

Abstract
Aberrant signal transduction contributes substantially to leukemogenesis. The Janus kinase 1 (JAK1) gene encodes a cytoplasmic tyrosine kinase that noncovalently associates with a variety of cytokine receptors and plays a nonredundant role in lymphoid cell precursor proliferation, survival, and differentiation. We report that somatic mutations in JAK1 occur in individuals with acute lymphoblastic leukemia (ALL). JAK1 mutations were more prevalent among adult subjects with the T cell precursor ALL, where they accounted for 18% of cases, and were associated with advanced age at diagnosis, poor response to therapy, and overall prognosis. All mutations were missense, and some were predicted to destabilize interdomain interactions controlling the activity of the kinase. Three mutations that were studied promoted JAK1 gain of function and conferred interleukin (IL)-3–independent growth in Ba/F3 cells and/or IL-9–independent resistance to dexamethasone-induced apoptosis in T cell lymphoma BW5147 cells. Such effects were associated with variably enhanced activation of multiple downstream signaling pathways. Leukemic cells with mutated JAK1 alleles shared a gene expression signature characterized by transcriptional up-regulation of genes positively controlled by JAK signaling. Our findings implicate dysregulated JAK1 function in ALL, particularly of T cell origin, and point to this kinase as a target for the development of novel antileukemic drugs.
Acute lymphoblastic leukemia (ALL) comprises a biologically heterogeneous group of clonal disorders that originate from the uncontrolled proliferation and expansion of immature lymphoblastic cells and are characterized by an extremely variable clinical outcome (1, 2). In recent years, substantial progress has been made toward understanding the molecular events contributing to malignant transformation. This has permitted the recognition of relevant prognostic factors and risk stratification, and has favored the implementation of therapeutic approaches based on cytogenetic and molecular lesions (2–5). Despite these accomplishments, long-term survival in adults with ALL remains largely unsatisfactory, making the design of novel antileukemic drugs tailored to specific biological targets a current priority (4, 6).
The four mammalian members of the JAK family (JAK1, JAK2, JAK3, and TYK2) are nonreceptor tyrosine kinases functioning as signal transducers to control cellular proliferation, survival, and differentiation (7, 8). JAK proteins associate constitutively with a variety of cytokine receptors lacking intrinsic kinase activity, and promote signal flow by phosphorylating tyrosyl residues of activated receptors to allow the recruitment and activation of STAT proteins. They share a complex multidomain structure characterized by a tyrosine kinase domain at the C terminus, which is flanked by a catalytically inactive pseudokinase domain with regulatory function. Their N-terminal half contains a FERM homology domain, which is implicated in receptor binding and possibly regulates the catalytic activity of the kinase, and an adjacent SH2-like domain. In contrast to their conserved structure, increasing experimental data indicate that JAK family members preferentially associate with a diverse subset of cytokine receptors, each differentially expressed by individual cell lineages and tissues, facilitating specificity in function (9). Among them, JAK1 plays an essential and nonredundant role in mediating biological responses induced by a specific subgroup of cytokines controlling lymphoid cell precursor development (10). Jak1 −/− mouse pups exhibit a thymus that is markedly reduced in size, which is associated with a severe decrease in cellularity. Jak1 loss of function is also associated with profound abnormalities in the B cell compartment caused by a block in differentiation at the pro–B/pre–B cell transition step, resulting in a deficit in the production of mature B lymphocytes (10). Based on its crucial role in lymphocyte proliferation, survival, and differentiation, we hypothesized that up-regulation of JAK1 signaling might contribute to malignancies of the lymphoid lineage. In this study, we report that somatic activating JAK1 mutations occur among adults with T cell precursors ALL, and are associated with poor response to therapy and overall prognosis.
RESULTS AND DISCUSSION
JAK1 mutation analysis in ALL
To explore possible contributions of somatic JAK1 gene mutations in ALL, genomic DNA samples from BM aspirates of adult subjects with B cell precursor ALL (B-ALL; n = 88) and T cell ALL (T-ALL; n = 38) obtained at diagnosis and before therapy were screened for mutations in the entire JAK1 coding region using denaturing HPLC (DHPLC). Direct sequencing of variant elution profiles allowed the identification of 37 intronic and exonic changes, including 9 nonsynonymous variants observed in 14 individuals (Table I and Table S1, available at http://www.jem.org/cgi/content/full/jem.20072182/DC1). Among the missense defects, genotyping of genomic DNAs from BM obtained during remission demonstrated the somatic origin of the 1535C>T (Ser512Leu), 1901C>A (Ala634Asp), and 2171G>A (Arg724His) changes in the leukemic clones (Fig. 1 A and Table I). To verify that the nonsynonymous substitutions identified in patients for whom nonleukemic DNA was not available were not gene variants occurring in the population, 335 population-matching control individuals were analyzed, and none harbored the 611A>T (Lys204Met), 2635C>A (Arg879Ser), 2635C>T (Arg879Cys), and 2636G>A (Arg879His) changes or other defects altering those codons. Although the T-ALL–restricted occurrence of three distinct substitutions affecting Arg879 (3/38 versus 0/335; Fisher's exact probability < 0.001) further supported the relevance of the substitution of this residue, we could not exclude that the 611A>T change might represent a private neutral variant. The two remaining missense changes, 184A>G (Ile62Val) and 1078C>T (Arg360Trp), were deemed nonpathogenic variants, as they were observed in nonleukemic cells of affected patients or in unaffected control subjects.
Table I.
List of nonsynonymous JAK1 changes identified in subjects with ALL
| Cohort and lineage | Number of cases |
Nucleotide substitution |
Exon | Amino acid substitution |
Domain | Mutation/ polymorphism |
|---|---|---|---|---|---|---|
| Adult ALL | ||||||
| B-ALL | 88 | |||||
| 1 | 184A>G | 2 | Ile62Val | FERM | Polymorphisma | |
| 1 | 611A>T | 5 | Lys204Met | FERM | Mutationb | |
| 1 | 1901C>A | 13 | Ala634Asp | Pseudokinase | Mutation | |
| 1 | 2171G>A | 15 | Arg724His | Pseudokinase | Mutationd | |
| T-ALL | 38 | |||||
| 1 | 184A>G | 2 | Ile62Val | FERM | Polymorphisma | |
| 1 | 1078C>T | 7 | Arg360Trp | FERM | Polymorphismc | |
| 1 | 1535C>T | 10 | Ser512Leu | SH2 | Mutationd | |
| 1 | 1901C>A | 13 | Ala634Asp | Pseudokinase | Mutationd | |
| 3 | 2171G>A | 15 | Arg724His | Pseudokinase | Mutatione | |
| 1 | 2635C>A | 18 | Arg879Ser | Kinase | Mutationb f | |
| 1 | 2635C>T | 18 | Arg879Cys | Kinase | Mutationb | |
| 1 | 2636G>A | 18 | Arg879His | Kinase | Mutationb | |
| Childhood ALL | ||||||
| B-ALL | 85 | |||||
| T-ALL | 49 | |||||
| 1 | 1957C>T | 13 | Leu653Phe | Pseudokinase | Mutationd |
This change was observed in unaffected individuals.
This change was not present among 335 population-matching control individuals.
This change was present at remission.
This change was not present at remission.
This change was not present at remission in the one individual analyzed.
This subject carried a concomitant 2171G→A change.
Figure 1.

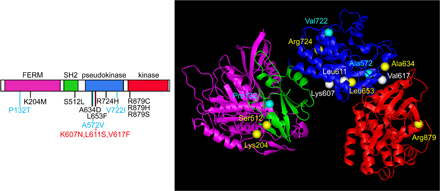
Somatic JAK1 mutations in ALL. (A) Representative electropherograms showing the occurrence of somatically acquired JAK1 mutations in subjects with T-ALL. In all cases, mutations were observed at diagnosis (top), but were undetectable during remission (bottom). (B) JAK1 domain structure and location of affected residues. The predicted amino acid substitutions resulting from the JAK1 mutations are positioned below the diagram of the protein with its functional domains indicated (left) and shown in JAK1 three-dimensional modeled structure (right). (C) Electropherograms showing the occurrence of mutations in a fraction of leukemic cells of two individuals with T-ALL. In both patients, the mutant allele constituted only a portion of the amplified fragment from BM obtained at diagnosis (blasts >70% of total cells; top). The heterozygous status of each subject for an intragenic polymorphic site (bottom) is shown for comparison.
All mutations occurred as heterozygous changes and affected conserved residues within the FERM, SH2, pseudokinase, and kinase domains (Fig. 1 B). In two cases, DHPLC profiles and electropherograms indicated that the mutant allele might be present in only a fraction of leukemic cells, suggesting that these lesions did not represent early events during leukemogenesis but were acquired during disease progression (Fig. 1 C). Remarkably, mutations were relatively common among individuals with T-ALL (18.4% of cases, 95% CI = 7.7–34.3%), whereas they occurred in only three patients with B-ALL (3.4% of cases, 95% CI = 0.7–9.6%; Table I). Such a difference in mutation prevalence between cohorts was statistically significant (Fisher's exact probability = 0.003). DHPLC screening performed on affected exons by using pooled DNAs excluded loss of the normal allele and a homozygous condition for a gene variant caused by mitotic recombination in all cases.
To investigate the prevalence of JAK1 mutations among pediatric ALL cases, genomic DNA from BM obtained at diagnosis was scanned for mutations in affected exons. No lesion was observed within the B-ALL cohort (n = 85), whereas a nonsynonymous 1957C>T transition (Leu653Phe) was identified in 1 of 49 subjects with T-ALL (2.0% of cases, 95% CI = 0.05–10.9%). This mutation was not observed in the BM obtained from the patient at the time of remission, indicating that it was somatically acquired in the leukemic cells.
Overall, these results indicated that JAK1 gene mutations occur in ALL and are more frequently observed among adult individuals with involvement of the T cell lineage.
Functional consequences of somatic JAK1 mutations
To examine the effects of the identified mutations on protein function, WT JAK1 or a mutant form (A634D, R724H, and R879C) was expressed transiently in JAK1-defective human fibrosarcoma U4C cells, and endogenous STAT1 phosphorylation was compared basally and after stimulation with IFN-γ (Fig. 2 A). Consistent with previous studies (10, 11), untransfected cells lacking functional JAK1 did not exhibit STAT1 phosphorylation in response to IFN-γ. All JAK1 mutants promoted an enhanced response to the ligand compared with WT JAK1. Of note, basal STAT1 phosphorylation was observed in cells expressing the A634D mutant, suggesting ligand-independent up-regulation of the kinase. Consistent with this, expression of the A634D mutant resulted in an essentially constitutive STAT1 transcriptional activation, whereas a statistically significant increase in STAT1 activity was observed in U4C cells expressing both the R724H and R879C mutants, basally and after stimulation (Fig. 2 B).
Figure 2.

Functional effects of leukemia-associated JAK1 mutations. (A) STAT1 phosphorylation assays. Basal and IFN-γ–stimulated endogenous STAT1 phosphorylation in JAK1-defective U4C cells transiently transfected with WT JAK1 or selected mutants. Blots are representative of three experiments. (B) STAT1 activation assays. Basal and IFN-γ–stimulated endogenous STAT1 transcriptional activity in JAK1-defective U4C cells transiently cotransfected with p-GAS-Luc and phRL-TK constructs, and WT JAK1 or a mutant allele. STAT1-induced luciferase gene expression levels were determined by measuring the luciferase activity (CPS, counts per second) normalized to the activity of the Renilla luciferase, using a dual-luciferase reporter assay system. Activity ratios are expressed as the mean of three replicates ± the SD. (C) Ba/F3 survival assays. WT or mutant Jak1-transduced Ba/F3 cells were grown in the absence of IL-3 (top) or with 0.5 or 5% WEHI-3B cell CM as source of IL-3 (bottom). Cell numbers (mean of three replicates ± the SD) were counted at the indicated time points (top) or at day 3 of culture (bottom). (D) Stat5, Akt, and ERK phosphorylation assays. Endogenous Stat5 Tyr694, Akt Ser473, and ERK1/2 Thr202/Tyr204 phosphorylation levels from lysates of Ba/F3 cells transduced with WT Jak1 or a mutant form and cultured without IL-3 (left) or with 2% WEHI-3B cell CM as the source of IL-3 (right). Activation of Stat5 (pStat5/Stat5), AKT (pAkt/Akt), and ERK1/2 (pERK/ERK) is expressed as a multiple of activation in untransduced cells. Blots are representative of at least three experiments performed. (E) BW5147 proliferation assays. WT or mutant Jak1 transduced BW5147 cells were grown in presence of 200 μg/ml dexamethasone and 50 μg/ml cyclosporine A, in absence (left) or with different concentrations (right) of IL-9. After 72 h, proliferation was measured after [3H]thymidine was added to the cultures (6 h). Data are shown as the means of three replicates ± the SD. For convenience, the amino acid changes affecting residues Ala633, Arg723, and Arg878 of the murine Jak1 protein (encoded by the pMX-Jak1-IRES-GFP constructs used to transduce the Ba/F3 and BW5147 cell lines) are indicated according to the homologous residues in human JAK1.
To further assess the ability of mutations to up-regulate signal flow, we transduced Ba/F3 cells with WT Jak1 or each of the selected mutants to evaluate whether their expression induced autonomous growth of cytokine-dependent cells. GFP-expressing Ba/F3 cells were selected by flow cytometry, cultured in 5 or 0.5% WEHI-3B cell conditional medium (CM) as a source of IL-3, as well as in absence of the cytokine, and counted to assess proliferation (Fig. 2 C). Three independent experiments indicated that expression of the A634D and, with less efficiency, R724H Jak1 mutants conferred IL-3–independent growth to cells, whereas cells expressing WT Jak1 or the R879C Jak1 mutant retained dependence on the cytokine for survival. Of note, cells expressing each of the three mutants exhibited enhanced growth in response to IL-3. Consistent with these findings, Ba/F3 cells expressing the A634D Jak1 mutant exhibited enhanced Stat5, Akt, and extracellular signal-regulated kinase (ERK) phosphorylation basally and after stimulation, whereas a higher phosphorylation level of these signal transducers in cells expressing the R724H Jak1 protein was observed in cultures maintained in the presence of IL-3 (Fig. 2 D). Notably, we did not observe Jak1 phosphorylation in parental and transduced Ba/F3 cells basally and cultured in 2% WEHI-3B cell CM; however, phosphorylation was appreciable in A634D and R724H Jak1-transduced cells after selection (7-d culture in absence of IL-3; unpublished data).
IL-9 protects the T cell lymphoma BW5147 cell line against dexamethasone-induced apoptosis (12), an effect that is dependent on Jak1-mediated Stat3 and Stat5 activation (13). To demonstrate the activating role of the ALL-associated JAK1 mutations in a different cellular system, BW5147 cells were transduced with WT Jak1 or each of the selected mutants to evaluate their effect on this stress response. GFP-expressing cells were isolated by flow cytometry, cultured in the presence of cyclosporine A and dexamethasone, with or without IL-9, and [3H]thymidine incorporation was determined to assess proliferation (Fig. 2 E). Three independent experiments documented that expression of the A634D and R879C Jak1 mutants, but not R724H Jak1, conferred increased growth to cells basally. Of note, whereas transduced cells exhibited comparable responses to high levels of IL-9, those expressing the A634D and R879C Jak1 mutants were more responsive to low levels of the cytokine. Expression of the A634D Jak1, but not that of the R879C mutant, was associated with enhanced phosphorylation of Jak1, Stat3, and Stat5 basally (unpublished data).
Overall, these data indicated that the three selected leukemia-associated JAK1 mutants are hypermorphs, with A634D Jak1 having a seemingly stronger effect, and that different mechanisms are likely to be involved in their cell context–related gain of function.
Molecular modeling of JAK1 and location of affected residues
To look at the structural causes resulting in JAK1 functional up-regulation, we generated a model of JAK1 structure because no crystallographic information was available for this protein. Energy-minimized models of each of the four domains were generated separately by homology to available crystallographic structures of proteins with similar sequences and overall fold. The quaternary arrangement of the four domains was then determined by superimposing the models on a predicted three-dimensional structure of JAK2 (14). According to the superimposed structure, Ala634 and Leu653 are placed on the surface of the pseudokinase domain involved in the interaction with the kinase domain (Fig. 1 B). Based on the evidence supporting a negative regulatory role of the pseudokinase domain on catalytic function of JAK proteins (15–17), the pathogenetic mechanism of the A634D and L653F changes is predicted to involve a looser interdomain interaction, relaxing inhibitory control on the kinase activity. Consistent with this hypothesis, substitution of two residues located in the pseudokinase domain at the interface with the kinase domain in JAK2 (Val617) and JAK3 (Ala572) promote increased catalytic activity basally (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20072182/DC1) (18–20). Our model also predicts that residues Lys204 and Ser512 would perturb the SH2–FERM interdomain interaction because they are located at the interface between these domains, approximately facing each other. This finding is noteworthy because it has been proposed that JAK1's SH2 domain does not function as a phosphotyrosyl-binding domain, but instead plays a structural role in stabilizing the conformation of the FERM domain (21), which mediates its association to cytokine receptors and exerts an as yet uncharacterized restraint on catalytic function (22, 23). The molecular mechanism through which these mutations affect JAK1 function remains to be explained. Structural and functional consequences were not obvious for the activating changes affecting residues Arg724 and Arg879.
Gene expression profile analysis in blasts with a mutated JAK1 allele
Total RNA was available from blasts of 5 JAK1 mutation-positive (S512L, A634D, and R724H) and 11 mutation-negative subjects of the T-ALL cohort. Unsupervised clustering based on 1,345 probe sets selected by nonspecific filtering clustered expression profiles of mutation-positive blasts into two clusters, suggesting contribution of JAK1 mutations to distinct major mechanisms of deregulation (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20072182/DC1). Notably, supervised gene expression analysis revealed a distinctive expression signature shared by leukemic cells with a mutated JAK1 gene (Fig. 3 A) based on the expression of 133 differentially expressed probe sets, consisting of 112 differentially expressed genes, the majority of them being overexpressed in JAK1 mutation-positive samples (Table S2). Among the up-regulated genes, those whose transcription was known to be positively modulated by JAK/STAT signaling, including IRF1, SOCS3, ISG15, ISGF3G, IFI44L, and IRF7, were overrepresented in all the JAK1 mutation-positive subjects, further supporting the gain-of-function role of the ALL-associated JAK1 lesions.
Figure 3.

Gene expression profiles and clinical relevance of somatic JAK1 mutations in adult T-ALL. (A) Supervised hierarchical clustering of gene expression profiles performed on blasts from 16 adult T-ALL patients, with (orange) or without (green) a JAK1 mutation. (B) Kaplan-Meier estimates of DFS (top) and OS (bottom) in subjects with (red) or without (black) a JAK mutation. Multivariate analysis confirmed the statistical significance of the reduced DFS and OV among JAK1 mutation-positive patients, and excluded a significant contribution of the more advanced age of these subjects.
Clinical relevance of somatic JAK1 mutations
The clinical relevance of JAK1 mutations within the adult T-ALL cohort was investigated. Although no statistically significant difference was observed in white blood cell counts, gender distribution, or association with specific chromosomal rearrangements, patients with a mutated JAK1 allele tended to have a more advanced age at diagnosis (median = 40.6 vs. 24.2; P < 0.01), which was consistent with the lower prevalence of mutations identified among children and adolescents with T-ALL included in the study. Comparison of the response to therapy between JAK1 mutation-positive and -negative patients indicated a higher percentage of cases exhibiting resistance to induction therapy in the former (43 vs. 20%), although this difference did not reach statistical significance caused by the relatively small size of the study cohort. Consistent with that finding, a statistically significant reduced disease-free survival (DFS; median =8.7 vs. 20.5 mo; P = 0.01) and overall survival (OS; median = 10.6 vs. 32.5; P < 0.01) was observed among JAK1 mutation-positive patients (Fig. 3 B). Multivariate analysis confirmed the statistical significance of these associations (DFS: HR= 6.20, 95% CI = 1.32–29.09, P = 0.02; OS: HR = 2.82, 95% CI = 1.07–7.48, P = 0.04), and excluded a significant contribution of patients' age (DFS: HR = 0.99, 95% CI = 0.93–1.05, P = 0.64; OS: HR= 1.01, 95% CI = 0.97–1.05, P = 0.60).
In the adult T-ALL cohort, the mutation status for NRAS, KRAS, NOTCH1, and PTEN was also assessed (unpublished data). Among the JAK1 mutation-positive cases, no defect was observed in the NRAS, KRAS, and PTEN genes, although such mutations had a relatively low prevalence in the entire adult T-ALL cohort (RAS genes: 11% of cases, 95% CI = 2.9–24.8%; PTEN: 14% of cases, 95% CI = 4.5–28.8%). In contrast, heterozygous mutations in NOTCH1 were observed in all JAK1 mutation-positive individuals. Given the high prevalence of NOTCH1 defects observed in the cohort (70% of cases, 95% CI = 53.0–84.1%), this association was not statistically significant (P = 0.06). This observation, however, suggests that activation of JAK1 and NOTCH1 transduction pathways might cooperate in T-ALL pathogenesis and/or progression. No significant difference in response to therapy or outcome was observed between NOTCH1 mutation-positive and -negative patients. Interestingly, NOTCH1 and PTEN mutations exhibited a mutually exclusive distribution because none of the five subjects carrying PTEN lesions had a NOTCH1 defect (Fisher's exact probability = 0.001).
Aberrant JAK1 function and leukemogenesis
Functional up-regulation of two members of the JAK family, JAK2 and JAK3, has recently been discovered in myeloproliferative disorders and other malignancies of the myeloid lineage (18–20, 24). The JAK2 V617F amino acid change occurs in the majority of polycythemia vera cases and in ∼50% of individuals with essential thrombocythemia or idiopathic myelofibrosis. The available data support the view that this recurrent change, which affects the pseudokinase domain of the protein, induces constitutive activation of the kinase and hypersensitivity to cytokines. Similarly, three JAK3 hypermorphic alleles promoting cytokine independence in Ba/F3 cells have been identified in acute megakaryoblastic myeloid leukemia. In this study, we showed that somatically acquired activating JAK1 mutations occur in ALL, particularly in adults, further emphasizing the importance of JAK-mediated signaling dysregulation in leukemogenesis and extending the spectrum of hematologic malignancies associated with aberrant activation of this signal transduction pathway. Even though the molecular mechanisms by which individual JAK1 mutations promote gain of function are likely to be diverse and remain to be fully characterized, modeling and biochemical data are consistent with the view that, similar to what has been observed for somatic leukemia-associated JAK2 and JAK3 defects, most mutations would interfere with the autoinhibitory control on the catalytic activity. For most mutations, this effect would be achieved by triggering local structural rearrangements in regions involved in interdomain interactions between the pseudokinase and kinase domains or the FERM and SH2 domains (Fig. S1).
JAK1 is expressed widely and participates in intracellular signaling elicited by class II cytokine receptors and receptors that use the gp130 or γc receptor subunit. Although the hematopoietic defects in Jak1 −/− mice were restricted to the lymphoid cell compartment as a result of an impaired response to IL-7 (10) and the present findings indicate a cell-context dependence of somatically acquired JAK1 mutations' contribution to leukemogenesis, we cannot exclude the involvement of this kinase in other malignancies. We speculate that a concomitant genetic event, including a mutation affecting other members of the JAK family, might synergize with the JAK1 defect to promote aberrant cell proliferation and/or survival in a cell-specific context. This hypothesis is currently under investigation.
Although 70–80% of pediatric patients with either B- or T-ALL have excellent long-term response to intensive combination chemotherapy, adult patients exhibit a less favorable outcome (4, 25). In B- ALL, such a poor prognosis has been associated in part with the presence of BCR/ABL or ALL1/AF4 gene rearrangements. In contrast, the unfavorable outcome of adult patients with T-ALL has not conclusively been attributed to any cytogenetic lesion, albeit the prognostic relevance of aberrant ERG and TLX1 gene expression and NOTCH1 mutations has recently been reported (26–28). The present work provides the first evidence that JAK1 gene defects are associated with a poor response to therapy, frequent relapse of the disease, and reduced OS, identifying such mutations as a novel informative prognostic marker occurring in a sizable proportion of adult T-ALL. Although studies on larger cohorts are required to determine more precisely the clinical relevance and prognostic value of JAK1 defects in adult and pediatric ALL, our findings provide a rationale for the development of novel therapeutic approaches tailored at interfering with JAK1 signaling, encouraging studies aimed at testing the efficacy and side effects of JAK1 inhibitors in the management of adult T-ALL patients.
MATERIALS AND METHODS
All cohorts and molecular analyses.
Cohorts studied included patients enrolled in the Gruppo Italiano Malattie Ematologiche dell'Adulto 0496 and 2000 (adults), and Associazione italiana ematologia ed oncologia pediatrica ALL 2000 (children and adolescents) clinical trials. Written informed consent for genetic analyses was obtained from all subjects according to the Declaration of Helsinki. BM mononuclear cells were isolated by density gradient centrifugation, and then cryopreserved. Genomic DNA was isolated in a standard fashion. Exons 26, 27, and 34 of the NOTCH1 gene were analyzed by direct sequencing, whereas the entire JAK1 and PTEN coding regions and exons 1 and 2 of NRAS and KRAS were screened by DHPLC (Wave 2100 System; Transgenomic) and sequencing. Primer sequences are available upon request. Gene expression profiling methodology is described in the Supplemental materials and methods (available at http://www.jem.org/cgi/content/full/jem.20072182/DC1).
Functional studies.
The mutations resulting in the A634D, R724H, and R879C changes were introduced by site-directed mutagenesis in a VSV-tagged JAK1 cDNA cloned in pRc/CMV vector provided by S. Pellegrini (Pasteur Institute, Paris, France). JAK1-defective human fibrosarcoma U4C cells (provided by S. Pellegrini) were maintained in DME supplemented with 10% heat-inactivated FCS and antibiotics. To evaluate endogenous STAT1 phosphorylation, after starvation (24 h), transiently transfected cells were stimulated with IFN-γ (1,000 U/ml, 15 min) or left unstimulated. Lysed samples were analyzed by 10% SDS-PAGE, transferred to a polyvinylidene difluoride membrane (Pierce Chemical Co.) and probed with anti–phospho-Stat1 (9171; Cell Signaling Technology), anti-Stat1 (9172; Cell Signaling Technology), and anti-Jak1 (3332; Cell Signaling Technology) antibodies. Endogenous STAT1 transcriptional activity was assessed by luciferase transactivation assays in cells cotransfected with a p-GAS-Luc construct, switched to serum-starvation medium (8 h), and then stimulated with IFN-γ (1,000 U/ml, 16 h) or left unstimulated. STAT1-induced luciferase expression was assessed and normalized using a dual luciferase reporter assay system (Promega) and a phRL-TK plasmid constitutively expressing the Renilla luciferase.
Each of the three leukemia-associated mutations was also introduced in the murine Jak1 cDNA cloned in the bicistronic retroviral vector pMX-IRES-GFP. Constructs were transfected into Phoenix or BOSC packaging cells to produce retroviruses, and murine Ba/F3 (maintained in RPMI 1640 medium containing 10% FCS, 1% l-glutamine, and 10% WEHI-3B cell CM) and BW5147 (maintained in Iscove-Dulbecco's medium supplemented with 10% FBS, 0.55 mM l-arginine, 0.24 mM l-asparagine, and 1.25 mM l-glutamine) cells were infected with retroviral supernatants. GFP-positive populations were purified by flow-cytometric sorting, and then expanded. Equal GFP expression levels of transduced cells were confirmed by FACS analysis (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20072182/DC1). Transduced Ba/F3 cells were cultured in the absence or presence of IL-3 (0.5 or 5% WEHI-3B cell CM) for assaying cytokine independence and response, and viable cells were counted by Trypan blue exclusion. Proliferation of transduced BW5147 cells cultured in the presence of 50 μg/ml cyclosporine A and 200 μg/ml dexamethasone, in the presence or absence of mIL-9, was determined using [3H]thymidine incorporation assay. In brief, 3,000 cells were seeded in 96-well plates (200 μl volume medium), and after 72 h, [3H]thymidine was added to the cultures for 6 h. Cells were then collected on microfilter plates, and thymidine incorporation was measured after addition of 25 μl of liquid scintillant. For Western blot studies, Ba/F3 cells were starved in RPMI 1640 medium containing 1% BSA (5 h), and then stimulated with 2% WEHI-3B cell CM (30 min) or left unstimulated, whereas BW5417 cells were starved in Iscove-Dulbecco's medium (12 h) and left unstimulated or stimulated with 100 U/ml mIL-9 for 15 min. Evaluation of Jak1, Akt, Stat3, Stat5, and ERK1/2 expression and phosphorylation levels was performed using anti–phospho-Jak1, anti-Jak1, anti–phospho-Akt, anti-Akt, anti–phospho-Stat3, anti-Stat3, anti–phospho-Stat5, anti-Stat5, anti–phospho-p44/42 ERK, and anti-p44/42 ERK antibodies (all from Cell Signaling Technology).
Structural models of the kinase, pseudokinase, and SH2 and FERM domains of JAK1 were generated as described in the online supplemental information section.
Statistical analysis.
Confidence intervals of proportions (at 95% level) were calculated based on the binomial distribution. The probabilities of OS and DFS were estimated using the Kaplan-Meier method. The log-rank test was used to compare treatment effect and risk factor categories. All tests were two-sided, accepting P ≤ 0.05 as indicating a statistically significant difference. Cox proportional hazard models, including age and JAK1 as variables, were used to perform multivariate analyses for OS and DFS. The SAS software (SAS Institute) was used for the analysis.
Online supplemental material.
Methodologies and approaches used to generate the JAK1 molecular modeling and to perform and analyze adult T-ALL leukemic cell gene expression profiles (including cited references) are provided in the Supplemental Materials and methods. Table S1 is a list of the intronic or synonymous JAK1 changes identified in the study. Table S2 is a list of differentially expressed genes in JAK1 mutation-positive versus mutation-negative adult subjects with T-ALL. Fig. S1 shows the JAK1 domain structure and the location of JAK1, JAK2, and JAK3 residues reported to be mutated in myeloproliferative disorders and leukemias. Fig. S2 shows the unsupervised hierarchical clustering of gene expression profiles of blasts from 16 adult T-ALL patients, with or without a JAK1 mutation. Fig. S3 shows the GFP expression levels of purified Ba/F3 and BW5417 cells transduced with a bicistronic retroviral vector pMX-Jak1-IRES-GFP coding for WT Jak1 or one of the three generated mutants. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20072182/DC1.
Supplemental Material
Acknowledgments
We are indebted to the patients who participated in the study and physicians of the GIMEMA network who referred the subjects and provided BM specimens for the study. Thanks are due to Sandra Pellegrini (Pasteur Institute, Paris, France) for providing the JAK1-defective U4C cell line and the construct encoding the human VSV-tagged JAK1 protein, and Romano Kroemer (Sanofi-Aventis, Centre de Recherche de Paris, Vitry-sur-Seine, France) for making available the coordinates of the JAK2 model.
This work was supported by grants from Ricerca Oncologica Project of Integrated Program “Multidimensional classification of lymphohematopoietic malignancies” (to M. Tartaglia) and Associazione Italiana per la Ricerca sul Cancro (AIRC; to R. Foa). EF is supported by a fellowship from Associazione ONLUS “Morgan Di Gianvittorio per la cura e la ricerca nei tumori e leucemie in età pediatrica.”
The authors declare no competing financial interests.
Abbreviations used: ALL, acute lymphoblastic leukemia; B-ALL, B cell precursor ALL; CM, conditional medium; DFS, disease-free survival; DHPLC, denaturing HPLC; ERK, extracellular signal-regulated kinase; OS, overall survival; T-ALL, T cell ALL.
E. Flex and V. Petrangeli contributed equally to this paper.
References
- 1.Armstrong, S.A., and A.T. Look. 2005. Molecular genetics of acute lymphoblastic leukemia. J. Clin. Oncol. 23:6306–6315. [DOI] [PubMed] [Google Scholar]
- 2.Pui, C.H., and W.E. Evans. 2006. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354:166–178. [DOI] [PubMed] [Google Scholar]
- 3.Yeoh, E.J., M.E. Ross, S.A. Shurtleff, W.K. Williams, D. Patel, R. Mahfouz, F.G. Behm, S.C. Raimondi, M.V. Relling, A. Patel, et al. 2002. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 1:133–143. [DOI] [PubMed] [Google Scholar]
- 4.Vitale, A., A. Guarini, S. Chiaretti, and R. Foa. 2006. The changing scene of adult acute lymphoblastic leukemia. Curr. Opin. Oncol. 18:652–659. [DOI] [PubMed] [Google Scholar]
- 5.Chiaretti, S., X. Li, R. Gentleman, A. Vitale, M. Vignetti, F. Mandelli, J. Ritz, and R. Foa. 2004. Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood. 103:2771–2778. [DOI] [PubMed] [Google Scholar]
- 6.Thomas, X. 2005. Emerging drugs for adult acute lymphoblastic leukaemia. Expert Opin. Emerg. Drugs. 10:591–617. [DOI] [PubMed] [Google Scholar]
- 7.Yamaoka, K., P. Saharinen, M. Pesu, V.E. Holt III, O. Silvennoinen, and J.J. O'Shea. 2004. The Janus kinases (Jaks). Genome Biol. 5:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schindler, C., D.E. Levy, and T. Decker. 2007. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282:20059–20063. [DOI] [PubMed] [Google Scholar]
- 9.Murray, P.J. 2007. The JAK-STAT signalling pathway: Input and output integration. J. Immunol. 178:2623–2629. [DOI] [PubMed] [Google Scholar]
- 10.Rodig, S.J., M.A. Meraz, J.M. White, P.A. Lampe, J.K. Riley, C.D. Arthur, K.L. King, K.C. Sheehan, L. Yin, D. Pennica, et al. 1998. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 93:373–383. [DOI] [PubMed] [Google Scholar]
- 11.Müller, M., J. Briscoe, C. Laxton, D. Guschin, A. Ziemiecki, O. Silvennoinen, A. Harpur, G. Barbieri, B.A. Witthuhn, C. Schindler, S. Pellegrini, A.F. Wilks, J.N. Ihle, G.R. Stark, and L.M. Kerr. 1993. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature. 366:129–135. [DOI] [PubMed] [Google Scholar]
- 12.Renauld, J.C., A. Vink, J. Louahed, and J. Van Snick. 1995. Interleukin-9 is a major anti-apoptotic factor for thymic lymphomas. Blood. 85:1300–1305. [PubMed] [Google Scholar]
- 13.Demoulin, J.B., C. Uyttenhove, E. Van Roost, B. DeLestre, D. Donckers, J. Van Snick, and J.C. Renauld. 1996. A single tyrosine of the interleukin-9 (IL-9) receptor is required for STAT activation, antiapoptotic activity, and growth regulation by IL-9. Mol. Cell. Biol. 16:4710–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giordanetto, F., and R.T. Kroemer. 2002. Prediction of the structure of human Janus kinase 2 (JAK2) comprising JAK homology domains 1 though 7. Protein Eng. 15:727–737. [DOI] [PubMed] [Google Scholar]
- 15.Chen, M., A. Cheng, F. Candotti, Y.J. Zhou, A. Hymel, A. Fasth, L.D. Notarangelo, and J.J. O'Shea. 2000. Complex effects of naturally occurring mutations in the JAK3 pseudokinase domain: evidence for interactions between the kinase and pseudokinase domains. Mol. Cell. Biol. 20:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindauer, K., T. Loerting, K.R. Liedl, and R.T. Kroemer. 2001. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 14:27–37. [DOI] [PubMed] [Google Scholar]
- 17.Saharinen, P., and O. Silvennoinen. 2002. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J. Biol. Chem. 277:47954–47963. [DOI] [PubMed] [Google Scholar]
- 18.James, C., V. Ugo, J.P. Le Couedic, J. Staerk, F. Delhommeau, C. Lacout, L. Garcon, H. Raslova, R. Berger, A. Bennaceur-Griscelli, et al. 2005. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 434:1144–1148. [DOI] [PubMed] [Google Scholar]
- 19.Levine, R.L., M. Wadleigh, J. Cools, B.L. Ebert, G. Wernig, B.J. Huntly, T.J. Boggon, I. Wlodarska, J.J. Clark, S. Moore, et al. 2005. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 7:387–397. [DOI] [PubMed] [Google Scholar]
- 20.Walters, D.K., T. Mercher, T.L. Gu, T. O'Hare, J.W. Tyner, M. Loriaux, V.L. Goss, K.A. Lee, C.A. Eide, M.J. Wong, et al. 2006. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 10:65–75. [DOI] [PubMed] [Google Scholar]
- 21.Radtke, S., S. Haan, A. Jorissen, H.M. Hermanns, S. Diefenbach, T. Smyczek, H. Schmitz-Vandeleur, P.C. Heinrich, I. Behrmann, and C. Haan. 2005. The Jak1 SH2 domain does not fulfill a classical SH2 function in Jak/STAT signaling but plays a structural role for receptor interaction and up-regulation of receptor surface expression. J. Biol. Chem. 280:25760–25768. [DOI] [PubMed] [Google Scholar]
- 22.Haan, C., H. Is'harc, H.M. Hermanns, H. Schmitz-Van De Leur, I.M. Kerr, P.C. Heinrich, J. Grotzinger, and I. Behrmann. 2001. Mapping of a region within the N terminus of Jak1 involved in cytokine receptor interaction. J. Biol. Chem. 276:37451–37458. [DOI] [PubMed] [Google Scholar]
- 23.Zhou, Y.J., M. Chen, N.A. Cusack, L.H. Kimmel, K.S. Magnuson, J.G. Boyd, W. Lin, J.L. Roberts, A. Lengi, R.H. Buckley, et al. 2001. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol. Cell. 8:959–969. [DOI] [PubMed] [Google Scholar]
- 24.Levine, R.L., M. Loriaux, B.J. Huntly, M.L. Loh, M. Beran, E. Stoffregen, R. Berger, J.J. Clark, S.G. Willis, K.T. Nguyen, et al. 2005. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 106:3377–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gokbuget, N., and D. Hoelzer. 2006. Treatment of adult acute lymphoblastic leukemia. Hematology (Am Soc Hematol Educ Program). 133–141. [DOI] [PubMed]
- 26.Baldus, C.D., T. Burmeister, P. Martus, S. Schwartz, N. Gokbuget, C.D. Bloomfield, D. Hoelzer, E. Thiel, and W.K. Hofmann. 2006. High expression of the ETS transcription factor ERG predicts adverse outcome in acute T-lymphoblastic leukemia in adults. J. Clin. Oncol. 24:4714–4720. [DOI] [PubMed] [Google Scholar]
- 27.Ferrando, A.A., D.S. Neuberg, R.K. Dodge, E. Paietta, R.A. Larson, P.H. Wiernik, J.M. Rowe, M.A. Caligiuri, C.D. Bloomfield, and A.T. Look. 2004. Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet. 363:535–536. [DOI] [PubMed] [Google Scholar]
- 28.Zhu, Y.M., W.L. Zhao, J.F. Fu, J.Y. Shi, Q. Pan, J. Hu, X.D. Gao, B. Chen, J.M. Li, S.M. Xiong, et al. 2006. NOTCH1 mutations in T-cell acute lymphoblastic leukemia: prognostic significance and implication in multifactorial leukemogenesis. Clin. Cancer Res. 12:3043–3049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}