

Abstract
Activation of protein kinase C (PKC) decreases the activity and cell surface expression of the predominant forebrain glutamate transporter, GLT-1. In the present study, C6 glioma were used as a model system to define the mechanisms that contribute to this decrease in cell surface expression and to determine the fate of internalized transporter. As was previously observed, phorbol 12-myristate 13-acetate (PMA) caused a decrease in biotinylated GLT-1. This effect was blocked by sucrose or by co-expression with a dominant-negative variant of dynamin 1, and it was attenuated by co-expression with a dominant-negative variant of the clathrin heavy chain. Depletion of cholesterol with methyl-β-cyclodextrin, co-expression with a dominant-negative caveolin-1 mutant (Cav1/S80E), co-expression with dominant-negative variants of Eps15 (epidermal-growth-factor receptor pathway substrate clone 15), or co-expression with dominant-negative Arf6 (T27N) had no effect on the PMA-induced loss of biotinylated GLT-1. Long-term treatment with PMA caused a time-dependent loss of biotinylated GLT-1 and decreased the levels of GLT-1 protein. Inhibitors of lysosomal degradation (chloroquine or ammonium chloride) or co-expression with a dominant-negative variant of a small GTPase implicated in trafficking to lysosomes (Rab7) prevented the PMA-induced decrease in protein and caused an intracellular accumulation of GLT-1. These results suggest that the PKC-induced redistribution of GLT-1 is dependent upon clathrin-mediated endocytosis. These studies identify a novel mechanism by which the levels of GLT-1 could be rapidly down-regulated via lysosomal degradation. The possibility that this mechanism may contribute to the loss of GLT-1 observed after acute insults to the CNS is discussed.
Introduction
A family of high affinity Na+-dependent glutamate transporters both ensures appropriate excitatory signaling and limits the excitotoxic potential of glutamate in the mammalian CNS. This family consists of five members; two of these transporters are enriched in astrocytes (GLT-1 and GLAST), two are enriched in neurons (EAAC1 and EAAT4), and the last is enriched in the retina (EAAT5) (for reviews, see Sims and Robinson, 1999; Danbolt, 2001). GLT-1 protein is enriched in astrocytic processes that sheath the synapse (Chaudhry et al., 1995), may represent up to 1% of total brain protein (Lehre and Danbolt, 1998), and is thought to be responsible for about 90% of forebrain glutamate transport activity (for reviews, see Robinson, 1999; Danbolt, 2001). Expression of GLT-1 is decreased in several animal models of neurodegenerative diseases, including amyotrophic lateral sclerosis (Trotti et al., 1999), traumatic brain injury (Rao et al., 1998), epilepsy (Samuelsson et al., 2000; Ingram et al., 2001) and also in brain tissue from patients with amyotrophic lateral sclerosis (Rothstein et al., 1995), epilepsy (Mathern et al., 1999), Alzheimer’s disease and Huntington’s disease (Lipton and Rosenberg, 1994; Li et al., 1997; for review, see Sheldon and Robinson, 2007). Therefore defining mechanisms that control either synthesis or degradation of GLT-1 has the potential to impact our understanding of both the physiology and pathology of glutamate.
The activities of many different plasma membrane proteins are regulated by changing the trafficking of these proteins to or from the plasma membrane. One of the classic examples involves agonist-dependent desensitization and internalization of G-protein coupled receptors (for reviews, see von Zastrow, 2003; Dhami and Ferguson, 2006). Relatively recent studies have shown that the activities of many of the neurotransmitter transporters are also regulated by similar mechanisms (for reviews, see Beckman and Quick, 2000; Blakely and Bauman, 2000; Robinson, 2002). For example, activation of PKC decreases cell surface expression of many of the monoamine transporters (serotonin, dopamine, and norepinephrine), at least one member of the GABA transporter family, and one of the glycine transporters (for review, see Robinson, 2002). In some cases, there is fairly convincing evidence that this redistribution is dependent upon clathrin. For example, the PKC-induced internalization of the dopamine transporter or the GAT1 subtype of GABA transporter depend at least in part on clathrin-mediated endocytosis (Daniels and Amara, 1999; Loder and Melikian, 2003; Wang and Quick, 2005; Sorkina et al., 2006). There is evidence that some transporters are found in a subcellular fraction that is enriched in cholesterol and operationally defined as a ‘lipid raft’ based on insolubility in 1% Triton or other mild detergents (for a recent review see, Allen et al., 2007). Furthermore, these lipid rafts and a protein enriched in this fraction, caveolin, may mediate endocytosis through a distinct pathway (for reviews, see Simons and Toomre, 2000; Allen et al., 2007). In fact, depletion or disruption of membrane cholesterol inhibits PKC-dependent redistribution of the norepinephrine transporter (Jayanthi et al., 2004).
The activity and cell surface expression of GLT-1 is regulated by various signaling molecules including PKC and scaffolding proteins (for reviews, see Danbolt, 2001; González and Robinson, 2004; Beart and O'Shea, 2007). Although Casado and colleagues originally suggested that activation of PKC increases activity in GLT-1-transfected HeLA cells (Casado et al., 1993), we were unable to replicate this result (Tan et al., 1999). In primary cultures derived from rat brain and Y-79 human retinoblastoma cells that endogenously express GLT-1, activation of PKC rapidly (within min) decreases GLT-1-mediated transport activity and reduces the amount of GLT-1 that is observed at the plasma membrane (Ganel and Crosson, 1998 Kalandadze et al., 2002; Zhou and Sutherland, 2004; Guillet et al., 2005). Similar effects have been observed in C6 glioma cells or Madin-Darby canine kidney cells transfected with GLT-1 (Carrick and Dunlop, 1999; Kalandadze et al., 2002; Zhou and Sutherland, 2004). The phorbol ester-dependent internalization of GLT-1 has been attributed to PKC because several of these studies have shown that inhibitors that are thought to be specific for PKC block the redistribution. In fact, we recently implicated the alpha subtype of PKC in this internalization (Gonzalez et al., 2005). Recent studies have shown that GLT-1 is enriched in the subcellular fraction that is commonly referred to as lipid rafts (Butchbach et al., 2004). Zhou and colleagues demonstrated that sucrose partially attenuates PKC-dependent decreases in GLT-1 mediated glutamate transport and that a dominant-negative variant of dynamin attenuates PKC-dependent clustering of GLT-1 in astrocytic processes (Zhou and Sutherland, 2004), but the mechanisms for the loss of cell surface GLT-1 have not been defined nor has it been determined if lipid rafts contribute to internalization of GLT-1.
In the present study, we examined the cellular machinery involved in the PMA-dependent loss of cell surface GLT-1 and also examined the fate of the transporter after internalization. Our results show that the loss of cell surface GLT-1 is blocked by sucrose or dominant-negative variants of clathrin and dynamin but is insensitive to dominant negative variants of Eps15. In addition, our data show that the PMA-dependent redistribution of GLT-1 is not affected by depletion of membrane cholesterol or by a dominant-negative variant of caveolin. Prolonged treatment with PMA resulted in a decrease in the levels of total GLT-1 protein, and this loss of GLT-1 was not observed in the presence of lysosomal inhibitors. Furthermore, this loss of GLT-1 was attenuated by co-expression with a dominant-negative variant of Rab7. These studies identify a novel mechanism by which the levels of GLT-1 protein can be rapidly regulated.
Experimental Procedures
Materials
Fetal bovine serum was obtained from Hyclone (Logan, UT, USA). All other tissue culture reagents were from Invitrogen (Carlsbad, CA). Tissue culture plates were from Corning (Corning, NY). Sulfo-NHS-biotin and Ultralink immobilized monomeric avidin were obtained from Pierce (Rockford, IL). Phorbol 12-myristate 13-acetate (PMA), ammonium chloride, chloroquine, methyl-β-cyclodextrin (MβCD), and anti-actin antibody were purchased from Sigma (St Louis, MO). Lactacystin was obtained from Calbiochem (La Jolla, CA). Geneporter transfection reagent was obtained from Gene Therapy Systems, Inc (San Diego, CA). Dr. Jeffrey D. Rothstein (Johns Hopkins University) generously provided polyclonal anti-GLT-1 and anti-EAAC1 antibodies directed against peptide sequence from the carboxyl termini of these transporters (Rothstein et al., 1994). The anti-GLT-1 antibody is specific for the splice variant of GLT-1 frequently referred to as GLT-1a or GLT-1 alpha (for recent discussions, see Reye et al., 2002; Chen et al., 2004). Mouse monoclonal anti-hemagglutinin (anti-HA) antibody was obtained from Roche Molecular Biochemicals (Indianapolis, IN). Monoclonal antibodies against green fluorescent protein (GFP) and Myc were obtained from BD Biosciences (Mountain View, CA). Chemiluminescence kits and rainbow molecular weight markers were purchased from Amersham (Arlington Heights, IL). Immobilon-P membrane was from Millipore (Bedford, MA).
The GLT-1 and myc-EAAC1 cDNA constructs in pcDNA3.1 have been described previously (Kalandadze et al., 2002; Sheldon et al., 2006). Control (D3Δ2) and dominant-negative mutants of Eps15 (EΔ95-295 and DIII) in pEGP-C2 vector were kindly provided by A. Benmerah (Benmerah et al., 1998; Benmerah et al., 1999). Caveolin 1 wild type and dominant negative mutant (S80E) in pcDNA were a kind gift from Dr J.E. Pessin (Shigematsu et al., 2003). Arf6/pLNX and dynamin/pcDNA mutants were generously provided by Drs M.M. Chou and M. Lemmon (University of Pennsylvania), respectively. Rab7/pCDM8.1 and hub/pCDM8.1 were kindly provided by M.S. Marks (University of Pennsylvania). The sequences of the rab7, caveolin, hub, Eps15, GLT-1, Myc-EAAC1, and their variants were verified by DNA sequencing.
Cell culture
C6 glioma cells were obtained from the American Type Tissue Collection (Rockville, MD) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL pencillin and 100 μg/mL streptomycin. Cells were maintained at 37°C in 5% CO2. These cells were passaged no more than 30 times. There was no apparent change in morphology, and there was no evidence that the effects reported in this study were dependent upon passage number.
Transient Transfection of C6 glioma cells
C6 glioma were grown on 10-cm plates to 40–60% confluence (which were split a day before transfection from an ~80% confluent plate). These cells were transfected with a glutamate transporter (GLT-1/myc-EAAC1) cDNA or co-transfected with a second cDNA using the cationic lipid Geneporter as per manufacturer’s instructions. When cells were transfected with only GLT-1, 10–12 μg of cDNA was used. When they were co-transfected with both GLT-1 and another cDNA, 3–4 μg of GLT-1 and 9–8 μg of another DNA was used. The ratio of Geneporter to cDNA was held constant with 5μl of Geneporter per μg of cDNA. After a 3–4 h incubation with the transfection mixture in 5 ml of DMEM, an additional 5 ml of modified medium (DMEM containing 20% FBS, 4 mM glutamine, 200 units/ml pencillin and 200 μg/ml streptomycin) was added to the mixture for an additional 16–20 h. Prior to the experiment cells were washed into DMEM containing (0.5%) bovine serum albumin for 2h.
Myc-tagged EAAC1 was only used as a positive control for some co-transfection experiments when there was no effect on GLT-1. For these studies we reduce the amount of cDNA to relatively low levels to reduce the likelihood that we have saturated cellular machinery required for constitutive recycling. We have previously found that compared to endogenous EAAC1, the same amount of myc-EAAC1 is expressed at the cell surface (30 and 34% respectively) (Fournier et al., 2004; Sheldon et al., 2006). We have also found that the rate of delivery to the cell surface is the same for both endogenous and myc-tagged EAAC1 (Fournier et al., 2004; Fournier and Robinson, 2006 González, Krizman-Genda, and Robinson, unpublished observations). Finally, both endogenous and myc-tagged EAAC1 are regulated by activation of PKC or PDGF receptor (Sheldon et al., 2006). Together these studies suggest that the cellular machinery required for constitutive trafficking of EAAC1 is not saturated under these conditions and suggest that the myc tag does not dramatically change constitutive trafficking. However, we acknowledge that it is theoretically possible that the myc tag could influence our results.
Biotinylation of cell surface proteins
Biotinylation of surface proteins was performed as described previously, with minor modifications (Davis et al., 1998; Susarla and Robinson, 2003). Cells were rinsed twice with ice-cold phosphate-buffered saline (PBS) containing 0.1 mM Ca2+ and 1 mM Mg2+ and incubated with 2 ml of NHS-biotin (1 mg/ml PBS containg Ca2+ and Mg2+) for 30 min at 4°C. Unreacted biotin was quenched by 100 mM glycine in PBS Ca2+/Mg2+. After cell lysis biotinylated proteins were extracted with Ultralink monomeric avidin-coated sepharose beads and extracted with sample buffer, three fractions of proteins were isolated: lysate, biotinylated (cell surface) and non-biotinylated (intracellular). They were diluted into identical total volumes so that the sum of the immunoreactivity in the biotinylated and non-biotinylated fractions should be equal to the amount of immunoreactivity in the lysate if the yield from the extraction is 100%.
Western blot analyses
Samples from biotinylation experiments (cell lysates, non-biotinylated and biotinylated fractions) were loaded onto 8% SDS-polyacrylamide gel. After separation and transfer, the polyvinylidene membranes were probed with antibodies against actin (1:5000), GLT-1 (1:10000), EAAC1 (1:75), or other antibodies (anti-Myc 1:500; anti-HA 1:500; anti-GFP 1:500). Immunoreactive bands were visualized using enhanced chemiluminescence. Several exposures of every blot were captured to ensure linearity of the signal. Immunoreactivity for each protein was quantified using Image software (National Institutes of Health). As has been previously reported (Haugeto et al., 1996), transporter immunoreactivity was observed at positions consistent with the presence of monomers and multimers. When both species were observed, they were quantitated separately. In all cases, there was no evidence that monomer or multimer was differentially affected by the treatments described in the current study. Therefore, data presented reflect the effects on the sum of the monomer and multimer immunoreactivities and are expressed as a percentage of control in the respective fraction.
Statistical Analyses
All data presented are the means ± SEM of n independent experiments. Differences between two groups were compared by Student’s t-test. When there were more than two groups, data were compared by ANOVA using Bonferroni post-hoc correction for multiple comparisons. A p < 0.05 was considered significant.
Results
Effects of sucrose on phorbol ester-induced redistribution of GLT-1
In the current study, C6 glioma were used as a model system to study the PKC-dependent redistribution of GLT-1. As is observed in primary astrocyte cultures, greater than 90% of GLT-1 immunoreactivity is observed in the biotinylated fraction in C6 cells transfected with GLT-1, and activation of PKC with PMA causes a decrease in the amount of biotinylated GLT-1 within 30 min (Kalandadze et al., 2002; Guillet et al., 2005). This suggests that transfected C6 glioma provide a reasonable system to study the mechanisms involved in PKC-induced redistribution of GLT-1. Several pathways have been implicated in endocytosis of plasma membrane proteins; many of these endocytic routes are differentiated based on their sensitivities to pharmacological agents or dominant-negative variants of proteins thought to support these different pathways (for reviews, see Conner and Schmid, 2003; Maldonado-Baez and Wendland, 2006; Roth, 2006). Since clathrin-mediated endocytosis is the best characterized and most extensively studied route of internalization for many membrane proteins and has been implicated in endocytosis of various neurotransmitter transporters (Daniels and Amara, 1999; Loder and Melikian, 2003; Sorkina et al., 2005; Wang and Quick, 2005), the possibility that this pathway may be involved was examined. Hypertonic sucrose is one of the classic strategies used to block clathrin-mediated endocytosis because it prevents formation of the clathrin lattice at the plasma membrane (Heuser and Anderson, 1989). C6 glioma were transiently transfected with GLT-1 cDNA, pretreated with vehicle or sucrose for 15 min, treated with vehicle or PMA (100 nM) for an additional 30 min, and then cell surface proteins were biotinylated with a membrane impermeant reagent. As has been previously observed, PMA decreased the amount of biotinylated GLT-1 as compared to control; this effect was abolished by pre-treatment with hypertonic sucrose (Fig. 1). In these studies, essentially no actin immunoreactivity was observed in the biotinylated fraction under any conditions (see figure legend). Chronic treatment with hypertonic sucrose induces GLT-1 expression in C6 glioma (Imura et al., 1999). In the present study, Western blotting of the lysate fraction revealed that sucrose had no effect on total GLT-1 expression (95 ± 8%, n=5, Western blot not shown). Together, these studies suggest that sucrose blocks the PKC-dependent redistribution of GLT-1 and that clathrin may be required for this redistribution.
Fig. 1.
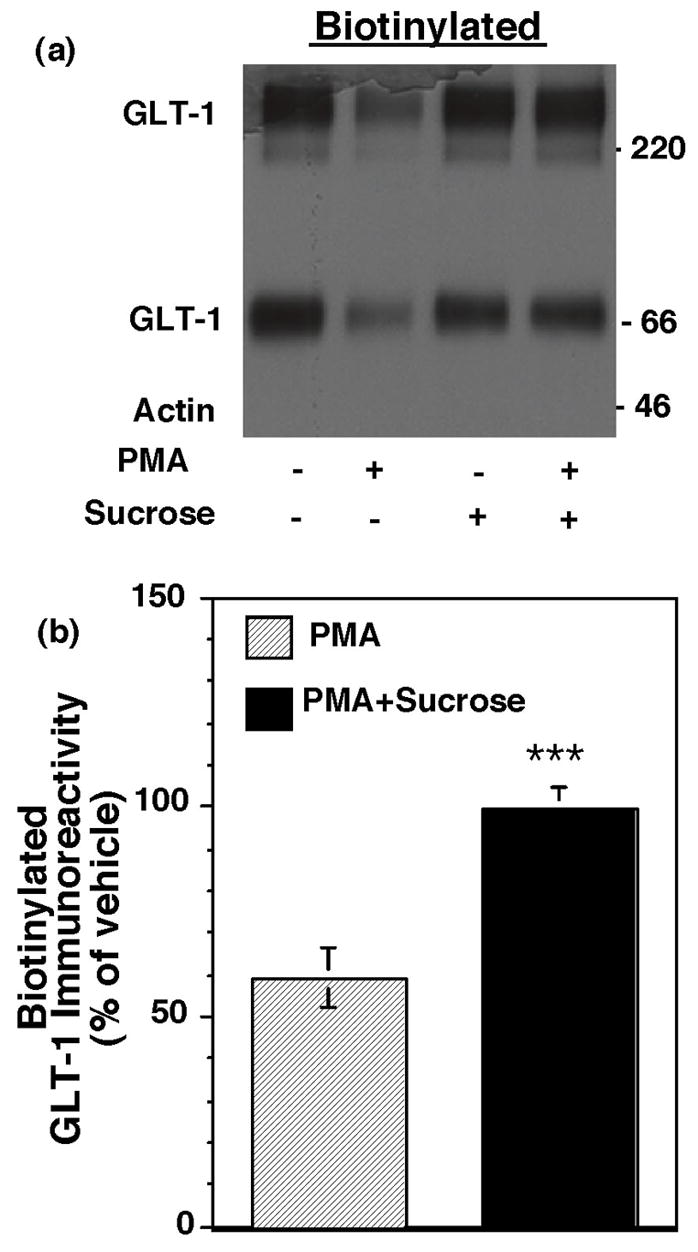
Effect of hypertonic sucrose on the PMA-induced decrease in biotinylated GLT-1. C6 cells transfected with GLT-1 cDNA were treated with hypertonic sucrose (0.45M) or vehicle for 15 min and then treated with PMA or vehicle (DMSO) for 30 min. After rapidly cooling to 4º C, cell surface proteins were biotinylated with a membrane impermeant reagent. Total protein, biotinylated proteins, and non-biotinylated proteins were analyzed by Western blot using antibodies directed against GLT-1 and actin. (a) Representative immunoblot of the biotinylated fraction; no actin was observed in the biotinylated fraction in this experiment. (b) Summary of effects of PMA in the presence and absence of sucrose on biotinylated GLT-1 (mean ± SEM of five independent experiments). The effects of PMA were expressed as a percentage of the corresponding vehicle control. In these studies, actin was essentially undetectable in the biotinylated fraction (mean = 5 ± 5% of total actin observed in the lysate fraction) and no treatment changed the amount of biotinylated actin (data not shown). There was no effect of sucrose on total GLT-1 (95 ± 8% of control) or biotinylated GLT-1 (95 ± 9 of control). ***indicates a p < 0.005 by unpaired Student’s t-test compared to the effects of PMA in the absence of sucrose.
Effects of dominant-negative variant of dynamin on phorbol ester-induced redistribution of GLT-1
The large GTPase dynamin is generally thought to be required for clathrin-dependent, and some forms of clathrin-independent, endocytosis (for reviews, see McNiven et al., 2000; Conner and Schmid, 2003). Through GTP binding and hydrolysis, dynamin pinches off membrane invaginations to form endocytic vesicles (Damke et al., 1994). To test the involvement of dynamin in the PMA induced redistribution of GLT-1, C6 cells were co-transfected with GLT-1 cDNA and wild-type (WT) dynamin, or a dominant-negative variant of dynamin that lacks GTPase activity (K44A), or pcDNA3.1. Sixteen to twenty h later, the effects of acute activation of PKC with PMA on GLT-1 cell surface expression were measured by biotinylation (Fig. 2). Compared to cells co-transfected with either pcDNA (data not shown) or WT-dynamin, the K44A dominant-negative variant of dynamin essentially abolished the PMA-induced decrease in biotinylated GLT-1. The total expression of GLT-1 was not different between cells co-transfected with WT or the K44A dominant-negative variant of dynamin (data not shown). Together these analyses provide strong evidence that the PMA-induced redistribution of GLT-1 is dynamin-dependent.
Fig. 2.
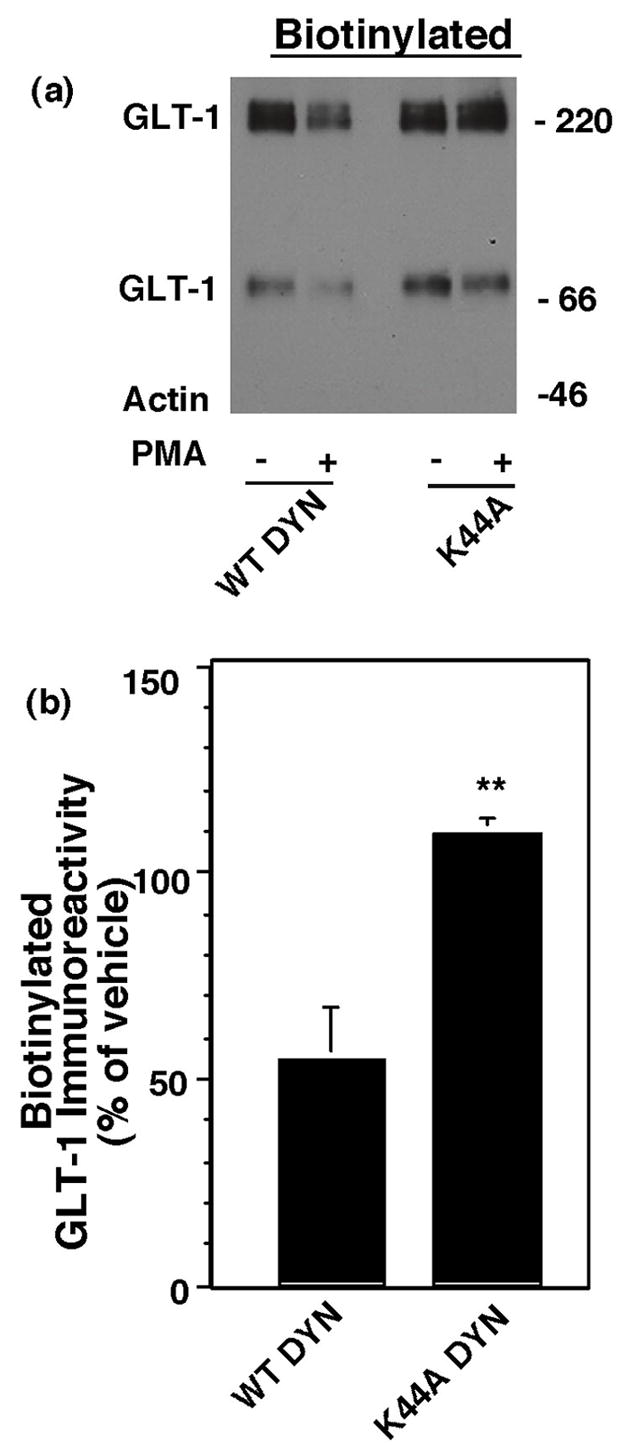
Effect of dominant-negative dynamin on the PMA-induced decrease in biotinylated GLT-1. C6 cells were cotransfected with GLT-1 and wild-type (WT) dynamin or dominant-negative dynamin (K44A). After expression of the transfected cDNAs (16–20 h), cells were treated with either vehicle (DMSO) or PMA (100 nM) for 30 min and cell surface GLT-1 was measured using a membrane impermeant biotinylating reagent. (a) Representative immunoblot of biotinylated GLT-1 with no actin biotinylated. (b) Summary of effects of PMA on biotinylated GLT-1 (mean ± SEM of five independent experiments). Data for the effects of PMA were expressed as a percentage of the corresponding control (no PMA). No biotinylated actin was detected in any of these experiments and no treatments increased the amount of biotinylated actin. The percentage of biotinylated GLT-1 was not significantly different in cells co-transfected with K44A compared to WT dynamin (99 ± 1% of wild type, mean ± SEM, n =3). Data were compared by unpaired Student’s t-test. ** indicates a p <0.02 compared to cells co-transfected with wild type dynamin.
Effects of a dominant-negative variant of clathrin on phorbol ester-induced redistribution of GLT-1
Since dynamin is required for both clathrin-dependent and at least some forms of clathrin-independent endocytosis (for reviews, see McNiven et al., 2000; Conner and Schmid, 2003), we examined the effects of a dominant-negative variant of clathrin heavy chain that competes with endogenous clathrin heavy chains for binding to the light chain subunits and disrupts formation of the clathrin lattice required for endocytosis (Liu et al., 1995; Liu et al., 1998). This variant of clathrin, frequently referred to as hub, or pcDNA was co-transfected with GLT-1 into C6 cells. This variant of clathrin contains a T7 epitope at the amino terminus that we used to verify that the product was expressed in C6 glioma; an immunoreactive band of ~66 kDa was detected (data not shown, replicated in three experiments). We found that co-expression with hub had no significant effect on total GLT-1 expression or on the percentage of GLT-1 that is found in the biotinylated fraction (see legend to Fig. 3). However, co-expression of hub significantly attenuated the PMA-induced decrease in biotinylated GLT-1 (see Fig. 3).
Fig. 3.
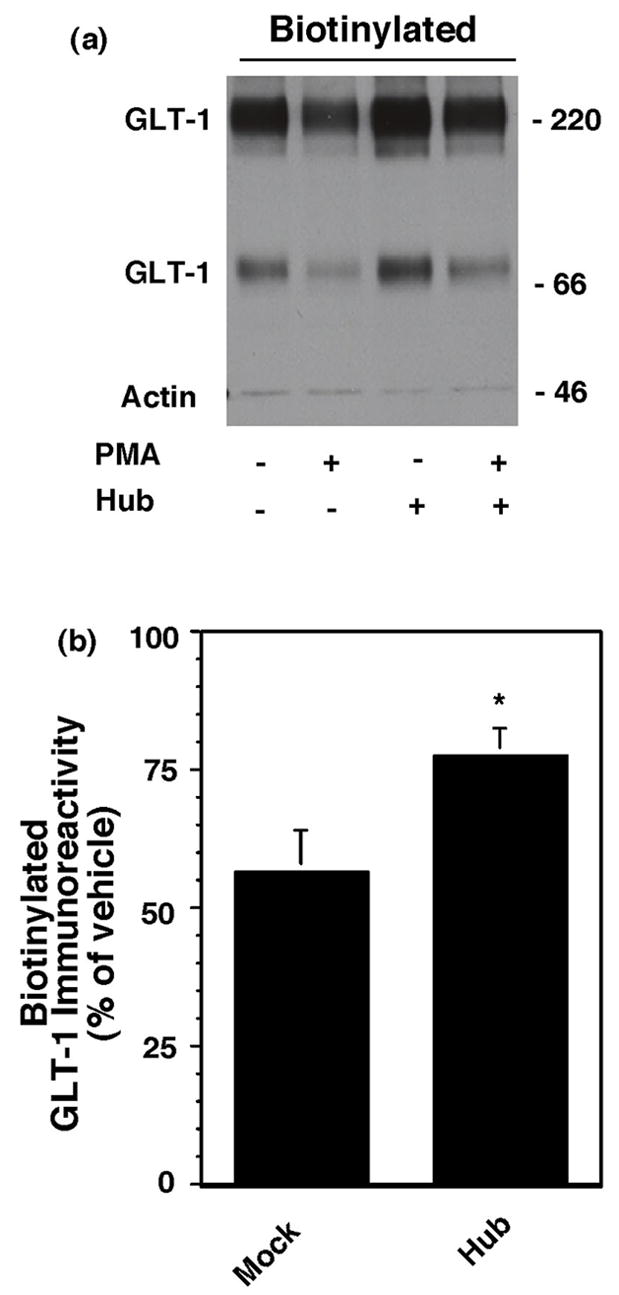
Effect of dominant-negative clathrin heavy chain (hub) on the PMA-induced decrease in biotinylated GLT-1. C6 cells were co-transfected with cGLT-1 and either pcDNA or T7 epitope tagged dominant negative clathrin heavy chain (Hub) construct. After being maintained overnight, cells were treated with vehicle (DMSO) or PMA for 30 min and biotinylated proteins were batch extracted using a membrane impermeant biotinylating reagent. (a) Representative western blot of GLT-1 immunoreactivity in the biotinylated fraction (note no actin was observed in this experiment). (b) Summary of effects of PMA on biotinylated GLT-1 in pcDNA transfected cells and hub transfected cells (mean ± SEM of six independent experiments). The percentage of actin detected in these experiments was 4 ± 3% of total (mean ± SEM), and it did not change under any of the conditions. Expression of total GLT-1 in cells co-transfected with hub was 114 ± 12% of that observed in cells co-transfected pcDNA. The percentage of biotinylated GLT-1 was not significantly different in cells co-transfected with hub (81 ± 5%) compared to cells co-transfected with pcDNA (76 ± 6%). * indicates a p<0.05 compared to the effects of PMA in cells co-transfected with pcDNA by unpaired Student’s t-test.
Effects of dominant-negative variants of Eps-15 on phorbol ester-induced redistribution of GLT-1
Clathrin-dependent endocytosis can be dependent on accessory proteins. We have recently implicated epidermal growth factor pathway receptor clone 15 (Eps15) in regulation of EAAC1 surface expression using dominant-negative variants of this protein (González et al., In Press.). Two different dominant-negative variants of Eps15 or a control construct were co-transfected with GLT-1. The first dominant-negative inhibitor, termed DIII, contains the carboxyl-terminal domain of Eps15 (529-897) and is thought to block clathrin-mediated endocytosis by competing with the binding of endogenous Eps15 to the adaptor protein AP-2 (Benmerah et al., 1998). The second dominant-negative variant of Eps15 (EΔ95/295) lacks second and third EH domains (Eps15-Homology), inhibiting the recruitment of AP-2 to the plasma membrane (Benmerah et al., 1999). The control construct (D3Δ2) used in these studies lacks amino acids 621-739 and all AP-2-binding sites and does not block clathrin-dependent endocytosis (Benmerah et al., 1998). All three of these constructs are epitope tagged with GFP, and antibodies against GFP were used to test for expression of these constructs after transfection. In C6 cells co-transfected with GLT-1 and pcDNA3.1, there was a robust cross-reacting band at ~200 kDa (Fig. 4a). However, this cross-reacting band was consistently resolved from the EΔ95/295 band at ~190 kDa. Finally, the two other variants of Eps15 (GFP-DIII & GFP-D3Δ2) were also observed in these same western blots (Fig. 4a). The levels of GLT-1 in cells lysates were not affected by either of the dominant-negative variants of Eps15 compared to the control non-functional construct (D3Δ2) (see legend to Fig. 4). We also found that neither dominant-negative variant of Eps15 affected the PMA-induced change in biotinylated GLT-1 (Fig. 4c). In a recent study, we found that the dominant-negative variants of Eps15 cause an increase in the steady state levels of biotinylated mycepitope tagged EAAC1, implicating Eps15 in endocytosis of EAAC1 (González et al., In Press.). Therefore, in a subset of these experiments Myc-tagged EAAC1 was co-expressed with EΔ95/295, and we found that it increased the amount of biotinylated myc-EAAC1 to ~130% of control (p < 0.01 by paired t-test, n=3). Therefore, we conclude that these constructs were expressed in C6 glioma and had an effect on the cell surface expression of a different protein in parallel experiments, providing strong evidence that the PMA-induced redistribution of GLT-1 is independent of Eps15.
Fig. 4.
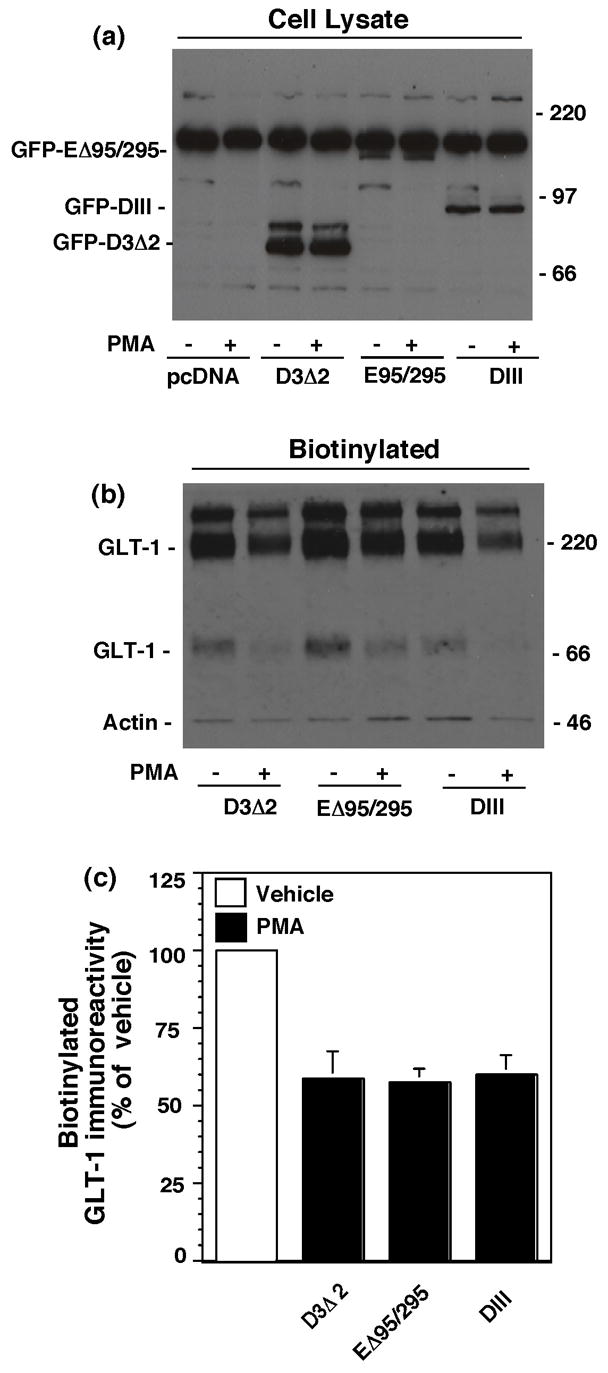
Effects of two different dominant-negative variants of Eps15 on the PMA-induced decrease in biotinylated GLT-1. C6 cells were co-transfected with cGLT-1 and one of the GFP tagged Eps15 constructs (D3Δ2, EΔ95/295 or DIII). After 16–20h, cells were treated with vehicle (DMSO) or PMA for 30 min and biotinylated proteins were batch extracted. (a) Representative western blot of lysates from cells transfected with GFP tagged Eps15 constructs to demonstrate expression of the Eps15 variants in C6 glioma using an anti-GFP antibody. (b) Representative western blot of biotinylated GLT-1 (note the faint band for actin immunoreactivity). (c) Summary of the effects of PMA on biotinylated GLT-1 in cells co-transfected with dominant-negative (EΔ95/295 or DIII) or control (D3Δ2) variants of Eps15 (mean ± SEM of six independent observations). In these studies, the percentage of actin biotinylated was 2 ± 1% under control conditions and did not change with any treatment. Expression of total GLT-1 in cells co-transfected with either of the dominant-negative variants of Eps15 was not different from that observed in C6 cells co-transfected with the control construct, D3Δ2 (EΔ95/295, 108 ± 12%; DIII, 89 ± 14%, expressed as a percentage of that observed in cells co-transfected with D3Δ2). The percentage of transporter biotinylated was also not affected by these constructs (EΔ95/295, 95 ± 3%; DIII, 98 ± 5%, expressed as a percentage of that observed in cells co-transfected with D3Δ2). There were no significant differences between groups as compared by ANOVA with Bonferroni post hoc correction.
Effects of cholesterol depletion or a dominant-negative variant of caveolin on phorbol ester-induced redistribution of GLT-1
There are endocytic pathways that are sensitive to depletion/disruption of membrane cholesterol and at least some forms of this endocytosis may depend on caveolin (for reviews, see Simons and Toomre, 2000; Allen et al., 2007). To determine if the PMA-induced redistribution of GLT-1 might be dependent upon cholesterol, we pretreated GLT-1-transfected C6 cells with MβCD (30 min) to deplete membrane cholesterol. Compared to vehicle-treated cells, MβCD had no effect on the PMA-induced reduction in biotinylated GLT-1 (Fig. 5a). In a recent study, we found that MβCD increases the steady state levels of EAAC1 and blocks endocytosis of EAAC1 measured using reversible biotinylation (González et al., In Press.). Therefore, in these same experiments, we examined the effects of MβCD on the levels of biotinylated EAAC1 that is endogenously expressed in C6 glioma and found that MβCD increased biotinylated EAAC1 (to 136 ± 6% on vehicle treated cells, p < 0.02), providing a control for the effectiveness of MβCD in these studies. We also examined the effect of co-expression of a dominant-negative variant of caveolin-1 that has been previously used to study endocytosis of the GLUT4 subtype of glucose transporter (Shigematsu et al., 2003). We found that compared to either pcDNA transfected (data not shown) or cells transfected with wild-type caveolin, the S80E variant of caveolin had no effect on the PMA-induced reduction in biotinylated GLT-1 (Fig. 5b). In other studies conducted in the laboratory, we have found that identical amounts of this variant of caveolin affects EAAC1 surface expression, suggesting that this construct is functional upon expression in C6 glioma. Together, these studies provide evidence that the PMA-induced redistribution of GLT-1 is independent of cholesterol and caveolin-1.
Fig. 5.
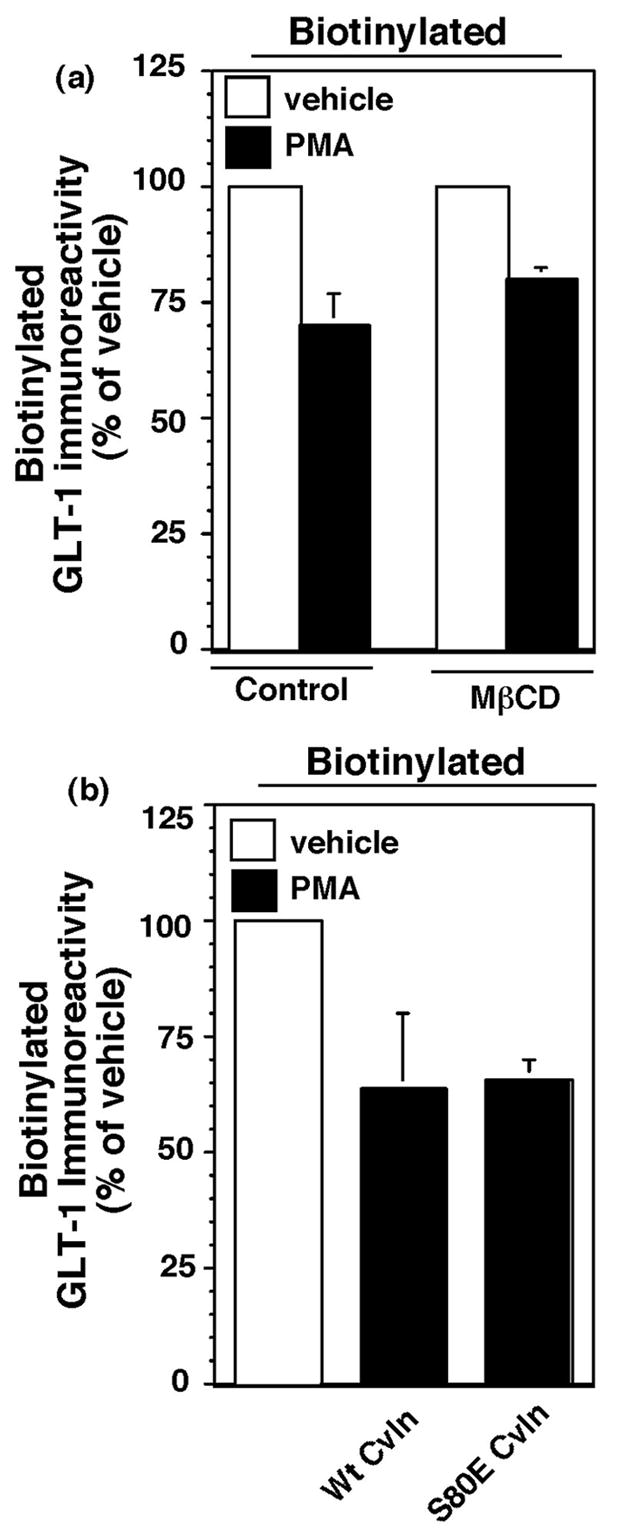
Effect of cholesterol depletion or co-expression with a dominant-negative variant of caveolin on the PMA-induced reduction in biotinylated GLT-1. (a) C6 cells were transfected with cGLT-1. After 16–20h, they were pretreated with MΔCD (10 mM) for 5 min and then treated with vehicle (DMSO) or PMA for 30 min. In these studies, no actin was detected in western blots of the biotinylated fractions. MßCD had no significant effect on total GLT-1 expression (86 ± 20 % of vehicle treated controls) nor on the percentage of biotinylated GLT-1 (79 ± 16 % of vehicle treated controls). MΔCD had no effect on the PMA-induced decrease in biotinylated GLT-1. Data are the mean ± SEM of three independent observations. (b) C6 cells were co-transfected with GLT-1 and either wild-type or the S80E mutant variant of caveolin. After 16–20h, cells were treated with vehicle or PMA (100 nM) for 30 min and then subjected to biotinylation and western blot analyses. Summary of the results from four independent experiments are presented. Data are expressed as the % of biotinylated transporter observed after treatment with PMA compared to the corresponding vehicle treated control. In these studies, biotinylated actin was 4 ± 2% of total actin, and it did not change with treatment. Co-expression with the S80E mutant of caveolin did not change the levels of GLT-1 expression observed in cell lysates (102 ± 14 %, expression of GLT-1 as a percentage of GLT-1 immunoreactivity in cells transfected with wild-type caveolin). There was no significant effect of the S80E mutant on the PMA-induced internalization of GLT-1.
Effect of a dominant-negative variant of Arf6 on phorbol ester-induced redistribution of GLT-1
Arf6 is a member of the Arf family of proteins that is thought to function as a molecular switch by cycling between active GTP-bound and inactive GDP-bound conformations at the plasma membrane. Arf6 has been implicated in the regulation of clathrin-dependent endocytosis, clathrin-independent endocytosis, recycling and actin reorganization (for reviews/discussions, see Donaldson, 2003; D'Souza-Schorey and Chavrier, 2006; Pellinen and Ivaska, 2006; Shultz et al., 2006; Gong et al., 2007). GTP hydrolysis by Arf6 is required for completion of internalization, while exocytosis is achieved by exchange of GTP for GDP. We examined the PMA-induced changes in biotinylated GLT-1 after co-transfection with wild-type, dominant-negative (T27N) or constitutively active (Q67L) variants of Arf6. None of these variants significantly affected the PMA-induced decrease in biotinylated GLT-1 (Fig. 6). Although these studies were not conducted with a positive control, we confirmed that the constructs result in expression of an immunoreactive band of the appropriate molecular weight upon transfection into HEK cells (data not shown). Therefore, it seems likely that the redistribution of GLT-1 is independent of Arf6.
Fig. 6.
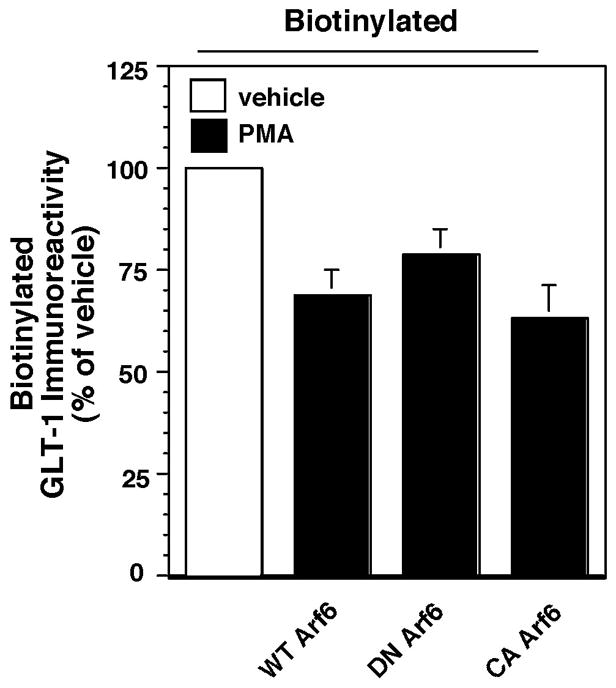
Effects of co-expression of variants of Arf6 on the PMA-induced redistribution of GLT-1. C6 cells were co-transfected with GLT-1 and wild-type, dominant-negative (DN) (T27N) or constitutively active (CA) (Q67L) variants of Arf6. After 16–20 h and treatment with PMA (100 nM) for 30 min, the levels of GLT-1 protein in the biotinylated fraction, in the non-biotinylated fraction, and in the total cell lysate were examined by western blot. In these studies, no actin was detected in the biotinylated fraction in any of the experiments and the levels of total GLT-1 protein were not significantly changed by either variant of Arf6 (DN Arf6, 103 ± 8 %; CA Arf6, 74 ± 28%, expressed as a % of that observed in cells co-transfected with wild-type Arf6). The proportion of GLT-1 on the plasma membrane was not significantly affected by either variant of Arf6 (DN Arf6, 103 ± 3 %; CA Arf6, 91 ± 4%; expressed as a % of that observed in cells co-transfected with wild-type Arf6). The data presented represent the summary of effects of PMA on biotinylated GLT-1 and is expressed as a percentage of the amount of immunoreactivity observed in cells transfected with the same cDNAs and treated with vehicle. Data are the mean ± SEM of five observations. There were no significant effect of either variant of Arf6 on the PMA-induced redistribution of GLT-1 (assessed by ANOVA).
Effects of prolonged activation of PKC on GLT-1 expression
After internalization, membrane proteins are either recycled back to the plasma membrane or targeted for degradation (for reviews see, Maxfield and McGraw, 2004; Melikian, 2004; Bernard et al., 2006). To determine if GLT-1 is targeted for degradation, the effects of long-term treatment with PMA on cell surface and total expression of GLT-1 were examined. C6 glioma were transfected with GLT-1 and treated with PMA for increasing periods of time. PMA caused a time-dependent decrease in the amount of biotinylated GLT-1 (Fig. 7b & d). PMA also caused a time-dependent decrease in total GLT-1 protein detected in cell lysates (Fig. 7a & c). There was no change in GLT-1 immunoreactivity with vehicle treatment (data not shown). Consistent with the notion that transporter is targeted for degradation with more prolonged treatment with PMA, there was a modest accumulation of non-biotinylated (intracellular) GLT-1 immunoreactivity at early times (15 min through 1 h), and at later time-points this immunoreactivity was not observed (data not shown). Although it seems unlikely that this decrease in transporter expression might be due to decreased transcription of GLT-1 introduced using pcDNA3.1 with cytomegalovirus promoter, the effects of a protein synthesis inhibitor, cycloheximide (10 μg/mL), were examined to determine if inhibition of transcription could cause a decrease in GLT-1 levels. In these studies cycloheximide had no significant effect on GLT-1 protein levels after 2 h (86 ± 8% of control in lysate) or 4 h (78 ± 8% of control in lysate). These results suggest that PKC activation initially causes an intracellular accumulation of GLT-1 and that prolonged treatment with PMA may be targeting GLT-1 for degradation.
Fig. 7.
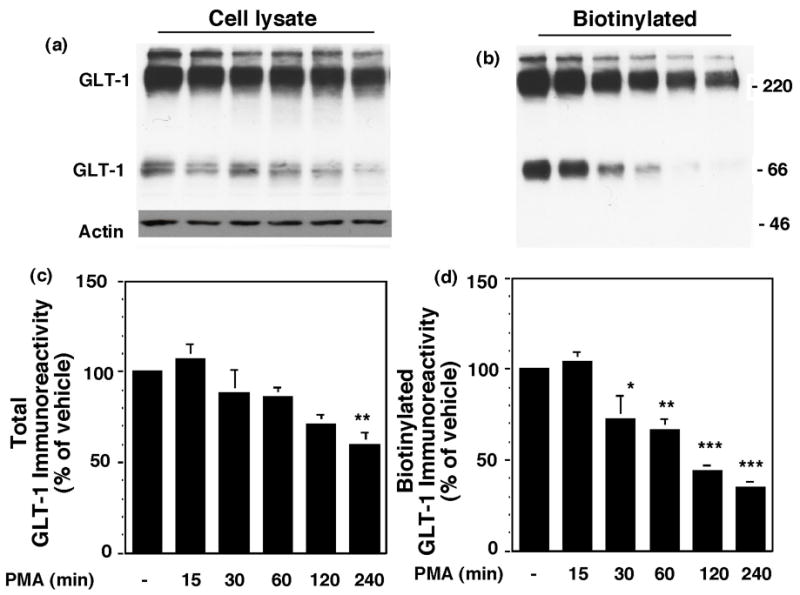
Effects of prolonged treatment with PMA on total and biotinylated GLT-1 immunoreactivity. C6 cells were transfected with GLT-1. After 16–20 h, they were treated with PMA or vehicle for 0–4 h before measuring cell surface proteins by biotinylation. Representative immunoblots of GLT-1 and actin immunoreactivity in total cell lysate (a) and biotinylated fractions (b). Summary of data from four independent experiments (mean ± SEM) are presented in (c) and (d). Since there was no change in GLT-1 immunoreactivity in any of the three fractions in vehicle treated cells (data not shown), data are presented as the percentage of immunoreactivity observed in un-treated cells (4 h time point). In these studies, no actin was detected in the biotinylated fractions. Data were compared by ANOVA using a Bonferroni post hoc correction. *p < 0.05, **p < 0.01, ***p < 0.001 compared to DMSO at 4 h time point.
In order to further investigate the mechanisms involved in this loss of GLT-1 immunoreactivity, the effects of either lysosomal or proteosomal inhibitors on this loss of GLT-1 were examined. GLT-1 transfected cells were pretreated with NH4Cl, chloroquine, or lactacystin for 15 min, followed by addition of PMA and incubation for 2 h. As was observed in the studies presented in Fig. 7, a two hour treatment with PMA caused a significant decrease in total GLT-1 protein (Fig. 8a). This effect was significantly attenuated by either NH4Cl or chloroquine, two inhibitors of lysosomal degradation (Heinzen et al., 1996; Bubeck et al., 2002), but not by lactacystin, an inhibitor of proteosomal degradation (Schmidt et al., 1999; Bubeck et al., 2002). In fact, in the presence of lysosomal inhibitors PMA had no significant effect on total GLT-1 expression (p > 0.05). This effect of lysosomal inhibitors was accompanied by a dramatic increase in non-biotinylated (intracellular) GLT-1 (Fig. 8c & d). However the lysosomal inhibitors had no effect on the decrease in biotinylated GLT-1 (data not shown). These studies suggest that GLT-1 is targeted for lysosomal degradation with prolonged activation of PKC.
Fig. 8.
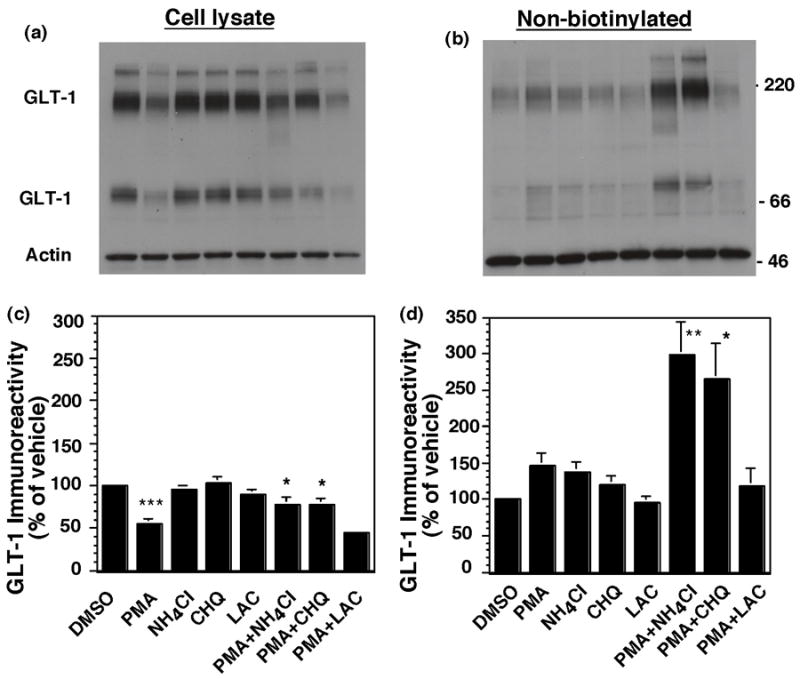
Effects of lysosomal or proteosomal inhibitors on the PMA-induced decrease in GLT-1 immunoreactivity. C6 glioma were transfected with GLT-1 (12 μg). After 16–20 h, cells were pretreated with NH4Cl (10 mM), chloroquine (CHQ, 50 μM) or lactacystin (LAC, 5 μM). After 15 min, cells were treated with PMA (100 nM) for 2 h and biotinylated GLT-1 was batch extracted. Representative immunoblots of cell lysate (a) and non-biotinylated fractions (b) are presented. Summary of effects on total GLT-1 immunoreactivity (c) or non-biotinylated (intracellular) GLT-1 immunoreactivity (d). Data are mean ± SEM of six independent experiments and are expressed as a percentage of that observed in cells treated with vehicle (DMSO). Data were compared by ANOVA using a Bonferroni post hoc correction. *p < 0.05, **p < 0.01 compared to cells treated with PMA.
The small GTPase Rab7 is thought to be important for trafficking of proteins from early sorting endosomes to late endosomes (Feng et al., 1995) and from late endosomes to lysosomes (Meresse et al., 1995; Bucci et al., 2000). Therefore, Rab7 is essential for maintenance of the perinuclear lysosome compartment by controlling aggregation and fusion of late endocytic structures to lysosomes. The potential involvement of Rab7 in the PMA-induced degradation of GLT-1 was examined by co-transfecting GLT-1 with an epitope (HA) tagged, dominant-negative variant of Rab7 (N125I). In these studies, expression of Rab7 was confirmed by probing cell lysate with an anti-HA antibody that revealed an immunoreactive band of 26 kDa that was not present in the cells co-transfected with pcDNA3.1 (data not shown). Since inhibition of internalization of GLT-1 might also be expected to decrease lysosomal degradation, the effects of PMA on redistribution and degradation of GLT-1 were examined at both a short time point (30 min) and a longer time point (2 h). Co-expression with the dominant-negative variant of Rab7 had no effect on the PMA-induced change in biotinylated transporter (see legend to Fig. 9). However, it significantly attenuated the PMA-induced loss of GLT-1 protein from the lysate observed after a 2 h incubation (Fig. 9a & 9c). It also caused a significant accumulation of GLT-1 in the non-biotinylated fraction after a 2 h treatment with PMA (Fig. 9 b & d). Together these results provide strong evidence that the dominant-negative variant of Rab7 inhibits the degradation of GLT-1 without affecting redistribution of GLT-1 from the plasma membrane to an intracellular compartment.
Fig. 9.
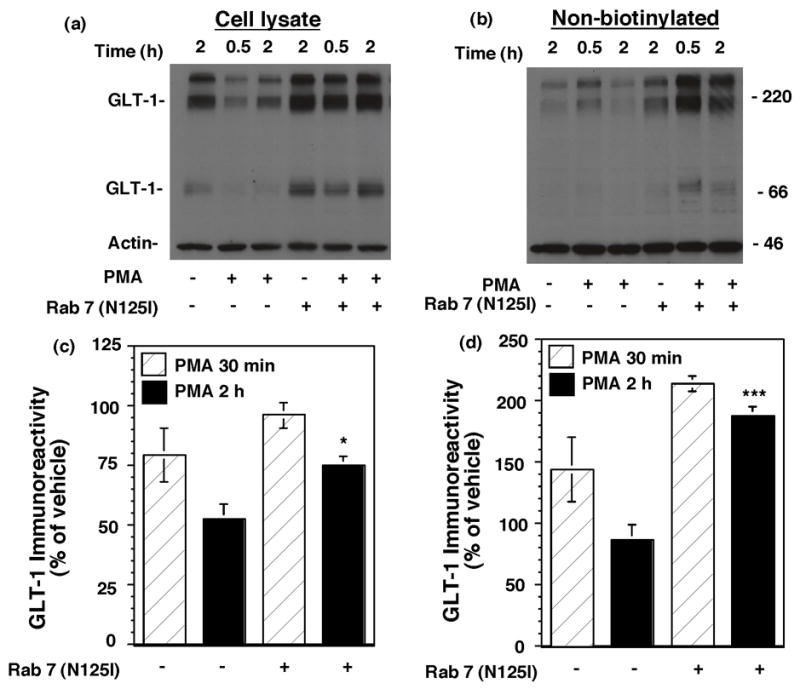
Effects of co-expression of a dominant-negative variant of rab7 on the PMA-induced loss of GLT-1 immunoreactivity. C6 cells were co-transfected with GLT-1 and empty vector or dominant negative rab7 (N125I). After 16–20h, cells were treated with either vehicle or PMA (100 nM) for 30 min or 2 h. GLT-1 immunoreactivity was measured after biotinylation. Representative immunoblots from cell lysate (a) and non-biotinylated (intracellular) fractions (b) are presented. (c) & (d) are the summary of results in cell lysate and intracellular fractions (mean ± SEM) from five independent experiments. Data were expressed as a percentage of control cells transfected with pcDNA and treated with vehicle. In these studies, the mean % of actin biotinylated was 13 ± 6%, and it was not significantly affected by any of the conditions. The dominant-negative variant of rab7 had no significant effect on the PMA-induced change in biotinylated transporter with either 30 min (pcDNA, 64 ± 11%; DN rab7, 82 ± 8%) or 2 h incubation with PMA (pcDNA, 32 ± 6%; DN rab7, 40 ± 6%); data were expressed as a percentage of the corresponding vehicle control and compared by paired t-test. *p < 0.05 and **p < 0.005 compared to cells co-transfected with pcDNA and treated with PMA for 2h; comparison was conducted by paired t-test.
Discussion
Previous studies have demonstrated that acute activation of PKC causes a redistribution of GLT-1 from the plasma membrane to an intracellular compartment. This effect has been observed in stably and transiently transfected C6 glioma, in primary astrocyte cultures induced to express GLT-1, and in co-cultures of neurons and astrocytes that express GLT-1 (Kalandadze et al., 2002; Zhou and Sutherland, 2004; Guillet et al., 2005). Using chimeras of EAAC1 and GLT-1, amino acids 475-517 of GLT-1 were found to be necessary for PKC-dependent redistribution (Kalandadze et al., 2002). Although serine-486 within this domain was also implicated in regulated redistribution of GLT-1, there no is evidence that this serine residue is phosphorylated (Kalandadze et al., 2002). In the present study, we began to explore the mechanisms involved in redistribution of GLT-1. We found that sucrose, a dominant-negative variant of dynamin, or a dominant-negative variant of clathrin blocked/attenuated the redistribution of GLT-1 from the plasma membrane. Although clathrin contributes to intracellular trafficking from trans-Golgi network (TGN) and endosomes (Liu et al., 1998; Royle, 2006), the simplest explanation is that this dominant-negative variant is affecting endocytosis. We also found that inhibitors of ‘lipid raft’/caveolin-dependent endocytosis had no effect on the redistribution of GLT-1. In addition, dominant-negative variants of Arf6 or Eps15, one of the clathrin-accessory proteins, had no effect on the redistribution of GLT-1. Together, these studies suggest that the effects of PKC on GLT-1 surface expression are dependent upon a clathrin-dependent endocytic pathway that is independent of cholesterol or the accessory protein Eps15. In addition, we report that longer term treatment with phorbol ester caused a time-dependent decrease in the amount of biotinylated transporter that was accompanied by a decrease in total transporter expression. This decrease in total transporter expression was completely blocked by inhibitors of lysosomal but not proteosomal degradation. This decrease in transporter expression was also attenuated by a dominant-negative variant of a small GTPase implicated in trafficking of proteins from early sorting endosomes through late endosomes to lysosomes (Feng et al., 1995; Meresse et al., 1995; Bucci et al., 2000). Together, these studies begin to define the cellular machinery required for redistribution of the predominant forebrain glutamate transporter and identify a mechanism by which total levels of this transporter might be controlled under physiologic or pathologic conditions. The current study was conducted using C6 glioma as a model system, we acknowledge that it is possible that the mechanisms involved in internalization of GLT-1 in primary astrocytes may be different from those used in this model system.
The activity and/or cell surface expression of several other neurotransmitter transporters is also affected by protein kinase C, including the dopamine transporter (Daniels and Amara, 1999; Melikian and Buckley, 1999; Sorkina et al., 2005), a subtype of GABA transporter (Corey et al., 1994; Quick et al., 1997), the serotonin transporter (Qian et al., 1997; Magnani et al., 2004), the norepinephrine transporter (Apparsundaram et al., 1998; Jayanthi et al., 2004), a subtype of glycine transporter (Gomeza et al., 1995; Sato et al., 1995), and one of the other subtypes of glutamate transporter, EAAC1 (Davis et al., 1998; Guillet et al., 2005). In some cases, these changes were originally documented as changes in the steady state levels of transporter at the plasma membrane. Since steady state levels are dependent upon both the rate at which transporters are delivered to the plasma membrane and the rate at which transporters are removed from the plasma membrane, it cannot be used to determine the rate of endocytosis, the rate of delivery, or both are changing in response to PKC activation. In some cases, it has been possible to measure the effects of PKC activation on both the rate of delivery of transporter to and the rate of removal of the transporter from the plasma membrane. PKC activation has been implicated in regulating endocytosis of some transporters (Wang and Quick, 2005), and both exocytosis and endocytosis of other transporters (Loder and Melikian, 2003; Fournier et al., 2004). Although we attempted to determine if PKC affects delivery or endocytosis of GLT-1 using biotinylating reagents as previously described (Fournier et al., 2004), the fact that essentially all (>90%) GLT-1 immunoreactivity is on the cell surface under baseline conditions precludes either of these measures from providing meaningful comparisons. For example, using a disulfide-containing biotinylating reagent to label cell surface proteins under conditions that halt constitutive trafficking (4º C) followed by rewarming for various periods of time, resulted in no accumulation of intracellular (unstripped with a membrane impermeant disulfide reducing reagent) biotinylated GLT-1 (data not shown, studies conducted in parallel with measures of EAAC1 trafficking to confirm that the procedure was being conducted appropriately). The simplest explanation of these data would suggest that GLT-1 is stable at the plasma membrane with little constitutive trafficking, however, one cannot formally exclude the possibility that GLT-1 is very rapidly recycling on and off the plasma membrane. Along the same lines, the current study clearly demonstrates that interfering with the function of a number of molecules normally and selectively involved in endocytosis block (or attenuate) the PMA-induced (PKC-dependent) redistribution of GLT-1. The simplest explanation of these data would seem to implicate PKC in the regulation of endocytosis of GLT-1, although we cannot exclude the possibility that PKC slows delivery of transporters that are rapidly recycling and that this rapid recycling is blocked by sucrose, dynamin, and a dominant-negative variant of clathrin.
In a separate study, we examined the effects of many of the same treatments/co-transfections on the constitutive trafficking of another member of the glutamate transporter family, EAAC1, in C6 glioma (González et al., In Press.). In this study, constitutive endocytosis of EAAC1 was slowed by depletion of cholesterol or by sucrose. In addition, the dominant-negative variants of Eps15 increased cell surface expression of myc-EAAC1, implicating Eps15 in constitutive endocytosis of EAAC1. In the present study, we confirmed that depletion of cholesterol or Eps15 had the predicted effects on EAAC1 (or myc-EAAC1), providing positive controls for these studies and strong evidence that the redistribution of GLT-1 is independent of cholesterol or Eps15. Therefore, in the same cellular system, EAAC1 trafficking is differentiated from GLT-1 trafficking based on the sensitivity to cholesterol depletion or co-expression with dominant-negative variants of Eps15. We have also found that caveolin is likely involved in constitutive endocytosis of EAAC1 (González, Krizman-Genda, and Robinson, unpublished observations), further differentiating GLT-1 and EAAC1 trafficking. There is evidence that regulated and constitutive endocytosis of the epidermal growth factor receptor are mediated by different processes (Confalonieri et al., 2000); therefore, it is possible that the differential effects of cholesterol and Eps15 variants are related to the fact that in the case of GLT-1 the trafficking is ‘regulated’, and in the case of EAAC1 the trafficking is ‘constitutive’. However, these studies provide evidence that in the same cellular milieu EAAC1 and GLT-1 can be trafficked by distinct mechanisms, implying that primary sequence differences between these transporters confer differences in this trafficking.
Proteins that redistribute from the plasma membrane to an internal pool are either recycled back to the plasma membrane or targeted for degradation. In some cases, it has been possible to directly determine if the proteins recycle back to the plasma membrane by labeling cell surface proteins with a disulfide-containing biotinylating reagent, stimulating redistribution, stripping the biotinylation reagent from the remaining cell surface proteins, and then determining if a strippable pool of transporter re-appears at the plasma membrane (Deken et al., 2003; Wang and Quick, 2005). Since we observe essentially no significant accumulation of intracellular transporter with short-term activation of PKC, these studies are not possible. Therefore, in the present study, we examined the effects of longer term treatment with phorbol ester and observed a time-dependent decrease in biotinylated GLT-1. This effect was accompanied by a loss of total GLT-1 expression that was consistent with the targeting of GLT-1 for lysosomal degradation. A similar pathway for degradation of the dopamine transporter has been defined (Daniels and Amara, 1999). These studies suggest that at least a portion of internalized GLT-1 is targeted for degradation after internalization. Others have shown that GLT-1 can be internalized in to co-expression with the variant of superoxide dismutase associated with amyotrophic lateral sclerosis (Vanoni et al., 2004). It will be interesting in future studies to determine if the cellular machinery contributing to phorbol ester-dependent internalization of GLT-1 also mediate internalization caused by this or other stimuli. Although several studies have shown that the acute phorbol ester-dependent internalization of GLT-1 is blocked by inhibitors of PKC (Kalandadze et al., 2002; Zhou and Sutherland, 2004; Guillet et al., 2005), we acknowledge that we have not shown that the degradation of GLT-1 is blocked by inhibitors of PKC. Therefore, it is possible that degradation is dependent upon interaction with other phorbol ester activated processes.
Several studies have demonstrated that the levels of GLT-1 protein are decreased in animal models of a variety of neurodegenerative diseases or in humans with these diseases (for reviews, see Danbolt, 2001; Beart and O'Shea, 2007; Sheldon and Robinson, 2007). In some cases, these effects occur quicker than one might expect for decreased transcription. For example, there is a decrease in GLT-1 protein after an ischemic insult. In several studies, decreased levels of GLT-1 have been documented at times as early as 3–6 h after an ischemic insult (20–50% decreases) with decreases as large as 85% 24 h after an insult (Torp et al., 1995; Martin et al., 1997; Pow et al., 2004; Chen et al., 2005). A similarly large decrease in GLT-1 is observed within a few hours of a fluid percussion insult that models traumatic brain injury (Yi et al., 2004). If one assumes that this effect is only dependent upon halting transcription and that GLT-1 is degraded by first order kinetics, the half-life would range from less than 6h to 10h. Although the half-life of GLT-1 or the other glutamate transporters have not been examined in vivo, there is evidence that the half-life of GLT-1 is longer than 24 h in astrocyte cultures (Zelenaia and Robinson, 2000). It also seems reasonable that the half-life of an extremely abundant protein that is critical for limiting excitotoxicity might be much longer than 9 hours. Therefore, although it is somewhat speculative at this time, it is possible that degradation of GLT-1 is activated under ischemic conditions (or other diseases). Except for one recent study, demonstrating that the carboxyl terminal tail of GLT-1 is cleaved by caspase 3 (Boston-Howes et al., 2006), very little is known about the mechanisms that might contribute to decreases in GLT-1 protein levels that are independent of transcription/translation. In this case, caspase 3 removes that tail from transporters prior to their removal from the plasma membrane. Since we found that activation of PKC caused an internalization of the transporter that preceded the loss of GLT-1 immunoreactivity, it seems unlikely that the effects observed in the present study can be attributed to effects of caspase 3.
In summary, we report that redistribution of GLT-1 from the plasma membrane to an intracellular compartment is blocked/attenuated by sucrose or dominant-negative variants of dynamin/clathrin. We also report that longer-term activation of PKC results in degradation of GLT-1 that is blocked by inhibitors of lysosomal (but not proteosomal) degradation. Together these studies define some of the cellular machinery involved in redistribution of GLT-1 and define a novel mechanism that could contribute to decreases in total GLT-1 levels under either physiologic or pathologic conditions.
Acknowledgments
This work was supported by NIH grant NS29868. The authors would also like to thank Dr. Marco González who provided helpful suggestions while these studies were being conducted for his help with the preparation of this manuscript. The authors would also like to thank Amanda Sheldon and Susannah Locke for their help with editing the manuscript.
Abbreviations
- DMEM
Dulbecco’s modified Eagle's medium
- EAAC1
excitatory amino acid carrier 1
- Eps15
epidermal-growth-factor receptor pathway substrate clone 15
- FBS
fetal bovine serum
- GLT-1
glutamate transporter 1
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nature Neurosci Rev. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Apparsundaram S, Schroeter S, Giovanetti E, Blakely RD. Acute regulation of norepinephrine transport:II. PKC-modulated surface expression of human norepinephrine transporter proteins. J Pharmacol Exp Ther. 1998;287:744–751. [PubMed] [Google Scholar]
- Beart PM, O'Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman ML, Quick MW. The ups and downs of neurotransmitter transporters. Neuroscientist. 2000;6:199–207. [Google Scholar]
- Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. Journal of Cell Science. 1999;112:1303–1311. doi: 10.1242/jcs.112.9.1303. [DOI] [PubMed] [Google Scholar]
- Benmerah A, Lamaze C, Bègue B, Schmid SL, Dautry-Varsat A, Cerf-Bensussan N. AP-2/Eps15 Interaction Is Required for Receptor-mediated Endocytosis. J Cell Biol. 1998;140:1055–1062. doi: 10.1083/jcb.140.5.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Decossas M, Liste I, Bloch B. Intraneuronal trafficking of G-protein-coupled receptors in vivo. Trends in Neuroscience. 2006;29:140–147. doi: 10.1016/j.tins.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Bauman AL. Biogenic amine transporters: regulation in flux. Curr Opin Neurobiol. 2000;10:328–336. doi: 10.1016/s0959-4388(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Boston-Howes W, Gibb SL, Williams EO, Pasinelli P, Brown RH, Jr, Trotti D. Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J Biol Chem. 2006;281:14076–14084. doi: 10.1074/jbc.M600653200. [DOI] [PubMed] [Google Scholar]
- Bubeck A, Reusch U, Wagner M, Ruppert T, Muranyi W, Kloetzel PM, Koszinowski UH. The glycoprotein gp48 of murine cytomegalovirusL proteasome-dependent cytosolic dislocation and degradation. J Biol Chem. 2002;277:2216–2224. doi: 10.1074/jbc.M104178200. [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchbach ME, Tian G, Guo H, Lin CL. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279:34388–34396. doi: 10.1074/jbc.M403938200. [DOI] [PubMed] [Google Scholar]
- Carrick T, Dunlop J. Protein kinase C modulation of the human excitatory amino acid transporter 2 subtype of glutamate transporter. Soc Neurosci Abs. 1999;25:426. [Google Scholar]
- Casado M, Bendahan A, Zafra F, Danbolt NC, Gimenez C, Kanner BI. Phosphorylation and modulation of brain glutamate transporters by protein kinase C. J Biol Chem. 1993;268:27313–27317. [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, Campagne MVL, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Chen JC, Hsu-Chou H, Lu JL, Chiang YC, Huang HM, Wang HL, Wu T, Liao JJ, Yeh TS. Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology. 2005;49:703–714. doi: 10.1016/j.neuropharm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci. 2004;24:1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Confalonieri S, Salcini AE, Puri C, Tacchetti C, Di Fiore PP. Tyrosine phosphorylation of Eps15 is required for ligand-regulated, but not constitutive, endocytosis. J Cell Biol. 2000;150:905–912. doi: 10.1083/jcb.150.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- Corey JL, Davidson N, Lester HA, Brecha N, Quick MW. Protein kinase C modulates the activity of a cloned γ-aminobutyric acid transporter expressed in xenopus oocytes via a regulated subcellular redistribution of the transporter. J Biol Chem. 1994;269:14759–14767. [PubMed] [Google Scholar]
- D'Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Daniels GM, Amara SG. Regulated trafficking of the human dopamine transporter. J Biol Chem. 1999;274:35794–35801. doi: 10.1074/jbc.274.50.35794. [DOI] [PubMed] [Google Scholar]
- Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci. 1998;18:2475–2485. doi: 10.1523/JNEUROSCI.18-07-02475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deken SL, Wang D, Quick MW. Plasma membrane GABA transporters reside on distinct vesicles and undergo rapid regulated recycling. J Neurosci. 2003;23:1563–1568. doi: 10.1523/JNEUROSCI.23-05-01563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami GK, Ferguson SSG. Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol Ther. 2006;111:260–271. doi: 10.1016/j.pharmthera.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Donaldson JG. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem. 2003;278:41573–41576. doi: 10.1074/jbc.R300026200. [DOI] [PubMed] [Google Scholar]
- Feng Y, Press B, Wandinger-Ness A. Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol. 1995;131:1435–1452. doi: 10.1083/jcb.131.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier KM, Robinson MB. A dominant-negative variant of SNAP-23 decreases the cell surface expression of the neuronal glutamate transporter EAAC1 by slowing constitutive delivery. Neurochem Int. 2006;48:596–603. doi: 10.1016/j.neuint.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Fournier KM, González MI, Robinson MB. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J Biol Chem. 2004;279:34505–34513. doi: 10.1074/jbc.M404032200. [DOI] [PubMed] [Google Scholar]
- Ganel R, Crosson CE. Modulation of human glutamate transporter activity by phorbol ester. J Neurochem. 1998;70:993–1000. doi: 10.1046/j.1471-4159.1998.70030993.x. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Zafra F, Olivares L, Gimenez C, Aragon C. Regulation by phorbol esters of the glycine transporter (GLYT1) in glioblastoma cells. Biochimica et Biophysica Acta. 1995;1233:41–46. doi: 10.1016/0005-2736(94)00249-o. [DOI] [PubMed] [Google Scholar]
- Gong Q, Weide M, Huntsman C, Xu Z, Jan LY, Ma D. Identification and characterization of a new class of trafficking motifs for controlling clathrin-independent internalization and recycling. J Biol Chem. 2007;282:13087–13097. doi: 10.1074/jbc.M700767200. [DOI] [PubMed] [Google Scholar]
- Gonzalez MI, Susarla BT, Robinson MB. Evidence that protein kinase Calpha interacts with and regulates the glial glutamate transporter GLT-1. J Neurochem. 2005;94:1180–1188. doi: 10.1111/j.1471-4159.2005.03330.x. [DOI] [PubMed] [Google Scholar]
- González MI, Robinson MB. Neurotransmitter transporters: Why dance with so many partners. Current Opinion in Pharmacology. 2004;4:30–35. doi: 10.1016/j.coph.2003.09.004. [DOI] [PubMed] [Google Scholar]
- González MI, Susarla BTS, Fournier KM, Sheldon AL, Robinson MB. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J Neurochem. doi: 10.1111/j.1471-4159.2007.04881.x. In Press. [DOI] [PubMed] [Google Scholar]
- Guillet BA, Velly LJ, Canolle B, Masmejean FM, Nieoullon AL, Pisano P. Differential regulation by protein kinases of activity and cell surface expression of glutamate transporters in neuron-enriched culture. Neurochem Int. 2005;46:337–346. doi: 10.1016/j.neuint.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Haugeto Ø, Ullensveng K, Levy LM, Chaudhry FA, Honore T, Neilsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infection and Immunity. 1996;64:796–809. doi: 10.1128/iai.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Anderson RGW. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura T, Shimohama S, Kageyama T, Kimura J. Selective induction of glial glutamate transporter GLT-1 by hypertonic stress in C6 glioma cells. Biochem Biophys Res Commun. 1999;265:240–245. doi: 10.1006/bbrc.1999.1655. [DOI] [PubMed] [Google Scholar]
- Ingram EM, Wiseman JW, Tessler S, Emson PC. Reduction of glial glutamate transporters in the parietal cortex and hippocampus of the EL mouse. J Neurochem. 2001;79:564–575. doi: 10.1046/j.1471-4159.2001.00612.x. [DOI] [PubMed] [Google Scholar]
- Jayanthi LD, Samuvel DJ, Ramamoorthy S. Regulated internalization and phosphorylation of the native norepinephrine transporter in response to phorbol esters. Evidence for localization in lipid rafts and lipid raft-mediated internalization. J Biol Chem. 2004;279:19315–19326. doi: 10.1074/jbc.M311172200. [DOI] [PubMed] [Google Scholar]
- Kalandadze A, Wu Y, Robinson MB. Protein kinase C activation decreases cell surface expression of the GLT-1 subtype of glutamate transporter. Requirement of a carboxyl-terminal domain and partial dependence on serine 486. J Biol Chem. 2002;277:45741–45750. doi: 10.1074/jbc.M203771200. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The Number of Glutamate Transporter Subtype Molecules at Glutamatergic Synapses: Chemical and Stereological Quantification in Young Adult Rat Brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56:901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurological disorders. N Engl J Med. 1994;330:613–621. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Liu SH, Marks MS, Brodsky FM. A dominant-negative clathrin mutant differentially affects trafficking of molecules with distinct sorting motifs in the class II major histocompatibility complex (MHC) pathway. J Cell Biol. 1998;140:1023–1037. doi: 10.1083/jcb.140.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SH, Wong ML, Craik CS, Brodsky FM. Regulation of clathrin assembly and trimerization defined using recombinant triskelion hubs. Cell. 1995;83:257–267. doi: 10.1016/0092-8674(95)90167-1. [DOI] [PubMed] [Google Scholar]
- Loder MK, Melikian HE. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J Biol Chem. 2003;278:22168–22174. doi: 10.1074/jbc.M301845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani F, Tate CG, Wynne S, Williams C. Partitioning of the serotonin transporter into lipid microdomains modulates transport of serotonin. J Biol Chem. 2004;279:38770–38778. doi: 10.1074/jbc.M400831200. [DOI] [PubMed] [Google Scholar]
- Maldonado-Baez L, Wendland B. Endocytic adaptors: recruiters, coordinators and regulators. Trends Cell Biol. 2006;16:505–513. doi: 10.1016/j.tcb.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J, Traystman RJ. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol. 1997;42:335–348. doi: 10.1002/ana.410420310. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, et al. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology. 1999;52:441–443. doi: 10.1212/wnl.52.3.453. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, McGraw TE. Endocytic recycling. Nature Reviews in Molecular Cell Biology. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- McNiven MA, Cao H, Pitts KR, Yoon Y. The dynamin family of mechanoenzymes: pinching in new places. Trends in Biochemical Sciences. 2000;25:115–120. doi: 10.1016/s0968-0004(99)01538-8. [DOI] [PubMed] [Google Scholar]
- Melikian HE. Neurotransmitter transporter trafficking: endocytosis, recycling, and regulation. Pharmacol Ther. 2004;104:17–27. doi: 10.1016/j.pharmthera.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meresse S, Gorvel JP, Chavrier P. The rab7 GTPase resides on a vesicular compartment connected to lysosomes. J Cell Sci. 1995;108(Pt 11):3349–3358. doi: 10.1242/jcs.108.11.3349. [DOI] [PubMed] [Google Scholar]
- Pellinen T, Ivaska J. Integrin traffic. J Cell Sci. 2006;119:3723–3731. doi: 10.1242/jcs.03216. [DOI] [PubMed] [Google Scholar]
- Pow DV, Naidoo T, Lingwood BE, Healy GN, Williams SM, Sullivan RK, O'Driscoll S, Colditz PB. Loss of glial glutamate transporters and induction of neuronal expression of GLT-1B in the hypoxic neonatal pig brain. Brain Res Dev Brain Res. 2004;153:1–11. doi: 10.1016/j.devbrainres.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Corey JL, Davidson N, Lester HA. Second messengers, trafficking-related proteins, and amino acid residues that contribute to the functional regulation of the rat brain GABA transporter GAT1. J Neurosci. 1997;17:2967–2979. doi: 10.1523/JNEUROSCI.17-09-02967.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VLR, Baskaya MK, Dogan A, Rothstein JD, Dempsey RJ. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J Neurochem. 1998;70:2020–2027. doi: 10.1046/j.1471-4159.1998.70052020.x. [DOI] [PubMed] [Google Scholar]
- Reye P, Sullivan R, Pow DV. Distribution of two splice variants of the glutamate transporter GLT-1 in the developing rat retina. J Comp Neurol. 2002;447:323–330. doi: 10.1002/cne.10218. [DOI] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: A focus on the GLT-1/EAAT2 subtype. Neurochem Int. 1999;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Robinson MB. Regulated trafficking of neurotransmitter transporters: Common notes different melodies. J Neurochem. 2002;80:1–11. doi: 10.1046/j.0022-3042.2001.00698.x. [DOI] [PubMed] [Google Scholar]
- Roth MG. Clathrin-mediated endocytosis before fluorescent proteins. Nature Reviews in Molecular Cell Biology. 2006;7:63–68. doi: 10.1038/nrm1783. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Royle SJ. The Cellular functions of clathrin. Cellular and Molecular Life Sciences. 2006;63:1823–1832. doi: 10.1007/s00018-005-5587-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson C, Kumlien E, Flink R, Lindholm D, Ronne-Engstrom E. Decreased cortical levels of astrocytic glutamate transport protein GLT-1 in a rat model of posttraumatic epilepsy. Neurosci Lett. 2000;289:185–188. doi: 10.1016/s0304-3940(00)01284-2. [DOI] [PubMed] [Google Scholar]
- Sato K, Adams R, Betz H, Schloss P. Modulation of a recombinant glycine transporter (GLYT1b) by activation of protein kinase C. J Neurochem. 1995;65:1967–1973. doi: 10.1046/j.1471-4159.1995.65051967.x. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Mertz K, Morgan jI. Regulation of heme oxygenase-1 expression by dopamine in cultured C6 glioma and primary astrocytes. Molecular Brain Research. 1999;73:50–59. doi: 10.1016/s0169-328x(99)00231-4. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007 doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon AL, Gonzalez MI, Robinson MB. A carboxyl-terminal determinant of the neuronal glutamate transporter, EAAC1, is required for platelet-derived growth factor-dependent trafficking. J Biol Chem. 2006;281:4876–4886. doi: 10.1074/jbc.M504983200. [DOI] [PubMed] [Google Scholar]
- Shigematsu S, Watson RT, Khan AH, Pessin JE. The Adipocyte Plasma Membrane Caveolin Functional/Structural Organization Is Necessary for the Efficient Endocytosis of GLUT4. J Biol Chem. 2003;278:10683–10690. doi: 10.1074/jbc.M208563200. [DOI] [PubMed] [Google Scholar]
- Shultz T, Nash-Livni N, Shmuel M, Altschuler Y. EFA6 regulates endosomal trafficking and affects early endosomes in polarized MDCK cells. Biochem Biophys Res Commun. 2006;351:106–112. doi: 10.1016/j.bbrc.2006.10.024. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nature Reviews in Molecular Cell Biology. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Sims KD, Robinson MB. Expression patterns and regulation of glutamate transporters in the developing and adult nervous system. Crit Rev Neurobiol. 1999;13:169–197. doi: 10.1615/critrevneurobiol.v13.i2.30. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Hoover BR, Zahniser NR, Sorkin A. Constitutive and protein kinase C induced internalization of the dopamine transporter is mediated by a clathrin-dependent mechanism. Traffic. 2005;6:157–170. doi: 10.1111/j.1600-0854.2005.00259.x. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A. RNA Interfernce screen reveals an essential role of Nedd4–2 in dopamine transporter ubiquitination and endocytosis. J Neurosci. 2006;26:8195–8205. doi: 10.1523/JNEUROSCI.1301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susarla BTS, Robinson MB. Rottlerin, an inhibitor of protein kinase Cδ (PKCδ), inhibits astrocytic glutamate transport activity and reduces GLAST immunoreactivity by a mechanism that appers to be PKCδ-independent. J Neurochem. 2003;86:635–645. doi: 10.1046/j.1471-4159.2003.01886.x. [DOI] [PubMed] [Google Scholar]
- Tan J, Zelenaia O, Correale D, Rothstein JD, Robinson MB. Expression of the GLT-1 subtype of Na+-dependent glutamate transporter: Pharmacological characterization and lack of regulation by protein kinase C. J Pharmacol Exp Ther. 1999;289:1600–1610. [PubMed] [Google Scholar]
- Torp R, Lekieffre D, Levy LM, Haug FM, Danbolt NC, Meldrum BS, Ottersen OP. Reduced postischemic expression of a glial glutamate transporter, GLT-1, in the rat hippocampus. Exp Brain Res. 1995;103:51–58. doi: 10.1007/BF00241964. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rolfs A, Danbolt NC, Brown RH, Hediger MA. SOD1 mutants linked to amyptrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:848. doi: 10.1038/12227. [DOI] [PubMed] [Google Scholar]
- Vanoni C, Massari S, Losa M, Carrega P, Perego C, Conforti L, Pietrini G. Increased internalisation and degradation of GLT-1 glial glutamate transporter in a cell model for familial amyotrophic lateral sclerosis (ALS) J Cell Sci. 2004;117:5417–5426. doi: 10.1242/jcs.01411. [DOI] [PubMed] [Google Scholar]
- von Zastrow M. Mechanisms regulating membrane trafficking of G protein-coupled receptors in the endocytic pathway. Life Sciences. 2003;74:217–224. doi: 10.1016/j.lfs.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Wang D, Quick MW. Trafficking of the plasma membrane g-aminobutyric acid transporter GAT1. Size and rates of an acutely recycling pool. J Biol Chem. 2005;280:18703–18709. doi: 10.1074/jbc.M500381200. [DOI] [PubMed] [Google Scholar]
- Yi J-H, Pow DV, Hazell AS. Early loss of the glutamate transporter splice-variant GLT-1v in rat cerebral cortex following lateral fluid-percusion injury. Glia. 2004;49:121–133. doi: 10.1002/glia.20099. [DOI] [PubMed] [Google Scholar]
- Zelenaia OA, Robinson MB. Degradation of glial glutamate transporter mRNAs is selectively blocked by inhibition of cellular transcription. J Neurochem. 2000;75:2252–2258. doi: 10.1046/j.1471-4159.2000.0752252.x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Sutherland ML. Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J Neurosci. 2004;24:6301–6306. doi: 10.1523/JNEUROSCI.1404-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]