

Abstract
Recent studies have demonstrated nuclear export by papillomavirus E1 proteins, but the requisite export sequence(s) for bovine papillomavirus (BPV) E1 were not defined. In this report we identify three functional nuclear export sequences (NES) present in BPV E1, with NES2 being the strongest in reporter assays. Nuclear localization of BPV1 E1 was modulated by over or under expression of the Crm1, the major cellular exportin, and export was strongly reduced by the Crm1 inhibitor, Leptomycin B, indicating that E1 export occurs primarily through a Crm1-dependent process. Consistent with the in vivo functional results, E1 bound Crm1 in an in vitro pulldown assays. In addition, sumoylated E1 bound Crm1 more effectively than unmodified E1, suggesting that E1 export may be regulated by SUMO modification. Lastly, an E1 NES2 mutant accumulated in the nucleus to a greater extent than wildtype E1, yet was defective for viral origin replication, implying that nucleocytoplasmic shuttling may be required to maintain E1 in a replication competent state.
Keywords: Nuclear export sequence, NES, CRM1, E1, sumoylation, NLS
Introduction
The papillomavirus (PV) E1 protein (PV-E1), one of the estimated eight different proteins produced by papillomaviruses during infection, constitutes the most promising target for the development of anti-papillomavirus therapies owing to its well characterized DNA helicase activity, essential for the maintenance and replication of the viral DNA. Comparative analyses among the PV-E1 proteins of numerous viral strains revealed the presence of three main domains within PV-E1, namely an N-terminal region of poorly defined activity, a spacer domain, and a C-terminal domain showing the typical features associated to DNA helicases, including DNA binding, ATP binding, and oligomerization (Wilson et al., 2002). Numerous reports have characterized the three- dimensional structure of the DNA binding domain for two PV-E1 proteins, those produced by the human papillomavirus 18 (HPV18-E1) and the bovine papillomavirus E1 (BPV-E1) (Auster and Joshua-Tor, 2004; Enemark et al., 2000; Enemark and Joshua-Tor, 2006; Enemark, Stenlund, and Joshua-Tor, 2002). Such studies have produced significant insights into the molecular characteristics mediating the enzymatic activity of these proteins. However, several aspects of the molecular mechanisms regulating the function of this viral protein remain enigmatic. Perhaps the most intriguing among these are the mechanisms governing the cellular localization of PV-E1. PV-E1 exerts its helicase role in the cell nucleus and therefore it must contain specific amino acid sequences that direct it to this cellular compartment. The presence of such sequences, known as Nuclear Localization Signals (NLSs), has been well characterized in both HPV-E1 and BPV-E1 (Leng and Wilson, 1994; Lentz et al., 1993; Yu et al., 2007), and a recent study has determined that the nuclear traffic of BPV-E1 is mediated by the classic nuclear import pathway involving the assembly of an importin α/β complex with the NLS of BPV-E1 (Bian et al., 2007). Thus, significant knowledge of the molecular mechanisms mediating the nuclear import of PV-E1 proteins is currently available.
Intriguingly, however, a recent report has indicated the presence in HPV11-E1 of a signal, known as Nuclear Export Signal (NES), that directs the protein out of the nucleus (Deng et al., 2004). The indicated NES appeared to trigger the efficient nuclear export of E1 mediated by the cellular nuclear export factor CRM1, thus preventing the nuclear accumulation of E1. Importantly, phosphorylation of residues located near the NES neutralized its activity and allowed nuclear accumulation of E1. The kinase responsible for this phosphorylation event was identified as a cyclin-dependent kinase (CDK), linking the cellular localization of the HPV11-E1 protein with the cell cycle of its host cell. Similarly, the presence of a NES in BPV-E1 has also been postulated in a recent study, although the specific location of this signal in BPV-E1 was not identified (Hsu, Mechali, and Bonne-Andrea, 2007). According to this study, and in sharp contrast with the NES identified in HPV11-E1, the putative BPV-E1 NES appears constitutively inactive, requiring the phosphorylation of Ser 283, also by a CDK, to become active. The presence of a NES in both HPV11-E1 and BPV-E1 suggests that nuclear shuttling may constitute an evolutionary conserved mechanism that plays an important role in maintaining and/or regulating the activity of PV-E1.
In this study, we have verified that the BPV-E1 protein is exported from the nucleus by an active mechanism that involves the nuclear export factor CRM1. Furthermore, we have mapped three nuclear export signals within BPV-E1, the strongest of which is fully dependent on CRM1 and is constitutively active both in reporter chimeric constructs and in the full-length BPV-E1 protein. The other two NES elements exert limited activity under the conditions tested, but appear to function by means of cellular nuclear export factors other than CRM1, therefore providing BPV-E1 with the ability to utilize alternative nuclear export pathways. Our data also indicates that nuclear shuttling may play an important role in keeping BPV-E1 in an active stage, as mutations affecting exclusively the activity of the strongest NES render the protein virtually unable to trigger the replication of a plasmid containing a viral origin of replication. Finally, we also present data indicating that sumoylation may play a regulatory role in nuclear shuttling by increasing BPV-E1's affinity toward CRM1, and redirect previous conclusions on the role of sumoylation for the nuclear localization of BPV-E1.
Results
The bovine papillomavirus E1 protein is actively exported from the cell nucleus
Like most DNA helicases, the papillomavirus E1 protein is targeted to the nucleus owing to the presence of a nuclear localization signal (NLS) on its primary structure (Leng and Wilson, 1994; Lentz et al., 1993; Yu et al., 2007). Interestingly, a recent study by Deng et al. reported the presence of a nuclear export signal (NES) in the human papillomavirus 11 (HPV11) E1 protein and postulated that phosphorylation of a nearby serine residue by cell-cycle dependent kinases regulated the accessibility of the NES, therefore linking the cellular localization of the HPV11 E1 viral helicase with the progression through the cell cycle of the host cell (Deng et al., 2004). Although in that study the authors indicated the absence of an NES in the equivalent region of the bovine papillomavirus E1 protein (BPV E1), we hypothesized that the function of BPV E1 could also be regulated by nuclear export potentially mediated by still unidentified NESs located elsewhere in the protein.
To verify if BPV E1 is actively exported from the cell nucleus, HeLa cells transfected with the pEGFP-E1 plasmid (which codes for a EGFP fusion of BPV E1, EGFP-E1) were partially permeabilized with digitonin, washed several times to eliminate both any residual cytosolic EGFP-E1 and the cellular soluble factors required for nucleocytoplasmic traffic, and incubated in transport buffer with or without reticulocyte extract. After incubation, the resulting supernatants were collected, immunoprecipitated with rabbit serum against GFP, and analyzed by immunoblotting using monoclonal antibodies to GFP. Incubation of the permeabilized HeLa cells with transport buffer alone did not allow the export of EGFP-E1 out of the nucleus as no EGFP-E1 was detected in the resulting supernatant (Fig. 1, lanes 1 and 2). In contrast, incubation of the permeabilized cells in transport buffer supplemented with reticulocyte extract resulted in the export of EGFP-E1 out of the nucleus, as EGFP-E1 was readily detected in the resulting supernatant and a significant decrease in the fraction of EGFP-E1 remaining in the nucleus was also observed (Fig. 1, lanes 3 and 4). Interestingly, under these conditions a high molecular weight form of EGFP-E1 was also detected in the supernatant (Fig. 1, asterisk); the identity of this form has not been established, but could represent sumoylated E1 (see below). Since reticulocyte extracts are rich in Ran, importins, and exportins, therefore providing an optimal source of nucleocytoplasmic traffic factors, the above data indicate that EGFP-E1 is actively transported from the nucleus as its export is dependent on cellular factors. Therefore, BPV E1, like HPV11 E1, must contain signals that allow its nuclear export.
Fig. 1. BPV E1 is actively exported out of the nucleus.
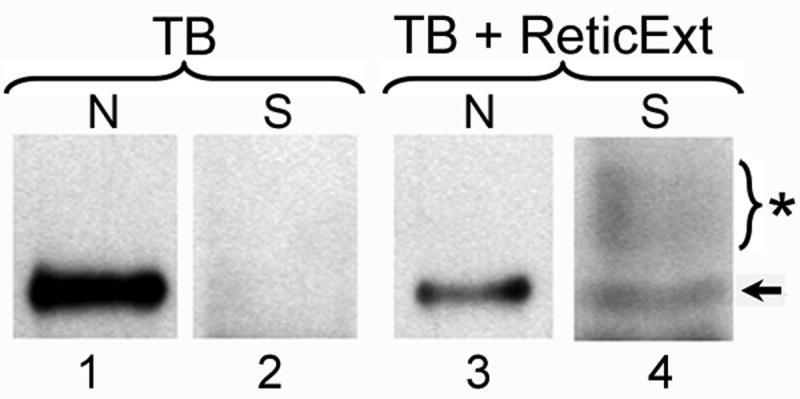
HeLa cells were transfected with an expression plasmid coding for an EGFP fusion of BPV E1 (EGFP-E1). 24 h post-transfection, the cells were partially permeabilized with digitonin, washed several times, and incubated in 1× Transport Buffer alone (TB, lanes 1 and 2) or 1× Transport Buffer supplemented with 20% rabbit reticulocyte extract (TB+ReticExt, lanes 3 and 4), for 1 hour at 37oC. The resulting supernatant (S) was collected and immunoprecipitated with anti-GFP antibodies (lanes 2 and 4), whereas the residual nuclei (N) of the permeabilized cells were solubilized and lysed in 2× Sample Buffer (lanes 1 and 3). Both, the immunoprecipitated samples and the solubilized nuclei were analyzed by SDS-PAGE and immunoblotting using anti-GFP antibodies. Arrow: EGFP-E1. Star: High molecular weight form(s) of EGFP-E1 (likely to correspond to sumoylated forms).
The nuclear export of BPV E1 is mainly regulated by CRM1
To identify putative sequences that could mediate the nuclear export of BPV E1, we scanned the primary structure of BPV E1 for the presence of consensus motifs known to serve as NESs for other proteins. We identified two leucine-rich NESs of the L(X2-3)L(X2-3)LXL type (where L represents leucine or any other large hydrophobic residue, namely valine, isoleucine, phenylalanine, or methionine), spanning residues 164-172 (NES1), and residues 409-418 (NES2) of BPV E1. Additionally, we also found a sequence exhibiting the L(X8)L(X3)L motif (where L specifically indicates leucine), a type of NES recently identified in the Cdk inhibitor p27 (Connor et al., 2003), spanning residues 140-153 (NES3) of BPV E1. Finally, we also identified another sequence related to the same L(X8)L(X3)L motif but containing methionine instead of leucine in the first position, spanning residues 262-275 (NES4) (Fig. 2A). All of the putative NES motifs indicated above are expected to mediate nuclear export by providing an interacting signal with the nuclear export receptor CRM1/Exportin 1. Therefore, we tested the ability of CRM1 to interact with BPV E1 using in vitro translated 35S-labeled BPV E1 and bacterially expressed purified GST-CRM1. The 35S-BPV E1 did not exhibit any significant binding to glutathione-sepharose beads when incubated in the presence of GST. However, when GST-CRM1 was used as bait, significant amounts of 35S-BPV E1 were co-purified with the beads (Fig. 2B). Furthermore, phosphordensitometry analysis indicated that the amount of 35S-BPV E1 recovered on the beads increased proportionally to the amount of GST-CRM1 loaded in the beads, therefore indicating a specific interaction between BPV E1 and CRM1 (Fig. 2C).
Fig. 2. BPV E1 contains four putative CRM1-dependent Nuclear Export Signals (NESs) and binds to CRM1.

(A) The presence in BPV E1 of consensus motifs known to serve as NESs for other proteins. The numbers to the left indicate the position of the first amino acid residue depicted in the primary sequence of the indicated protein. Similar motifs found in other proteins and the known consensus motifs are presented for comparison. The name given to each NES identified in BPV E1 is indicated in parenthesis. (B) Pull-down assays performed with purified GST or GST-CRM1 and in vitro translated BPV E1. The pull-downs were performed using different amounts of bait, as indicated on top of each lane. The samples were analyzed by SDS-PAGE and quantified by phosphordensitometry. (C) Quantitative data indicating the percentage of input BPV E1 that remained bound to the beads after the pull-down procedure presented in B.
Since the in vitro binding assay suggested that CRM1 could act as an exportin for the nuclear export of BPV E1, we sought to determine if CRM1 affected the cellular localization of BPV E1. To this end we employed Leptomycin B (LMB), a fungal cytotoxin that acts as an inhibitor of CRM1-mediated nuclear export by disrupting the interaction of CRM1 with its cargo through the modification of a cysteine residue located in the cargo binding pocket in CRM1 (Kudo et al., 1999; Wolff, Sanglier, and Wang, 1997). CHO-K1 cells transfected with pEGFP-E1 were incubated for 6 hours in the presence of cyclohexamide with or without LMB, and the cellular localization of EGFP-E1 was determined by fluorescence microscopy. As a control for these experiments we used the plasmid pRev1.4(NES3)GFP (Henderson and Eleftheriou, 2000), which codes for a fusion of the HIV-Rev protein with GFP (Rev-GFP). In this plasmid, the NES in Rev was inactivated by alanine substitutions of the central leucines, and an active copy (free of mutations) of the NES was inserted in the junction between Rev and GFP. The Rev-GFP fusion protein produced upon transfection of the plasmid in mammalian cell lines shuttles continuously between the nucleus and the cytosol, owing to the strong NLS and NES present on the Rev sequence, and exhibits a characteristic cytoplasmic and nucleolar localization (Fig. 3A, untreated), this last due to the ability of Rev to interact with nucleolar components. Furthermore, the nuclear import of Rev-GFP can be significantly decreased by treatment with actinomycin D (ActD)(Fig. 3A, +ActD), and its nuclear export is fully abolished by LMB (Fig. 3A, +ActD+LMB). In several parallel transfections performed with either pRev1.4(NES3)GFP or pEGFP-E1 we observed that LMB treatment produced a complete re-localization of Rev-GFP to the nucleus, more specifically to the nucleolus, and LMB treatment produced a substantial but incomplete redistribution of EGFP-E1 to the nucleus (Fig. 3B). Biochemical fractionation experiments (Fig. 3C.) revealed that there was a low molecular weight form derived from the EGFP-E1 that was present in the cytoplasmic fraction and likely accounted for the residual cytoplasmic fluorescence. These results suggest that E1 export is mediated primarily through a CRM1-dependent pathway.
Fig. 3. LMB significantly decreases the nuclear export of BPV E1.
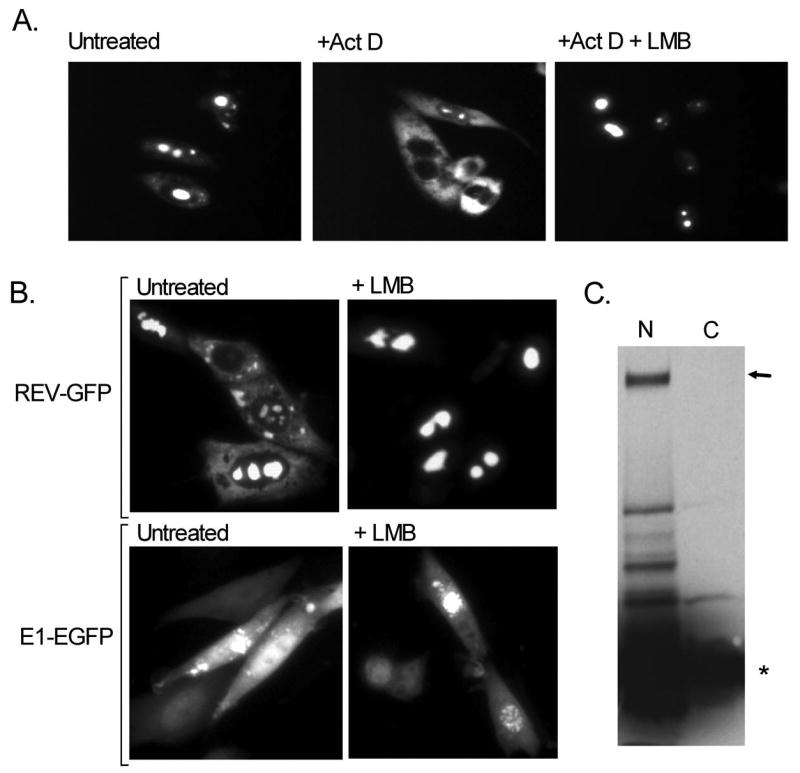
CHO-K1 cells were transfected with pEGFP-E1 or with the control plasmid pRev1.4(NES3)GFP, and 16 h post-transfection the cells were incubated for 6 hours in the presence of cyclohexamide with or without Leptomycin B (LMB) and actinomycin D (Act D), and the cellular localization of the EGFP fusion proteins produced was determined by fluorescence microscopy or biochemical fractionation. (A) Cellular localization of the mutant REV-GFP protein encoded by plasmid pRev1.4(NES3)GFP with GFP (Rev-GFP) upon treatment with Act D and LMB. (B) Effect of LMB treatment on the cellular localization of E1-GFP, as compared with the effect of LMB treatment on the cellular localization of REV-GFP. (C) Biochemical fractionation of LMB-treated CHO cells into nuclear (N) and cytoplasmic (C) fractions. Both fractions were resolved by SDS-PAGE and immunoblotted with anti-GFP. The arrow indicates full-length EGFP-E1, and the star indicates a low molecular weight species present in both the nuclear and cytoplasmic fractions. This low molecular weight species is slightly larger than EGFP and is likely EGFP with a small portion of E1.
If CRM1 is the primary exporter of E1, then fluctuations in the cellular amount of CRM1 should exert substantial effects on the nuclear export of BPV E1. This could be tested by cotransfecting mammalian cells with pEGFP-E1 and pcDNA3-CRM1, a previously reported plasmid construct that over-expressions CRM1 and leads to a concomitant cytoplasmic redistribution of nuclear proteins containing CRM1-dependent NESs (Boyle et al., 1999). Alternatively, transfecting a plasmid coding for a hairpin-forming sequence complementary to sequences in CRM1 should induce an RNAi response against CRM1 leading to decreased CRM1 levels and a subsequent increase in nuclear accumulation of proteins containing CRM1-dependent NESs. To develop a plasmid capable of inducing RNAi against CRM1 upon transfection, we selected two sequences derived from the human CRM1 gene transcript (accession number NM_003400), and developed oligonucleotides containing inverted repeats of the selected sequences. The inverted repeats were designed to produce hairpins with a 19-nucleotide stem and a 9-nucleotide loop (Supplementary Fig. 1A). The oligonucleotides were cloned into the pSilencer™ 4.1-CMV puro vector (Ambion), producing the plasmids pSIL-CRM1A (containing a sequence derived from the junction between exons 2 and 3 in CRM1) and pSIL-CRM1B (containing a sequence derived from the 3′ end of exon 3 in CRM1) (Supplementary Fig. 1A). As a control, we also generated a plasmid containing a scrambled sequence forming a 19-nucleotide stem lacking significant similarity to any known transcript, denominated pSIL-Scrambled. To evaluate the effectiveness of these constructs in inducing RNAi against CRM1 and therefore down-regulating the cellular levels of CRM1, CHO-K1 cells were co-transfected with the pSilencer constructs and the pRev1.4(NES3)GFP plasmid, and the cellular localization of Rev-GFP was evaluated in the transfected cells approximately 60 h post-transfection. This relatively long incubation time after transfection was essential to induce a significant decrease in the cellular amount of CRM1 due to the previously reported high stability of this protein (Fornerod et al., 1997). The large majority of the CHO-K1 cells transfected with pRev1.4(NES3)GFP alone or in conjunction with the pSIL-CRM1B or the pSIL-Scrambled plasmids exhibited a predominantly cytosolic distribution of Rev-GFP upon ActD treatment (Supplementary Fig. 1B, +ActD, +ActD+pSIL-CRM1B, +ActD+pSIL-Scrambled, and 1C). In contrast, the large majority of the cells co-transfected with pSIL-CRM1A exhibited a mostly nucleolar distribution of Rev-GFP, even upon treatment with ActD (Supplementary Fig. 1B, +ActD+pSIL-CRM1A, and 1C), therefore indicating that the pSIL-CRM1A effectively induced an RNAi response directed against CRM1 which resulted in diminished nuclear export of CRM1-dependent cargo.
Next, we tested the ability of CRM1 to induce changes in the cellular distribution of E1-EGFP upon conditions of over and under-expression. Initial co-transfection experiments in CHO-K1 were largely unsuccessful due to the toxic effects induced by the over-expression of BPV E1. This intrinsic feature of BPV E1 was implied by the observation that by 60 h post-transfection the only cells displaying significant expression of BPV E1 also exhibited a largely disrupted morphology characterized by a round shape, lack of cytoplasmic extensions, and an apparent lack of cytoplasm as the nucleus seemed to occupy the full volume of the cell. To overcome this difficulty, we opted for a sequential transfection strategy. Briefly, HeLa cells were transfected with either pcDNA3-CRM1, pSIL-CRM1A, or pSIL-Scrambled, and maintained in culture for 36 h, the cells transfected with the pSilencer-based plasmids being maintained in the presence of puromycin to select for transfected cells. Then, the cells were re-transfected with the pEGFP-E1 or pREV1.4(NES3)GFP plasmids, treated with cyclohexamide (CHX) and/or ActD, fixed, and analyzed by fluorescence microscopy 24 h after the second transfection. In these experiments we consistently observed that over-expression of CRM1 led to a homogeneous distribution of BPV E1 throughout the cell (Fig. 4, +pcDNA3-CRM1), making the nucleus unapparent. In sharp contrast, under-expression of CRM1 consistently led to a substantial decrease in the cytosolic signal observed, and a significant increase in the nuclear signal produced (Fig. 4, +pSIL-CRM1A+ActD). Both profiles were substantially different from the normal distribution of E1-EGFP observed in cells co-transfected with the scrambled siRNA, in which E1-EGFP appeared predominantly nuclear but exhibited also significant cytosolic distribution (Fig. 4, +pSIL-SCRAM+ActD). The efficacy of this approach to induce over- and under-expression of CRM1 was indicated by the cellular distribution of Rev-GFP in the cells co-transfected with pREV1.4(NES3)GFP. As expected, Rev-GFP appeared almost exclusively cytoplasmic in cells over-expressing CRM1 even in the absence of ActD (Fig. 4, +pcDNA3-CRM1), was mostly cytosolic upon ActD treatment in cells transfected with the scrambled siRNA (Fig. 4, +pSIL-SCRAM +ActD), and displayed a predominantly nucleolar distribution in cells co-transfected with pSIL-CRM1A even upon ActD treatment (Fig. 4, +pSIL-CRM1A +ActD). Altogether, these observations demonstrated that the cellular distribution of BPV E1 correlated closely with the cellular abundance of CRM1, therefore supporting the conclusion that CRM1 is the main nuclear export factor for BPV E1.
Fig. 4. The cellular distribution of BPV E1 is dictated by the abundance of CRM1.

HeLa cells were transfected with either pcDNA3-CRM1 or pSIL-CRM1A to produce either an increase or a substantial decrease in the cellular levels of CRM1, respectively. A third group of cells was transfected with pSIL-SCRAM, a pSIL-based plasmid containing a scrambled sequence known not to target any specific cellular gene. Thirty-six hours post-transfection the cells were transfected with pEGFP-E1 or pREV1.4(NES3)GFP plasmids, treated with cyclohexamide (CHX) and/or ActD, fixed, and analyzed by fluorescence microscopy 24 h after the second transfection. The plasmid used in the first transfection is indicated on top of each panel, whereas the plasmid used in the second transfection is indicated on the left. The cells shown are representative of the cellular localization observed for the indicated proteins under the conditions described.
BPV E1 contains three functional leucine-rich nuclear export signals
Next, we evaluated the functional activity of the individual putative NESs identified in BPV E1. For these experiments, the NES element located between the BamHI and AgeI sites on the parental pREV1.4(NES3)GFP plasmid was replaced with each of the putative NESs identified in BPV E1. To prevent constraints associated with the length of the NES expressed between the BamHI and AgeI sites, an adaptor sequence was introduced between the BamHI/AgeI sites on pREV1.4(NES3)GFP. The adaptor was designed to introduce mutations in the original BamHI and AgeI sites, therefore inactivating them (Fig. 5A, unmarked horizontal lines above the sequence), while providing a new set of BamHI and AgeI sites in the middle section of the adaptor (Fig. 5A, marked horizontal lines above the sequence). Next, complementary oligonucleotides coding for the putative E1 NES elements were cloned into the modified pREV1.4(NES3)GFP (designated pREV/GFP/Adpt). The resulting constructs were transfected into HeLa cells and the nuclear export activity of each putative NES was evaluated by counting the number of cells exhibiting only nuclear, both nuclear and cytoplasmic, and only cytoplasmic distribution of the GFP reporter in the absence and presence of ActD and LMB, according to the method previously reported by Henderson and Eleftheriou (Henderson and Eleftheriou, 2000). While the reporter encoded by pREV/GFP/Adpt exhibited exclusively nuclear localization, three out of the four putative NESs identified in BPV E1, namely NES2, NES3, and NES4, had the ability to induce some cytosolic localization of the reporter protein (Fig. 5B). Although for all these NESs the cytosolic localization of the reporter increased upon ActD treatment (Table 1), the percentage of cells exhibiting some cytosolic fluorescence remained low even upon ActD treatment, with NES2 being the one that induced the highest percentage of cells displaying some cytosolic fluorescence. This indicated that the strengths of the BPV E1 NESs were weak compared to the Rev-NES and other previously reported NESs (Henderson and Eleftheriou, 2000). Quantitative comparison by the scoring system developed by Henderson and Eleftheriou confirmed the qualitative observations (Table 1 and Fig. 5B). Interestingly, LMB treatment completely inhibited the activity of NES2 but had no little or no effect on the activity of both NES3 and NES4 (Table 1 and Fig. 5B).
Fig. 5. NES2, the strongest NES in BPV E1, is sensitive to the CRM1-inhibitory activity of LMB.

(A) Nucleotide and amino acid sequences of the region in pREV/GFP/Adpt where the putative nuclear export signals (NESs) identified in BPV E1 were introduced to test their activity. The unlabeled lines on top of the nucleotide sequence indicate the original cloning sites used to introduce the REV NES in the parental pREV1.4(NES3)GFP construct. Those cloning sites were mutated and the REV NES eliminated by the introduction of the linker sequence depicted between the mutated BamHI and AgeI. This linker extends the sequence between REV and GFP, thus preventing constraints associated to the length of the NES expressed, and contains new BamHI and AgeI sites. Thus, the pREV/GFP/Adpt vector allows the expression of REV-NES-GFP fusion proteins by directional cloning of the different NESs between the BamHI and AgeI sites. (B) HeLa cells were transfected with the different constructs coding for the REV-NES-GFP fusion proteins, and the cellular localization of the encoded proteins was determined by fluorescence microscopy. The figure summarizes the data obtained for the different NESs identified in BPV E1. The cells shown are representative of the localization of each REV-NES-GFP. NES activity was measured and rated according to the method described by Henderson and Eleftheriou (Henderson and Eleftheriou, 2000), which gives an activity value between 0 and 9+ to a putative NES, with 0 being a sequence that lacks any NES activity, and 9+ being the strongest NES possible. LMB Sensitivity indicates whether LMB treatment prevented the nuclear export of the reporter carrying the indicated putative NES.
Table 1.
Intracellular distribution of NES constructs
| Construct | CHX Alone | CHX + ActD | CHX + ActD+ LMB | NES Activity* | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nuc | N/C | Cyt | Nuc | N/C | Cyt | Nuc | N/C | Cyt | ||
| pREV4.1(NES3)EGFP | 31 | 61 | 8 | 3 | 32.5 | 64.5 | 97.5 | 2.5 | 0 | 5+ |
| pREV/EGFP/Adpt-1 | 99 | 1 | 0 | 97.9 | 2.1 | 0 | 97.5 | 2.5 | 0 | 0 |
| MVM-NS2 | 80 | 20 | 0 | 70.5 | 29.5 | 0 | 88.5 | 11.5 | 0 | 1+ |
| NES1 | 98 | 2 | 0 | 95 | 5 | 0 | 97.5 | 2.5 | 0 | 0 |
| NES1M | 96.5 | 3.5 | 0 | 91.5 | 8.5 | 0 | 93 | 6.5 | 0.5 | 0 |
| NES2 | 74 | 26 | 0 | 41 | 59 | 0 | 96 | 4 | 0 | 2+ |
| NES2M | 96.5 | 3.5 | 0 | 94.5 | 5.5 | 0 | 95 | 5 | 0 | 0 |
| NES3 | 78.5 | 21.5 | 0 | 67 | 33 | 0 | 76 | 24 | 0 | 1+ |
| NES3M | 97.5 | 2.5 | 0 | 92.5 | 7.5 | 0 | 89.5 | 10.5 | 0 | 0 |
| NES4 | 85.5 | 14.5 | 0 | 69 | 30.5 | 0.5 | 76 | 24 | 0 | 1+ |
| NES4M | ND | ND | ND | ND | ND | ND | ND | ND | ND | 0 |
The NES activity is given according to the measuring method developed by Henderson and Eleftheriou (Henderson and Eleftheriou, 2000).
Nuc: Nuclear; N/C: Nuclear and cytosolic; Cyt: Cytosolic; ND: Not determined; CHX: cycloheximide; ActD: actinomycin D; LMB: leptomycin B.
To confirm that the conserved hydrophobic residues in the BPV E1 NESs were responsible for their function, we introduced double alanine substitutions for two of the bulky hydrophobic residues in both E1 NES2 and NES3, and the resulting mutants were transfected into both Cos1 cells (Fig. 6) and HeLa cells (data not shown). In both cell lines, alanine replacements of two of the residues postulated to constitute part of the NESs identified in NES2 and NES3 fully eliminated their activity, therefore confirming their functional role as constituents of the NESs identified.
Fig. 6. The conserved hydrophobic residues in the BPV E1 NESs are responsible for their function.

Two hydrophobic residues considered functional components of nuclear export signals NES2 and NES3 were substituted by alanine residues in their respective pREV/GFP/Adpt constructs. The effect of these replacements on the cellular localization of the GFP reporter was assessed by transfection of the mutated constructs into COS1 cells followed by fluorescence microscopy analysis. NES1 is presented as a negative control. The sequences shown correspond to the actual sequence coded by the nucleotide sequence placed between the BamHI and the AgeI sites on the pREV/GFP/Adpt vector (between proline and glycine, see figure 5). WT: Wild-type sequence. MU: Mutated sequence. The residues substituted with alanine are indicated by bars.
NES2, the strongest NES in BPV E1, is CRM1-dependent
The observation that LMB inhibited the nuclear export activity mediated by NES2 but not the export mediated by NES3 or NES4 suggested that perhaps NES2 was the only NES in BPV-E1 that used CRM1 as exportin. To test if the functional NESs identified worked in a CRM1-dependent manner, we transfected the pREV/GFP/Adpt constructs containing the different NESs into HeLa cells either on their own or together with the pcDNA3-CRM1 plasmid, treated the cells with either CHX+ActD or CHX+ActD+LMB, and determined the percentage of cells exhibiting nuclear, nuclear and cytosolic, and cytosolic fluorescence. As expected, both of the previously characterized CRM1-dependent NESs tested, MVM-NS2 and the HIV-Rev NES contained in pREV4.1(NES3)GFP, exhibited a significant increase in cytoplasmic fluorescence upon CRM1 over-expression (Fig. 7). However, out of the three functional NESs identified in BPV E1, only NES2 exhibited a substantial increase in cytosolic localization upon CRM1 over-expression (Fig. 7A), with over 50% of the cells exhibiting exclusively cytoplasmic fluorescence when the cells were treated with CHX+ActD (Fig. 7B). For NES3 and NES4, there were no cells exhibiting an exclusively cytoplasmic E1 signal after CRM1 over-expression, and in fact the percentage of cells exhibiting any cytosolic signal decreased slightly. Furthermore, for the NES2 construct, LMB treatment led to a fully nuclear distribution of the reporter, whereas for NES3 and NES4 LMB treatment did not exert significant effects (data not shown). Altogether, these data indicate that the nuclear export activity of NES2 is CRM1-dependent, whereas the nuclear export activity of NES3 and NES4 is insensitive to CRM1 levels and appears to be mediated by other exportin(s).
Fig. 7. NES2, the strongest NES in BPV E1, is CRM1-dependent.

HeLa cells were transfected with the different pREV/GFP/Adpt-NES constructs either alone or in combination with the pcDNA3-CRM1 plasmid. The transfected cells were treated with CHX and ActD and analyzed by fluorescence microscopy. The effect of over-expressing CRM1 on the cellular localization of the different REV-NES-GFP fusion proteins was quantified by counting the number of cells exhibiting nuclear, nuclear and cytosolic, and cytosolic fluorescence. (A) Representative images of cells expressing REV NES, BPV E1 NES2, and BPV E1 NES3 with and without over-expressed CRM1. (B) Quantitative data indicating the cellular distribution of the different REV-NES-GFP constructs with or without over-expressed CRM1. The data is presented as percentage of cells exhibiting either exclusively nuclear, nuclear and cytoplasmic, or exclusively cytoplasmic fluorescence.
Inactivation of NES2 in the context of E1 protein increases the nuclear distribution of E1 and negatively affects replication activity
The above results implicated NES2 as a significant CRM1-dependent export motif, and we next sought to confirm this within the context of E1 itself. The use of EGFP fusions allowed us to determine the cellular localization of both wild type and the NES 2 mutant form of BPV E1 (referred to as NES2M). The double alanine replacement mutant of NES2 (I413 and L416), which inactivates NES2 in the pREV/GFP/Adpt construct, also produced a dramatic change in the cellular distribution of the EGFP-E1 fusion. While the WT BPV E1 exhibited significant nuclear and cytoplasmic distribution, the NES2M mutant exhibited a substantial increase in nuclear localization accompanied by a marked decrease in cytoplasmic localization (Fig. 8A). Therefore, inactivation of NES2 markedly decreases the export of BPV E1 thus resulting in increased BPV E1 nuclear localization, in agreement with our previous observation that NES2 is the strongest NES in BPV E1.
Fig. 8. Inactivation of NES2 affects the replication activity of BPV E1.
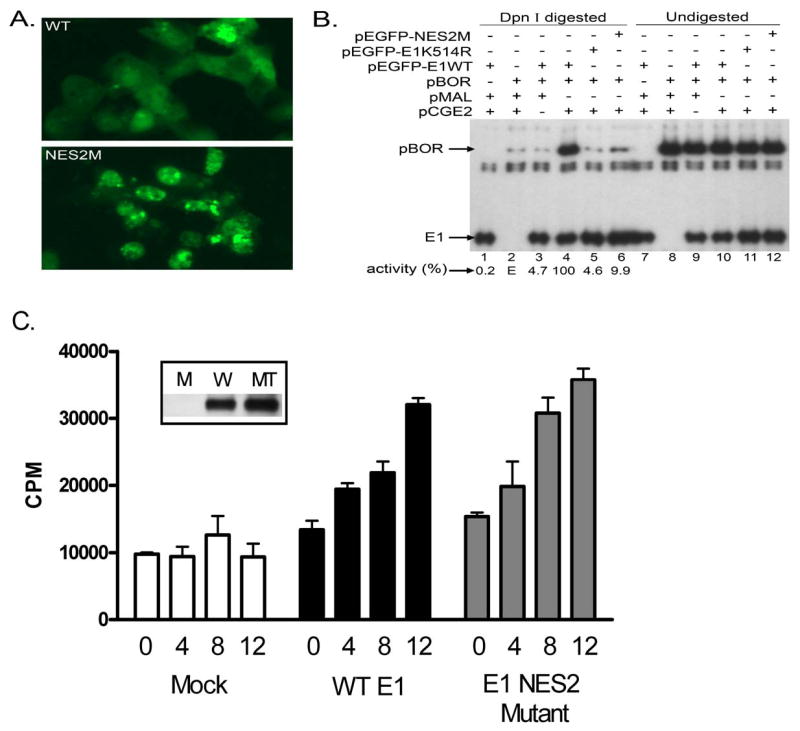
CHO-K1 cells were transfected with an expression plasmid for EGFP fusions of either the wild type (WT), a NES2 mutant (NES2M), or a K514R mutant of BPV E1, either alone or in combination with different permutations of the plasmids pBOR, pMAL (control empty plasmid), and pCGE2 (coding for the BPV E2 protein). The transfected cells were either analyzed by fluorescence microscopy or processed for DNA purification, digestion of methylated DNA, and PCR amplification in the presence of 32P-dCTP to detect replicated DNA. (A) Cellular localization of WT and NES2M BPV E1. (B) Transient replication assays. The location of the PCR products derived from pBOR and E1 are indicated. (C) In vitro DNA replication assay. Shown are the results using the amounts (in μl) of in vitro translated wild-type E1 (WT), NES2 mutant E1, or a Mock sample as indicated. The Mock samples were performed with an in vitro translation reaction primed with DNA which did not encode E1. Values shown are the average of either 2 or 3 experiments. The inset shows an anti-E1 immunoblot of the in vitro translation reactions (M is the mock reaction, W is the wild-type E1, and MT is the NES2 mutant E1).
Functionally, BPV E1 carries out its helicase activity in the nucleus of the cell, and several previous reports have indicated that regulation of the nuclear localization of BPV E1 is a critical mechanism to modulate its activity. Therefore, we sought to determine if the altered nuclear export of the NES2 mutant E1 would exert any effect on BPV E1 replication activity. We performed transient BPV DNA replication assays by co-transfecting CHO-K1 cells with an expression plasmid for BPV E2 (pCGE2), a plasmid carrying the BPV origin of replication (pBOR), and an expression plasmid for EGFP fusions of either the wild type or the NES2 mutant form of BPV E1. Replicated pBOR was quantified using a modification of a previously reported PCR based method (Titolo et al., 1999). To ensure that any replication differences observed among samples were not due to transfection differences, we also measured the amount of E1 expression plasmid in each group of transfected cells in the same set of PCR reactions. Interestingly, even though there was increased nuclear localization by the NES2M mutant, there was decreased replication of the pBOR plasmid (Fig. 8B). As similar transfection efficiencies were achieved among all samples tested, as demonstrated by the E1 control, the diminished replication activity observed in cells transfected with the NES2M mutant suggests that either nuclear shuttling is important to maintain the activity of BPV E1, or the substitutions introduced in the NES2M mutant negatively affect the enzymatic activity of BPV E1. Importantly, although the activity of the NES2M mutant in the transient replication assays was approximately 1/10th of the activity of the WT protein, it was consistently more active than the K514R mutant, a previously described BPV E1 mutant (see below).
To evaluate intrinsic replication activity of the NES2 mutant, in vitro replication assays were performed (Fig. 8C). For these studies, both the WT and mutant E1 proteins were synthesized in vitro to avoid any issues with differences in modifications or purification properties that might result from their somewhat different intracellular distributions. In contrast to the in vivo results, NES2M was as active as WT E1, indicating that these two amino acid changes did not disrupt E1 activities necessary for DNA replication. Consequently, the in vivo replication deficit appears to be related to reduce nuclear-cytoplasmic shuttling.
Sumoylation enhances the interaction of BPV E1 with CRM1
BPV E1 is post-translationally modified by the covalent attachment of SUMO (Rangasamy and Wilson, 2000), a small ubiquitin-related modifier, and this modification is enhanced by the SUMO ligase activity exerted by proteins belonging to the PIAS family (Rosas-Acosta et al., 2005). Although a previous report indicated that a non-sumoylatable form of BPV E1 containing a Lys to Arg substitution at position 514 (referred to as the K514R mutant) displayed a predominantly cytoplasmic localization (Rangasamy et al., 2000), more recent extensive experimentation using confocal microscopy has demonstrated that the non-sumoylatable mutant is almost exclusively nuclear and is enriched in close proximity to the nuclear envelop, exhibiting a more dotted appearance than the wild-type form (Fig. 9A). These findings still support a role for SUMO in the regulation of the cellular distribution of BPV E1 albeit different to the one previously suggested. To determine if sumoylation affects the binding of BPV E1 to CRM1, we performed in vitro sumoylation reactions and assessed the ability of the sumoylated and un-sumoylated forms of BPV E1 to bind CRM1 using pull-down assays. Interestingly, sumoylation appeared to enhance the interaction of BPV E1 with CRM1 as the amount of un-sumoylated BPV E1 detected in the pull-downs performed with the sumoylated samples was larger than that detected in pull-downs performed with un-sumoylated samples (compare the intensity of bands 1 and 2, Fig. 9B). Furthermore, sumoylated BPV E1 was efficiently pulled-down by CRM1 (arrow 3, Fig 9B), and the fraction of sumoylated BPV E1 recovered constituted a larger fraction of the initial amount present than for the un-sumoylated BPV E1 (Fig. 9C). Altogether, these findings indicate that, at least under the experimental conditions employed in our in vitro binding assays, sumoylation enhances the binding of BPV E1 to CRM1. This finding is consistent with the observation that wild-type BPV E1 exhibits a more abundant cytoplasmic distribution than the K514R mutant.
Fig. 9. Sumoylation enhances the interaction of BPV E1 with CRM1.

(A) Cellular localization of EGFP fusions of the E1 K514R mutant (upper panel) and wild type E1 (lower panel), as observed by confocal microscopy. (B) Pull-down assays performed with 4 μg of purified GST or GST-CRM1 and 2 μL of in vitro translated BPV E1 either un-modified or modified by in vitro sumoylation. The pull-downs were performed in triplicate and all the samples are shown. The samples were analyzed by SDS-PAGE and quantified by phosphordensitometry. Arrows 1 and 2 indicate Crm1-associated unmodified E1 from the unsumoylated and sumoylated input samples, respectively. Arrow 3 indicates Crm1-associated sumoylated E1. (C) Quantitative data indicating the percentage of input unmodified (1 and 2) or sumoylated BPV E1 (3) that remained bound to the beads after the pull-down procedure, as determined by phosphordensitometry of the triplicate samples presented in B. The numbers in parenthesis indicate the location of the bands measured in each case, as shown in B.
Discussion
In this study we demonstrate that, similar to its human papillomavirus counterpart HPV E1, the bovine papillomavirus helicase protein BPV E1 shuttles between the nucleus and the cytoplasm. Furthermore, we provide evidence for one strong export signal, spanning residues 409-418 (NES2), whose export activity is mediated by the nuclear export factor CRM1. Importantly, mutations affecting the CRM1-dependent nuclear export signal result in a substantial increase in the nuclear accumulation of BPV E1, but decreased replication activity as assessed by an in vivo replication assay. Since the intrinsic replication activity of the NES2 mutant appears normal in an in vitro replication assay, the in vivo results suggest a requirement for nuclear-cytoplasmic shuttling for maintenance of E1 activity. Additionally, we identified two other nuclear export signals within BPV E1, spanning residues 140-153 (NES3) and 262-275 (NES4). These latter two element have only weak activity in the context of a reporter protein, and their export activity is independent of the nuclear export factor CRM1. We also present data supporting an altered but still nuclear localization for the BPV E1 K514R mutant, a previously reported mutation known to affect the sumoylation and replicative function of BPV E1, and demonstrate that sumoylation enhances the interaction of BPV E1 with CRM1 in an in vitro assay, therefore supporting a role for BPV E1 sumoylation in regulating the cellular localization of BPV E1.
In contrast with the previous report related to the nuclear export of HPV11 E1, which identified only one NES in HPV11 E1, in this study we identified three functional NESs in BPV E1 by using REV-GFP chimeras. Although the actual contribution to the nuclear export of BPV E1 mediated by the two weak NESs identified (NES3 and NES4) could not be accurately defined based on the data presented, our results suggests that NES2 is the main contributor to the nuclear export of BPV E1. First, NES2 is the only NES acting via a CRM1-dependent nuclear export pathway as the activity of NES3 and NES4 in our functional assay was not affected by the cellular concentration of CRM1 nor by LMB treatment. Thus, NES3 and NES4's nuclear export activity is mediated by another yet unidentified exportin. This result was rather surprising as the sequence motif exhibited by NES3 and NES4 correspond to that present in p27, which has been previously reported to act on a CRM1-dependent manner (Connor et al., 2003). Second, RNAi directed against CRM1 resulted in a significant increase in the nuclear localization of BPV E1. Third, LMB treatment, a well established inhibitor of CRM1-mediated nuclear export, also resulted in a substantial increase in BPV E1's nuclear accumulation. Finally, mutations affecting NES2 also resulted in almost exclusive nuclear localization of BPV E1. Thus, NES3 and NES4 appear to provide a very limited contribution to the nuclear export of BPV E1, in agreement with their weak nuclear export activity measured in the REV-GFP chimeras. However, it is also possible that NES3 and NES4 may be substantially more active upon BPV E1 phosphorylation, therefore providing BPV E1 with alternative phosphorylation-dependent nuclear export pathways, as discussed below. In either case, the existence of alternative nuclear export pathways for BPV E1 emphasizes the biological relevance of this process for BPV E1 function.
The role of CRM1 in the nuclear export of BPV E1 herein reported is consistent with previous reports by other groups. Work by the Broker-Chow group (Deng et al., 2004) mapped a nuclear export sequence (NES) in the HPV11 E1 protein which functions in a CRM1-dependent way. The NES identified in HPV11 E1 was constitutively active and was inhibited by phosphorylation of a nearby Ser residue. Importantly, the sequence motif mediating HPV11 E1's nuclear export was mapped to a well conserved region of the protein that is nevertheless absent in the equivalent region of BPV E1, thus leading the authors to postulate the absence of a CRM1-dependent nuclear export mechanism for BPV E1. However, work by the Bonne-Andrea group demonstrated that BPV E1 was actively exported from the nucleus, and although an NES motif in BPV E1 was not mapped, they presented evidence indicating that BPV E1 export occurred via a CRM1-dependent mechanism (Hsu, Mechali, and Bonne-Andrea, 2007). According to the data presented in that study, and in sharp contrast with HPV11 E1, the nuclear export of BPV E1 did not seem constitutively active but instead required the phosphorylation of Ser283 by an active cyclin A-Cdk2 complex. In our present study we did not evaluate the potential regulatory role exerted by phosphorylation on the nuclear export of BPV E1. However, one of the functional NESs identified, NES4 which encompasses residues 262-275, is located just upstream of Ser283. Although NES4 is one of the two weakest NES elements identified in BPV E1, it is possible that phosphorylation of Ser283 may increase substantially its activity. Likewise, though NES2, which spans residues 409-418, is more active than NES4 when expressed in the REV-GFP chimeric constructs, it is possible the phosphorylation could enhance its activity even further. Direct testing of the effect of phosphorylation on the strength of NES2 and the other two functional NESs identified will be required to determine if these NESs are affected by phosphorylation as suggested by previous publications (Hsu, Mechali, and Bonne-Andrea, 2007) (Deng et al., 2004).
Mechanisms dictating the cellular localization of BPV E1 have been postulated to be the main contributors to the regulation of BPV E1 function (Bian et al., 2007; Hsu, Mechali, and Bonne-Andrea, 2007; Yu et al., 2007). Previous studies focused on the regulation of the nuclear localization of BPV E1 and HPV E1 have shown a tight correlation between the abundance of the E1 helicase in the nucleus and the replication of plasmids containing a papillomavirus origin of replication (Deng et al., 2004; Yu et al., 2007). Importantly, this was not the case for the NES2 mutant developed in this study. The BPV E1 NES2M mutant protein, containing the I413A/L416A substitutions, which inactivate NES2 as determined with the REV-GFP chimera, appeared to accumulate in the nucleus up to a higher concentration than the wild type protein. However, the NES2M mutant exhibited a significantly reduced replication activity compared to that of the wild type protein in a transient in vivo BPV DNA replication assay, even though this mutant E1 protein appeared functionally normal for replication activity in vitro. Consistent with the in vitro results, the NES2M mutations introduced on BPV E1 are located on an externally exposed alpha helical structure in BPV E1, and do not appear to be located near regions required for single or double-stand DNA binding or protein oligomerization, based on the crystallographic data reported by Enemark and Joshua-Tor (Enemark and Joshua-Tor, 2006). In addition, a previously constructed deletion mutant missing 4 amino acids within the NES2 motif (residues 412-415) had wild-type DNA binding and ATP binding activity, though was also severely impaired for transient replication similar to NES2M (Ludes-Meyers, 1995). Thus, the region mutated in NES2M appears to have little or no direct contribution to E1 enzymatic function, which suggests that continuous nuclear import and export of BPV E1 may be an important requirement to maintain its activity. One hypothetical scenario that could explain this requirement is that the continuous shuttling of BPV E1 may prevent its sequestration in replication-inactive nuclear subdomains. The existence of such subdomains is suggested by the dotted nuclear structures containing BPV E1 observed in the NES2M mutant.
Previous studies by our group had indicated that the nuclear localization of BPV E1 was dependent upon sumoylation. Now we have re-evaluated those findings and have concluded that sumoylation affects the sub-nuclear localization and cellular distribution of BPV E1 but is not required for its nuclear localization, as the K514R mutant (previously shown to be non-sumoylatable in vivo) is localized in the nucleus. Importantly, we have observed that BPV E1 sumoylation enhances BPV E1's interaction with CRM1 in in vitro pull-down assays. The increase in the amount of unmodified BPV E1 retained by CRM1 in the presence of sumoylated BPV E1 may be due to oligomerization of the SUMO-modified form of BPV E1 with unsumoylated forms of BPV E1. Thus, sumoylation may enhance the nuclear export of BPV E1, as suggested by the observed cellular distribution of the WT and the K514R mutant proteins. Sumoylation is already known to play a role in the nuclear export of other proteins: the nuclear export of the Dictyostelium discoideum MEK1 protein is dependent upon sumoylation (Sobko, Ma, and Firtel, 2002), the intranuclear targeting and nuclear export of the adenovirus E1B-55K protein are regulated by sumoylation (Kindsmuller et al., 2007), SUMO conjugation regulates the nuclear export of the putative tumor suppressor protein TEL (Wood et al., 2003), and cycles of SUMO conjugation and de-conjugation appear to regulate the nuclear shuttling of hnRNPs (Vassileva and Matunis, 2004). Most proteins exported from the nucleus using a CRM1-dependent pathway establish weak unstable interactions with CRM1 (Kutay and Guttinger, 2005), and NESs able to establish stable complexes with CRM1 appear trapped in a complex with CRM1 at the nuclear pore complex (Engelsma et al., 2004). This is to our knowledge the first direct demonstration that sumoylation stabilizes the interaction of CRM1 with a nuclear shuttling protein. This finding supports the possibility that cycles of sumoylation and de-sumoylation may play a role in nuclear shuttling as previously postulated by Vassileva and Matunis (Vassileva and Matunis, 2004), as a sumoylated protein would likely remain bound to CRM1 until de-sumoylated by a SUMO-protease. This is further supported by previous reports indicating the localization of different components of the sumoylation system, including SUMO conjugating and SUMO de-conjugating enzymes, and ligases, to the nuclear pore complex (Hang and Dasso, 2002; Panse et al., 2003; Pichler et al., 2002; Takahashi et al., 2000; Zhang, Saitoh, and Matunis, 2002). Further studies aimed at clarifying the role played by sumoylation in the regulation of traffic across the nuclear pore complex are likely to reveal intriguing aspects of the biological events dictating this process and have the potential to reveal novel targets for therapeutic approaches to infectious diseases and cancer.
Materials and Methods
Cell lines, transfections, fluorescence microscopy, and confocal fluorescence microscopy
HeLa and Cos1 cells were maintained in 1× DMEM supplemented with 10% FetalPlex™ (Gemini BioProducts, West Sacramento, CA). CHO-K1 cells were maintained in 1× Ham's F12K supplemented with 10% FetalPlex™. All transfections were performed using Lipofectamine™ 2000 (Invitrogen Corp., Carlsbad, CA), according to the manufacturer's protocol. Fluorescence microscopy was performed with a Nikon Eclipse TS 100 microscope (Nikon Inc., Melville, NY) using an X-Cite™ 120 Fluorescence Illumination System (EXFP America Inc., Plano, TX) and a Nikon Digital Sight DS 2 Mv camera (Nikon Inc.). Confocal fluorescence microscopy was performed on a Zeiss Axiovert 135 microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY) with a CARV confocal module (Zeiss Micro-Imaging and Atto Bioscience) and images were collected at 0.75-μm intervals using Zeiss AXIOVISION 3.1.
RNAi and Reporter Constructs
Candidate sequences to develop effective RNAi constructs to human CRM1 (NM_003400) were obtained from Ambion (Ambion, Austin, TX). The sequences obtained contained additional sequence elements, including inverted repeats designed to generate hairpins with a 19-nucleotide stem and a 9-nucleotide loop, and sequences for cloning into the pSilencer™ 4.1-CMV puro vector (Ambion). The sequences of the oligonucleotides used to develop the two different RNAi constructs are indicated below. pSIL-CRM1A construct: Forward primer, 5′ GATCCGGAGC CCAGCAAAGA ATGGTTCAAG AGACCATTCT TTGCTGGGCT CCTTA 3′; Reverse primer, 5′ AGCTTAAGGA GCCCAGCAAA GAATGGTCTC TTGAACCATT CTTTGCTGGG CTCCG 3′. pSIL-CRM1B construct: Forward primer, 5′ GATCCGGAGC ATCCTGATGC TTGGTTCAAG AGACCAAGCA TCAGGATGCT CCTTA 3′; Reverse primer, 5′ AGCTTAAGGA GCATCCTGAT GCTTGGTCTC TTGAACCAAG CATCAGGATG CTCCG 3′. To map the nuclear export sequences (NESs) contained within BPV E1, we introduced an adaptor sequence between the BamHI and AgeI sites on the plasmid pREV1.4(NES3)GFP developed by Henderson and Eleftheriou (Henderson and Eleftheriou, 2000). The adaptor sequence was produced by annealing the complementary primers Adpt.FW (5′ GATCGACCAC CTGTACCACT GGATCCGCGG GGACCGGTAG ATTGCAAC 3′) and Adpt.RV (5′ CCGGGTTGCA ATCTACCGGT CCCCGCGGAT CCAGTGGTAC AGGTGGTC 3′). The adaptor sequence introduced mutations in the original BamHI and AgeI sites, contained a sequence coding for the sequence surrounding the NES contained in the REV-HIV protein, and provided a new set of BamHI and AgeI sites. The resulting plasmid produced was denominated pREV/GFP/Adpt and was used for the subsequent expression of all the putative NESs identified within BPV E1. To this end, sequences coding for the putative NESs were introduced into BamHI/AgeI digested pREV/GFP/Adpt as complementary oligonucleotides recreating sticky BamHI and AgeI ends upon annealing (precise sequences available on request). Constructs containing substitutions of specific bulky hydrophobic in the putative NESs were developed in a similar manner.
Nuclear export assay
Nuclear export assays were performed following a combination of the procedures described by Connor et al. (Connor et al., 2003) and Love et al. (Love, Sweitzer, and Hanover, 1998). Briefly, HeLa cells were plated on 6 cm Petri dishes at a density of 1 × 106 cells per dish and transfected 24 hours later with the plasmid pEGFP-E1 (coding for a fusion of EGFP and the BPV E1 protein) using Lipofectamine™ 2000. Twenty four hours post-transfection the cells were washed twice with 1× Transport Buffer (1× TB; 20 mM Hepes pH 7.8, 2 mM EGTA, 110 mM Potassium Acetate, 2 mM Magnesium Acetate), and permeabilized by incubation for 5 min at room temperature in 1 mL of 1× TB supplemented with Digitonin at 50 μg/mL, a 1:200 dilution of Protease Inhibitor Cocktail P8340 (Sigma-Aldrich Co., Saint Louis, MO), and 10 μM MG-132. The cells were washed twice (1 min per wash at room temperature) with 5 mL of 1× TB, and incubated in 1 mL of 1× TB supplemented with a 1:500 dilution of Protease Inhibitor Cocktail P8340, 5 mM ATP, 2 mM GTP, 5 mM Creatine Phosphate, 20 U/mL Creatine Phosphokinase, and 2 mM DTT, with or without 20% Reticulocyte Extract (from the TnT® T7 in vitro translation system, Promega Corp., Madison, WI). One hour later, the resulting supernatant (exported fraction) was collected and immunoprecipitated using 5 μL of anti-GFP rabbit polyclonal antibody and 40 μL of Protein G-Plus bead slurry (both from Santa Cruz Biotechnology Inc., Santa Cruz, CA). The permeabilized cells were washed once more with 1× TB, and collected in 500 μL of 2× Sample Buffer (100 mM Tris pH 6.8, 20% glycerol, 8% SDS, 0.02% bromophenol blue, 4% β-mercaptoethanol). The resulting cell lysate and the immunoprecipitated samples were resolved on a 10% SDS-PAGE gel, transferred to an Immobilon™ PVDF membrane (Millipore Corp., Billerica, MA), and probed with an anti-GFP monoclonal antibody (Santa Cruz Biotechnology Inc.).
Pull-down and in vitro sumoylation assays
Pull down assays and in vitro sumoylation assays were performed as previously reported (Rosas-Acosta et al., 2005). For pull down analysis of in vitro sumoylated samples, upon in vitro sumoylation the full in vitro sumoylation reaction volume was diluted in 1× TBS supplemented with 5 mM MgCl2, 0.05% Tween 20, and 5 mg/mL BSA, and incubated for 3 hours at 4°C with 20 μL of packed Glutathione-Sepharose beads and 4 μg of the indicated purified protein (either GST or GST-CRM1) in a final volume of 500 μL. The beads were washed four times with 1× TBS supplemented with 0.05% Tween 20 and 5 mM MgCl2, the remaining supernatant was discarded, and the beads were resuspended in 25 μL of 4× Sample Buffer, resolved by SDS-PAGE on a 10% gel, blotted into an Immobilon™ PVDF membrane, and analyzed by phosphordensitometry as previously described (Rosas-Acosta et al., 2005).
Biochemical fractionation
CHO-K1 cells were transfected with pEGFP-E1 as for the nuclear export assay and harvested 24 hr post-transfection. Four hr prior to harvest, leptomycin B (LMB) and cycloheximide were added to the medium at a final concentrations of 20 ng/ml and 10 ng/ml, respectively. Cells were harvested by scaping into ice-cold PBS, pelleted, and washed 1 time with 1 ml of ice-cold PBS. The washed pellet was resuspended in 200 μl of hypotonic buffer (10 mM Tris-HCl, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 0.25% Triton X100), incubated 10 min on ice, and then lysed by 25 strokes with a Dounce homogenizer. The lysed cells were centrifuged for 4 min at 2000 × g and the supernatant was removed as the cytosolic fraction. The nuclear pellet was washed 1 time with 500 μl of hypotonic buffer, resuspended in 150 μl of hypotonic buffer, and lysed by addition of 50 μl of 4X SDS sample buffer to yield the nuclear fraction. The fractionation efficiency was evaluated by examination of endogenous RanGAP and was greater than 90% (not shown).
Transient replication assays
The transient HPV DNA replication assays were performed following a modification of the procedures previously described by Titolo et al. (Titolo et al., 2003; Titolo et al., 2000). Briefly, approximately 1 × 106 CHO-K1 cells were plated on 6 well plates. Twenty four hours later, the cells were transfected with 4 μL of Lipofectamine™ 2000 and 2 μg of different combinations of plasmids encoding, respectively, E1 (pEGFP-E1WT), a previously reported mutant form of E1 containing a Lys to Arg substitution at residue 514 (pEGFP-E1K514R), a mutant form of E1 containing Ala substitutions affecting NES2 (pEGFP-NES2M), E2 (pCGE2), a plasmid containing the HPV ori region (pBOR), and an empty plasmid lacking mammalian promoters (pMAL-c2E). The pCGE2, pBOR, and the various E1 plasmids were transfected at a ratio of 0.2:1:1, respectively. Seven hours post-transfection, the cells were treated with 500 μL of tissue-culture trypsin, washed with complete Ham's F12K, and plated in 10 cm Petri dishes. The cells were subsequently incubated at 37°C and 5% CO2 for another 65 hours. At 72 h post-transfection the cells were collected by trypsinization and washed in ice cold 1× PBS. The wet cell pellets obtained were stored at -20°C until used for DNA purification. DNA was purified using the QIAamp DNA Blood Midi purification kit (QIAGEN Inc, Valencia, CA), as indicated by the manufacturer. Purified DNA was eluted off the spin column by two rounds of elution using 300 μL of elution buffer each. Approximately equal DNA concentrations of total genomic DNA (∼250 ng/μL) were obtained for all samples as determined by spectrophotometry. Replicated pBOR plasmid was detected by PCR amplification of a region containing 5 DpnI restriction sites. To this end, 21 μL of the purified genomic DNA were digested with 10 U of DpnI for 16 h at 37°C in a final volume of 25 μL. Then, 10 μL of the digestion mix (∼25 ng gDNA) were used as template for the PCR reaction. Four primers were used in each PCR reaction, two amplifying a ∼507 bp region void of DpnI sites in the E1 gene contained in the pEGFP-E1 plasmids, and two amplifying a ∼952 bp region containing the indicated 5 DpnI restriction sites in the pBOR plasmid. The sequences of the E1 primers were 5′ TCGGTATGATAGCCAGGATGAGG 3′ and 5′ CTTCTTTAGGAGTTCGAAACTCGCC 3′, and the sequences of the pBOR plasmid primers were 5′ TTCTTCGGGGCGAAAACTCTCAAGG 3′ and 5′ GATTACGAATTCGAGCTCGGTACCC 3′. Each primer was added at a final concentration of 0.5 μM, and each PCR reaction had a final volume of 50 μL. To allow the detection and precise quantification of the amplified products, 5 μCi of [α-32P] dCTP were added to each reaction. The PCR conditions used consisted of an initial denaturation at 95°C for 3 min, followed by 23 cycles of denaturation at 95°C for 15 sec, annealing at 55°C for 30 sec, and extension at 72°C for 1 min. The use of 23 cycles of PCR amplification was deemed optimal during initial standardizations. After the final cycle, the samples were incubated at 72°C for 10 min, and kept at 4°C. The PCR products were analyzed on a 1 mm thick 3.5 % acrylamide gel containing 1× TBE buffer, by loading 3 μl of the PCR products per lane. The gel was fixed in a 10 % acetic acid solution for 10 min, dried, and analyzed by phosphordensitometry as previously described (Rosas-Acosta et al., 2005).
In vitro replication assay
Assays were performed using in vitro translation reactions as the source of E1 proteins as previously described (Amin et al., 2000). In vitro translation was performed using the TnT T7 Quick Coupled Transcription/Translation kit (Promega Corp.). The cell extracts for the replication assay were prepared by the procedure of Wang and Wang (Wang and Wang, 1999). Replication assays were performed for 90 min at 37°C, and the extent of DNA synthesis was monitored by scintillation counting of 32P-dCMP incorporated into TCA precipitated nucleic acid. Results shown are the average of 2-3 experiments for each sample.
Supplementary Material
Supplementary Fig. 1. The plasmid pSIL-CRM1A efficiently decreases cellular CRM1 activity. (A) Sequences incorporated into the pSilencer™ 4.1-CMV puro vector and the hairpins produced by each sequence upon transcription. The exons within the CRM1 gene targeted by each construct are indicated under the construct's name. (B) Evaluation of the effectiveness of the different pSIL constructs to decrease the cellular levels of CRM1. The assessment performed involved a functional assessment rather that an overall measure of total CRM1 in cell extracts. To this end, cells were transfected with the reporter construct pRev1.4(NES3)GFP coding for the REV-GFP fusion protein described in the paper. The nuclear export of REV-GFP is fully dependent on CRM1, and in the presence of Act D is more efficient than its nuclear import. Transfection with pSIL-CRM1B or a pSIL construct carrying a scrambled sequence failed to alter the cellular localization of REV-GFP. In contrast, transfection with pSIL-CRM1A produced a significant increase in the number of cells exhibiting nuclear localization of the REV-GFP reporter, thus indicating that pSIL-CRM1A had efficiently decreased cellular CRM1 levels. (C) Quantification of the data presented in (B). The data is presented as percentage of cells exhibiting almost exclusively nuclear (Nuc>Cyt), almost exclusively cytosolic (Nuc<Cyt), or equal nuclear and cytosolic (Nuc=Cyt) distribution of the REV-GFP reporter. In each case, over 500 cells were counted.
Acknowledgments
We sincerely acknowledge Dr. S. Swaminathan, University of Texas Medical Branch, Galveston, Texas, for the pcDNA3-CRM1 plasmid; Dr. B. Henderson, The University of Sydney, Australia, for the pRev(1.4)-GFP and pRev1.4(NES3)-GFP plasmids; and Dr. B.R. Cullen, Duke University, for the GST-Crm1 plasmid. We thank Adeline Deyrieux and Ashley Stone for assisting with the in vitro DNA replication assay. This work was supported by grant CA89298 from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amin AA, Titolo S, Pelletier A, Fink D, Cordingley MG, Archambault J. Identification of domains of the HPV11 E1 protein required for DNA replication in vitro. Virology. 2000;272(1):137–150. doi: 10.1006/viro.2000.0328. [DOI] [PubMed] [Google Scholar]
- Auster AS, Joshua-Tor L. The DNA-binding domain of human papillomavirus type 18 E1 - Crystal structure, dimerization, and DNA binding. J Biol Chem. 2004;279(5):3733–3742. doi: 10.1074/jbc.M311681200. [DOI] [PubMed] [Google Scholar]
- Bian XL, Rosas-Acosta G, Wu YC, Wilson VG. Nuclear import of bovine papillomavirus type 1 E1 protein is mediated by multiple alpha importins and is negatively regulated by phosphorylation near a nuclear localization signal. J Virol. 2007;81(6):2899–2908. doi: 10.1128/JVI.01850-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle SM, Ruvolo V, Gupta AK, Swaminathan S. Association with the cellular export receptor CRM 1 mediates function and intracellular localization of Epstein-Barr virus SM protein, a regulator of gene expression. J Virol. 1999;73(8):6872–6881. doi: 10.1128/jvi.73.8.6872-6881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor MK, Kotchetkov R, Cariou S, Resch A, Lupetti R, Beniston RG, Melchior F, Hengt L, Slingerland JM. CRM1/Ran-mediated nuclear export of p27Kip1 involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell. 2003;14:201–213. doi: 10.1091/mbc.E02-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng WT, Lin BY, Jin G, Wheeler CG, Ma TL, Harper JW, Broker TR, Chow LT. Cyclin/CDK regulates the nucleocytoplasmic localization of the human papillomavirus E1 DNA helicase. J Virol. 2004;78(24):13954–13965. doi: 10.1128/JVI.78.24.13954-13965.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enemark EJ, Chen G, Vaughn DE, Stenlund A, Joshua-Tor L. Crystal structure of the DNA binding domain of the replication initiation protein E1 from papillomavirus. Mol Cell. 2000;6(1):149–158. [PubMed] [Google Scholar]
- Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442(7100):270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- Enemark EJ, Stenlund A, Joshua-Tor L. Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J. 2002;21(6):1487–1496. doi: 10.1093/emboj/21.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelsma D, Bernad R, Calafat J, Fornerod M. Supraphysiological nuclear export signals bind CRM1 independently of RanGTP and arrest at Nup358. EMBO J. 2004;23(18):3643–52. doi: 10.1038/sj.emboj.7600370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornerod M, Vandeursen J, Vanbaal S, Reynolds A, Davis D, Murti KG, Fransen J, Grosveld G. The human homologue of yeast crm1 is in a dynamic subcomplex with can/nup214 and a novel nuclear pore component nup88. EMBO J. 1997;16(4):807–816. doi: 10.1093/emboj/16.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J, Dasso M. Association of the human SUMO-1 protease SENP2 with the nuclear pore. J Biol Chem. 2002;277(22):19961–19966. doi: 10.1074/jbc.M201799200. [DOI] [PubMed] [Google Scholar]
- Henderson BR, Eleftheriou A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp Cell Res. 2000;256(1):213–224. doi: 10.1006/excr.2000.4825. [DOI] [PubMed] [Google Scholar]
- Hsu CY, Mechali F, Bonne-Andrea C. Nucleocytoplasmic shuttling of bovine papillomavirus E1 helicase downregulates viral DNA replication in S phase. J Virol. 2007;81(1):384–394. doi: 10.1128/JVI.01170-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindsmuller K, Groitl P, Hartl B, Blanchette P, Hauber J, Dobner T. Intranuclear targeting and nuclear export of the adenovirus E1B-55K protein are regulated by SUMO1 conjugation. Proc Natl Acad Sci USA. 2007;104(16):6684–6689. doi: 10.1073/pnas.0702158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA. 1999;96(16):9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutay U, Guttinger S. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 2005;15(3):121–4. doi: 10.1016/j.tcb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Leng X, Wilson VG. Genetically defined nuclear localization signal sequence of bovine papillomavirus E1 protein is necessary and sufficient for the nuclear localization of E1-Beta-galactosidase fusion proteins. J Gen Virol. 1994;75(9):2463–2467. doi: 10.1099/0022-1317-75-9-2463. [DOI] [PubMed] [Google Scholar]
- Lentz MR, Pak D, Mohr I, Botchan MR. The E1 replication protein of bovine papillomavirus type 1 contains an extended nuclear localization signal that includes a p34cdc2 phosphorylation site. J Virol. 1993;67:1414–1423. doi: 10.1128/jvi.67.3.1414-1423.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DC, Sweitzer TD, Hanover JA. Reconstitution of HIV-1 rev nuclear export: independent requirements for nuclear import and export. Proc Natl Acad Sci USA. 1998;95(18):10608–13. doi: 10.1073/pnas.95.18.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludes-Meyers JH. Dissertation. Texas A&M Univeristy; College Station, Texas: 1995. [Google Scholar]
- Panse VG, Kuster B, Gerstberger T, Hurt E. Unconventional tethering of Ulp1 to the transport channel of the nuclear pore complex by karyopherins. Nature Cell Biol. 2003;5(1):21–27. doi: 10.1038/ncb893. [DOI] [PubMed] [Google Scholar]
- Pichler A, Gast A, Seeler JS, Dejean A, Melchior F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002;108(1):109–120. doi: 10.1016/s0092-8674(01)00633-x. [DOI] [PubMed] [Google Scholar]
- Rangasamy D, Wilson VG. Bovine papillomavirus E1 protein is sumoylated by the host cell Ubc9 protein. J Biol Chem. 2000;275(39):30487–30495. doi: 10.1074/jbc.M003898200. [DOI] [PubMed] [Google Scholar]
- Rangasamy D, Woytek K, Khan SA, Wilson VG. SUMO-1 modification of bovine papillomavirus E1 protein is required for intranuclear accumulation. J Biol Chem. 2000;275:37999–38004. doi: 10.1074/jbc.M007777200. [DOI] [PubMed] [Google Scholar]
- Rosas-Acosta G, Langereis MA, Deyrieux A, Wilson VG. Proteins of the PIAS family enhance the sumoylation of the papillomavirus E1 protein. Virology. 2005;331(1):190–203. doi: 10.1016/j.virol.2004.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobko A, Ma H, Firtel RA. Regulated SUMOylation and ubiquitination of DdMEK1 is required for proper chemotaxis. Developmental Cell. 2002;2(6):745–756. doi: 10.1016/s1534-5807(02)00186-7. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Mizoi J, Toh-e A, Kikuchi Y. Yeast Ulp1, an Smt3-specific protease, associates with nucleoporins. Yeast. 2000;128(5):723–725. doi: 10.1093/oxfordjournals.jbchem.a022807. [DOI] [PubMed] [Google Scholar]
- Titolo S, Brault K, Majewski J, White PW, Archambault J. Characterization of the minimal DNA binding domain of the human papillomavirus E1 helicase: Fluorescence anisotropy studies and characterization of a dimerization-defective mutant protein. J Virol. 2003;77(9):5178–5191. doi: 10.1128/JVI.77.9.5178-5191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titolo S, Pelletier A, Pulichino AM, Brault K, Wardrop E, White PW, Cordingley MG, Archambault J. Identification of domains of the human papillomavirus type 11 E1 helicase involved in oligomerization and binding to the origin. J Virol. 2000;74:7349–7361. doi: 10.1128/jvi.74.16.7349-7361.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassileva MT, Matunis MJ. SUMO modification of heterogeneous nuclear ribonucleoproteins. Mol Cell Biol. 2004;24(9):3623–3632. doi: 10.1128/MCB.24.9.3623-3632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang HY. A rapid preparation of extracts for DNA replication in vitro. Rad Res. 1999;151(1):59–62. [PubMed] [Google Scholar]
- Wilson VG, West M, Woytek K, Rangasamy D. Papillomavirus E1 proteins: Form, function, and features. Virus Genes. 2002;24(3):275–290. doi: 10.1023/a:1015336817836. [DOI] [PubMed] [Google Scholar]
- Wolff B, Sanglier JJ, Wang Y. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol. 1997;4(2):139–47. doi: 10.1016/s1074-5521(97)90257-x. [DOI] [PubMed] [Google Scholar]
- Wood LD, Irvin BJ, Nucifora G, Luce KS, Hiebert SW. Small ubiquitin-like modifier conjugation regulates nuclear export of TEL, a putative tumor suppressor. Proc Natl Acad Sci USA. 2003;100(6):3257–3262. doi: 10.1073/pnas.0637114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Lin BY, Deng WT, Broker TR, Chow LT. Mitogen-activated protein kinases activated the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J Virol. 2007;81(10):5066–5078. doi: 10.1128/JVI.02480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Saitoh H, Matunis MJ. Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol Cell Biol. 2002;22(18):6498–6508. doi: 10.1128/MCB.22.18.6498-6508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. The plasmid pSIL-CRM1A efficiently decreases cellular CRM1 activity. (A) Sequences incorporated into the pSilencer™ 4.1-CMV puro vector and the hairpins produced by each sequence upon transcription. The exons within the CRM1 gene targeted by each construct are indicated under the construct's name. (B) Evaluation of the effectiveness of the different pSIL constructs to decrease the cellular levels of CRM1. The assessment performed involved a functional assessment rather that an overall measure of total CRM1 in cell extracts. To this end, cells were transfected with the reporter construct pRev1.4(NES3)GFP coding for the REV-GFP fusion protein described in the paper. The nuclear export of REV-GFP is fully dependent on CRM1, and in the presence of Act D is more efficient than its nuclear import. Transfection with pSIL-CRM1B or a pSIL construct carrying a scrambled sequence failed to alter the cellular localization of REV-GFP. In contrast, transfection with pSIL-CRM1A produced a significant increase in the number of cells exhibiting nuclear localization of the REV-GFP reporter, thus indicating that pSIL-CRM1A had efficiently decreased cellular CRM1 levels. (C) Quantification of the data presented in (B). The data is presented as percentage of cells exhibiting almost exclusively nuclear (Nuc>Cyt), almost exclusively cytosolic (Nuc<Cyt), or equal nuclear and cytosolic (Nuc=Cyt) distribution of the REV-GFP reporter. In each case, over 500 cells were counted.