

Abstract
Objective
To examine the feasibility of using blood-derived smooth muscle cells (BD-SMCs) as a target for to deliver therapeutic proteins.
Materials and Methods
Mononuclear cells (MNC) were isolated from peripheral blood. The outgrowth colonies from MNC culture were differentiated into BD-SMCs in media containing platelet-derived growth factor BB. Phenotypic characterization of BD-SMCs was assessed by immunocytochemistry. Cell proliferation, gene transfer efficiency with a retroviral vector, apoptosis, and the biological activity of the transduced gene product from the BD-SMCs were evaluated in vitro and in vivo in comparison with vascular derived SMC (VSMCs).
Results
BD-SMCs stained positive for SMC markers. No significant difference was observed between BD-SMCs and VSMCs in cell proliferation, migration, adhesiveness, and gene transfer efficiency. After BD-SMCs were transduced with a retroviral vector carrying the secreted alkaline phosphatase gene (SEAP), 174 ± 50 μg biologically active SEAP was produced per 106 cells over 24 hrs. After injecting 5×106 cells expressing SEAP intravenously into rabbits, SEAP concentration increased significantly in the circulation from 0.14 ± 0.04 μg/ml to 2.34 ± 0.16 μg/ml 3 days after cell injection (P<0.01, n=3). Circulating levels of SEAP decreased to 1.76 μg /ml one week later and remained at this level up to 8 weeks, then declined to pre-cell injection level at 12 weeks. VSMC in vivo gene expression data were equivalent.
Conclusion
BD-SMCs have similar characteristics to mature VSMCs, and can be used as a novel target for gene transfer to deliver a therapeutic protein.
Clinical relevance
Cell-based therapy strategies offer the potential to correct a wide spectrum of inherited and acquired human diseases. Translation to a clinical trial will require a detailed pre-clinical study to understand the characteristics of the isolated cells. BD-SMC are practical and effective targets for ex vivo genetic engineering. They are obtained with ease by phlebotomy, eliminating the need for surgical tissue explantation. This study tested the suitability of BD-SMC in vivo as a target for gene therapy. The outcome of the study has direct application in progenitor cell-based therapy.
Keywords: smooth muscle cells, blood derived progenitor cells, gene transfer, gene expression in vivo
Introduction
Progenitor cells outgrown from peripheral blood mononuclear cells (PB-MNCs) have been detected in adult peripheral blood of a variety of mammalian species including human 1,2, dog 3, rabbit 2,4, guinea pig 2,5, rat6, and mouse 7. Endothelial progenitor cells (EPC) isolated from peripheral blood have been differentiated into endothelial cells (EC) 8 in culture. Smooth muscle progenitor cells have also been isolated from PB-MNCs and differentiated into cells positive for smooth muscle cell (SMC) markers in vitro in the presence of platelet-derived growth factor BB (PDGF-BB) 9,10.
Ex vivo gene therapy strategies utilize autologous cells that are genetically modified prior to re-implantation. In principal, this approach offers the potential to correct a wide spectrum of inherited and acquired human diseases. Genetically modified mesenchymal cells such as fibroblasts 11 and myoblasts 12, as well as vascular SMCs 13 have been reported as vehicles to deliver recombinant proteins in vivo. However surgery is required to explant tissue or a vessel from which the cells can be enzymatically harvested.
Blood derived autologous cells hold promise as an alternative cell resource for cell therapy or as cellular vehicles that can be genetically engineered to deliver therapeutic enzymes. EPC from blood have been used to promote patency in tissue-engineered small-diameter blood vessels 14 and to deliver anticoagulants 15. Isolating cells from a peripheral blood specimen obviates the need for surgery. SMC can serve as a candidate for delivery of a therapeutic protein because the rapid proliferation and multilayer formation of SMC allow more exogenous gene product forming units per unit volume than ECs, which grow as a monolayer. SMCs secrete larger amounts of extracellular matrix that enable cells to adhere more firmly to the graft than ECs 16. This report explores the potential to use SMC differentiated from progenitor cells isolated from blood (BD-SMCs) as a target for gene therapy. The behavior of BD-SMCs in vitro was compared with mature vascular SMC (VSMC) digested from surgically excised vein. The behavior of BD-SMC is then further explored in vivo following retroviral gene transduction as compared with VSMC. No significant difference between BD-SMCs and VSMCs was found.
Materials and Methods
Cell isolation and culture
The animal protocol was approved by the University of Miami Animal Care and Use Committee. Peripheral blood (20 ml) was collected from the ear artery of male New Zealand White rabbits into tubes (Tyco Healthcare Group, Mansfield, MA) containing 15% ethylenediaminetetraacetic acid (EDTA). The mononuclear cells (PB-MNCs) were isolated by density gradient centrifugation with Histopaque-1077 (Sigma) and plated in a fibronectin (0.1mg/ml, BD Bioscience, Bedford, MA) coated well with MCDB 131 medium (Invitrogen) supplemented with 20% fetal bovine serum (FBS) and EGM™-2 SingleQuots (Cambrex, San Diego, CA) that contains growth factors (hydrocortisone, hEGF, VEGF, hFGFB, R3-IGF-1, hEGF, GA-1000, ascorbic acid, heparin and gentamicin/amphotericin-B). To promote colony formation, MNC were also cultured in the same medium described above plus 100 ng/ml stromal cell-derived factor-1α (SDF-1 α) (R&D Systems, Minneapolis MN) or 10 nM Fluvastatin (Toronto Research Chemicals Inc., ON, Canada) for 2 weeks. Media were changed 3 days after initial plating to remove the unattached cells. After cultured for 2 weeks, colony-forming cells were passed and subsequently cultured in MCDB 131 supplemented with PDGF-BB (50 ng/mL, R&D Systems) and 20% FBS to induce SMC differentiation. The resultant BD-SMCs were then cultured in Williams' medium (Invitrogen) containing 20% FBS (no more PDGF-BB) for further characterization.
To examine if the BD-SMCs are terminally differentiated, BD-SMCs were grown to confluence in αMEM supplemented with 10% FBS, 7×10−3 β-Glycerophosphate (β-GP), 0.2 mM ascorbic acid, and 10nM dexamethasone (Sigma-Aldrich) for 14 days to allow osteoblast differentiation. For EC differentiation, out-growth cells or BD-SMC were cultured in MCDB131 supplemented with VEGF and 20% FBS for 14 days.
Rabbit VSMCs were isolated from surgically harvested jugular veins by collagenase digestion as described previously 17. The VSMCs were then cultured in Williams' medium (Invitrogen) containing 20% FBS, L-glutamine (2 mM), and antibiotics. Both BD-SMC and VSMC were studied and used at passage of 2−5.
Phenotypic characterization of BD-SMC
Morphological appearance and immunohistochemistry were used to define smooth muscle cell phenotype. Primary antibodies were used against CD34 (Santa Cruz Biotechnology, Santa Cruz, Calif), α smooth muscle actin (αSMA) (Calbiochem, La Jolla, CA), calponin (Sigma, Saint Louis, Missouri), and smooth muscle heavy chain (SMHC) (Dako Corp, Carpenteria, Calif). A nonspecific isotype-matched IgG was used as a negative control. Primary antibodies were detected with the DAKO LASB 2 Kit (DAKO Corp., Carpinteria, Calif) which uses biotinylated secondary antibody, streptavidin peroxidase and its enzyme substrate diaminobenzidine tetrahydrochloride (DAB; Vector Laboratories). For determination of acetylated low density lipoprotein (Ac-LDL) uptake as a marker for endothelial cells, cells were incubated with Dil-Ac-LDL (10 μg/ml) (Biomedical Technologies Inc, Stoughton, MA) for 4 h at 37°C.
Cell proliferation assay
Both BD-SMCs and VSMCs at a similar passage were plated at a density of 1×104 cells per well in a 24-well plate and incubated 24 hours with serum-free Williams' medium for growth arrest. The cells were then cultured with Williams' media plus 20% FBS. The cell number in each well was counted continuously for 8 days, along with trypan blue staining to eliminate dead cells. Cell growth doubling time was calculated with the formula: DT=t/ln(N1/N2), where t is the time for the cells to grow from cell number counted at time 1 (N1) to cell number at time 2 (N2). Cells grow exponentially during day 3 and day 5 so the cells numbers were counted at Day 3 and day 5 for the calculation of the doubling time with t=48 hrs in the equation.
Cell migration assay
A modified Boyden-chamber assay was used to study SMC migration 18. BD-SMC and VSMC were plated on top of transwell filter inserts (Costar Corning, NY) inside a 24-well plate. After cultured in serum free medium for 48 hrs, 5% FBS was added to the medium in the lower chamber, followed with overnight incubation. The total cells and the cells that had migrated to the lower surface were counted separately to calculate the migration rate. All assays were run in triplicate.
Apoptosis assay
Cells from passage 10 were grown in complete media overnight in 6 well plates at a seeding density of 5×105 per well. The cells were washed twice with PBS and fixed in 4 % paraformaldehyde for 30 min. The fixed cells were rinsed twice with PBS and incubated in 1 μg/ml 4’-6-Diamidino-2-phenylindole (DAPI) solution (Sigma) for 30 min. Apoptotic cells were identified under fluorescence microscope by their white condensed pyknotic nuclei . The percent of apoptotic cells was calculated as apoptotic cells versus total cells.
Retroviral vectors and cell transduction
The replication-incompetent murine leukemia virus (MuLV) derived viral vectors, pseudotyped with the vesicular stomatitis virus G envelope glycoprotein (VSV-G), were used to mediate gene transfer into the SMC 19. LPCSEAP is a retroviral vector carrying the human secreted alkaline phosphatase (SEAP) gene whose expression is controlled by the cytomegalovirus (CMV) promotor. Plasmid pLPCSEAP was generated by inserting the Hind III/Xba I fragment of pSEAP2 Basic (Clontech, Mountain View, CA) into vector pLPCX (Clontech) Hind III/Avr II sites. Viral vector supernatants with titers of 1 − 5 × 106 colony forming unit per ml (cfu/ml) were generated as previously described 20 from producer cell lines 293/GPG/G1nBgSvNa and 293/GPG/pLPCSEAP for transfer of genes encoding for nuclear-localized β–galactosidase (β-gal) and SEAP, respectively. Transduction of BD-SMC and VSMC with viral vectors G1nBgSvNa and LPCSEAP was performed using the multiplicity of infection (MOI) of approximate 100. The transduced cell populations were named SMC/lacZ and SMC/SEAP, respectively, after drug selection with either G418 (Gibco BRL) or puromycin, respectively.
Cell retention assay
A pulsatile in vitro flow circuit as described 20 was used to evaluate retention of cells grown on the surface of a polytetrafluoroethylene (PTFE) graft. BD-SMCs and VSMCs, transduced with lacZ gene, were grown on the fibronectin coated PTFE graft (5-mm internal diameter) for 48 hrs, and then exposed to a pulsatile flow circuit of MCDB 131 medium for 60 minutes at 500 ml/min with a shear stress of 6.1 dyne/cm2. After this flow exposure, the graft segments were removed from the pump and rinsed with PBS. Both pre-flow and post-flow grafts were fixed with 10% formaldehyde. The grafts were stained with X-gal to visualize the seeded cells as described 16. The number of residual cells were counted by use of a microscope and expressed as cell density in cells per millimeters squared.
SEAP quantification
SEAP concentration in collected samples was evaluated using the ELISA based Great EscAPe Detection Kit (Clontech, Mountain View, CA). Samples include the conditioned medium from cell cultures and the serum from the rabbits. After centrifugation at 12,000 × g for 2 min to remove any debris, samples (15 μl) were mixed with dilution buffer (45 μl) in a 96-well plate and were incubated at 65°C for 30 min to eliminate the endogenous alkaline phosphatase activity, followed by addition of assay buffer (60 μl) and incubation for 5 min at room temperature. The disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2'-(5'-chloro)tricyclo[3.3.1.1 3,7]decan}-4-yl) phenyl phosphate (CSPD) substrate (1.25 mM, 60 μl/well) in chemiluminescence enhancer buffer was added to each sample, and incubated for 10 min at room temperature. The intensity of the chemiluminescent signal was determined by a luminometer (Veritas™ Microplate Luminometer, Sunnyvale, California).
In vivo cell infusion
Confluent cells (5. 0 ×106 cells) in culture were trypsinized, washed 2 times with phosphate-buffered saline (PBS), and then resuspended in 10 ml PBS. Rabbits were anesthetized with a mixture of ketamine (35 mg/kg) and xylazine (10 mg/kg). The ear vein was catheterized, and cells suspension was infused. Blood samples were obtained before infusion, then 3 days after cell infusion, and weekly thereafter. The serum was collected and frozen for SEAP analysis. All specimens were analyzed together at the end of the experiment.
Statistical analysis
Experimental values were expressed as mean ± standard deviation. Statistically significant differences between groups were compared using 2-tailed Student t-test. Significance was attributed to a p value of less than 0.05.
Results
Colony formation of PB MNCs
MNCs (2.3 ± 0.52 ×107, n=26) were isolated from 20 ml rabbit blood and cultured in a fibronectin coated plate. After 2-week culture of the attached MNCs (Fig 1a), 2/3 of the cultures resulted in outgrowth colonies (1−35) (Fig. 1b). The attached MNCs stained positive for progenitor markers CD 34 (Fig 1d) and CD 133 (data not shown), and are also positive for EC markers: acetylated-LDL uptake (Fig. 1e) and lectin binding (Fig. 1f).
Fig. 1. In vitro differentiation of PB-MNC into SMC-like cells.
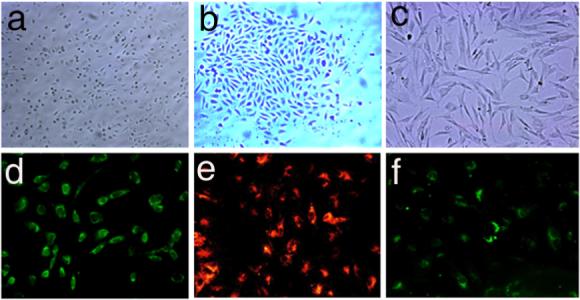
A few attached PB-MNCs grew out (a) and formed colonies after 2 weeks in culture (b). They were differentiated into SMC-like cells (c) after cultured in medium containing PDGF-BB for 14 days. The outgrowth colony cells were immunostained with FITC conjugated antibody against CD34 (d). Fluorescent Dil-ac-LDL uptake (e) and lectin binding (f) showed EPC characteristic of the PB-MNC (original magnification: ×100).
To increase the efficiency of colony formation from the PB-MNCs, MNCs were cultured in the presence of SDF-1 or Fluvastatin. Both have been reported to increase EPC survival 21-23. More attached MNCs were observed when they were cultured in the presence of SDF-1 or Fluvastatin (201 ± 55 and 172 ± 37 cells/mm2, respectively) as compared to that without treatment (118 ± 34 cells/mm2, P<0.05, n=3) (Fig. 2), but colony formation did not increase (data not shown). Neither SDF-1 nor Fluvastatin induced colony formation if the same batch of blood did not generate a colony after regular culture. Treating the no-colony-yield rabbits with granulocyte-colony stimulating factor (G-CSF) resulted in 5-fold increase in white blood cells and 1-fold increase in MNC, but did not induce colony formation.
Fig. 2. Effect of statin and SDF-1 on MNCs.

A). PB-MNCs were cultured in MNC culture medium with additional of a). control: no additive, b) SDF-1, c) Fluvastatin. The attached MNCs were stained with Dil-ac-LDL at the 10th day of culture and visualized under fluorescence microscopy. B). Relative MNC number in the 3 groups described in A. * indicates p<0.05 vs control (n=3) (original magnification: ×200).
Differentiation and Characterization of BD-SMC
To differentiate the outgrowth colony forming cells (OG-CFC) into SMCs, the cells were cultured in medium containing PDGF-BB (50ng/ml). OG-CFCs grew into a spindle-shaped appearance (Fig. 1c) with a “hill and valley” morphology suggestive of SMC after being cultured in PDGF-BB for 14 day. These cells were named blood derived SMCs (BD-SMCs).
The differentiated BD-SMCs were immunostained with antibodies against SMC markers. The original OG-CFCs were negative for α-SMA (Fig. 3a), calponin (Fig. 3e), and smooth muscle heavy chain (SMHC) (Fig. 3i). After being cultured with PDGF-BB enriched medium for 14-days, the resultant BD-SMCs stained positively for α-SMA (Fig. 3b), calponin (Fig. 3f) and MHC (Fig. 3g), similar to the control VSMCs (Fig. 3 c, g, k ) while ECs were stained negatively for all these SMC markers (Fig 3d, h, and I). The expression of α-SMA and SMHC was confirmed at the mRNA level by RT-PCR analysis of total RNA isolated from BD-SMC using EPC as a negative control. BD-SMCs were negative for CD 34 immunostaining (data not shown). They, like VSMCs, did not uptake acetylated-LDL or bind lectin (data not shown). These data indicate the EPC characteristics were lost after the cells differentiated into BD-SMCs in the presence of PDGF-BB.
Fig. 3. Characterization of BD-SMC by immunostaining.
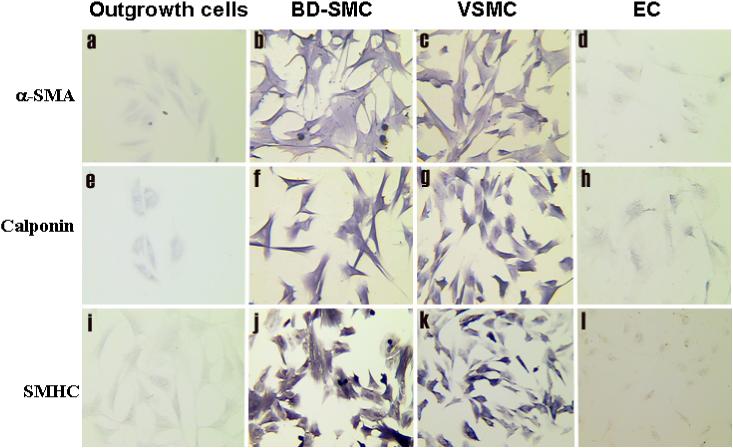
The outgrowth colony cells from MNC culture (a, e, I) and the differentiated BD-SMCs (b, f, j) were immunostained with antibodies against SMC markers: smooth muscle α-actin (α-SMA, upper panel), calponin (middle panel), and smooth muscle heavy chain (SMHC, lower panel). Vascular SMC (c, g, k) and EC (d, h, l) isolated from jugular vein were similarly stained as a positive and negative control, respectively (original magnification: ×100).
To examine if BS-SMCs can be further differentiated, BD-SMCs were cultured for 2 weeks in a medium that induces progenitor cell differentiation into either EC or osteoblasts. The resultant cells were negative for DiI-Ac-LDL uptaking and von Kossa staining, a characteristic for osteoblasts (data not shown). The data suggest BD-SMCs are terminally differentiated.
Proliferation and migration of BD-SMC
BD-SMCs had similar growth rates as VSMC of the same passage (Fig 4a). The doubling times for BD-SMC and VSMC at the early passages are 19.2 ± 1.2 hours and 19.1 ± 0.6 hours, respectively (P>0.05, n=3). SMC mobility was analyzed with a modified Boyden chamber assay. The percentage of BD-SMC migrating through a porous membrane (29 ± 3%) was not significantly different to VSMC (31 ± 4%, n=3, P=0.70) (P>0.05, n=3) (Fig. 4b).
Fig. 4. Proliferation and migration of SMC.
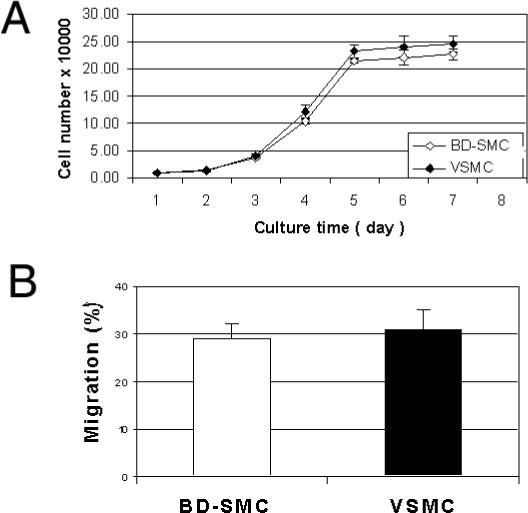
A). Cell proliferation was measured by counting the cell number daily after the culture medium was switched from serum free to 20% FBS. B) Migration in a Boyden chamber. The percentage of migration was expressed as the ratio of migrated cells on bottom side to the total number of cells on both sides. No significant difference in either proliferation or migration was observed between BD-SMC and VSMC (P>0.05, n=3).
The BD-SMCs can be passed approximately 10 passages. After that the cells gradually stop growing. The percentage of apoptotic BD-SMCs at passage 10 (11±2 %) is significantly higher than that at passage 3 (4±2%, n=3, P<0.05). The increase of apoptotic cells in BD-SMCs at different passages is similar to that of VSMCs (Fig. 5).
Fig. 5. SMC apoptosis.

A. DAPI staining was used to detect apoptotic cells (arrow points at white condensed pyknotic nuclei) after BD-SMC (a, c) or VSMC (b, d) were passed for 3 (top panel) and 10 (bottom panel) passages (original magnification: ×400).
B. The percentage of apoptotic BD-SMCs at passage 10 (11±2 %) is significantly higher than that at passage 3 (4±2%, n=3, p<0.05). The increase of apoptotic cells in BD-SMCs at different passages is similar to that of VSMCs
Cell retention
To study the adhesiveness of BD-SMCs, cells were seeded on a PTFE graft and exposed to in vitro flow for 60 minutes. Most of the BD-SMCs were retained on the surface of PTFE grafts after exposure to this flow. The cell retention rate of BD-SMCs (92.5 ±1.5) is not significantly different from that of VSMCs (89.1 ± 2.6%, n=3, P>0.05).
Transduction efficiency
To study the feasibility of using BD-SMCs as a target for genetic engineering, BD-SMCs were transduced in vitro with VSV-G pseudo-typed MuLV vector carrying the lacZ gene. The efficiency of retroviral transduction of BD-SMCs (90±2%, n=3) was similar to that of VSMC (86±1%, P>0.05, n=3) (Fig. 6A). Retroviral transduction does not affect the SMC character of BD-SMC. They were still α-actin and calponin positive after the transduction.
Fig. 6. Retroviral transduction efficiency and Stability of transgene expression.
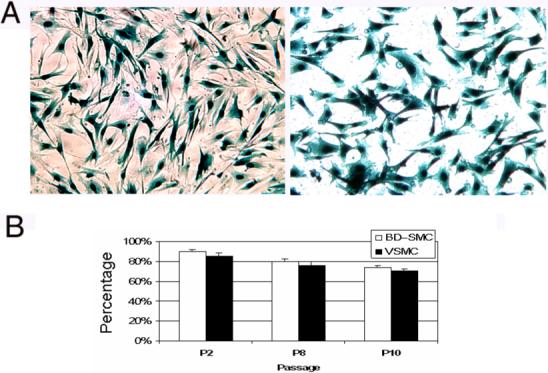
A. BD-SMCs (left) and VSMCs (right) have similar transduction efficiency with MuLV vector bearing lacZ gene. The cells with blue-stained nucleus are the ones successfully transduced at passage 3 (P3) (original magnification: ×100). B. The number of β-gal positive cells in the LacZ transduced BD-SMCs and VSMCs decreased slightly after passages, but no significantly difference between the groups was observed (P>0.05, n=3).
The stability of transgene expression was determined by examining the percentage of β-galactosidase positive cells after passages of transduced SMCs. The percentage of β-galactosidase positive cells slightly decreased from 90% at passage 2 to 80% at passage 10 (Fig. 6B). No significant difference on the transgene expression after the passages was observed between BD-SMCs and VSMC.
Gene expression in vitro and in vivo
SEAP was used as a marker to analyze the expression of transgene expression from BD-SMCs both in vitro and in vivo. The concentration of SEAP in the conditioned media of the BD-SMCs/SEAP cells was 173 ± 32 μg/106 cells/24 hrs, which is similar with that of VSMC (213 ± 16 μg/106 cells/24 hrs, p=0.70, n=3)(Fig.7A).
Fig. 7. Transgene expression in vitro and in vivo.

A. SEAP concentration in the cell culture media of BD-SMCs and VSMCs, which have been retrovirally transduced with SEAP gene. B. SEAP concentrations in blood after IV injection of BD-SMC/SEAP (open squares) and VSMC/SEAP (filled squares) into rabbits.
After intravenous infusion of the BD-SMCs/SEAP (5×106 cells per animal) into a rabbit, SEAP was detected in the blood. At 3 days post-injection, the circulating SEAP concentration increased to 2.34 ± 0.63 μg/ml from 0.14 ± 0.04 μg/ml before cell injection (P<0.01, n=3). Circulating levels of SEAP decreased to 1.76 μg /ml one week later and remained at this level up to 8 weeks, then declined to pre-cell injection SEAP level (0.12 ± 0.04 μg/ml) at 12 weeks. Similar SEAP expression pattern was detected from blood of 3 rabbits injected with VSMCs/SEAP (Fig. 7B).
Discussion
This study explored the potential of BD-SMCs as a target for gene therapy compared with VSMCs. VSMCs are effective targets for intravascular gene therapy strategies; but a surgical operation to explant a vein is required to isolate autologous VSMC for ex vivo manipulation. A practical alternative is to isolate progenitor cells from circulating blood and differentiate them into smooth muscle like cells. In humans, BD-SMC will have a significant advantage over VSMC by eliminating the need to harvest mature vessels. Simple phlebotomy and a gradient centrifugation will result in PB-MNC that then can be cultured and differentiated into BD-SMC. These procedures can be automated and completed in a closed system, eliminating contamination. In contrast, vein harvesting depends on tissue availability and requires surgery. Dissociation of cells from vein needs more hands-on manipulation, and more chance for contamination. Blood derived cells may be kept in the undifferentiated stage, which will have progenitor characters and be passed for more generations, and yield more cells for therapeutic use. In contrast, VSMC can only be passed 5−7 passages, and yield limited number of cells. In this study, the feasibility of using BD-SMCs from putative smooth muscle progenitor cells as a target cell for gene transfer and expression in vivo was demonstrated.
BD-SMCs are less traumatic to obtain compared to VSMC from a vessel that needs to be surgically explanted. Simple phlebotomy and a gradient centrifugation will result in PB MNCs that can be cultured to yield outgrowth colony forming cells (OG-CFC). However the number of outgrowth colonies reported in the literature is variable. Griese et al reported that the number of colonies ranged between 5 and 30 per 50 ml blood from rabbit 24. Simper et al reported 6 to 8 colonies per MNC sample from healthy young men and women grew out 9. None of them reported failure to obtain a colony from the blood. Ojeda-Uribe et al reported that no outgrown stromal progenitor cells were obtained from culture of PB MNCs of 17 patients with solid tumors and hematological malignancies 25. Our study shows that there was a 33% chance that no colony would grow out from the attached PB-MNCs isolated from 20 ml of rabbit blood after 2-week culture. Colony formation would be more likely if higher volumes of blood (50−100 ml) could be drawn, as is possible in the human being.
Colony formation could be affected by many factors. Age, sex, health situation, diet, habits, and menstrual status impact the number of circulating progenitor cells. Culture medium (growth factors, composition) will also influence BD-SMC yield. It has been reported that statins and SDF-1 increase the number of adherent EPCs 21-23. In the present study, the attached MNCs did increase 1.7 fold and 1.5 fold in the presence of SDF-1 or statin, respectively in comparison with untreated MNC group (Fig 2). However, adding SDF-1 or Fluvastatin into the culture medium did not induce colony formation from rabbit blood that did not initially have outgrowth MNC colonies. Treating the animal with G-CSF prior to phlebotomy resulted in an increase in white blood cells and MNC, but failed to yield colony formation. G-CSF treatment may increase only the hematopoietic progenitor cells, but not the number of mesenchymal stem cells in blood. Interestingly, the rabbits that initially did not yield any colony from their MNC culture generated more than 30 colonies after they were housed at our animal facility for more than one year (data not shown). Why some rabbits failed to yield out growth MNC colonies from a phlebotomy specimen is unknown. The composition of cells recovered from the buffy coat after the gradient centrifugation may vary each time. It is not clear what kind of progenitor cells out-growing from the attached cells to form the colony. Anecdotally a blood sample from a dog that did not yield colonies following PB MNC culture resulted in approximately 100 colonies from 50 ml blood one month after implantation of a stent–graft into its abdominal aorta. Perhaps arterial injury may induce the mobilization of these MNC progenitor cells into the circulation. Study of the mechanism of MNC mobilization will benefit the therapeutic model proposed within our project, and may provide insight into disease evolution and repair of injury.
OG-CFCs have multi-differentiation potential. Depending on the culture conditions, they are able to be differentiated into osteoblasts, reticular cells, lipocytes, chondrocytes, and fibroblasts 2,6,26. OG-CFCs were differentiated into SMC by culturing them in medium containing PDGF-BB. OG-CFCs initially do not express classic SMC markers but do express EPC makers: CD34, LDL-uptaking, and lectin binding (Fig 1d, e, f), which are absent from mature SMC. After exposure to PDGF-BB for two weeks, OG-CFCs expressed the classic SMC markers (Fig 3) and no longer expressed the endothelial markers (LDL-uptake and CD34, data not shown). This indicates they differentiated into mature SMC.
Krebsbach et al reported that bone marrow stromal cells used as vehicles for gene delivery formed new bone in an in vivo osteogenic assay 27. In the present study, culturing BD-SMC with either osteoblasts or EC inducing medium for two weeks did not alter BD-SMCs phenotype. The resultant BD-SMCs did not stain for the markers of osteoblasts or EC but remained positive for SMC markers. This indicates that the BD-SMCs are stable and are terminally differentiated cells.
The growth profile of differentiated BD-SMCs is similar to VSMC. They proliferate readily in vitro (Fig 4a). The doubling time of BD-SMC is about 19 hrs, and they were able to be extended to 10 passages in this study. There are approximately 3 − 4 doubling times during the interval of 3−4 days between two passages. This agrees with previous report that smooth muscle outgrowth cells had a capacity for extended growth in culture over 40 population doublings 9.
The adhesiveness of BD-SMC is also similar to VSMC. This property is important for cell seeding on grafts to deliver therapeutic proteins. Our previously studies 16,20 demonstrated that VSMC can improve endothelial cell retention on polytetrafluoroethylene grafts in vivo. A stent-graft suffused with engineered VSMC is a potential vehicle for therapeutic gene delivery into the blood stream 13.
The differentiated BD-SMCs can be efficiently transduced with retroviral vectors (Fig 6a). Production of the transgene from BD-SMC is high, and similar to production of the same transgene from VSMC (Fig 7a). The expression of transduced genes from BD-SMCs is stable in vitro for at least 10 passages (Fig 6b). The in vivo expression of the transduced marker gene SEAP from BD-SMCs lasted 8 weeks after the transduced cells were injected IV into the circulation. These data demonstrated that the engineered BD-SMCs continuously secreted the product of transduced gene into the circulation. Subsequently, circulating SEAP levels declined to a background level at the 12th week of the study (Fig. 7b). The gene expression pattern of BD-SMCs is similar to that of VSMCs both in vitro and in vivo. These results agree with the data reported by Clowes' group stating that gene expression from genetically engineered VSMC peaks at day 10 and returns to background levels 1 month after cell implantation 28. The reasons for the lack of persistent high level SEAP secretion are unknown. Potential causes include clearance of the injected cells, inactivation of the viral promoter 29,30 and development of anti-SEAP antibodies, as in the case of factor VIII expression in canine 31.
BD-SMC can be used for graft engineering through cell seeding on a graft to achieve sustained drug delivery. They can also be directly implanted subcutaneously to act as a depot for designed protein expression. Engineered SMC can also be injected IV as reported in this study. A weakness of IV infusion of cells is the inability to retrieve the injected cells. The seeded SMC on a graft could potentially compromise small diameter lumena (<5 mm diameter). For these smaller vessels, transduction of SMC with genes that can regulate the proliferation of SMC could be a way to control the growth of implanted SMC. We have demonstrated that seeding eNOS gene transduced SMC on PTFE graft had minimal neointima growth32. SMC could be transduced with a suicide gene (e.g. thymidine kinase gene33) before injection, so that the engineered cells inside the body can be eliminated by administration of a drug (ganciclovir).
In conclusion, a range of MNC outgrowth colonies can be isolated from peripheral blood. A third of the time no outgrowth colonies were isolated. SMCs differentiated from these putative smooth muscle progenitor cells have similar characteristics as mature SMC isolated from a vessel. They can continuously secrete retrovirally engineered SEAP in vivo for up to 8 weeks. They can be used as a novel cell target for gene transfer to deliver therapeutic proteins without having to do surgery to harvest donor tissue.
Acknowledgement
This study was supported by NIH R21 HL076356 (HY), VA Merit Review Grant #2539.03 (DE) and American Heart Association Fellowship Award 0525521B (ZY).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paul J. Establishment of permanent cell strains from human adult peripheral blood. Nature. 1958;182:808. doi: 10.1038/182808a0. [DOI] [PubMed] [Google Scholar]
- 2.Kuznetsov SA, Mankani MH, Gronthos S, Satomura K, Bianco P, Robey PG. Circulating skeletal stem cells. J Cell Biol. 2001;153:1133–1140. doi: 10.1083/jcb.153.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huss R, Lange C, Weissinger EM, Kolb HJ, Thalmeier K. Evidence of peripheral blood-derived, plastic-adherent cd34(-/low) hematopoietic stem cell clones with mesenchymal stem cell characteristics. Stem Cells. 2000;18:252–260. doi: 10.1634/stemcells.18-4-252. [DOI] [PubMed] [Google Scholar]
- 4.Wan C, He Q, Li G. Allogenic peripheral blood derived mesenchymal stem cells (mscs) enhance bone regeneration in rabbit ulna critical-sized bone defect model. J Orthop Res. 2006;24:610–618. doi: 10.1002/jor.20119. [DOI] [PubMed] [Google Scholar]
- 5.Luria EA, Panasyuk AF, Friedenstein AY. Fibroblast colony formation from monolayer cultures of blood cells. Transfusion. 1971;11:345–349. doi: 10.1111/j.1537-2995.1971.tb04426.x. [DOI] [PubMed] [Google Scholar]
- 6.Wu GD, Nolta JA, Jin YS, Barr ML, Yu H, Starnes VA, et al. Migration of mesenchymal stem cells to heart allografts during chronic rejection. Transplantation. 2003;75:679–685. doi: 10.1097/01.TP.0000048488.35010.95. [DOI] [PubMed] [Google Scholar]
- 7.Piersma AH, Ploemacher RE, Brockbank KG, Nikkels PG, Ottenheim CP. Migration of fibroblastoid stromal cells in murine blood. Cell Tissue Kinet. 1985;18:589–595. doi: 10.1111/j.1365-2184.1985.tb00702.x. [DOI] [PubMed] [Google Scholar]
- 8.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–966. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 9.Simper D, Stalboerger PG, Panetta CJ, Wang S, Caplice NM. Smooth muscle progenitor cells in human blood. Circulation. 2002;106:1199–1204. doi: 10.1161/01.cir.0000031525.61826.a8. [DOI] [PubMed] [Google Scholar]
- 10.Deb A, Skelding KA, Wang S, Reeder M, Simper D, Caplice NM. Integrin profile and in vivo homing of human smooth muscle progenitor cells. Circulation. 2004;110:2673–2677. doi: 10.1161/01.CIR.0000139842.15651.B2. [DOI] [PubMed] [Google Scholar]
- 11.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector [see comments]. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 12.Yao SN, Smith KJ, Kurachi K. Primary myoblast-mediated gene transfer: Persistent expression of human factor ix in mice. Gene Ther. 1994;1:99–107. [PubMed] [Google Scholar]
- 13.Eton D, Yu H, Wang Y, Raines J, Striker GE, Livingstone A. Endograft technology: A delivery vehicle for intravascular gene therapy. J Vasc Surg. 2004;39:1066–1073. doi: 10.1016/j.jvs.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 14.Kaushal S, Amiel GE, Guleserian KJ, Shapira OM, Perry T, Sutherland FW, et al. Functional small-diameter neovessels created using endothelial progenitor cells expanded ex vivo. Nat Med. 2001;7:1035–1040. doi: 10.1038/nm0901-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griese DP, Achatz S, Batzlsperger CA, Strauch UG, Grumbeck B, Weil J, et al. Vascular gene delivery of anticoagulants by transplantation of retrovirally-transduced endothelial progenitor cells. Cardiovasc Res. 2003;58:469–477. doi: 10.1016/s0008-6363(03)00266-9. [DOI] [PubMed] [Google Scholar]
- 16.Yu H, Dai W, Yang Z, Kirkman P, Weaver FA, Eton D, et al. Smooth muscle cells improve endothelial cell retention on polytetrafluoroethylene grafts in vivo. J Vasc Surg. 2003;38:557–563. doi: 10.1016/s0741-5214(03)00334-3. [DOI] [PubMed] [Google Scholar]
- 17.Eton D, Terramani TT, Wang Y, Takahashi AM, Nigro JJ, Tang L, et al. Genetic engineering of stent grafts with a highly efficient pseudotyped retroviral vector. J Vasc Surg. 1999;29:863–873. doi: 10.1016/s0741-5214(99)70214-4. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Eton D, Zheng F, Livingstone AS, Yu H. Effect of tissue plasminogen activator on vascular smooth muscle cells. J Vasc Surg. 2005;42:532–538. doi: 10.1016/j.jvs.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 19.Yu H, Eton D, Wang Y, Kumar S, Tang L, Terramani T, et al. High efficiency in vitro gene transfer into vascular tissues using a pseudotyped retroviral vector without pseudotransduction. Gene Ther. 1999;6:1876–1883. doi: 10.1038/sj.gt.3301019. [DOI] [PubMed] [Google Scholar]
- 20.Yu H, Wang Y, Eton D, Rowe VL, Terramani TT, Cramer DV, et al. Dual cell seeding and the use of zymogen tissue plasminogen activator to improve cell retention on polytetrafluoroethylene grafts. J Vasc Surg. 2001;34:337–343. doi: 10.1067/mva.2001.114817. [DOI] [PubMed] [Google Scholar]
- 21.Lataillade J-J, Clay D, Dupuy C, Rigal S, Jasmin C, Bourin P, et al. Chemokine sdf-1 enhances circulating cd34+ cell proliferation in synergy with cytokines: Possible role in progenitor survival. Blood. 2000;95:756–768. [PubMed] [Google Scholar]
- 22.De Falco E, Porcelli D, Torella AR, Straino S, Iachininoto MG, Orlandi A, et al. Sdf-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood. 2004;104:3472–3482. doi: 10.1182/blood-2003-12-4423. [DOI] [PubMed] [Google Scholar]
- 23.Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, et al. Hmg-coa reductase inhibitors (statins) increase endothelial progenitor cells via the pi 3-kinase/akt pathway. J Clin Invest. 2001;108:391–397. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griese DP, Ehsan A, Melo LG, Kong D, Zhang L, Mann MJ, et al. Isolation and transplantation of autologous circulating endothelial cells into denuded vessels and prosthetic grafts: Implications for cell-based vascular therapy. Circulation. 2003;108:2710–2715. doi: 10.1161/01.CIR.0000096490.16596.A6. [DOI] [PubMed] [Google Scholar]
- 25.Ojeda-Uribe M, Brunot A, Lenat A, Legros M. Failure to detect spindle-shaped fibroblastoid cell progenitors in pbpc collections. Acta Haematol. 1993;90:139–143. doi: 10.1159/000204395. [DOI] [PubMed] [Google Scholar]
- 26.Tondreau T, Meuleman N, Delforge A, Dejeneffe M, Leroy R, Massy M, et al. Mesenchymal stem cells derived from cd133-positive cells in mobilized peripheral blood and cord blood: Proliferation, oct4 expression, and plasticity. Stem Cells. 2005;23:1105–1112. doi: 10.1634/stemcells.2004-0330. [DOI] [PubMed] [Google Scholar]
- 27.Krebsbach PH, Zhang K, Malik AK, Kurachi K. Bone marrow stromal cells as a genetic platform for systemic delivery of therapeutic proteins in vivo: Human factor ix model. J Gene Med. 2003;5:11–17. doi: 10.1002/jgm.292. [DOI] [PubMed] [Google Scholar]
- 28.Osborne WR, Ramesh N, Lau S, Clowes MM, Dale DC, Clowes AW. Gene therapy for long-term expression of erythropoietin in rats. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8055–8058. doi: 10.1073/pnas.92.17.8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer TD, Rosman GJ, Osborne WR, Miller AD. Genetically modified skin fibroblasts persist long after transplantation but gradually inactivate introduced genes. Proc Natl Acad Sci U S A. 1991;88:1330–1334. doi: 10.1073/pnas.88.4.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kay MA, Baley P, Rothenberg S, Leland F, Fleming L, Ponder KP, et al. Expression of human alpha 1-antitrypsin in dogs after autologous transplantation of retroviral transduced hepatocytes. Proc Natl Acad Sci U S A. 1992;89:89–93. doi: 10.1073/pnas.89.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallo-Penn AM, Shirley PS, Andrews JL, Tinlin S, Webster S, Cameron C, et al. Systemic delivery of an adenoviral vector encoding canine factor viii results in short-term phenotypic correction, inhibitor development, and biphasic liver toxicity in hemophilia a dogs. Blood. 2001;97:107–113. doi: 10.1182/blood.v97.1.107. [DOI] [PubMed] [Google Scholar]
- 32.Yu H, Dai W, Yang Z, Romaguera RL, Kirkman P, Rowe VL. Neointimal hyperplasia on a cell-seeded ptfe graft is promoted by transfer of tissue plasminogen activator gene and inhibited by transfer of nitric oxide synthase gene. J Vasc Surg. 2005;41:122–129. doi: 10.1016/j.jvs.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 33.Chang MW, Ohno T, Gordon D, Lu MM, Nabel GJ, Nabel EG, et al. Adenovirus-mediated transfer of the herpes simplex virus thymidine kinase gene inhibits vascular smooth muscle cell proliferation and neointima formation following balloon angioplasty of the rat carotid artery. Mol Med. 1995;1:172–181. [PMC free article] [PubMed] [Google Scholar]