

Abstract
Adiponectin and leptin are two adipokines secreted by white adipose tissue that regulate insulin sensitivity. Previously we reported that adiponectin but not leptin release depends on GGA-coated vesicle formation, suggesting that leptin and adiponectin may follow different secretory routes. Here we have examined the intracellular trafficking pathways that lead to the secretion of these two hormones. While adiponectin and leptin displayed distinct localization in the steady-state, treatment of adipocytes with brefeldin A inhibited both adiponectin and leptin secretion to a similar level, indicating a common requirement for class III ADP-ribosylating factors and an intact Golgi apparatus. Adiponectin secretion was significantly reduced by endosomal inactivation in both 3T3L1 and rat isolated adipocytes, whereas this treatment had no effect on leptin secretion. Importantly, endosomal inactivation completely abolished the insulin stimulatory effect on adiponectin release in rat adipocytes. Confocal microscopy studies revealed colocalization of adiponectin with endogenous rab11 a marker for the recycling endosome, and with expressed rab5-GFP mutant (rab5Q75L) a marker for the early endosome compartment. Colocalization of adiponectin and rab5Q75L was increased in endosome inactivated cells. Consistent with these findings adiponectin secretion was reduced in cells expressing mutants of Rab11 and Rab5 proteins. In contrast, expression of an inactive (kinase dead) mutant of Protein Kinase D1 moderately but significantly inhibited leptin secretion without altering adiponectin secretion. Taken together, these results suggest that leptin and adiponectin secretion involve distinct intracellular compartments and that endosomal compartments are required for adiponectin but not for leptin secretion.
Keywords: adipokine, adiponectin, leptin, secretion, trafficking, adipocyte
INTRODUCTION
Adiponectin and leptin are two hormones secreted by white adipose tissue that regulate insulin sensitivity and energy balance [1–3]. Adiponectin, functions in vivo as an insulin sensitizer [4–7], reducing glucose production by the liver [8] and enhancing fatty acid oxidation in skeletal muscle [6]. Leptin acts in the hypothalamus to regulate food intake and energy expenditure (reviewed in [9–11]) and also activates fatty acid oxidation in skeletal muscle through the stimulation of the enzyme AMP-dependent kinase (AMPK) [12].
Serum levels of leptin correlate with adipose cell mass and BMI [13–16]. Leptin levels are also increased by feeding and decreased by fasting. Leptin synthesis and secretion is stimulated by insulin [17–20], glucocorticoids [18, 21–23], glycolytic substrates and amino acids [24] and inhibited by β-adrenergic agonists [25–27]. Insulin is thought to increase both transcription of the leptin gene and secretion [18]. Like leptin, adiponectin secretion is stimulated by insulin [28, 29] and reduced in the fasting state. However, in contrast to leptin, adiponectin serum levels are negatively correlated with obesity and fat cell size [15], and peroxisome proliferative activated receptor (PPAR) gamma agonists increase adiponectin production and secretion [30–32]. Adiponectin is known to be secreted in different oligomeric forms [33, 34], with the highest oligomeric form being the most biologically active [33, 34].
While substantial information exists on the physiological effects of leptin and adiponectin, the precise intracellular compartmentalization and trafficking pathways leading to the secretion of these two hormones and the molecular components that mediate the traffic of these adipokines are still poorly understood. Our previous observations [35] and the work by others [36] support the hypothesis that adiponectin is secreted following synthesis at the endoplasmic reticulum, and processing in the Golgi/trans-Golgi network. We have recently reported that at the trans-Golgi network, formation and exit of adiponectin-containing vesicles is dependent on the formation of Golgi localizing γ-ear adaptor 1 (GGA1) coated vesicles, while the secretion of leptin is independent of GGA1 coat formation [35]. These results suggested that these two hormones may be located in distinct intracellular compartments and may follow distinct trafficking pathways. Indeed, the intracellular compartmentalization of leptin in adipocytes has remained controversial. While several studies [17, 37] have reported that in rat adipocytes a substantial amount of leptin is localized in the endoplasmic reticulum in the steady state, other studies suggest that the majority of leptin is accumulated in distinct small intracellular vesicle compartments [38]. Recent fluorescence microscopy studies provided evidence that a portion of leptin is localized in the endoplasmic reticulum, Golgi apparatus and also in small intracellular vesicular vesicles [37].
In the present study, we aimed to elucidate the intracellular compartmentalization and trafficking pathways of these two hormones in 3T3L1 and isolated rat adipocytes. We report here that adiponectin and leptin have distinct intracellular localization in their steady state and adiponectin but not leptin release occurs via endosomal compartments in both 3T3L1 and isolated rat adipocytes. Moreover, insulin-mediated stimulation of adiponectin release is completely blocked by endosomal inactivation in isolated rat adipocytes suggesting that insulin acts at the level of endosomes to enhance adiponectin release. Further, we found that expression of a kinase dead mutant of the enzyme protein kinase D1 selectively inhibits leptin but not adiponectin release. Taken together, these data support the hypothesis that these two proteins take divergent trafficking pathways at the trans-Golgi network, and thus, while adiponectin release is dependent on intact recycling and early endosome compartments, leptin secretion is independent of the endosomal system.
MATERIALS AND METHODS
Materials and antibodies
Brefeldin A, DAB, BSA were obtained from Sigma.Anti-myc antibody (Clone 9E11) was obtained from Santa Cruz Biotechnology, anti-HA antibody was from Sigma, anti-rab5 and anti-rab 11 were from BD Biosciences, anti-adiponectin antibody was a gift from Dr. P. Scherer (Albert Einstein, NY), and anti-transferrin receptor was obtained from Zymed. Transferrin-HRP was acquired from Accurate Chemical & Scientific Corp. Dulbecco’s Modified Eagle Media, Opti-Minimum Essential Media, Fetal Bovine Serum, Calf Serum and Trypsin were purchased from Invitrogen. Collagenase type I was obtained from Worthington pharmaceuticals.
Generation of DNA constructs
The construct encoding for human adiponectin was generated by RT-PCR and subcloned in the pcDNA3 vector (Invitrogen) incorporating a myc tag at the C-terminus. The clone coding for the mouse leptin was subcloned into the pcDNA 3 vector (Invitrogen) and an HA (Haemagglutinin) tag was added in frame in the C-terminus. Identity and positioning of the clones was confirmed by sequencing. The plasmid encoding for GFP tagged syntaxin 6 protein was a gift from Dr. J. Pessin (SUNY, Stonybrook). Plasmids encoding the GFP tagged wild type or K618N mutant of the protein kinase D1 were provided by Dr. V. Malhotra (UCSD). Plasmids encoding the GFP-rab5 wild type, Q79L and S34N mutants were provided by Dr. B. Ceresa (University of Oklahoma). Plasmids encoding the GFP-rab11 Q70L and S25N mutants were provided by Dr. M.McCaffrey (University College Cork).
Animals and adipocyte isolation
Sprague-Dawley rats (160–200g) were obtained from Charles River. Animals were housed in a 12 hr light/dark cycle and fed ad libitum. Animals were euthanized by carbon dioxide asphyxiation and the epidydimal adipose pads were quickly removed. Isolation of adipocytes was performed as previously described [39]. The experimental protocol was examined and approved by the Kansas State University Institutional Committee for the Use of animals.
Cell culture and transient transfection of 3T3L1 adipocytes was performed as previously reported [35].
Pull-down Assay and Western Blot Analysis was performed as described [35].
Immunofluorescence and image analysis
Cells were washed in phosphate buffered saline (PBS) and were fixed, permeabilized and immunostained as described [35].
Inactivation of the endosomal compartments
Endosomal ablation of 3T3-L1 cells was performed as described previously [40]. Endosomal ablation of isolated rat adipocytes was performed with a minor modification. Briefly, after isolation adipocytes were incubated in Krebs-ringer solution for 1.5 h at 37°C. Cells were then incubated in Krebs-ringer solution containing Tfn-HRP (20 µg/ml) for 1.5 h at 37°C. Cells were then washed three times with isotonic citrate buffer (150 mM NaCl, 200 mM Sodium Citrate, pH= 5.0) for 10 minutes each time and once with Krebs-ringer solution. Krebs-ringer solution containing 200µg/ml DAB and 0.04% H2O2 pH=7.2, was then added to the cells. Following a 60 min. incubation at 37°C in the dark, the reaction was stopped by washing with warm Krebs-Ringer containing 0.5% BSA. Cells were then incubated in Krebs-Ringer for 2 hours. When indicated insulin was added at a final concentration of 20 nM.
Sucrose gradient centrifugation of adipocytes
Cells were lysed in ice-cold HES buffer (0.25M glucose, 20 mM HEPES, pH 7.4, 1 mM EDTA and 150 mM NaCl) containing protease inhibitors: 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml aprotinin, 1 µg/ml leupeptin, and 1 µg/ml pepstatin. Cells were homogenized with 5 strokes on a glass Dounce homogenizer. Homogenates were centrifuged at 2,095 × g for 15 min. and the supernatants were collected and loaded onto a discontinuous sucrose gradient containing: 1.5M, 1.3M, 1.1M, 1.0M, 0.9M, 0.8M, 0.7M, 0.6M and 0.5M sucrose in HES. Samples were then centrifuged at 75,000 × g for 24h at 4°C and 0.5 ml fractions recovered from the top of the tube. Equal volumes of each fraction were separated by SDS-PAGE and transferred to nitrocellulose filters for immunoblotting.
Determination of adipokine secretion by ELISA
ELISA kits specific for adiponectin or leptin were obtained from R&D Systems (Minnesota). Prior to the ELISA, floating cells were removed by a brief centrifugation at 500 × g for 5 min and an aliquot of the supernatant was used for the quantification. In parallel, an aliquot of the whole cell lysate was also quantified. Adipokine secretion was calculated as a percentage of total adipokine content as follows: adipokine in the media/ total adipokine (= media + lysate) × 100. To detect adiponectin-myc a modified sandwich ELISA protocol was used with an antihuman adiponectin antibody (DY1065 from R&D Systems at a final concentration of 2µg/ml) and an antibody anti-myc conjugated to biotin (Sigma, Cat. №: B7554) as a capture antibody.
Statistical Analysis
A one way ANOVA was performed using SAS statistical software. Statistical significance was considered if p<0.05.
RESULTS
Adiponectin and leptin secretion require an intact Golgi apparatus
Our previous findings demonstrated the requirement of the formation of GGA1-coated vesicles at the trans-Golgi network for adiponectin release but not for leptin release [35]. This suggested that the intracellular trafficking pathways for the secretion of these hormones may be different. To test this hypothesis we determined by confocal fluorescence microscopy the steady-state intracellular compartmentalization of these two hormones in adipocytes. Differentiated 3T3L1 cells were electroporated with either a myc-tagged adiponectin or HA-tagged leptin constructs to allow for the detection of these proteins using the tag specific antibodies. This was especially important for leptin, since in our hands, the endogenous expression in 3T3L1 cells is very low. After expression of these proteins, cells were fixed permeabilized and immunostained with anti-tag and antibodies against well known intracellular markers. We found that while adiponectin-myc was localized in the perinuclear region [35] and significantly colocalized with syntaxin 6 (Fig1A panels a–c), leptin displayed no colocalization with syntaxin 6 (Fig. 1B panels a–c). Leptin distribution overlapped with the endoplasmic reticulum marker BIP (Fig 1B, panels d–f) whereas the majority of adiponectin-myc was not colocalized with this marker (Fig. 1A. panels d–f).
Fig. 1. Localization of adiponectin and leptin in 3T3L1 adipocytes.

Fully differentiated adipocytes were electroporated with a construct coding for either adiponectin-myc (panel A) or leptin-HA (panel B). Cells were then fixed, permeabilized, and immunostained with a specific antibody anti-myc and a secondary antibody conjugated to Texas red (A, panels a and d) , or a primary anti-HA antibody and a secondary Texas red-conjugated antibody (B, panels a and d) or with an antibody specific for the trans-Golgi network marker syntaxin 6 (A and B, panel b), the ER marker BIP (A and B, panel e). The merged images are shown in the right panels. The yellow color in the merged images (A and B, panels c and f) indicate colocalization. Images were obtained with a Zeiss 510 META confocal microscope.
Despite having substantially different steady-state localization, it is possible that both hormones traffic through the same secretory pathway. To examine whether a functional Golgi apparatus is required for leptin secretion, fully differentiated 3T3L1 adipocytes expressing leptin-HA were either left untreated or treated with the fungal metabolite brefeldin A (BFA) for 2 hrs. Since BFA inhibits guanidyl nucleotide exchange factors that act on ARF-GTP binding proteins in the Golgi [41], this inhibition causes a block in the anterograde Golgi trafficking and thereby causes the collapse of the Golgi stacks back into the endoplasmic reticulum [42, 43]. Following treatment, the amount of secreted leptin-HA (or adiponectin as control) in the conditioned media was measured by ELISA. As shown in Fig.2A. Brefeldin A significantly inhibited the secretion of both adiponectin and leptin-HA to 53.0 ± 6.4% and 65.0 ± 6.6%, respectively, in 3T3L1 adipocytes. Similar results were obtained in isolated rat adipocytes treated in vitro with BFA (Fig 2.B), where an inhibition of 61.0 ±3.5 % and 64.0 ± 8.4% was seen for endogenous adiponectin and leptin respectively. These findings demonstrate that blockage of class I ADP-ribosylating factors (ARFs) in the secretory pathway significantly inhibits secretion of both hormones and suggest that both leptin and adiponectin traffic through the Golgi and trans-Golgi network.
Fig. 2. Adiponectin and leptin secretion are inhibited by BFA.

A) Secretion in 3T3L1 adipocytes. Adiponectin secretion (white bars): Fully differentiated 3T3L1 adipocytes were trypsinized and replated in 12-well dishes to distribute the same number of cells per dish. Two hours later the medium was changed to DMEM without serum in the absence (control) or presence of 5 µg/ml BFA. Cells were incubated for additional 4hrs, and an aliquot of media and cell lysate were then taken for adiponectin content quantification by ELISA as indicated in the methods section. Leptin secretion (black bars): Fully differentiated adipocytes were electroporated with a construct coding for leptin-HA and replated in 12-well dishes. 18 h following the electroporation the media was changed to DMEM without BFA, or to DMEM supplemented with 5 µg/ml BFA. Four hours later, media and whole cell lysates were obtained and an aliquot quantified by ELISA as described in the methods section. The amount of secreted adipokine (expressed as % of total adipokine) was calculated as described in the methods section and compared to the value obtained in control cells (no BFA added). The graphs represent the percentage of adiponectin or leptin secreted vs. control adipocytes (−BFA) ± S.E. obtained from three independent experiments (n=18). B) Secretion in isolated rat adipocytes: Isolated rat adipocytes were obtained as described in the methods section. Equal volumes of packed cells were distributed in separate tubes and incubated in Krebs-Ringer buffer in the absence or presence of 5 µg/ml BFA for 2 hours. A small aliquot of medium was taken to independently determine the amount of adiponectin and leptin by ELISA. Cell lysates were prepared and an aliquot quantitated for the amount of each adipokine by ELISA. The amount of secreted adipokine (expressed as % of total adipokine content) was calculated as described in the methods section and compared to the value obtained for control adipocytes (no BFA treated). The graphs represent the percentage of adiponectin or leptin secreted vs. control adipocytes (−BFA) ± S.E. obtained from three independent experiments, each performed in triplicate. Differences were considered significantly different when p < 0.001 and is indicated by an asterisk.
Adiponectin but not leptin secretion requires intact endosomes
We have previously reported that adiponectin release requires the function of GGA1 adaptor proteins [35]. Since GGA proteins have been reported to direct traffic of selective cargo from the trans-Golgi network to the endosomes [44], we hypothesized that adiponectin secretion may occur via the endosomal system. To test this hypothesis, fully differentiated 3T3L1 adipocytes and rat isolated adipocytes were subjected to the endosomal inactivation procedure described by Livingstone et al. [40]. As a control, a subset of cells were incubated in the presence of HRP-conjugated transferrin (Tfn-HRP) but without the substrate DAB. Following the endosome inactivation procedure, both the control and endosome inactivated cells were lysed and fractionated to equilibrium in a sucrose gradient ultracentrifugation as described in the methods section. The fractions were separated by SDS-PAGE transferred to a nitrocellulose membrane and immunoblotted with the endosomal markers: transferrin receptor, rab5 (an early endosome marker) and rab 11 (a recycling endosome). As shown in Fig. 3A, in control cells where the DAB was omitted, transferrin receptor, rab 5 and rab 11, were revealed in light to medium sucrose density fractions. Under conditions where cells were incubated in the presence of the HRP substrates DAB and H2O2, endosomal inactivation was made evident by the presence of these proteins in the heaviest fractions (pelleted material). For 3T3L1 cells (Fig 3A, top panel) a nearly complete endosomal inactivation was observed. To confirm that the endosome inactivation procedure did not affect other non-endosome compartments, samples were immunoblotted with an antibody for the trans-Golgi network marker syntaxin 6. The localization of syntaxin 6 was unchanged after the endosomal inactivation In addition, the endosomal inactivation procedure did not alter the overall cell morphology or affect the intracellular steady-state localization of adiponectin-myc and leptin-HA in these cells (data not shown). These results were reproduced in rat isolated adipocytes (Fig. 3A lower panel).
Fig. 3. Secretion of adiponectin but not leptin is inhibited by endosomal inactivation.

A) Distribution of membranes containing transferrin receptor, rab5, rab11 and syntaxin 6 (syn6) in control cells (−) or cells subjected to endosomal inactivation (+). Fully differentiated 3T3L1 cells (top panel) or rat isolated adipocytes (bottom panel) were subjected to the endosomal inactivation procedure as described in the methods section. As a control, a subset of cells was subjected to the same procedure without the substrates DAB and H2O2. Total internal membranes were prepared and separated on a discontinuous sucrose gradient as described in the methods section. Fractions of 0.5 ml were collected from the top of the tube. For each fraction, an aliquot of 50 µl was separated by SDS/PAGE, transferred to a nitrocellulose filter and immunoblotted with specific antibodies to detect transferring receptor, rab5, rab11 and syntaxin 6 as indicated. Fraction 13 was the pellet at the bottom of the gradient. B) Secretion of adiponectin (top) and leptin (bottom) in 3T3L1cells is inhibited by endosomal inactivation. Cells were subjected to the endosome inactivation technique as described in the methods section using different dosages of DAB as indicated. As a control, a subset of cells were either left untreated or treated with H2O2 only, DAB only, or transferrin-HRP only as indicated. After endosomal inactivation, cells were incubated in serum free DMEM for 4 hours at 37°C. At that time an aliquot of media was harvested and whole cell lysates prepared and an aliquot of each was quantitated by ELISA for the presence of adiponectin. The graphs display the amount of secreted adiponectin ± S.E. obtained from three independent experiments, each experiment was performed in triplicate. Differences were considered significantly different when p< 0.05 and is indicated with an asterisk. C) Secretion of adiponectin but not leptin in isolated rat adipocytes is inhibited by endosomal inactivation. Isolated epidydimal rat adipocyte cells were obtained by collagenase digestion as described in the methods section. An equal volume of packed cells were incubated in Krebs-Ringer buffer containing transferrin-HRP. A subset of cells were then treated with DAB and H2O2 as described in the methods section to inactivate the endosomal compartments. A subgroup of adipocytes was further incubated in the absence or presence of 20 nM insulin at 37°C for 2 hrs. After this time, the conditioned media and a cell lysate were obtained and quantitated with adiponectin or leptin specific ELISA kits. The amount of adiponectin and leptin secretion was calculated as a % of total adipokine content as indicated in the methods section. The results shown are the secretion percentage values respective to the secretion measured in control cells (no endosomal inactivation) without insulin and are the mean ± S.E., representing data from three independent experiments and each experiment was performed in triplicate. Differences were considered significantly different when p < 0.05 and is indicated with an asterisk.
We next determined whether adiponectin and leptin secretion are affected by endosome inactivation in 3T3L1 adipocytes. As shown in Fig. 3B (top panel) endosomal ablation significantly reduced the amount of adiponectin secreted. This effect was DAB dose dependent (Fig. 3B, top panel). In contrast, leptin HA secretion in was not affected (Fig.3B, lower panel). To determine whether endosomes are important for adiponectin or leptin secretion in rat-isolated adipocytes and to investigate whether insulin-stimulated release of these hormones is dependent on the endosomal system, the same experiments were performed in isolated rat adipocytes. Cells were incubated with transferrin-HRP and either vehicle or DAB. Following the ablation, a subset of cells were either left untreated or treated with 20 nM of insulin and incubated for 2 hrs. Aliquots of media were then taken and quantitated by ELISA for adiponectin or leptin. As shown in Fig. 3C, insulin stimulated adiponectin release by 1.5 fold (+INS group) whereas endosomal inactivation (+DAB) significantly inhibited adiponectin secretion. Interestingly, insulin-mediated activation of adiponectin release was completely abolished by endosomal inactivation (Fig 3C, +DAB+INS group). These results suggest that insulin stimulates adiponectin release by increasing membrane traffic through the endosomal system. Insulin caused a small but significant increase of leptin secretion (Fig 3C, +INS group) but in contrast to adiponectin, leptin secretion was not affected by endosomal inactivation (Fig. 3C, +DAB group). Further, insulin-mediated increase in leptin secretion was not affected by endosomal ablation (Fig.3C, +DAB+INS group).
Recycling and early endosomes are involved in adiponectin secretion
We next identified which endosomal compartments participate in adiponectin trafficking. We analyzed by confocal fluorescence microscopy the localization of adiponectin-myc and of endogenous rab11 and rab 5 proteins or expressed GFP-tagged wild type and constitutively active/inactive mutants of rab5. We found that adiponectin-myc partially colocalized with the endogenous rab 5 (data not shown) or with expressed wild type GFP-rab5 (Fig 4A, panels a–c). Adiponectin-myc could be detected in enlarged early endosomes generated by the expression of the constitutively active form of rab5 (rab5Q79L) (Fig.4A. panels d–f). Increased colocalization of adiponectin-myc with GFP-Rab Q79L was observed in cells subjected to the endosomal inactivation (Fig 4B, panels d–f) compared to cells with functional endosomes (Fig.4B, panels a–c). We also found significant colocalization between adiponectin-myc and endogenous rab11 (Fig 4A, panels g–i).
Fig. 4. Adiponectin colocalizes with endosomal markers.

A) Fully differentiated adipocytes were coelectroporated with a construct coding for adiponectin-myc and either a construct coding for a GFP-tagged wild type rab5 (panels a–c), or constitutive active mutant of rab5 (Rab5Q79L) (panel d–f). Cells were then fixed, permeabilized, and immunostained with a specific antibody for myc and a secondary antibody conjugated to Texas red (panels a, d and g), or for endogenous Rab11 and a secondary antibody conjugated to Alexa-488 (panel h)). The merged images are shown in the right panels (c–i). Images were obtained using a Zeiss 510 META confocal microscope. B) A fraction of adiponectin-myc is retained in the early endosome compartment after endosomal inactivation. Fully differentiated adipocytes expressing adiponectin-myc and the GFP-tagged rab5 Q75L protein were subjected to the endosomal inactivation. Upper panels (a to c) show the control group without DAB, lower panels (d–f) show the endosome inactivated cells. Cells were then fixed, permeabilized, and immunostained with a specific antibody for myc and a secondary antibody conjugated to Texas red (panels a, and d). The merged images are shown in the right panels (c–f). Images were obtained using a Zeiss 510 META confocal microscope. C) Recombinant GST- GGA1 protein precipitates vesicles containing adiponectin and rab 11. Pull down assays were performed with recombinant GST or GST-GGA1VHS recombinant protein as described in the methods section. The pelleted samples and supernatants (SN) were separated by SDS-PAGE transferred to a nitrocellulose membrane and immunoblotted with an antibody specific for adiponectin, rab5 or rab11. A representative blot of five independent experiments is shown.
These results suggested that the recycling and early endosome compartments may form part of the secretory pathway for adiponectin in adipocytes. To confirm these results, we conducted biochemical and functional studies in 3T3L1 adipocytes. We have previously reported that a fraction of intracellular vesicles containing adiponectin can be precipitated with recombinant GGA1 protein fused to GST [35]. We conducted pull down assays of 3T3L1 intracellular membranes using either GST or GST-GGA1 recombinant proteins immobilized to glutathione-sepharose beads as we have described [35]. The pelleted samples were washed in PBS, separated by SDS-PAGE, transferred to a nitrocellulose membrane and immunoblotted with specific antibodies to detect endogenous adiponectin, rab5 and rab 11. The results displayed in Fig. 4C. show that, a substantial amount of rab11 was readily detected selectively in the precipitates containing adiponectin and the GST-GGA1 but not in the pellets of samples incubated with control GST recombinant protein. This result is consistent with the amount of colocalization between adiponectin-myc and rab11 detected in the immunofluorescence studies. However, we were unable to detect endogenous rab 5 in the GGA1 precipitates of the pull down assay (Fig.4B). The absence of rab5 in the precipitates could also originate from low sensitivity of the assay, since only a fraction of adiponectin-containing vesicles precipitated with GGA1. To complement these results, we performed functional studies to determine whether the expression of mutant rab 5 or rab11 proteins could affect the amount of secreted adiponectin in 3T3L1 adipocytes. As a control, we measured the secretion of leptin in the conditioned medium of these cells. The results expressed as a percentage of adiponectin or leptin secreted vs. total adipokine present in the cell was compared to that obtained in cells expressing either adipokine or an empty vector. As shown in Fig 5A the over expression of either rab11 mutant moderately (~25%) but significantly inhibited the secretion of adiponectin without affecting leptin release. A greater inhibition of adiponectin (~30% with rab5Q75L, ~64% with rab5S34N) was observed in cells expressing mutants of rab5. As expected, no change in leptin secretion was observed with either rab11 or rab5 mutants (Fig.5). Taken together, these results support the hypothesis that both the recycling and early endosome compartments participate in the secretion of adiponectin.
Fig. 5. Expression of rab5 and rab11 mutants inhibit adiponectin secretion in 3T3L1 cells.
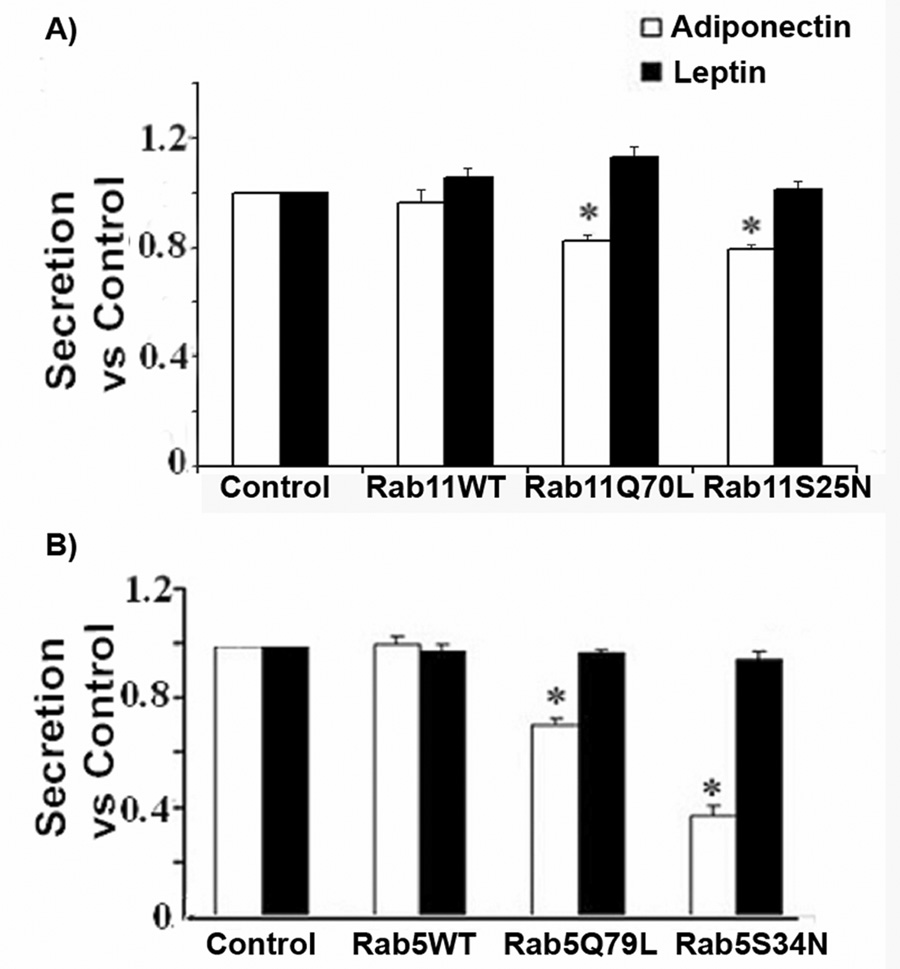
Fully differentiated 3T3L1 cells were electroporated with a construct expressing adiponectin-myc (white bars) or leptin-HA (black bars) and either an empty vector, a wild type or constitutive active/inactive mutants of rab 11 (Panel A) or rab5 (Panel B). Following expression of the proteins, media and whole cell lysates were harvested and an aliquot of each were quantified by ELISA as described in the methods section. The amount of secretion is shown as the ratio to the secretion obtained in control adipocytes expressing each adipokine and the empty vector ± S.E. The graph represents data from three independent experiments and 18 independent observations. Differences were considered significantly different when p < 0.05 and is indicated with an asterisk. Expression of adiponectin-myc, leptin-HA and rab proteins were confirmed by western blot analysis (data not shown). For this ELISA we utilized an adiponectin capture and detection antibodies, respectively, as described before [35].
Leptin but not adiponectin traffic uses protein kinase D1
Our results are consistent with the hypothesis that adiponectin and leptin take divergent trafficking pathways en route to the plasma membrane. Recent evidence in the literature suggests that the serine/threonine Protein Kinase D1, regulates fission at the trans-Golgi network of transport vesicles that deliver cargo to the plasma membrane [45, 46]. We hypothesized that if leptin secretion occurs directly from the TGN to the plasma membrane, its secretion would be inhibited by mechanisms that block activation of this enzyme. To test this hypothesis, we expressed a wild-type form of a GFP-tagged PKD1 or a GFP-kinase-dead mutant incorporating the substitution K618N in 3T3L1 adipocytes. This mutation renders the protein catalytically inactive but allows it to function as a dominant interfering mutant blocking the activation of the endogenous PKD1 [45, 47]. The localization of PKD1 proteins in the Golgi/TGN compartment was confirmed by confocal fluorescent microscopy and compared to the localization of adiponectin-myc. The results shown in Fig.5A demonstrate that GFP-PKD1 and GFP-PKD1-K618N localize in a perinuclear compartment (Fig.6A, panels b and e) and in close proximity to adiponectin-myc (Fig.6A, panels c and f). These results are consistent with the TGN localization of the expressed GFP-PKD1 proteins. We then determined whether expression of these PDK1 constructs would alter the secretion of co-expressed adiponectin-myc or leptin-HA. While we did not detect any change in the secretion of adiponectin-myc by expressing either of the GFP-PKD1 constructs (Fig.6B, white bars) there was a moderate but significant reduction in the amount of secreted leptin-HA in cells selectively expressing the PKD1 mutant (Fig.6B, black bars).
Fig. 6. Secretion of leptin, but not adiponectin is inhibited by a mutant of PKD1.

A) Colocalization of adiponectin and PKD1. Fully differentiated adipocytes were co-electroporated with a construct coding for adiponectin-myc and either a construct coding for a GFP-tagged wild type PKD (panels a–c), or a construct coding for the PKD1 dominant interfering mutant PKD1-K618N (panel d–f). Cells were then fixed, permeabilized and immunostained with a specific antibody anti-myc and a secondary antibody conjugated to Texas red (panels a and d). The merged images are shown in the right panels. Images were obtained using a Zeiss 510 META confocal microscope. B) A PKD1 mutant inhibits leptin but not adiponectin secretion in 3T3L1 cells. Cells were co-electroporated with a vector expressing adiponectin-myc or leptin-HA and either an empty vector, a construct coding for PKD1 wild type or a construct coding for the mutant PKD1-K618N. Following expression of the proteins, the conditioned media and whole cell lysates were obtained and an aliquot was taken for the adipokine quantification by ELISA as described in the methods section. The secreted adipokine was calculated in each group as a percentage of total adipokine content and compared to the secretion obtained in the control group expressing adipokine plus the empty vector. The graph shows adipokine secretion vs. control ± S.E, representing data from three independent experiments, each experiment performed in triplicate. Differences were considered significantly different when p<0.05 and is indicated with an asterisk. Adiponectin: White bars. Leptin: black bars.
DISCUSSION
In addition to its important role as a lipid storage depot, adipocytes play a key role in the control of energy balance and insulin sensitivity through the secretion of various hormones. Several studies have documented that secretion of these proteins occurs by both constitutive and regulated mechanisms. For example, it is well documented that insulin stimulates the secretion of leptin [17], adiponectin [48], and adipsin [49, 50]. The specific intracellular compartmentalization and trafficking of these hormones is poorly understood. In this report, we examined and compared the intracellular secretory pathways of two critical adipocyte hormones, adiponectin and leptin.
Biochemical and fluorescence microscopy studies have previously demonstrated that neither leptin [51] nor adiponectin [48] localize intracellularly with the well-studied intracellular compartment containing the insulin-responsive glucose transporter Glut4. Furthermore, the precise intracellular steady-state distribution of leptin in adipocytes has remained somewhat controversial. While initial studies [17] had reported that a substantial amount of leptin is found in the cortical endoplasmic reticulum of rat isolated adipocytes, other biochemical studies [38] had found that the distribution of leptin-containing vesicles separated by equilibrium sucrose density centrifugation was not consistent with that of the localization of endoplasmic reticulum markers. To reconcile these differences and to investigate the intracellular trafficking pathways required for leptin secretion in adipocytes, we first compared the intracellular steady-state distribution of expressed leptin-HA with that of another adipokine, adiponectin-myc in 3T3L1 adipocytes. Here we report that these hormones have distinct steady state localizations, whereas adiponectin is localized within the TGN overlapping with the marker syntaxin6, leptin is predominantly localized in close proximity of the endoplasmic reticulum marker BIP. Thus our results support those found by Barr et.al. [17] in rat adipocytes, and confirm that in the steady-state the majority of adiponectin and leptin are located in distinct intracellular compartments.
Despite displaying a distinct intracellular localization, treatment with brefeldin A, inhibited the secretion of both adipokines in either cultured adipocytes or rat isolated adipose cells (by 58% and 35% in 3T3L1 and 39% and 36% in rat adipocytes for adiponectin and leptin secretion respectively). This finding suggests that both adiponectin and leptin traffic via Golgi/TGN. Interestingly however, in both cell types, BFA did not completely inhibit the secretion of these adipokines, indicating that the short term release of these hormones occurs partially through a BFA insensitive mechanism, and most probably, a post-TGN compartment which we hypothesize could represent the site for regulated secretion.
GGA adaptor proteins are important for the formation of adiponectin-containing vesicles at the TGN of adipocytes [35]. Since GGA proteins participate in the TGN to endosome traffic, we hypothesized that adiponectin secretion but not leptin secretion would involve the endosomal compartment(s). To test this hypothesis, we conducted biochemical experiments to selectively inactivate the endosomes of 3T3L1 adipocytes following the procedure first described by Livingstone et al., 1996 [40]. We also adapted this technique to isolate rat adipocytes. To our knowledge this is the first time that this technique has been applied to these cells. With this technique we achieved a complete inactivation of the endosomal compartments in 3T3L1 cells and a substantial (60%) inactivation of the endosomes in isolated rat adipocytes. Several reasons could account for this difference. First, transferrin-HRP internalization may be slower or less efficient than in 3T3L1 cells or alternatively, rat adipocytes may contain a larger endosomal compartment than 3T3L1 adipocytes which may require higher amounts of Tfn-HRP and DAB to achieve a complete inactivation. Attempts to increase the time of capture of transferrin-HRP or DAB and H2O2 substrate concentrations did not result in an increase in endosome inactivation in these cells. Furthermore, higher concentrations of H2O2 resulted in decreased viability of adipocytes (not shown). Nevertheless, our findings in rat isolated adipocytes corroborated the results obtained in the cell line 3T3L1. Endosomal inactivation profoundly reduced the amount of adiponectin secreted while leptin release remained unaffected., indicating that adiponectin but not leptin secretion requires functional endosomes.
To identify the endosomal compartments involved in adiponectin secretion we used confocal immunofluorescence and secretion studies in 3T3L1 adipocytes expressing adiponectin-myc and GFP-tagged wild type and/or constitutively active/inactive mutants of the endosomal markers rab11 and rab5. Immunofluorescence studies revealed adiponectin-myc distributed in close proximity to the endogenous rab 11 and rab5 proteins. in addition rab11 was detected in precipitated adiponectin-containing vesicles. Furthermore, over expression of a constitutively active or inactive forms of either rab11 or the rab5 proteins, modestly but significantly, inhibited adiponectin-myc release in 3T3L1 cells without affecting leptin release. Taken together, these findings strongly suggest that intact endosomal compartments are required for adiponectin but not leptin secretion. In agreement with these results, Gould’s group recently reported that a mutant of rab11 (rab11S25N) inhibited both basal and insulin-stimulated adiponectin secretion in 3T3L1 cells [52]. In line with this, our results in rat adipocytes strongly support the idea that insulin-stimulation of adiponectin release may be mediated via insulin-mediated activation of the endosomal membrane recycling. Similarly to adiponectin, adipsin, a serine protease released by adipocytes is also released via TGN to endosome traffic [50]. While the acute effects of insulin on leptin secretion have been reported to act at the level of secretion [18], our data support a model where insulin may increase secretion of adiponectin and leptin in different ways. It remains to be established whether a similar or distinct insulin intracellular signaling cascades are utilized to control secretion of each adipokine.
To further identify molecular regulators for leptin secretion we tested whether leptin secretion is dependent on Protein Kinase D1 activity, and enzyme that regulates the formation of TGN-derived vesicles en route to the plasma membrane [46, 53]. Over expression of a dominant interfering mutant PKD1 K618N exhibited a moderate but significant decrease in leptin release. This suggests the possibility that PKD1 may be involved in regulating at least partially, the trafficking of leptin-containing vesicles. Further experiments are required to test whether the PKD1 activity is required for the insulin-stimulated effect on leptin secretion.
Taken together, our results demonstrate that adiponectin and leptin are secreted through distinct intracellular trafficking pathways and suggest that adipocytes rely on different avenues for the constitutive and regulated secretion of these adipokines.
ACKNOWLEDGEMENTS
This work was supported by a JFA from the American Diabetes Association, N.I.H. grant P20-RR17708 and a research grant from Diabetes UK to S.M., N.I.H. Grant P20-RR017686 to the Confocal Microfluorometry and Microscopy Core at Kansas State University and N.I.H grants RR16475 and AI052206 and AES 08-162-J08 to S.K.C.We are thankful to Dr. J. Pessin (SUNY Stonybrook), Dr. V.Malhotra (UCSD), Dr. M. McCaffrey (University College Cork, Ireland) and Dr. B. Ceresa (University of Oklahoma) for providing the cDNAs used in this manuscript and to Dr. P. Schrerer (Albert Einstein, NY) for the anti-adiponectin antibody.
Abbreviations
- ARF
ADP-ribosylating factor
- BFA
Brefeldin A
- BSA
Bovine Serum Albumin
- DAB
3,3′-Diaminobenzidine
- GGA1
Golgi localizing γ-adaptin ear homology domain
- ARF
binding protein
- GST
Glutathione-S-transferase
- HRP
Horse-Radish Peroxidase
- PDK1
protein kinase D1
- PPAR
Peroxisome Proliferator-Activated receptor-γ agonist
- Tfn
Transferrin
- HRP
Horse radish peroxidase
- TGN
Trans-Golgi network
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Bradley R, Cleveland K, Cheatham B. The adipocyte as a secretory organ: mechanisms of vesicle transport and secretory pathways. Recent Prog Horm Res. 2001;53:329–358. doi: 10.1210/rp.56.1.329. [DOI] [PubMed] [Google Scholar]
- 2.Guerre-Millo M. Adipose tissue and adipokines: for better or worse. Diabetes Metab. 2004;30:13–19. doi: 10.1016/s1262-3636(07)70084-8. [DOI] [PubMed] [Google Scholar]
- 3.Klaus S. Adipose tissue as a regulator of energy balance. Curr Drug Targets. 2004;5:241–250. doi: 10.2174/1389450043490523. [DOI] [PubMed] [Google Scholar]
- 4.Yamanuchi T, Kamon J, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman M, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura A, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nature medicine. 2001;7:941–945. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 5.Berg A, Combs T, Du X, Brownlee M, Scherer P. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat.Med. 2001;7:847–853. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 6.Fruebis J, Tsao T, Javorschi S, Ebbets-Reed D, Erickson M, Yen F, Bihain B, Lodish H. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci, USA. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pajvani UB, Scherer PE. Adiponectin: systemic contributor to insulin sensitivity. Curr Diab Rep. 2003;3:207–213. doi: 10.1007/s11892-003-0065-2. [DOI] [PubMed] [Google Scholar]
- 8.Combs T, Berg A, Obici S, Scherer P, Rossetti L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J. Clin. Invest. 2001;108:1875–1881. doi: 10.1172/JCI14120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marti A, Berraondo B, Martinez JA. Leptin: physiological actions. J Physiol Biochem. 1999;55:43–49. [PubMed] [Google Scholar]
- 10.Moran O, Phillip M. Leptin: obesity, diabetes and other peripheral effects--a review. Pediatr Diabetes. 2003;4:101–109. doi: 10.1034/j.1399-5448.2003.00017.x. [DOI] [PubMed] [Google Scholar]
- 11.Trayhurn P. Leptin--a critical body weight signal and a "master" hormone? Sci STKE. 2003;169:1–3. doi: 10.1126/stke.2003.169.pe7. [DOI] [PubMed] [Google Scholar]
- 12.Minokoshi Y, Kim Y, Peroni O, Fryer L, Muller C, Carling D, Kahn B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 13.Couillard C, Mauriege P, Imbeault P, Prud'homme D, Nadeau A, Tremblay A, Bouchard C, Despres JP. Hyperleptinemia is more closely associated with adipose cell hypertrophy than with adipose tissue hyperplasia. Int J Obes Relat Metab Disord. 2000;24:782–788. doi: 10.1038/sj.ijo.0801227. [DOI] [PubMed] [Google Scholar]
- 14.Houseknecht KL, Baile CA, Matteri RL, Spurlock ME. The biology of leptin: a review. J Anim Sci. 1998;76:1405–1420. doi: 10.2527/1998.7651405x. [DOI] [PubMed] [Google Scholar]
- 15.Diamond FB, Jr., Cuthbertson D, Hanna S, Eichler D. Correlates of adiponectin and the leptin/adiponectin ratio in obese and non-obese children. J Pediatr Endocrinol Metab. 2004;17:1069–1075. doi: 10.1515/jpem.2004.17.8.1069. [DOI] [PubMed] [Google Scholar]
- 16.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England journal of medicine. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 17.Barr VA, Malide D, Zarnowski MJ, Taylor SI, Cushman SW. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology. 1997;138:4463–4472. doi: 10.1210/endo.138.10.5451. [DOI] [PubMed] [Google Scholar]
- 18.Bradley RL, Cheatham B. Regulation of ob gene expression and leptin secretion by insulin and dexamethasone in rat adipocytes. Diabetes. 1999;48:272–278. doi: 10.2337/diabetes.48.2.272. [DOI] [PubMed] [Google Scholar]
- 19.Kolaczynski JW, Nyce MR, Considine RV, Boden G, Nolan JJ, Henry R, Mudaliar SR, Olefsky J, Caro JF. Acute and chronic effects of insulin on leptin production in humans: Studies in vivo and in vitro. Diabetes. 1996;45:699–701. doi: 10.2337/diab.45.5.699. [DOI] [PubMed] [Google Scholar]
- 20.Saladin R, De Vos P, Guerre-Millo M, Leturque A, Girard J, Staels B, Auwerx J. Transient increase in obese gene expression after food intake or insulin administration. Nature. 1995;377:527–529. doi: 10.1038/377527a0. [DOI] [PubMed] [Google Scholar]
- 21.Papaspyrou-Rao S, Schneider S, Petersen R, Fried S. Dexamethasone increases leptin expression in humans in vivo. The Journal of clinical endocrinology and metabolism. 1997;82:1635–1637. doi: 10.1210/jcem.82.5.3928. [DOI] [PubMed] [Google Scholar]
- 22.Lee MJ, Wang Y, Ricci M, Sullivan S, Russell C, Fried S. Acute and chronic regulation of leptin synthesis, storage and secretion by insulin and dexamethasone in human adipose tissue. Am. J. Phys. Endocrinol. Metab. 2007;292:E858–E864. doi: 10.1152/ajpendo.00439.2006. [DOI] [PubMed] [Google Scholar]
- 23.Russell CD, Petersen RN, Rao SP, Ricci MR, Prasad A, Zhang Y, Brolin RE, Fried SK. Leptin expression in adipose tissue from obese humans: depot-specific regulation by insulin and dexamethasone. The American journal of physiology. 1998;275:E507–E515. doi: 10.1152/ajpendo.1998.275.3.E507. [DOI] [PubMed] [Google Scholar]
- 24.Cammisotto PG, Gelinas Y, Deshaies Y, Bukowiecki LJ. Regulation of leptin secretion from white adipocytes by insulin, glycolytic substrates and aminoacids. Am. J. Physiol. Endocrinol Metab. 289:E166–E171. doi: 10.1152/ajpendo.00602.2004. [DOI] [PubMed] [Google Scholar]
- 25.Trayhurn P, Duncan JS, Rayner DV, Hardie LJ. Rapid inhibition of ob gene expression and circulating leptin levels in lean mice by the beta 3- adrenoceptor agonists BRL 35135A and ZD2079. Biochemical and biophysical research communications. 1996;228:605–610. doi: 10.1006/bbrc.1996.1704. [DOI] [PubMed] [Google Scholar]
- 26.Ricci M, Lee M, Russell C, Wang Y, Sullivan S, Schneider S, Brolin R, Fried S. Isoproterenol decreases leptin release from rat and human adipose tissue through posttranscriptional mechanisms. American journal of physiology. 2005;288(4):E798–E804. doi: 10.1152/ajpendo.00446.2004. [DOI] [PubMed] [Google Scholar]
- 27.Donahoo WT, Jensen DR, Yost TJ, Eckel RH. Isoproterenol and somatostatin decrease plasma leptin in humans: a novel mechanism regulating leptin secretion. The Journal of clinical endocrinology and metabolism. 1997;82:4139–4143. doi: 10.1210/jcem.82.12.4434. [DOI] [PubMed] [Google Scholar]
- 28.Motoshima H, Wu X, Sinha MK, Hardy VE, Rosato EL, Barbot DJ, Rosato FE, Goldstein BJ. Differential regulation of adiponectin secretion from cultured human omental and subcutaneous adipocytes: effects of insulin and rosiglitazone. The Journal of clinical endocrinology and metabolism. 2002;87:5662–5667. doi: 10.1210/jc.2002-020635. [DOI] [PubMed] [Google Scholar]
- 29.Halleux CM, Takahashi M, Delporte ML, Detry R, Funahashi T, Matsuzawa Y, Brichard SM. Secretion of adiponectin and regulation of apM1 gene expression in human visceral adipose tissue. Biochemical and biophysical research communications. 2001;288:1102–1107. doi: 10.1006/bbrc.2001.5904. [DOI] [PubMed] [Google Scholar]
- 30.Combs T, Wagner J, Berger J, Doebber T, Wang W, Zhang B, Tanen M, Berg A, O'Rahilly S, Savage D, Chatterjee K, Weiss S, Larson P, Gottesdiener K, Gertz B, Charron M, Scherer P, Moller D. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology. 2002;143:998–1007. doi: 10.1210/endo.143.3.8662. [DOI] [PubMed] [Google Scholar]
- 31.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–2099. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 32.Yu J, Javorschi S, Hevener A, Kruszynska Y, Norman R, Sinha M, Olefsky J. The effect of thiazolidinedinones on plasma adiponectin levels in normal, obese and type 2 diabetic subjects. Diabetes. 2002;51:2968–2974. doi: 10.2337/diabetes.51.10.2968. [DOI] [PubMed] [Google Scholar]
- 33.Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. The Journal of biological chemistry. 2003;278:9073–9085. doi: 10.1074/jbc.M207198200. [DOI] [PubMed] [Google Scholar]
- 34.Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, Utzschneider KM, Kahn SE, Olefsky JM, Buchanan TA, Scherer PE. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. The Journal of biological chemistry. 2004;279:12152–12162. doi: 10.1074/jbc.M311113200. [DOI] [PubMed] [Google Scholar]
- 35.Xie L, Boyle D, Sanford D, Scherer PE, Pessin JE, Mora S. Intracellular trafficking and secretion of adiponectin is dependent on GGA-coated vesicles. The Journal of biological chemistry. 2006;281:7253–7259. doi: 10.1074/jbc.M511313200. [DOI] [PubMed] [Google Scholar]
- 36.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. The Journal of biological chemistry. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 37.Cammisotto PG, Gelinas Y, Deshaies Y, Bukowiecki LJ. Regulation of leptin secretion from white adipocytes by insulin, glycolytic substrates, and amino acids. American journal of physiology. 2005;289:E166–E171. doi: 10.1152/ajpendo.00602.2004. [DOI] [PubMed] [Google Scholar]
- 38.Roh C, Roduit R, Thorens B, Fried S, Kandror K. Lipoprotein lipase and leptin are accumulated in different secretory compartments in rat adipocytes. The Journal of biological chemistry. 2001;276:35990–35994. doi: 10.1074/jbc.M102791200. [DOI] [PubMed] [Google Scholar]
- 39.Weber T, Joost J, Simpson I, Cushman S. Methods for assessment of glucose transport activity and the number of glucose transporters in isolated rat adipose cells and membrane fractions. In: Kahn CR, Harrison LC, editors. The Insulin receptor. Vol 2. New York: Liss; 1988. pp. 171–187. [Google Scholar]
- 40.Livingstone C, James D, Rice J, Hanpeter D, Gould G. Compartment ablation analysis of the insulin-responsive glucose transporter (GLUT4) in 3T3-L1 adipocytes. Biochem J. 1996;315:487–495. doi: 10.1042/bj3150487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morinaga N, Tsai SC, Moss J, Vaughan M. Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7- like domain. Proc Natl. Acad Sci. USA. 1996;93:12856–12860. doi: 10.1073/pnas.93.23.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klausner RD, Donaldson JG, Lippincott SJ. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chardin P, McCormick F. Brefeldin A: The Advantage of Being Uncompetitive. Cell. 1999;97:153–155. doi: 10.1016/s0092-8674(00)80724-2. [DOI] [PubMed] [Google Scholar]
- 44.Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol. 2004;5:23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- 45.Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V. Protein Kinase D regulates the Fission of Cell surface destined transport carriers from the Trans-Golgi Network. Cell. 2001;104:409–420. doi: 10.1016/s0092-8674(01)00228-8. [DOI] [PubMed] [Google Scholar]
- 46.Ghanekar Y, Lowe M. Protein kinase D: activation for Golgi carrier formation. Trends in cell biology. 2005;15:511–514. doi: 10.1016/j.tcb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Iglesias T, Rozengurt E. Protein Kinase D activation by mutations within its pleckstrin homology domain. The Journal of biological chemistry. 1998;273:410–416. doi: 10.1074/jbc.273.1.410. [DOI] [PubMed] [Google Scholar]
- 48.Bogan JS, Lodish HF. Two compartments for insulin-stimulated exocytosis in 3T3-L1 adipocytes defined by endogenous ACRP30 and GLUT4. J Cell Biol. 1999;146:609–620. doi: 10.1083/jcb.146.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitagawa K, Rosen BS, Spiegelman BM, Lienhard GE, Tanner LI. Insulin stimulates the acute release of adipsin from 3T3- L1 adipocytes. Biochem Biophys Acta. 1989;1014:83–89. doi: 10.1016/0167-4889(89)90244-9. [DOI] [PubMed] [Google Scholar]
- 50.Millar C, Meerloo T, Martin S, Hickson G, Shimwell N, Wakelam M, James D, Gould G. Adipsin and the glucose transporter GLUT4 traffic to the cell surface via independent pathways in adipocytes. Traffic. 2000;1:141–151. doi: 10.1034/j.1600-0854.2000.010206.x. [DOI] [PubMed] [Google Scholar]
- 51.Roh C, Thoidis G, Farmer S, kandror K. Identification and characterization of leptin-containing intracellular compartment in rat adipose cells. American journal of physiology. 2000;279:E893–E899. doi: 10.1152/ajpendo.2000.279.4.E893. [DOI] [PubMed] [Google Scholar]
- 52.Clarke M, Ewart MA, Santy LC, Prekeris R, Gould GW. ACRP30 is secreted from 3T3-L1 adipocytes via a Rab11-dependent pathway. Biochemical and biophysical research communications. 2006;342:1361–1367. doi: 10.1016/j.bbrc.2006.02.102. [DOI] [PubMed] [Google Scholar]
- 53.Maeda Y, Beznoussenko G, Van Lint J, Mironov A, Malhotra V. Recruitment of protein kinase-D to the trans-Golgi network via the first cysteine-rich domain. EMBO J. 2001;20:5980–5990. doi: 10.1093/emboj/20.21.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]