supernatants. The protein concentration in HeLa cell lysates was adjusted to 1 mg protein/mL lysate and TNF-α levels are expressed in pg/mL cell lysate/mg cell protein.
DISCUSSION
Among all the available inducible gene expression systems, the Tet-regulated system has been developed extensively due to several advantages it has over other systems. Despite refinements, however, the system still has the problems of basal leakiness and low induction of the gene, although several researchers have been successful in overcoming these problems to a greater extent.14–17 While several groups have incorporated both the components of the Tet system (rTA and TRE) into one vector/plasmid,14,15,17 a recent report16 suggests that the Tet system (adenoviral vectors) works optimally when the two components are incorporated separately into separate vectors. The aim of our study was therefore to achieve tight regulation for our gene RAGE in cell lines by optimizing the two-plasmid Tet-On system, namely, the regulatory and the response plasmids.
The original response plasmid from Clontech (BD Biosciences), pTRE2, does not have the EGFP reporter gene, and the TRE (TetCMVmin) promoter/Tet-inducible promoter, has a basal effect in the absence of the inducer (see references 23 – 25 , our unpublished observations); i.e., this promoter is active in the absence of dox. A newer version of the response plasmid, pTRE-tight (Clontech), does not have the reporter gene, nor a selection marker to allow for making stable mammalian cell lines. In the pTRE-tight-EGFP plasmid (Clontech), the MCS has been replaced with EGFP,26 making cloning the gene of interest very difficult. In addition, these plasmids do not have any mammalian selection markers. In our system, we wanted to have the EGFP reporter gene in the response plasmid only, and internal ribosome entry site (IRES) between the gene and EGFP, as IRES enables translation of both EGFP and the gene from the same bicistronic mRNA. We also wanted to use a drug/antibiotic resistance gene as a mammalian selection marker, in addition to having tight regulation and cell-type specificity for our gene expression. To achieve these aims, we modified the pIRESE2-EGFP plasmid (Clontech). The MCS of the pIRES2-EGFP has various cloning sites, giving several options for cloning, and has the EGFP reporter gene and IRES between EGFP and MCS. It also has the neomycin resistance (Neor) gene, which can be used as a mammalian selection marker. Our own observations and the published literature23–25 suggest that the main reason for the basal leakiness in the Tet-system is the intrinsic activity of the CMV minimal promoter in the TRE of the response plasmid. To avoid basal leakiness, Zabala et al. (2004)14 substituted CMVm in the TRE with the very weak liver-specific promoter. Although they were successful in reducing basal expression of the system to an almost undetectable level, they suggested that reducing basal expression could also lead to a decrease in the amount of the final induced protein. In addition, this approach is suitable for liver-specific cells only; it would be very difficult to find such suitable weak promoters for most other cells. We wanted to use a much tighter Tet-responsive element to prevent the basal leakiness without using any other weak promoter. We therefore replaced the CMV promoter in pIRES2-EGFP with TREtight (Clontech; TREtight is a modified Tet-responsive element, which consists of seven direct repeats of 36-bp sequence that contains the 19-bp tet-operator sequence, and this is fused to CMVm promoter). The gene RAGE was cloned at the MCS of this plasmid (pTREtight-RAGE-EGFP). This modification gave very tight regulation of RAGE expression when co-transfected with pTet-On/pCMV-rtTA, and we found negligible RAGE and EGFP expression in both HeLa and Neuro2a cells in the absence of the inducer dox. EGFP expression allowed easy microscopic assessment of the transfection efficiency and also allowed us to assess whether the transfection was homogenous in the cells.
To achieve cell-type specificity, we constructed a regulatory plasmid with a neuronal-specific promoter, NSE, regulating the expression of rtTA (reverse transactivator). The regulatory plasmid pTet-On (Clontech) did not have any restriction site for this modification.27 Therefore, again we modified the pIRES2-EGFP plasmid to obtain a neuron-specific regulatory plasmid. EGFP was removed from this plasmid before cloning rtTA at the MCS downstream of the NSE promoter (plasmid named as pNSE-rtTA). We removed the EGFP before these modifications, since we did not want to have a green fluorescent reporter gene in any of our regulatory plasmid. Our response plasmid already had EGFP, and induction of the gene required the cell lines to be transfected with both the plasmids either by transient or stable transfection. Presence of both the plasmids was therefore needed for gene expression. EGFP expression indicates that the gene is being expressed and both the plasmids are present in the cell. Since both plasmids have the neomycin resistance gene and EGFP reporter is only in the response plasmid, a double-stable cell line (having both the plasmids in one cell) is one that exhibits neomycin resistance and EGFP expression when induced with dox. In agreement with Lee et al.,16 we also found that the ratio of regulatory and response plasmid is important and that a 1:10 nm ratio of regulatory to response plasmid gave optimal results. Co-transfection of HeLa cells and Neuro2a cells with either set 1 or set 2 plasmids showed that set 2 expresses RAGE in Neuro2a cells only, indicating that our system exhibits strict cell-type specificity. Both the set 1 and set 2 showed negligible basal expression of RAGE and EGFP in the absence of dox, and showed high induciblity of the gene (142-fold). There are three immunoglobulin-like extracellular domains in full-length RAGE: an N-terminal V-type (variable) region, followed by two C-type (constant) regions. The V region is important for ligand binding and contains two glycosylation sites. Full-length RAGE also contains a transmembrane and a cytoplasmic domain. This cytoplasmic domain is important for RAGE-mediated signal transduction, and for fully functional RAGE, all three domains are essential.1 Secretory RAGE (sRAGE) is C-truncated and lacks transmembrane and cytoplasmic domains; it can bind to ligands, but does not lead to signal transduction. V-truncated RAGE lacks the V-type domain and hence is incapable of binding ligands. The induced RAGE in our system was full length. We observed two bands on Western blots of 55 and 50 kda, corresponding to the size of the glycosylated and non-glycosylated full-length RAGE protein.1 Following RAGE induction, we observed increased levels of TNF-α and caspase-8 in the presence of AGEs. TNF-α has been shown to be a downstream effector of the AGE-RAGE interaction,1,28 and caspase-8 is a proximal effector protein of the TNF-α receptor family cell death pathway.5 The amount of TNF-α secreted by HeLa cells was higher than that secreted by Neuro2a cells. We also detected TNF-α in cell lysates (membrane bound) of HeLa cells. HeLa cells express about 3,000 molecules of TNFR1/cell, and the specific binding of TNF-α to its receptors on HeLa cells is very rapid.29,30 TNFR1 (TNF-α receptor 1) and its ligand TNF-α also exhibit positive feedback. This may well explain the abundant amount of TNF-α we detected in the cell lysates of HeLa cells compared to Neuro2a cells following induction of RAGE in the presence of AGEs. The presence of TNF-α in both secretory and membrane-bound forms and caspase-8 following the induction of RAGE in the presence of AGEs indicates that the induced RAGE is functional.
In conclusion, we have modified the existing Tet-On system for the gene RAGE. Our system showed negligible basal leakiness and high induction of the gene RAGE. This system can be used for any other gene of interest and also can be modified to provide further additional cell-type specificity by replacing the NSE promoter with an alternative promoter. This system is highly suitable to study the effect of any gene in a specific cell type, in mixed cell populations or mixed primary cultures.
Acknowledgments
We thank our close colleagues in the Faculty of Medical and Health Sciences, University of Auckland: Dr. Debbie Young (Department of Molecular Medicine and Pathology, University of Auckland) for providing the plasmids pIRES2-EGFP and pAAV/NSE-luc and Dr. Stephanie Hughes (Department of Pharmacology, University of Auckland) for providing pTet-on and pTRE-tight plasmids. We also thank Dr Shi-Fang Yan (Division of Surgical Science, Columbia University, New York) for a gift of full-length RAGE cDNA (in pcDNA-3 plasmid) This work was funded by a University of Auckland Research Committee (UARC) Postdoctoral Fellowship, the Maurice and Phyllis-Paykel Trust, and the Medical School Foundation (Faculty of Medical and Health Sciences, University of Auckland).
REFERENCES
- 1.Lue LF, Yan SD, Stern DM, Walker DG. Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr Drug Targets CNS Neurol Disord 2005;4:249–266. [DOI] [PubMed] [Google Scholar]
- 2.Ma L, Nicholson LF. Expression of the receptor for advanced glycation end products in Huntington’s disease caudate nucleus. Brain Res 2004;1018:10–17. [DOI] [PubMed] [Google Scholar]
- 3.Dalfo E, Portero-Otin M, Ayala V, Martinez A, Pamplona R, Ferrer I. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J Neuropathol Exp Neurol 2005;64:816–830. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands, Bio-chim Biophys Acta 2000;1498:99–111. [DOI] [PubMed] [Google Scholar]
- 5.Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, et al. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J Neurosci 2001;21:2247–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 1992;89:5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gossen M, Freundlieb S, Bender G, Muller G,Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995;268:1766–1769. [DOI] [PubMed] [Google Scholar]
- 8.Fussenegger M, Morris RP, Fux C, Rimann M, von Stockar B, Thompson CJ, et al. Streptogramin-based gene regulation systems for mammalian cells. Nat Biotechnol 2000;18:1203–1208. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, DeMayo FJ, Tsai SY, O’Malley BW. Ligand-inducible and liver-specific target gene expression in transgenic mice. Nat Biotechnol 1997;15:239–243. [DOI] [PubMed] [Google Scholar]
- 10.Christopherson KS, Mark MR, Bajaj V, Godowski PJ. Ecdysteroid-dependent regulation of genes in mammalian cells by a Drosophila ecdysone receptor and chimeric transactivators. Proc Natl Acad Sci USA 1992;89:6314–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.No D, Yao TP, Evans RM. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc Natl Acad Sci USA 1996;93:3346–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivera VM, Clackson T, Natesan S, Pollock R, Amara JF, Keenan T, et al. A humanized system for pharmacologic control of gene expression. Nat Med 1996;2:1028–1032. [DOI] [PubMed] [Google Scholar]
- 13.Liberles SD, Diver ST, Austin DJ, Schreiber SL. Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc Natl Acad Sci USA 1997;94:7825–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zabala M, Wang L, Hernandez-Alcoceba R, Hillen W, Qian C, Prieto J, et al. Optimization of the Tet-on system to regulate interleukin 12 expression in the liver for the treatment of hepatic tumors. Cancer Res 2004;64:2799–2804. [DOI] [PubMed] [Google Scholar]
- 15.Zhang XY, Su BL, Li H, Bai R, Xu ZH, Li CC. A single tetracycline-regulated vector devised for controlled insulin gene expression. Chin Med Sci J 2004;19:266–269. [PubMed] [Google Scholar]
- 16.Lee YB, Glover CP, Cosgrave AS, Bienemann A, Uney JB. Optimizing regulatable gene expression using adenoviral vectors. Exp Physiol 2005;90:33–37. [DOI] [PubMed] [Google Scholar]
- 17.Berenjian S, Akusjarvi G. Binary AdEasy vector systems designed for Tet-ON or Tet-OFF regulated control of transgene expression. Virus Res 2006;115:16–23. [DOI] [PubMed] [Google Scholar]
- 18.Gossen M, Bujard H. Efficacy of tetracycline-controlled gene expression is influenced by cell type: commentary. Biotechniques 1995;19:213–216; discussion 216–217. [PubMed] [Google Scholar]
- 19.BD Biosciences Clontech Laboratories I. Technical Info: pTRE- Tight Vector.
- 20.Munch G, Gasic-Milenkovic J, Dukic-Stefanovic S, Kuhla B, Heinrich K, Riederer P, et al. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp Brain Res 2003;150:1–8. [DOI] [PubMed] [Google Scholar]
- 21.Kuhla B, Loske C, Garcia De Arriba S, # Schinzel R, Huber J, Munch G. Differential effects of “Advanced glycation end-products” and beta-amyloid peptide on glucose utilization and ATP levels in the neuronal cell line SH-SY5Y. J Neural Transm 2004;111:427–439. [DOI] [PubMed] [Google Scholar]
- 22.Yan SF, Ramasamy R, Naka Y, Schmidt AM. Glycation, inflammation, and RAGE: A scaffold for the macrovascular complications of diabetes and beyond. Circ Res 2003;93:1159–1169. [DOI] [PubMed] [Google Scholar]
- 23.Ackland-Berglund CE, Leib DA. Efficacy of tetracycline-controlled gene expression is influenced by cell type. Biotechniques 1995;18:196–200. [PubMed] [Google Scholar]
- 24.Bohl D, Salvetti A, Moullier P, Heard JM. Control of erythropoietin delivery by doxycycline in mice after intramuscular injection of adeno-associated vector. Blood 1998;92:1512–1517. [PubMed] [Google Scholar]
- 25.Kramer MG, Barajas M, Razquin N, Berraondo P, Rodrigo M, Wu C, et al. In vitro and in vivo comparative study of chimeric liver-specific promoters. Mol Ther 2003;7:375–385. [DOI] [PubMed] [Google Scholar]
- 26.BD Biosciences Clontech Laboratories I. Technical Info: pTRE-Tight-EGFP, 2003.
- 27.BD Biosciences Clontech Laboratories I. Technical Info: pTet-On Vector. 2000.
- 28.Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Munch G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J Neurochem 2003;87:44–55. [DOI] [PubMed] [Google Scholar]
- 29.Haas E, Grell M, Wajant H, Scheurich P. Continuous autotropic signaling by membrane-expressed tumor necrosis factor. J Biol Chem 1999;274:18,107–18,112. [DOI] [PubMed] [Google Scholar]
- 30.Drimal D, Drimal J, Drimal J, Jr. The regulation of human adrenomedullin (AM) and tumor necrosis factor alpha (TNF-alpha) receptors on human epithelial carcinoma (HeLa) cells. The role of AM secretion in tumor cell sensitivity. Neoplasma 2006;53:144–149. [PubMed] [Google Scholar]