

Abstract
Previous mouse and clinical studies demonstrate a link between Th2 intestinal inflammation and induction of the effector phase of food allergy. However, the mechanism by which sensitization and mast cell responses occurs is largely unknown. We demonstrate that interleukin (IL)-9 has an important role in this process. IL-9–deficient mice fail to develop experimental oral antigen–induced intestinal anaphylaxis, and intestinal IL-9 overexpression induces an intestinal anaphylaxis phenotype (intestinal mastocytosis, intestinal permeability, and intravascular leakage). In addition, intestinal IL-9 overexpression predisposes to oral antigen sensitization, which requires mast cells and increased intestinal permeability. These observations demonstrate a central role for IL-9 and mast cells in experimental intestinal permeability in oral antigen sensitization and suggest that IL-9–mediated mast cell responses have an important role in food allergy.
Allergic responses in the gastrointestinal (GI) tract were relatively uncommon several decades ago; however, recent studies have demonstrated that food allergies now affect 2–6% of the population (1, 2). Clinical and experimental analyses suggest that initiation of food-induced intestinal anaphylactic responses is regulated by numerous inflammatory mediators, including Th2-cytokines. Indeed, peripheral blood and intestinal tissue from patients with food allergy contain elevated numbers of activated T cells, which correlate with elevated levels of Th2 cytokines and the degree of GI inflammation and dysfunction (3, 4). Furthermore, in vitro allergen-stimulated T cells and T cell clones generated from food allergic patients produce Th2-cytokines (IL-4, -5, and -13) (5). These cytokines activate immunological pathways associated with the onset of allergic reactions, including Th2 cell differentiation, IgE synthesis, mast cell and eosinophil recruitment, and activation.
IL-9 is a pleiotropic cytokine involved in Th2 inflammatory reactions (6). Transgenic expression of IL-9 in the lung promotes Th2-mediated allergic pulmonary disease characterized by elevated Th2 cytokines and immune pathology (mucus hypersecretion) and bronchial hyperresponsiveness (7, 8). In vitro studies demonstrate that IL-9 enhances IL-4–induced IgE production (9–11), and airway epithelial cell–derived chemokine expression (CCL11/eotaxin-1, CCL3/MIP-1α, CCL2/MCP1, CCL7/MCP-3, and CCL12/MCP-5). IL-9 has also been implicated in the regulation of mast cell recruitment and effector function (6). Transgenic expression of IL-9 in the lung promotes pulmonary mastocytosis (7, 8) and IL-9 stimulation of mast cells induces histamine release and promotes mast cell protease, IL-6, and FcεRIα expression (12, 13). Although IL-9 has been implicated in the regulation of several Th2 processes, the contribution of this cytokine to oral antigen–induced intestinal allergic responses has not been explored.
The molecular basis underlying the causality of food antigen sensitization in susceptible individuals is not currently understood. One predisposing factor that has long been suspected for GI diseases is impaired barrier function, termed “leaky gut” (14, 15). First-degree relatives of inflammatory bowel disease (IBD) patients have increased intestinal permeability in the absence of clinical symptoms (16–19). Food allergy patients also have increased intestinal permeability, which correlates with the severity of their clinical symptoms (14). Although constitutive abnormalities in intestinal permeability have not been consistently observed in food allergic individuals, it is postulated that environmental events, including infection and stress, may alter intestinal permeability and promote food antigen sensitization (15).
In this study, we evaluate the roles of IL-9 and mast cells in the oral antigen–sensitization and effector phases of experimental intestinal anaphylaxis. We demonstrate that an IL-9–stimulated, mast cell–mediated increase in intestinal permeability is central to the induction of oral antigen sensitization.
RESULTS
Experimental intestinal anaphylaxis is IL-9 dependent
The temporal expression of IL-9 mRNA in the jejunum after oral OVA challenge was evaluated by quantitative PCR analysis in OVA-sensitized mice that had received one and three oral OVA or saline intragastric (i.g.) challenges. Jejunal IL-9 mRNA expression was up-regulated in WT mice after 1 (∼200-fold) and 3 (∼150-fold) oral OVA challenges as compared with saline-challenged mice (1 i.g. challenge, 200.8 ± 115.1-fold change in IL-9/GADPH ratio; 3 i.g. challenges, 142.5 ± 29.3-fold change in IL-9/GADPH ratio; P < 0.05 and P < 0.01, respectively). Thus, oral antigen–induced intestinal anaphylaxis is associated with increased intestinal IL-9 mRNA expression.
To begin to elucidate the contribution of IL-9 to oral antigen–induced intestinal anaphylaxis, we used IL-9–deficient mice (IL-9−/−) and WT BALB/c mice. i.g. challenges of OVA to OVA-sensitized WT mice induced the intestinal anaphylaxis phenotype (intestinal mastocytosis, mast cell activation, and diarrhea; Fig. 1). WT mice started developing diarrhea acutely after the third i.g. challenge with >75% of the mice suffering from diarrhea after the seventh challenge (Fig. 1 A). Food antigen–induced diarrhea is driven by electrogenic Cl− secretion (20, 21). To confirm abnormalities in Cl− secretory activity in our experimental intestinal anaphylaxis model, we measured basal intestinal epithelial short-circuit current (Isc) responses to cholinergic stimulation. We show that induction of experimental intestinal anaphylaxis (9 i.g. challenges) induced altered basal Isc (basal Isc [μÅ/cm2] −0.30 ± 8.4 vs. 101.0 ± 13.3; mean Isc ± the SEM; P < 0.05; Isc of the jejunum of OVA-sensitized saline– vs. OVA-treated WT mice; n = 3 and 7 mice per group, respectively). Furthermore, we demonstrate significant concentration-dependent increase in Isc responses to methacholine in mice with experimental intestinal anaphylaxis as compared with control animals (ΔIsc [μÅ/cm2] at 100 μM methacholine; 18.3 ± 6.8 vs. 58.3 ± 10.0; mean ΔIsc ± the SEM; P < 0.05; ΔIsc of the jejunum of OVA-sensitized saline– vs. OVA-treated WT mice; n = 3 and 7 mice per group, respectively). Diarrhea in the OVA-challenged and -sensitized WT mice was also noted by direct observation of the colon and cecum after the ninth i.g. challenge; the liquid stool observed after OVA challenge of WT mice contrasts with the solid pellets seen in the distal colon of saline-challenged WT and OVA-challenged IL-9−/− mice (unpublished data). Notably, intestinal mast cell and serum mMCP-1 levels were also significantly elevated compared with saline-challenged, OVA-sensitized WT mice (Fig. 1, B–E). In contrast, oral antigen–induced intestinal anaphylaxis was attenuated in IL-9−/− mice. Typically, in any one experiment, 1/6 OVA-challenged, OVA-sensitized IL-9−/− mice would have evidence of diarrhea 45–60 min after i.g. OVA challenge. No more than 20% of IL-9−/− mice developed diarrhea after 9 i.g. OVA challenges (Fig. 1 A). Consistent with the reduction in intestinal anaphylaxis, intestinal mast cell and serum mMCP-1 levels were significantly lower in oral antigen–challenged OVA-sensitized IL-9−/− mice than in WT mice (Fig. 1, B, C, and E). To determine whether the ablation of diarrhea in IL-9−/− mice was caused by an attenuated OVA-CD4+ Th2 response, we examined splenic cytokine production and levels of OVA-specific IgE in OVA-challenged, OVA-sensitized WT and IL-9−/− mice. These levels were equivalent in WT and IL-9−/− mice (Table S1, available at http://www.jem.org/cgi/content/full/jem.20071046/DC1, and Fig. 1 F).
Figure 1.

Oral antigen–induced intestinal anaphylaxis is attenuated in IL-9–deficient mice. Diarrhea occurrence (A) and mean number of mast cells per high power field (HPF; B) in the intestine of OVA-sensitized and subsequently i.g. saline- or OVA-challenged BALB/c WT and IL-9−/− mice. (C and D) Photomicrograph of CAE–stained jejunal sections from OVA-sensitized and -challenged BALB/c IL-9−/− and WT mice. Serum mouse mast cell protease-1 (E) and serum OVA-specific IgE (F) in saline- or OVA-sensitized, OVA-challenged BALB/c WT and IL-9−/− mice. Mean number of mast cells per HPF (G) and mast cell progenitor numbers (H) in the intestine under basal conditions in BALB/c WT and IL-9−/− mice. (A) Data are represented as the percentage of diarrhea occurrence over the number of OVA challenges. (B, E, and F) Data are represented as the mean ± the SEM; 4–5 mice per group from n = 3 experiments. (C and D) Photomicrograph, 10× magnification; insert, 40× magnification. Saline/OVA indicate saline-sensitized i.g. OVA-challenged mice and OVA/OVA indicate OVA-sensitized i.g. OVA-challenged mice. (G) Data represented as the mean ± the SEM; 4–5 mice per group from n = 4 experiments. (H) Data represented as the mean ± the SEM; 4 mice per group. Bars: (capped) 100 μm; (uncapped) 10 μm.
The inhibition of intestinal mastocytosis in oral antigen–challenged IL-9−/− mice led us to examine basal intestinal mast cell levels in WT and IL-9−/− mice. These were decreased twofold in IL-9−/− mice compared with WT mice (Fig. 1 G). In contrast, the basal level of mast cell progenitors was similar in the intestines of IL-9−/− mice and WT mice (Fig. 1 H).
Intestinal overexpression of IL-9 promotes intestinal mastocytosis
In an attempt to delineate the IL-9–regulated inflammatory pathways associated with oral antigen–induced intestinal anaphylaxis, we took a transgenic approach using the intestine-specific promoter of the rat fatty acid–binding protein (iFABPp) gene. This promoter has been extensively used to direct the expression of genes specifically in enterocytes of the small intestine (22, 23). LightCycler PCR analysis using murine IL-9–specific primers revealed an increase in the intestinal mIL-9 mRNA expression in the iFABPp-IL-9Tg mice compared with WT mice (Fig. 2 A). To confirm tissue-specific expression of IL-9, we performed PCR analysis on multiple tissues, including lung, kidney, liver, jejunum, ileum, and colon. IL-9 expression was only detected in the small intestine (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071046/DC1). Furthermore, IL-9 protein in serum and intestinal tissue was significantly elevated in iFABP-IL-9Tg mice compared with WT mice (Fig. 2, B and C). To assess the consequence of IL-9 expression in the small intestine, we performed histological analysis on the small intestine from WT and iFABPp-IL-9Tg mice. The gross morphology and architecture of the small intestine of iFABPp-IL-9Tg mice were similar in appearance to those of WT mice (Fig. 3, C and D). Furthermore, epithelial cell subpopulation numbers (goblet cell, enteroendocrine, Paneth, and enterocytes) and intestinal epithelial cell proliferation in the small intestine were equivalent between WT and iFABPp-IL-9Tg mice (unpublished data). Mast cell levels, however, were significantly elevated in iFABPp-IL-9Tg mice compared with WT mice (Fig. 3 A). Mast cells were predominantly localized to intraepithelial, intercryptic, and lamina propria regions of the small intestine of IL-9 intestinal transgenic mice (Fig. 3, A and D). We observed no difference in mast cell levels in the trachea and kidney between WT and iFABPp-IL-9Tg mice, confirming intestinal specificity (unpublished data). The level of mMCP-1 in the serum of iFABPp-IL-9Tg mice was approximately sixfold greater than that of WT mice (Fig. 3 B). In contrast, levels of mast cell progenitors in the small intestine and other tissues, including lung, spleen, and BM of iFABPp-IL-9Tg and WT mice were similar (Fig. 3 E). To determine the effect of intestinal expression of IL-9 on other intestinal immune parameters, we examined CD4+, CD8+ T cell, B (B220) cell, regulatory T cell (CD4+, CD25+, CD45RBlow, and FoxP3+), and DC levels in the mesenteric LN (MLN) of iFABPp-IL-9Tg and WT mice (Table S2). We observed no difference in the number or percentages of these cells in the draining MLNs of iFABPp-IL-9Tg and WT mice and no difference in T cell activation status (CD44, CD62L, and CD69) or DC subpopulations between groups. Finally, we examined serum IL-4 and IFNγ and total Ig levels in WT and iFABPp-IL-9Tg mice and observed no significant differences between groups (IL-4, 103.9 ± 28.8 vs. 134.2 ± 47.4 ng/ml; IFNγ, 717.4 ± 134.3 vs. 736.4 ± 133.1 ng/ml; mean ± the SD; n = 4–5 mice per group, WT and iFABPp-IL-9Tg, respectively; unpublished data). Collectively, these studies reveal that overexpression of IL-9 in the small intestine selectively promotes intestinal mastocytosis and mast cell activation.
Figure 2.

Increased systemic and intestinal IL-9 in iFABPp-IL-9Tg mice. (A) Quantitative PCR analysis of IL-9 mRNA expression in the jejunum and IL-9 protein levels in the sera (B) and jejunum (C) of WT and iFABPp-IL-9Tg mice. Open circles in A represent individual mice. Data are represented as the mean ± the SEM; 4–5 mice per group from n = 3 experiments.
Figure 3.

Intestinal mastocytosis in iFABPp-IL-9Tg mice. Localization of mean number of mast cells in the small intestine (A) and serum mMCP-1 in WT and iFABPp-IL-9Tg mice (B). (C and D) Photomicrograph of CAE-stained jejunum sections of BALB/c WT (C) and iFABPp-IL-9Tg mice (D). (E) Mast cell progenitor levels in the intestine, lung, spleen and BM of iFABPp-IL-9Tg and BALB/c WT mice. (A–E) Data represented as the mean ± the SEM; 4–5 mice per group from n = 4 experiments. A is a pictorial representation of localization determination. (C and D) Photomicrograph, 10× magnification; insert, 40× magnification. (E) Data represented as the mean ± the SEM; 4 mice per group. Bars: (capped) 100 μm; (uncapped) 10 μm.
Intestinal overexpression of IL-9 induces an experimental intestinal anaphylaxis transcriptome and phenotype
To gain a further understanding of the consequence of elevated intestinal IL-9 and mast cells on the intestine, we performed a genome-wide expression profile analysis using Affymetrix oligonucleotide chips on small intestinal RNA from WT and iFABPp-IL-9Tg mice. Using a criterion of 2-fold change, we identified 176 genes altered in the iFABPp-IL-9Tg mice (Table S3, available at http://www.jem.org/cgi/content/full/jem.20071046/DC1). Out of these transcripts, 126 were up-regulated and 52 were down-regulated. Functional classification of the altered transcripts revealed a significant predominance of mast cell–associated genes, including phospholipase A2, group IVC (26.9-fold), carbonic anhydrase 3 (12.7-fold), mMCP-2 (7.4-fold), mMCP-1 (6.8-fold), and Fc receptor IgE, high-affinity α polypeptide (FcεRI; Fig. 4 B). To validate the whole genome–wide findings, we performed real-time PCR analysis on select genes. Levels of mMCP-1 (∼50-fold), mMCP-2 (∼100-fold), mMCP-4 (∼10-fold), and FcεR1 (∼100-fold) in the small intestine of iFABPp-IL-9Tg mice were significantly elevated compared with WT mice (Fig. 4 A). Notably, we observed no increase in the levels of mMCP-5 mRNA expression (Fig. 4 A). These studies demonstrate that IL-9 overexpression up-regulates mast cell gene expression in the intestine. Previous whole genome–wide analysis of oral antigen–induced intestinal anaphylaxis (24) demonstrated that the most up-regulated genes associated with oral antigen–induced intestinal anaphylaxis were the mast cell–associated genes mMCP-2, mMCP-1, and Fc receptor, as well as IgE, high-affinity α polypeptide, and Th2-immunity genes, including RELMβ and small proline-rich protein 2A (Fig. 4 B) (24). Remarkably, comparison of the up-regulated genes in these studies and the up-regulated genes in the intestine of syngeneic iFABPp-IL-9Tg mice revealed a similar profile (Fig. 4 B). Thus, intestinal expression of IL-9 is sufficient to induce a genetic profile that overlaps with that observed in oral antigen–induced intestinal anaphylaxis.
Figure 4.

Overexpression of IL-9 in the intestine induces features of an intestinal anaphylaxis genotype. (A) Quantitative PCR analysis of mast cell gene mRNA expression in the jejunum of iFABPp-IL-9Tg and BALB/c WT. Results are expressed as the gene/GADPH ratio in respect to fold change over BALB/c WT. Gene expression was normalized to GADPH expression in each individual sample. Circles represent individual mice and black line represents mean value in each group (B) Genome-wide expression gene profile comparative analysis of iFABPp-IL-9Tg and BALB/c WT mice compared with OVA-sensitized, OVA-challenged BALB/c WT mice (6). Values represent fold increase over respective control. The complete dataset is available at the NCBI gene expression Omnibus (http://www.ncbi.nlm.gov) accession no. GSE10658.
Oral antigen–induced intestinal anaphylaxis in WT mice is associated with increased intestinal permeability (24). The similarities in the intestinal phenotype and gene profile between iFABPp-IL-9Tg mice and WT mice led us to examine this parameter in WT and iFABPp-IL-9Tg mice (Fig. 5). Resistance, which is a measure of tissue permeability, was significantly decreased in iFABPp-IL-9Tg mice compared with WT mice, indicating increased intestinal permeability (Fig. 5 A). To confirm altered epithelial cell barrier function in iFABPp-IL-9Tg mice, we examined intestinal permeability by analyzing FITC-dextran and HRP transport in jejunum segments ex vivo. Compared with control mice, iFABP-IL-9Tg jejunum had increased intestinal permeability to FITC-dextran and HRP (Fig. 5, B and C). To exclude the possibility that the increased intestinal permeability is not caused by mechanical stress of mast cells, we examined intestinal permeability in vivo. We confirmed our ex vivo analysis demonstrating in vivo increased intestinal permeability in iFABPp-IL-9Tg mice as compared with WT mice (unpublished data).
Figure 5.

Overexpression of IL-9 in the intestine induces features of an intestinal anaphylaxis phenotype including mast cell–dependent increased intestinal permeability and intravascular leakage. Transepithelial resistance (A) and intestinal permeability measured by FITC-dextran (B) and horseradish peroxidase (HRP; C) transport in jejunal segments ex vivo for iFABPp-IL-9Tg and BALB/c WT mice, serum mouse mast cell protease-1 and mean number of mast cells per high power field (hpf; D) and intestinal permeability measured by FITC-dextran (E) and HRP (F) transport in jejunal segments ex vivo for iFABPp-IL-9Tg and BALB/c WT mice treated with control or the mast cell stabilizing agent cromolyn sodium. (G) Percentage of hematocrit before and after 4× or 6× i.g. saline or OVA challenges of OVA-sensitized BALB/c WT mice. Percentage of hematocrit in iFABPp-IL-9Tg mice compared with BALB/c WT (H) and Evans blue extravasation in the jejunum and colon of iFABPp-IL-9Tg and BALB/c WT mice (I). (B) Data represents genes found to be up-regulated from profile analysis. (C–G) Data represented as the mean ± the SEM; 4–5 mice per group. (I) Data represents Evans blue concentration in jejunum and colon normalized per milligram of tissue protein. Black line represents mean value in each group.
Because previous studies using models of parasitic infestations demonstrated a role for mast cells in intestinal permeability (25, 26), the observed intestinal mastocytosis in iFABPp-IL-9Tg mice led us to assess the role of mast cells in intestinal permeability in these mice. Initially, we treated iFABPp-IL-9Tg and WT mice with the anti–c-kit (ACK2) (27) neutralizing antibody and demonstrated that mast cell depletion abrogated intestinal permeability in iFABPp-IL-9Tg mice (unpublished data). c-kit, however, is also expressed on interstitial cells of Cajal, which play a central role in the regulation of intestinal epithelial cell barrier function (28, 29). To confirm that the increased intestinal permeability in iFABPp-IL-9Tg mice is mast cell mediated, we treated mice with the mast cell stabilizer cromolyn. i.p. administration of cromolyn reduced serum mMCP-1 levels, confirming mast cell stabilization (Fig. 5 D). Notably, reduction in mast cell activity correlated with a reduction in HRP and dextran-FITC intestinal permeability in iFABPp-IL-9Tg mice (Fig. 5, E and F). Collectively, these studies indicate that intestinal permeability in iFABPp-IL-9Tg mice is mast cell mediated.
A major manifestation of food allergy, cardiovascular dysfunction, is primarily caused by increased vascular leakage (VL) (30–32). Initially, we assessed whether multiple oral OVA challenges induced VL in our experimental model of intestinal anaphylaxis. As VL causes hemoconcentration, we measured venous hematocrit in mice with oral antigen–induced intestinal anaphylaxis. Previous studies have demonstrated an increase in hematocrit during systemic anaphylaxis in humans, rats, dogs, and mice (33–36). We show that oral sensitization and oral antigen challenge induces a significant increase in hematocrit after four and six challenges (Fig. 5 G). We next examined naive iFABPp-IL-9Tg and WT mice and found that hematocrit levels in iFABPp-IL-9Tg mice were significantly greater than those observed in WT mice, suggesting increased VL (Fig. 5 H). To confirm VL, we measured Evans blue extravasation in the tissue of naive iFABPp-IL-9Tg and WT mice. 3 h after i.v. injection, Evans blue concentration in the small intestine of iFABPp-IL-9Tg mice was significantly greater than in WT mice. Notably, increased VL directly correlated with intestinal mastocytosis and IL-9 transgene expression in the jejunum (Fig. 5 I). These studies demonstrate that intestinal expression of IL-9 was sufficient to promote an intestinal anaphylaxis-like phenotype.
Intestinal expression of IL-9 is sufficient to predispose to intestinal anaphylaxis
The intestinal anaphylaxis phenotype in iFABPp-IL-9Tg mice prompted us to determine whether overexpression of IL-9 in the intestine increases susceptibility to oral antigen–induced intestinal anaphylaxis. We performed i.g. OVA challenge in OVA-sensitized WT and iFABPp-IL-9Tg mice. Susceptibility to intestinal anaphylaxis in iFABPp-IL-9Tg mice was significantly increased as compared with WT mice (Fig. 6 A). Greater than 80% of OVA-sensitized iFABPp-IL-9Tg mice developed diarrhea by the third OVA challenge compared with <25% of WT mice (Fig. 6 A). The increased acute diarrhea in iFABPp-IL9-Tg mice correlated with increased intestinal mast cell numbers and serum mMCP-1 levels compared with WT mice (Fig. 6, B and C). Notably, we observed no significant difference in OVA-specific IgE (Fig. 6 D), suggesting that the increased susceptibility was not caused by enhanced antigen-specific Th2 immunity. Remarkably, OVA challenge of unsensitized iFABPp-IL-9Tg mice induced diarrhea in 25% of iFABPp-IL-9Tg mice by challenge 6, and >80% by challenge 9 (Fig. 6 A). In contrast, i.g. OVA challenges of unsensitized WT mice did not induce diarrhea. We confirmed that the persistence of diarrhea in unsensitized iFABPp-IL-9Tg mice was antigen specific by administering consecutive i.g. OVA challenges to iFABPp-IL-9Tg mice and WT mice until they developed diarrhea, and then administering i.g. BSA (Fig. 6 E). BSA failed to induce acute diarrhea in iFABPp-IL-9Tg mice. Notably, subsequent i.g. OVA challenge reinduced diarrhea (Fig. 6 E). These results and the observation that unsensitized WT mice did not develop diarrhea demonstrate that diarrhea in unsensitized iFABPp-IL-9Tg mice was not solely the result of an osmotic load in the gut, but rather an antigen-specific acute immunological response.
Figure 6.

Overexpression of IL-9 in the intestine increases susceptibility to oral antigen–induced intestinal anaphylaxis. Diarrhea occurrence (A), mean number of mast cells per high power field (hpf; B), serum mouse mast cell protease-1 (C), and serum antigen-specific IgE (D) in OVA- or saline-sensitized and subsequently OVA-challenged BALB/c WT and iFABPp-IL-9Tg mice. (E) Diarrhea occurrence in OVA-challenged BALB/c WT and iFABPp-IL-9Tg mice and subsequently challenged with BSA. (A) Data represented as the percentage of diarrhea occurrence over number of OVA challenges. (B–D) Data are represented as the mean ± the SEM; 4–5 mice per group from n = 3 experiments. (E) Data are represented as the percentage of diarrhea occurrence over the number of OVA challenges, and then subsequent BSA challenge.
Intestinal anaphylaxis in unsensitized iFABPp-IL-9Tg mice is dependent on STAT6 and IL-4Rα pathways
IgE-mediated anaphylaxis is IL-4Rα dependent (37). IL-4 and -13 signal through IL-4R α-chain via STAT6 to promote CD4+ Th2-differentiation and IgE antibody production and to exacerbate the effector phase of anaphylaxis by increasing target cell responsiveness to vasoactive mediators (37). To confirm that oral antigen–induced intestinal anaphylaxis is mediated by STAT6 and IL-4Rα–dependent pathways, IL-4Rα and STAT6−/− mice were i.p. sensitized to OVA and subsequently received repeated i.g. OVA challenges. We demonstrate that oral antigen–induced intestinal anaphylaxis was dependent on STAT6 and IL-4Rα expression (Table S4, available at http://www.jem.org/cgi/content/full/jem.20071046/DC1). Notably, the absence of intestinal anaphylaxis in these mice was linked to diminished intestinal mast cell and serum mMCP-1 levels (Table S4) and OVA-specific IgE (Fig. S2). To determine if OVA-induced intestinal anaphylaxis in unsensitized iFABPp-IL-9Tg mice was dependent on an IL-4Rα–STAT6–IgE–mediated pathway, we crossed the iFABPp-IL-9Tg mice onto IL-4Rα– and STAT6-deficient (BALB/c) backgrounds. OVA-induced intestinal anaphylaxis (diarrhea) in naive iFABPp-IL-9Tg mice was ablated in the absence of IL-4Rα and STAT6 (Table I). Thus, intestinal anaphylaxis in iFABPp-IL-9Tg mice is largely dependent on IL-4Rα-, STAT6-, and mast cell/IgE pathways.
Table I.
Experimental intestinal anaphylaxis in iFABPp-IL-9Tg mice is IL-4Rα and STAT6 dependent
| Experimental group | Treatment | % Diarrhea occurrence (on day 9) |
Mast cells/HPF (mean ± SEM) |
Serum mMCP-1 (ng/ml; mean ± SEM) |
|
|---|---|---|---|---|---|
| BALB/c WT | 0.68 ± 0.07 | 27.46 ± 3.90 | |||
| BALB/c WT | Vehicle | OVA | 0/12 | 0.13 ± 0.01 | 23.91 ± 3.64 |
| BALB/c WT | OVA | OVA | 14/14 | 31.29 ± 0.57 | 16,260.57 ± 4,817.16 |
| IFABPp-IL-9Tg | 7.20 ± 0.33 | 152.50 ± 1.65 | |||
| IFABPp-IL-9Tg | Vehicle | OVA | 15/15 | 28.68 ± 0.82 | 19,319.34 ± 3,961.13 |
| IFABPp-IL-9Tg | OVA | OVA | 12/16 | 97.52 ± 2.19 | 83,399.48 ± 7,880.93 |
| IFABPp-IL-9Tg (IL-9Tg)/ STAT6−/− | 2.87 ± 0.31 | 37.90 ± 1.60 | |||
| IL-9Tg/STAT6−/− | Vehicle | OVA | 0/9 | 2.93 ± 0.21 | 870.30 ± 191.92 |
| IL-9Tg/STAT6−/− | OVA | OVA | 1/8 | 11.91 ± 0.66 | 1,168.17 ± 268.46 |
| IFABPp-IL-9Tg (IL-9Tg)/IL-4Rα−/− | 2.75 ± 0.01 | 26.20 ± 0.90 | |||
| IL-9Tg/IL-4Rα−/− | Vehicle | OVA | 0/10 | 3.04 ± 0.14 | 100.26 ± 15.01 |
| IL-9Tg/IL-4Rα−/− | OVA | OVA | 0/9 | 8.05 ± 0.19 | 808.07 ± 154.09 |
Diarrhea, mean number of mast cells per high power field of view and serum mMCP-1 for iFABPp-IL-9Tg and BALB/c WT mice deficient in IL-4Rα or STAT6 following intraperitoneal sensitization with OVA or saline and nine intragastric OVA or saline challenges.
Intestinal expression of IL-9 predisposes to oral antigen sensitization
The demonstration that oral antigen challenge induced intestinal anaphylaxis in unsensitized iFABPp-IL-9Tg mice and that this response depends on Th2-signaling led us to hypothesize that oral antigen challenge of iFABPp-IL-9Tg mice promotes sensitization rather than oral tolerance. To test this hypothesis, we orally challenged WT and iFABPp-IL-9Tg mice with OVA and examined intestinal IL-4 and CD4+ IL-4+ T cell and OVA-specific IgG1 levels (Fig. 7, A–C). Oral antigen challenge of WT mice induced no significant change in intestinal IL-4 mRNA or protein levels compared with naive WT mice (Fig. 7 A and not depicted). Furthermore, we observed no significant difference in the level of jejunal OVA-specific IgG1 (Fig. 7 C). In contrast, repeated oral antigen challenge of iFABPp-IL-9Tg mice increased the level of intestinal IL-4 mRNA and protein (Fig. 7 A and not depicted). This was associated with increased levels of intestinal OVA-specific IgG1 and total IgE (Fig. 7, C and D). To examine whether the IL-4–producing cells included CD4+ T cells, we used flow cytometry to evaluate anti-CD3/CD28–stimulated lamina propria (LP) cells from the small intestine of OVA-challenged WT and iFABPp-IL-9Tg mice. The percentage of CD4+ IL-4+ T cells in the LP of iFABPp-IL-9Tg mice was ∼2–3 fold greater than in WT mice (Fig. 7 B). Thus, intestinal expression of IL-9 appears to predispose to oral antigen–induced CD4+ Th2-type sensitization.
Figure 7.

Overexpression of IL-9 in the intestine increases local Th2 responses after OVA i.g. challenge. IL-4 protein levels in jejunal lysates (A) and percentage of CD4+ IL-4+ cells (B) in the lamina propria of the jejunum of BALB/c WT and iFABPp-IL-9Tg mice after i.g. OVA challenges. Antigen-specific IgG1 (C) and total IgE protein levels (D) in jejunal lysates from BALB/c WT and iFABPp-IL-9Tg mice under basal conditions and after 5 i.g. OVA challenges. (A) Data are expressed as protein level in picograms/milliliter per milligram protein. In A, each circle represents an individual mouse, and the black line represents the mean value in each group. (A–D) Data are represented as the mean ± the SEM; 4–5 mice per group from at least n = 2 experiments. Dotted line depicts the detection limit.
Intestinal IL-9 mast cell–mediated intestinal permeability (i.e., leaky gut) predisposes to oral antigen sensitization
We next evaluated the involvement of mast cell–mediated intestinal permeability in oral antigen sensitization and predisposition to intestinal anaphylaxis in iFABPp-IL-9Tg mice. To distinguish the involvement of mast cells in the sensitization and effector phases of intestinal anaphylaxis, we blocked mast cell activity during the initial i.g. sensitization stage, and reconstituted mast cell activity to evaluate effector function. To accomplish this, we used the mast cell stabilizer cromolyn that was previously shown to block mast cell activity and abrogate intestinal permeability in iFABPp-IL-9Tg mice (Fig. 5, D–F). Mice were treated with vehicle or cromolyn for 3 d to block intestinal permeability and were subsequently challenged i.g. with OVA while continuing cromolyn treatment for another 6 d. The control and cromolyn-treated iFABPp-IL-9Tg mice received six additional i.g. OVA challenges in the absence of cromolyn over the following 2 wk (Fig. 8 A). Consistent with our previous observations, i.g. OVA challenge of vehicle-treated iFABPp-IL-9Tg mice induced intestinal anaphylaxis (Fig. 8 B). However, OVA challenge of cromolyn-treated iFABPp-IL-9Tg mice did not induce intestinal anaphylaxis (Fig. 8 B). Studies performed to determine whether the attenuation of intestinal anaphylaxis was caused by inhibition of oral sensitization demonstrated elevated levels of IgE in OVA-challenged, control-treated, iFABPp-IL-9Tg mice compared with OVA-challenged, cromolyn-treated, iFABPp-IL-9Tg mice (Fig. 8 C). To confirm that the lack of diarrhea occurrence in these mice was not caused by cromolyn-suppression of mast cell activity during the effector phase, we performed passive sensitization in control- and cromolyn-treated iFABPp-IL-9Tg mice. After the ninth i.g. OVA challenge, mice were i.v. administered anti–TNP-IgE, and 24 h later they were challenged i.v. with BSA-TNP and body temperature, an indicator of systemic anaphylaxis, was measured over 60 min (Fig. 8 D at 20 min). Administration of BSA-TNP i.v. to naive iFABPp-IL-9Tg mice did not affect body temperature (Fig. 8 D). In contrast, i.v. administration of BSA-TNP to naive iFABPp-IL-9Tg mice that had previously been injected i.v. with IgE anti-TNP induced a significant decrease in body temperature. Importantly, BSA-TNP administration to vehicle- or cromolyn-treated, OVA-challenged iFABPp-IL-9 mice that received IgE anti-TNP also induced a rapid body temperature decrease, demonstrating functional mast cell effector activity (Fig. 8 D). Thus, lack of diarrhea occurrence in cromolyn-treated iFABPp-IL-9Tg mice was not caused by cromolyn-suppression of mast cell activity during the effector phase. Furthermore, these studies indicate that mast cell–mediated intestinal permeability promotes oral antigen sensitization and subsequent predisposition to intestinal anaphylaxis.
Figure 8.

Treatment with mast cell stabilizing agent cromolyn sodium blocks intestinal permeability and protects against antigen sensitization. (A) Experimental regimen. Diarrhea occurrence (B), total serum IgE for iFABPp-IL-9Tg and BALB/c WT mice treated with control or the mast cell stabilizing agent cromolyn sodium and subsequently OVA-challenged mice (C). (D) Passive anaphylaxis (maximum temperature decrease over 20 min) in iFABPp-IL-9Tg mice treated with control or the mast cell stabilizing agent cromolyn sodium. After the 9th OVA i.g. challenge, the mice were i.v. administered IgE-anti-TNP and subsequently i.v. injected withTNP-BSA. (A–D) Data represented as the mean ± the SEM; 4–5 mice per group. (A) Data are represented as the percentage of diarrhea occurrence over the number of OVA challenges in iFABPp-IL-9Tg and BALB/c WT mice treated with control or the mast cell stabilizing agent cromolyn sodium.
DISCUSSION
Our studies demonstrate a nonredundant role for IL-9 in the induction of the effector phase of intestinal anaphylaxis. The observed comparable levels of oral antigen–specific IgE and IL-4, -5, and -13 in IL-9−/− and WT mice suggest that the reduction in disease is not caused by an attenuated Th2 response. Consistent with this observation, previous studies have demonstrated no role for IL-9 in the development and differentiation of CD4+ Th2 T cells or antigen-driven antibody responses (38). Attenuation of oral antigen–induced intestinal anaphylaxis in IL-9−/− mice was linked to a reduction in intestinal mast cell number and degranulation, which is required for development of the effector phase of intestinal anaphylaxis (24). Our studies in IL-9–deficient and IL-9 transgenic mice demonstrate that IL-9 is critical for the induction of intestinal mastocytosis.
The molecular basis of IL-9–mediated intestinal mastocytosis is not fully elucidated. Previous studies show that IL-9 is insufficient to induce mast cell growth and differentiation of mast cell progenitors, but can enhance stem cell factor–dependent mast cell growth (39). Consistent with this, we observed no difference in intestinal mast cell progenitor levels among IL-9 deficient, iFABPp-IL-9Tg and WT mice, suggesting that IL-9 promotes recruitment of mature mast cells from other tissues or enhances mature mast cell survival. However, we did not observe an alteration in mature mast cell levels in tissues other than the intestine (unpublished data), and IL-9 has previously been shown to be insufficient to support survival of mature mast cells (39). An alternate explanation is that IL-9 may promote intestinal mastocytosis via enhancing intestinal mast cell maturation and that immune mechanisms independent of IL-9 critically maintain intestinal mast cell progenitor number.
We also demonstrate that IL-9 transgene–induced intestinal mastocytosis is reduced but can occur in the absence of IL-4Rα and STAT6 (Table S4). Moreover, intestinal mast cell levels in iFABPp-IL-9Tg mice backcrossed onto the IL-4Rα or STAT6 backgrounds were significantly higher than those observed in IL-4Rα or STAT6 deficient mice. Consistent with this observation, previous studies have demonstrated intestinal mastocytosis in the combined absence of IL-4- and IL-13, and that this was dependent on IL-9 (40). Importantly, although mast cell levels in iFABPp-IL-9Tg/IL-4Rα–deficient or iFABPp-IL-9Tg/STAT6–deficient mice were elevated compared with the IL-4Rα– or STAT6-deficient mice, the levels were reduced by ∼50% compared with iFABPp-IL-9Tg mice that express IL-4Rα and STAT6 normally. These studies suggest that optimal intestinal mastocytosis requires factors dependent on IL-4Rα and STAT6 signaling in our model. IL-4 has previously been shown to promote proliferation and prime human and murine mast cells for enhanced survival and IgE-mediated cytokine and chemokine production (41–45). Furthermore, antibody neutralization of IL-3 and -4 has been shown to block Nippostrongylus brasiliensis–induced intestinal mastocytosis by 85–90% (46). To confirm that the intestinal mastocytosis in iFABPp-IL-9Tg mice is not caused by increased IL-4 signaling, we examined the Th2 immune profile and IL-4Rα expression on intestinal mast cells in WT and iFABPp-IL-9Tg mice and observed no difference (unpublished data). We speculate that IL-9 is a potent inducer of intestinal mastocytosis; however, IL-9 may act additively or synergistically with other factors, including IL-3 and -4, to induce an optimal intestinal mast cell response. Furthermore, the importance of a particular mast cell stimulatory factor or signaling pathway may vary in different models. Consistent with this possibility, STAT6 signaling has been shown to enhance mastocytosis in some systems but suppress it in others (47, 48).
Our demonstration that IL-9–induced mastocytosis is associated with elevated mMCP-1 and -2 (enzymes associated with mucosal mast cells) suggests that IL-9 selectively stimulates generation of intestinal mucosal mast cells. Consistent with this, we observed a significant increase in interepithelial and intercryptic mast cells in the iFABP-IL-9Tg mice and experimental studies in cIL-9Tg mice have demonstrated IL-9–induced mastocytosis associated with elevated MCP-1 and -2 (49). Furthermore, in vitro studies demonstrate that IL-9 stimulates mouse BM–derived mast cells to express high steady-state levels of mMCP-1 and -2 transcripts (50, 51). Observations that mast cell lines, human CD34+ cord blood cells, and 12-wk–cultured mast cells all express IL-9Rα mRNA suggest this may be a direct effect of IL-9 on mast cells (39, 52).
The effector phase of intestinal anaphylaxis has been shown to be mediated via mast cell–, FcεRI-, and IgE-dependent pathways (24). Consistent with this observation, we show that intestinal anaphylaxis in iFABPp-IL-9Tg mice was dependent on mast cells and IL-4Rα and STAT6 signaling. Importantly, murine systemic anaphylaxis can also occur via a mast cell, FcεRI, and IgE-independent pathway, via IgG antibody, macrophages, FcγRIII, and PAF (53–55). Recent studies that used IL-9 transgenic mice in which IL-9 is constitutively expressed in all tissues (cIL-9Tg mice) (56), IL-9R−/−, and models of systemic anaphylaxis have demonstrated that IL-9/-9R signaling can potentiate, but is not essential for, systemic anaphylaxis (57). In contrast, we demonstrate that oral antigen–induced intestinal anaphylaxis is critically dependent on IL-9. A possible explanation for these contrasting findings is the distinct molecular pathways central to the induction of systemic and intestinal anaphylaxis, or alternatively, that mast cell/IgE-mediated systemic anaphylaxis is regulated by IL-9–independent pathways. To begin to discriminate between these hypotheses, we examined active systemic anaphylaxis in WT and IL-9−/− mice. i.v. OVA challenge of OVA-sensitized WT and IL-9−/− mice induced systemic anaphylaxis (Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20071046/DC1). Furthermore, we observed an equivalent increase in serum mMCP-1 and histamine in WT and IL-9−/− mice, indicating that activation of the mast cell–dependent pathway can occur in the absence of IL-9 (Fig. S3, B and C). Collectively, these studies demonstrate that IL-9 plays a critical role in mast cell/IgE–mediated intestinal anaphylaxis; however, mast cell/IgE–mediated systemic anaphylaxis can occur in the absence of IL-9/IL-9R signaling. Interestingly, we observed elevated levels of serum histamine in WT and IL-9−/− mice during active systemic anaphylaxis, but not during intestinal anaphylaxis. This is consistent with our prior studies demonstrating that mast cell–mediated systemic anaphylaxis is histamine-dependent, whereas, intestinal anaphylaxis is mediated by PAF and serotonin (24, 55). These studies also demonstrate that differential mast cell–derived mediators (histamine and PAF/serotonin) exacerbate the clinical symptoms of systemic and intestinal anaphylaxis.
Induction of intestinal allergic responses, including OVA-induced intestinal anaphylaxis in WT mice, is dependent on antigen sensitization with adjuvant and subsequent multiple-antigen challenges (24, 58, 59). Remarkably, intestinal overexpression of IL-9 was sufficient to predispose mice to intestinal anaphylaxis in the absence of systemic sensitization. Notably, we show that the development of intestinal anaphylaxis in the absence of systemic sensitization was associated with development of an antigen-specific CD4+ Th2 cell response and dependent on CD4+ Th2-signaling pathways (IL-4Rα and STAT6). These studies suggest that intestinal overexpression of IL-9 promotes the generation of antigen-specific CD4+ Th2 cell responses. Consistent with this observation, recent investigations using a model of Leishmania major infection demonstrated a role for IL-9 in the promotion of a detrimental Th2-type intestinal response (60). Although in this study the mode of IL-9 action was not defined, we demonstrate that IL-9 promotion of an oral antigen–specific CD4+ Th2 cell response is primarily via induction of mast cell–mediated intestinal permeability. Notably, mast cell–mediated intestinal permeability has been shown to be primarily regulated via IL-4 and -13 (26). The increased intestinal permeability in iFABPp-IL-9Tg mice occurred in the absence of elevated IL-4 and -13. The mechanism of mast cell–induced increase in intestinal permeability that leads to oral antigen sensitization and predisposes to intestinal anaphylaxis is not yet fully elucidated. Previous studies using cIL-9Tg mice have demonstrated a mast cell–mediated increase in intestinal epithelial permeability (25). It remained possible that the IL-9–induced intestinal mastocytosis and increased intestinal permeability were mediated via an indirect mechanism, possibly through stimulation of intestinal epithelial cells. To test whether IL-9 could act directly on intestinal epithelial cells, we examined IL-9Rα expression on the human intestinal epithelial cell line (CaCO2bbe) by flow cytometry. We did not detect any IL-9Rα expression on CaCO2bbe cells (unpublished data). Consistent with this observation, we observed no effect of IL-9 stimulation on CaCO2bbe cell TER and ΔIsc to cholinergic stimulation (unpublished data). Mast cells generate several mediators that promote an increase in intestinal permeability, including histamine, serotonin, eicosanoid mediators (leukotrienes [LTB4 and LTC4] and prostaglandins [PGD2 and PGE2]), the cytokines IL-4 and TNFα, and mast cell proteases (mMCP-1 and -2) (25, 26, 61). Previous studies using a helminth infection model of intestinal permeability have demonstrated a role for mMCP-1 in increased mucosal leakiness via degradation of the tight junction protein occludin (25). Interestingly, gene chip analysis revealed a significant increase in the expression of phospholipase A2 gene in the intestine of iFABPp-IL-9Tg mice as compared with WT mice. PLA2 catalyzes the hydrolysis of the sn-2 position of membrane glycerophospholipids to liberate arachidonic acid, which is a precursor of eicosanoids including prostaglandins, leukotrienes, and PAF. Notably, we have previously demonstrated a role for serotonin and PAF in intestinal diarrhea in an IgE-mast cell–dependent model of intestinal anaphylaxis (62). Defining the molecular mechanisms involved in IL-9/mast cell–mediated increase in intestinal permeability will be important for understanding of oral sensitization and subsequent development of intestinal anaphylaxis.
The demonstration that increased intestinal permeability in iFABPp-IL-9Tg mice predisposes to oral antigen sensitization suggests that a constitutive defect in barrier function could predispose to oral sensitization and subsequent development of food allergy. Impairment of intestinal barrier function has been implicated as a critical determinant in the predisposition to several GI diseases, including IBD and food allergy (63, 64). Indeed, patients with atopic diseases, including food allergy, have increased intestinal permeability (15, 65, 66). Furthermore, increased intestinal permeability in IBD is predictive of clinical relapse and 10–25% of first-degree relatives of patients with IBD have increased intestinal permeability (64). Recently, a mutation in the caspase recruitment domain family member 15/nucleotide binding oligomerisation domain 2 3020insC has been linked to increased intestinal permeability in quiescent Crohn's disease patients and their first-degree relatives (63). Genetic mapping studies in both humans and mice demonstrated a linkage between the atopic phenotype and the IL-4 and -9 gene (67). Furthermore, gene–gene interactions between IL-4Rα and -9 SNPs, particularly IL4RA Q576R and IL-9R rs731476, have been observed in asthma-related diseases (68).
An alternate explanation to a congenital abnormality in intestinal permeability predisposing to oral sensitization and food allergy is that perturbations of environmental factors, such as stress, may induce intestinal barrier dysfunction and leaky gut. Notably, recent clinical studies have demonstrated an increase in food protein sensitivity in pediatric liver transplant patients treated with the immunosuppressive tacrolimus (69, 70). Tacrolimus, a calcineurin inhibitor, increases intestinal permeability by uncoupling mitochondrial oxidative phosphorylation, leading to a disruption in intercellular junctional integrins (71, 72). Two recent studies examining heart and liver transplant patients taking tacrolimus revealed they have an increased risk of developing food allergy (73, 74). Experimental investigations have also provided corroborative evidence supporting a role for intestinal barrier dysfunction and leaky gut, predisposing to oral sensitization and subsequent development of food allergy. Malnutrition increased β-lactoglobulin–specific IgE and intestinal anaphylactic responses in guinea pigs, demonstrating an effect of environmental factors in intestinal permeability and oral antigen sensitization (75, 76).
Other environmental factors, including intestinal infection that leads to elevated levels of IL-9, mast cells and increased intestinal barrier dysfunction, may also predispose to oral antigen sensitization. Notably, L. major and Helicobacter pylori infection has been shown to be associated with the up-regulation of IL-9 expression (60, 77–79). Consistent with these observations, mast cells and mast cell–derived mediator chymase are elevated in experimental leishmaniasis and human H. pylori–associated gastritis (60, 80). Notably, H. pylori gastric infection has been shown to positively correlate with food allergy (81).
Previous investigations using nematode infestation and systemic and intestinal anaphylaxis models have demonstrated a role for mast cells in the end-stage effector phase of disease (20, 24–26). We provide corroborative data supporting a role for mast cells in exacerbation of the intestinal anaphylactic phenotype. Notably, the role of mast cells in the end-stage effector phase of disease is dependent on IgE and an established antigen-specific CD4+ Th2-type response (24). In this study, we demonstrate that mast cells also promote the development of an antigen-specific CD4+ Th2-type response. Furthermore, we show that the mast cell–dependent development of the antigen-specific CD4+ Th2-type response was regulated via an indirect mechanism involving intestinal epithelial permeability. Importantly, these studies have identified a role for mast cells in oral antigen sensitization. Interestingly, patients with systemic mastocytosis often present with intestinal manifestations, and some of these patients have impaired small intestinal absorption (82). Clinical studies have demonstrated that cromolyn is a successful treatment modality for intestinal symptoms of systemic mastocytosis (83). A major draw back of cromolyn is its poor absorption properties, thus it is not unreasonable to speculate that the ability of cromolyn to successfully treat the intestinal symptoms in systemic mastocytosis may be attributable, at least in part, to increased intestinal permeability (84).
In conclusion, we demonstrate a central role for IL-9 in the regulation of oral antigen–induced intestinal anaphylaxis and identify a previously unappreciated role for mast cell–induced intestinal permeability in oral antigen sensitization and predisposition to intestinal anaphylaxis. These studies demonstrate the importance of intestinal barrier function in oral antigen sensitization and identify a role for IL-9–driven mast cells in this process.
MATERIALS AND METHODS
Mice
6–8-wk-old IL-9–deficient mice (N10 BALB/c), as previously described (38), were a gift from A. McKenzie (Medical Research Council, Laboratory of Molecular Biology, Cambridge, UK). BALB/c mice were obtained from the National Cancer Institute. All mice were maintained in a barrier facility, and animals were handled under Institutional Animal Care and Use Committee–approved protocols (from Cincinnati Children's Hospital Medical Center).
Generation of transgenic mice
cDNA was a gift from A. McKenzie. The IL-9 cDNA was amplified by PCR using oligonucleotides containing BamHI sites (5′-GGATCCATGTTGGTGACATACATCCTTGC-3′ and 3′-GGATCCTCATGGTCGGCTTTTCTGCC-5′) and the 446-bp fragment containing the entire coding region of the murine IL-9 cDNA was ligated into pCR2.1 TOPO TA cloning vector. The IL-9 cDNA was digested with BamHI, and the IL-9 DNA was ligated into the BamHI site of the PBSIF1178-hGHpgkNeo plasmid, which contained a 3.5-kb EcoRI fragment containing nucleotides −1,178 to 28 of rat Fabpi promoter linked to nucleotides 3–2,150 of the human growth hormone (hGH) gene (except for its 5′ regulatory sequences). The transgene plasmid was propagated in Escherichia coli DH5α cells, and the transgene fragment was liberated from the vector sequences by EcoRI endonuclease digestion and gel electrophoresis, and then purified using the QIAEX DNA extraction kit (QIAGEN). After extensive dialysis, 5 μg of the linearized fragment was co-electroporated with 5 μg of circular neomycin resistance plasmid (pMC1Neo; Stratagene) into BALB/c embryonic stem cells, a gift from B. Ledermann (University of Zurich, Zurich, Switzerland). Positive selection was performed with G418 for 10 d, and 7 surviving clones were screened for integration of the transgene by PCR. Of 3 Tg-positive embryonic stem colonies, 1 was injected into 3.5-d-old blastocysts from C57BL/6 mice and implanted into pseudopregnant females. Chimeric mice were bred with WT BALB/c females and germline (white) mice genotyped to identify positive transgenic mice. Heterozygous-positive Tg mice were crossed to WT BALB/c mice for 2 generations to remove any possible random modifications caused by tissue culture. Transgenic mice were identified by Southern blot analyses after restriction fragment digestion with BamHI, using the hGH genomic fragment to ensure specificity for identification of the transgene. Mice transgenic for IL-9 were also identified by PCR using a forward primer (P1; 5′-GGATCCATGTTGGTGACATACATCCTTGC-3′) specific for the IL-9 cDNA in the transgenic construct and a reverse primer specific for hGH (P2; 5′-GTGAGCTGTCCACAGGACC-3′); the transgenic band was ∼484 bp. STAT6- or IL-4Rα–deficient mice expressing the iFABPp-IL-9Tg were generated by mating STAT6 or IL-4Rα–deficient mice of the BALB/c background (85–87) with iFABPp-IL-9 mice and subsequently mating iFABPp-IL-9+ F1 mice with STAT6- or IL-4Rα–deficient mice. The resulting F2 mice were screened by PCR analysis for the presence of the IL-9 transgene and for the homozygous deficiency of the STAT6 or IL-4Rα gene using the aforementioned primers. Control mice were matched to WT mice derived from both original backgrounds.
Experimental intestinal anaphylaxis.
6–8-wk-old mice were sensitized twice, 2 wk apart, with 50 μg of OVA (grade V, A-5503; Sigma-Aldrich) in the presence of 1 mg of aluminum potassium sulfate adjuvant (alum, AIK[SO4]2-12H2O; A-7210; Sigma-Aldrich) in sterile saline or sterile saline by i.p. injection. 2 wk later, mice were held in the supine position three times a week (every other day) and orally administered 250 μl OVA (50 mg) in saline. Before each i.g. challenge, mice were deprived of food for 3–4 h with the aim of limiting antigen degradation in the stomach. Challenges were performed with i.g. feeding needles (01–290-2B; Thermo Fisher Scientific). Diarrhea was assessed by visually monitoring mice for up to 1 h after i.g. challenge. Mice demonstrating profuse liquid stool were recorded as diarrhea-positive animals. Sometimes multiple observers blinded to the experimental protocol scored the occurrence of diarrhea. To stabilize mast cell activity, mice were administered i.p. 100 mg/kg (200 μl) cromolyn sodium (Sigma-Aldrich) every 12 h (final volume 200 μl) for 2.5 d. Mast cell stabilization was determined by serum mMCP-1 levels.
Active systemic anaphylaxis.
WT and IL-9−/− mice were immunized with OVA/alum (50 μg OVA/1 mg alum [200 μl]) or alum alone on day 0 and 7. On day 14, mice were i.v. challenged with 1 mg OVA in 200 μl saline. Rectal temperatures were measured with a Digital Thermocouple Thermometer (model BAT-12; Physitery Instruments, Inc.) just before challenge, and then at 5, 10, 15, 30, 45, and 60 min after OVA challenge. As controls, naive WT mice were i.v. administered anti-IgE (clone EM95; 10 μg/200 μl saline), isotype control (clone: GL117; 10 μg/200 μl saline), or OVA (grade V; 1 mg/200 μl saline). Additional WT and IL-9−/− mice were i.p. injected with OVA/alum and subsequently challenged i.g. with OVA until 100% of the WT experienced diarrhea. After the eighth i.g. challenge, development of diarrhea was confirmed and serum was collected for histamine analysis. 48 h after the eighth i.g. challenge, the mice were injected i.v. with OVA (100 μg/200 μl saline) and their rectal temperatures were monitored to demonstrate systemic anaphylaxis. 15 min after i.v. OVA challenge, serum was taken for histamine analysis.
Ussing chambers.
1-cm segments of mucosa were mounted in U2500 Dual-Channel Ussing chambers (Warner Instruments) that exposed 0.30 cm2 of tissue to 10 ml of Krebs buffer. Agar-salt bridges and electrodes were used to measure the potential difference. After a 15-min equilibrium period, basal Isc, representing the net ion flux at baseline, and tissue resistance (TER), a measure of tissue permeability, were determined. Every 50 s, the tissues were short circuited at 1 V (EC 800 Epithelial Cell Voltage Clamp; Warner Instruments), and the Isc was monitored continuously. In addition, every 50 s, the clamp voltage was adjusted to 3 mV for 5 s to allow calculation of tissue resistance using Ohm's law. After a second 15-min period, concentration-dependent changes in Isc (ΔIsc) were determined for the cumulative addition of methacholine to the serosal side of the stripped mucosa. In some experiments, after equilibrium period and baseline potential difference and resistance had been established, FITC-dextran (2.2 mg/ml, molecular mass 4.4 kD; Sigma-Aldrich) was added to the mucosal reservoir. Medium (0.25 ml out of 10 ml) was removed from the serosal reservoir and replaced with fresh medium every 20 min over a period of 180 min for measurement of FITC-dextran. The concentration of HRP was measured by a kinetic enzymatic assay. In brief, 120 μl of sample were added to 800 μl of phosphate buffer containing 0.003% H2O2 and 80 μg/ml o-dianisidine (Sigma-Aldrich), and the enzymatic activity was determined from the rate of increase in optical density at 460 nm during a 1.5-min period. The luminal-to-serosal flux was calculated using a standard formula and expressed as nanograms/milliliter. FITC-dextran concentration was determined from analysis of standard curve of dextran-FITC using a FLX800 96-well microplate fluorescence reader (excitation, 490 nm; emission, 530 nm; BioTek Instruments).
Solutions and drugs.
Krebs buffer contained 4.70 mM KCl, 2.52 mM CaCl2, 118.5 mM NaCl, 1.18 mM NaH2PO4, 1.64 mM MgSO4, and 24.88 mM NaHCO3 on each side. The tissues were allowed to equilibrate for 15 min in Krebs buffer containing 5.5 mM glucose. All reagents were obtained from Sigma-Aldrich unless stated otherwise.
Ribonuclease protection assay
Jejunum RNA was obtained using TRIzol reagent (Life Technologies, Inc.) following the manufacturer's protocol. The ribonuclease protection assay was performed by making a radioactive probe from the mCk-1b multiprobe template (RiboQuant Multi-Probe RPA System; BD Biosciences). RNA from OVA- and saline-challenged BALB/c WT mice was hybridized overnight with the radioactive probe, purified, and finally run on an urea-acrylamide gel at 75 W as described in the RiboQuant protocol from BD Biosciences.
Northern blot analysis
RNA was extracted from the lung, kidney, liver, jejunum, ileum, and colon tissue using TRIzol reagent following the manufacturer's protocol. 20 μg of total RNA was used for Northern blot analysis, as previously described (88).
Mononuclear cell (MNC) preparation and MCp assessment
Mice were killed by CO2 asphyxiation, and the small intestine, lungs, spleen, and BM were harvested. The entire organs were removed except for BM, in which case a single femur was taken from each mouse. Individual tissues from 2 mice were pooled, placed in 20 ml RPMI 1640 complete (RPMI 1640 containing 100 U/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml gentamicin, 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 10% heat-inactivated fetal calf serum), and processed essentially as previously described (89). In brief, the intestines (flushed out and rinsed twice in HBSS) were finely chopped with a scalpel blade and transferred separately to 50-ml plastic tubes with 30 ml RPMI 1640 complete plus 1 mg/ml collagenase type 4 (Worthington Biochemical Corp.). There were 3 enzymatic digestions performed for ∼20 min each at 37°C. The undigested tissue clumps were collected after each digestion period and were subjected to another enzymatic digestion, whereas the liberated cells were pelleted, resuspended in 44% Percoll (Sigma-Aldrich), overlayed on a 67% Percoll layer, and spun at 400 g for 20 min at 4°C. The cell collection procedure for BM and spleen omitted the digestion steps. BM was extruded from one femur of each animal using a 25-gauge syringe and 5–10 ml complete RPMI 1640. Spleen cells were obtained from crushed whole spleens suspended in complete RPMI 1640. The collected cells were pelleted and resuspended in 44% Percoll before centrifugation over 67% Percoll as described. The MNCs were harvested from the interfaces of the three digestions of the lung and intestine, pooled by a separate tissue source, and washed in complete RPMI 1640. The numbers of viable cells were determined by trypan blue dye exclusion with a hemocytometer. Cells were serially diluted in complete RPMI 1640, and 100-μl samples of the MNC dilutions were added to each well of standard 96-well flat-bottomed microtiter plates (Corning). Typically, 24 wells were plated for each cell concentration. Intestinal or BM MNCs were plated starting at 5,000–10,000 cells/well, and lung or spleen MNCs starting at 20,000–40,000 cells/well. Next, each well received 100 μl γ-irradiated (30 Gy) splenic feeder cells plus cytokines (recombinant mouse IL-3 at 20 ng/ml and recombinant mouse stem cell factor at 100 ng/ml). The cultures were incubated in humidified 37°C incubators with 5% CO2 for 12 to 14 d, and positive wells containing mast cell colonies were identified and counted with an inverted microscope. The MC colonies were easily distinguished as large colonies of nonadherent small- to medium-sized cells. The MCp concentration is expressed as the number of MCps per 106 MNCs isolated from the tissue. The number of MCps/tissue is derived by multiplying the concentration of MCps by the MNC yield/organ.
ELISA measurements.
mMCP-1 and histamine serum levels were measured by ELISA according to the manufacturer's instructions (Moredun Scientific and Beckman Coulter). ELISA determined serum and jejunal OVA-specific IgE. In brief, plates were coated for 2 h with 100 μl of anti-IgE antibody (EM-95; 10 μg/ml; BD Biosciences) and blocked with 200 μl of 10% FBS (diluted in sterile PBS) before adding serial dilutions of plasma samples (100 μl/well). After overnight incubation, plates were washed and biotinylated OVA was added (2.5 μg/ml, 100 μl/well). After 1 h of incubation, 1 μg/ml streptavidin-HRP (Biosource) was added. Before the initiation of each step, plates were washed with 0.05% Tween-20 in PBS. Finally, after a 1-h incubation, 100 μl of substrate (TMB Substrate Reagent Set; BD Biosciences) was added. Colorimetric reaction was stopped with 1 M H2SO4 and was quantified by measuring optical density with an ELISA plate reader at 450 nm. Jejunal OVA-specific IgG1 levels were determined by ELISA. In brief, wells were coated with 100 μg/ml OVA and blocked with 10% FBS in PBS. The wells were washed with 0.05% Tween-20 in PBS and 100 μl of plasma samples (diluted 1/100) were added and incubated for 2 h at room temperature. Plates were washed and 0.5 μg/ml HRP-conjugated anti–mouse IgG1 (X56; BD Biosciences) or biotin-conjugated rat anti–mouse IgG1 (A85-1; BD Biosciences), followed by streptavidin and 1 μg/ml HRP conjugate (Biosource), was added. 100 μl of substrate (TMB Substrate Reagent Set; BD Biosciences) was added. Colorimetric reaction was stopped with 1 M H2SO4 and was quantified by measuring optical density with an ELISA plate reader at 450 nm. IL-4, -5, -9, -13, and IFNγ in jejunum was measured by ELISA according to the manufacturer's instructions (BD Biosciences and R&D Systems). For jejunal lysate samples, whole jejunum was excised and snap frozen at −70°C. Frozen jejunum sections were mechanically disrupted and suspended in 1 ml of PBS containing protease inhibitors. The jejunal pellet was vigorously vortexed, the suspension was centrifuged at 12,000 g for 10 min, and supernatant was removed, aliquoted, and stored at −20°C until analysis. Tissue protein samples were quantitated using a BCA protein assay kit following the manufacturer's instructions (Pierce Chemical Co.). In vivo IL-4 and IFNγ levels were determined by IVCCCA, as previously described (90). In brief, iFABPp-IL-9Tg and BALB/c WT mice were i.v. injected with biotinylated rat IgG neutralizing monoclonal antibody anti–mouse IL-4 (BVD-1D11 [10 μg/mouse] and biotinylated rat IgG neutralizing monoclonal antibody anti–mouse IFNγ [R4-6A2; 10 μg/mouse]) and bled 24 h later. Serum levels of IL-4 and IFNγ were determined by ELISA, as previously described (90).
Intestinal mast cell quantification.
Jejunum tissue was collected 7–10 cm distal to the stomach, whereas ileum and colon samples were collected 1 cm proximal or distal of the cecum. All samples were fixed in 10% formalin and processed by standard histological techniques. The 5-μm tissue sections were also stained for mucosal mast cells with chloroacetate esterase (CAE) activity, as described elsewhere (24) and lightly counterstained with hematoxylin. At least four random sections per mouse were analyzed. Quantification of stained cells was performed by counting the number of chloroacetate-positive cells from 25–50 fields of view (magnification 40×).
Microarray hybridization.
After TRIzol purification, RNA was repurified with phenol-chloroform extraction and ethanol precipitation. Purified RNA from four WT and four iFABPp-IL-9Tg mice were then pooled together and processed at Cincinnati Children's Hospital Medical Center Affymetrix Gene Chip Core facility, using the murine MOE430_2, a whole genome expression chip encoding 45,101 genes as previously described by the manufacturer (Affymetrix). Differences between WT and iFABPp-IL-9 mice were also determined using the GeneSpring software (Silicon Genetics). Data were normalized to WT mice, and genes were screened for a greater than twofold change over saline. A further description of the methodology, according to MIAME (minimum information about a microarray experiment) guidelines are available at www.mged.org/Workgroups/MIAME/miame.html. The complete dataset is available at the National Center for Biotechnology Information gene expression Omnibus (http://www.ncbi.nlm.gov) accession no. GSE10658.
Lightcycler PCR
BALB/c WT and iFABPp-IL-9Tg mice were obtained and killed. Intetinal samples were harvested. RNA was isolated from intestinal samples and cDNA was generated by standard procedures. The RNA samples (500 ng) were subjected to reverse transcription analysis using Iscript reverse transcription (Bio-Rad Laboratories) according to manufacturer's instructions. GAPDH, mMCP -1, -2, -4, -5, and FCεR1α were quantified by real-time PCR using the LightCycler instrument and LightCycler FastStart DNA Master SYBR Green I as a ready-to-use reaction mix (Roche). Results were then normalized to GAPDH amplified from the same cDNA mix and expressed as fold induction compared with the controls. cDNAs were amplified using the following primers; GAPDH forward 5′-TGGAAATCCCATCACCATCT-3′, reverse 5′-GTCTTCTGGGTGGCAGTGAT-3′; mIL-9 forward 5′-AGCTGCTTGTGTGTCTCTCCGTC-3′, reverse 5′-TCACCCGATGGAAAACAGGCAA-3′, mMCP-1, -2, and -4 forward 5′-GCTGGAGCTGAGGAGATTATTG-3′, mMCP-1 reverse 5′-GATTAAAAACAGCATACATGGGAG-3′, mMCP-2 reverse 5′-CCTCTCCTTCGAACCGTTCTTA-3′, mMCP-4 reverse 5′-GAGGCCTGTAAAAACTATTGGCA-3′; and mMCP-5 forward 5′-GAACTACCTGTCGGCCTGCAG-3′, mMCP-5 reverse 5′-TTCAGTAGCATGATGTGCGTGGAC-3′, and FcεR1α forward 5′-TTAGCACCTGAAGGTGCAGGGG-3′ and FcεR1α reverse 3′-ACATGAGTGGCCTTTGACAGTG-5′. mIL-4 primers were used as described by the manufacturer (PPM3013A; SuperArray Frederick). Quantitative expression data from each gene of interest was normalized to GAPDH expression, and then expression in IL-9 Tg mice was compared with expression in WT mice.
FACS analysis
Single-cell suspensions from indicated organs were washed with FACS buffer (PBS/1% FCS) and incubated with combinations of the following antibodies (all antibodies were obtained from BD Biosciences unless indicated): PerCP anti–mouse CD4 (l3T4) (RM4-5); PE anti–mouse CD8a (53–6.7); APC anti–mouse CD62L (MEL-14); FITC anti–mouse CD44 (IM7), APC anti–mouse CD25 (PC61), PE anti–mouse CD45RB (16A), and FITC anti–mouse FoxP3 (FJK-16S); PE anti–mouse B220 (RA3-6B2), FITC anti–mouse CD23 (B3B4) and PE-Cy7 anti–mouse IgM (R6-60.2); APC anti–mouse CD11c (HL3), APC-Cy7 anti–mouse Gr-1 (RB6-8C5) and PE-Cy7 anti–mouse CD11b (M1/70); PE anti–mouse CD4 (L3T4), and PE-Cy7 anti–mouse IL-4 (BVD6; ebioscience). The following antibodies were used as appropriate isotype controls: PerCP rat IgG2a (R35-95), PE rat IgG2a, (53–6.7), APC rat IgG2a (R35-95); FITC rat IgG2a (R35-95), PerCP rat IgG2a, (R35-95), APC rat IgG1, (R3-34), PE rat IgM (R4-22); and FITC rat IgG2a, (R35-95), PE rat IgG2a (R35-95), FITC rat IgG2a (R35-95), and PE-Cy7 rat IgG2a (R35-95), respectively. 7-amino-actinomycin-D was used to identify nonviable cells (BD Biosciences). Cells were analyzed on FACSCalibur (BD Biosciences) and analysis was performed using FlowJo software.
Vascular permeability
Peripheral blood samples were collected in EDTA Microtainer tubes (Becton Dickinson) by retroorbital bleeding. Automated total cell counts and differential counts were performed according to manufacturer's instructions (Thermo Fisher Scientific). Evan's blue tissue extravasation was performed as previously described (91). In brief, mice received i.v. Evan's blue dye in (20 mg/kg) PBS, and 3.5 h later, mice were anesthetised i.p. with 20 mg/kg pentobarbital, and heart perfusion was performed (10 ml PBS arterial perfusion). Jejunum and colon were harvested, and Evan's blue extravasation was measured in OD at 650 nm. Tissue protein levels were quantified using a BCA protein assay kit, following the manufacturer's instructions (Pierce Chemical Co.).
Mononuclear cell isolation from jejunal tissue
Approximately 5 cm of jejunum was excised and flushed with 1 ml of calcium- and magnesium-free HBSS (CMF-HBSS). The jejunum was dissected longitudinally and placed in 5 ml CMF-HBSS and shaken vigorously for 30 s at room temperature to remove luminal debris. Tissue was then incubated in CMF-HBSS containing 10% FCS, 25 mM Hepes, and 5 mM EDTA for 10 min at 37°C and shaken in 5-min intervals to remove epithelia and intraepithelial lymphocytes. The tissue was then washed and incubated in CMF-HBSS to block any remaining EDTA activity. The remaining tissue was cut into small pieces and incubated with incomplete RPMI 1640 supplemented with Collagenase A (2.4 mg/ml) for 30 min at 37°C. The cell suspension was filtered using sterile gauze, washed in incomplete RPMI 1640, and centrifuged, and the remaining pellet was resuspended in RPMI 1640 + 10% FCS. MNC suspension was used in in vitro stimulation assays.
In vitro stimulation of jejunum MNCs
MNCs were plated at 5 × 105 cells/ml for 6 h in the presence of 10 ng/ml IL-2 (BD Biosciences) and αCD3/αCD28 (5 and 1 μg/ml, respectively; BD Biosciences) and monensin (eBioscience) to block intracellular protein transport. MNCs were examined for CD4 and IL-4 expression by flow cytometry as previously described (89).
Passive anaphylaxis model
Mice were primed i.v. with 10 μg of IgEαTNP and then challenged i.v. 24 h later with 100 ng TNP-BSA. The severity of the anaphylactic shock was assessed by change in rectal temperature using rectal probe (Model BAT-12; Physitemp) as previously described (34, 92).
Statistical analysis
Data are expressed as the mean ± the SEM, unless otherwise stated. Statistical significance comparing different sets of mice was determined by Student's t test. In experiments comparing multiple experimental groups, statistical differences between groups were analyzed using the one-way analysis of variance nonparametric and a Bonferroni post test. P < 0.05 was considered significant. All analyses were performed using Prism 4.0 software.
Online supplemental material
Fig. S1 shows multitissue analysis of IL-9 mRNA expression in WT and iFABPp-IL-9Tg mice. Fig. S2 shows OVA-specific IgE in OVA-sensitized and nonsensitized saline- and OVA-challenged IL-4Rα– and STAT6 factor–deficient and iFABP-IL-9Tg/IL-4Rα–and STAT6 factor–deficient mice. Fig. S3 shows active systemic anaphylaxis in WT and IL-9−/− mice. Table S1 shows splenocyte cytokine production from OVA-sensitized OVA-challenged IL9−/− and BALB/c WT mice. Table S2 shows characterization of mesenteric LN immune profile in WT and iFABPp-IL-9Tg mice. Table S3 is a gene profile analysis of the small intestine of WT and iFABPp-IL-9Tg mice. Table S4 shows experimental intestinal anaphylaxis in OVA-sensitized and nonsensitized saline- and OVA-challenged IL-4Rα– and STAT6 factor–deficient mice. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071046/DC1.
Supplemental Material
Acknowledgments
The authors would like to thank Anne Prins for her histopathology expertise, Dan Friend for guidance on mast cell histochemistry, and Andrea Lippelman and Lisa Roberts for their editorial assistance. The authors would also like to thank Amal Assa'ad, Nives Zimmermann, Yoshi Yamada and Ariel Muntz for helpful discussions.
This work was supported in part by Fulbright New Zealand MoRST award (E.E. Forbes), Australia National Health and Medical Research Council Program grant (224207; P.S. Foster, K.I. Matthaei, and S.P. Hogan), Cincinnati Children's Hospital Medical Center Trustee grant 2005, and the American Academy of Allergy and Asthma and Immunology Interest Section Award 2007 (S.P. Hogan).
The authors have no conflicting financial interests.
Abbreviations used: CAE, chloroacetate esterase; CMF, calcium- and magnesium-free; GI, gastrointestinal; hGH, human growth hormone; IBD, inflammatory bowel disease; i.g., intragastric; Isc, short-circuit current; MLN, mesenteric LN; MNC, mononuclear cell; VL, vascular leakage.
E.E. Forbes and K. Groschwitz contributed equally to this paper.
References
- 1.Sampson, H.A. 1999. Food allergy. Part 1: immunopathogenesis and clinical disorders. J. Allergy Clin. Immunol. 103:717–728. [DOI] [PubMed] [Google Scholar]
- 2.Sampson, H.A. 2003. Food Allergy. J. Allergy Clin. Immunol. 111:S540–S547. [DOI] [PubMed] [Google Scholar]
- 3.Eigenmann, P.A. 2002. T lymphocytes in food allergy: overview of an intricate network of circulating and organ-resident cells. Pediatr. Allergy Immunol. 13:162–171. [DOI] [PubMed] [Google Scholar]
- 4.Eigenmann, P.A., S.K. Huang, D.G. Ho, and H.A. Sampson. 1996. Human T cell clones and cell lines specific to ovomucoid recognize different domains and consistently express IL-5. Adv. Exp. Med. Biol. 409:217. [DOI] [PubMed] [Google Scholar]
- 5.Turcanu, V., S.J. Maleki, and G. Lack. 2003. Characterization of lymphocyte responses to peanuts in normal children, peanut-allergic children, and allergic children who acquired tolerance to peanuts. J. Clin. Invest. 111:1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hauber, H.P., C. Bergeron, and Q. Hamid. 2004. IL-9 in allergic inflammation. Int. Arch. Allergy Immunol. 134:79–87. [DOI] [PubMed] [Google Scholar]
- 7.Temann, U.A., P. Ray, and R.A. Flavell. 2002. Pulmonary overexpression of IL-9 induces Th2 cytokine expression, leading to immune pathology. J. Clin. Invest. 109:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Temann, U.A., G.P. Geba, J.A. Rankin, and R.A. Flavell. 1998. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J. Exp. Med. 188:1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLane, M.P., A. Haczku, M. van de Rijn, C. Weiss, V. Ferrante, D. MacDonald, J.C. Renauld, N.C. Nicolaides, K.J. Holroyd, and R.C. Levitt. 1998. Interleukin-9 promotes allergen-induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. Am. J. Respir. Cell Mol. Biol. 19:713–720. [DOI] [PubMed] [Google Scholar]
- 10.Vink, A., G. Warnier, F. Brombacher, and J.C. Renauld. 1999. Interleukin 9–induced in vivo expansion of the B-1 lymphocyte population. J. Exp. Med. 189:1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dugas, B., J.C. Renauld, J. Pene, J.Y. Bonnefoy, C. Peti-Frere, P. Braquet, J. Bousquet, J. Van Snick, and J.M. Mencia-Huerta. 1993. Interleukin-9 potentiates the interleukin-4-induced immunoglobulin (IgG, IgM and IgE) production by normal human B lymphocytes. Eur. J. Immunol. 23:1687–1692. [DOI] [PubMed] [Google Scholar]
- 12.Louahed, J., Y. Zhou, W.L. Maloy, P.U. Rani, C. Weiss, Y. Tomer, A. Vink, J. Renauld, J. Van Snick, N.C. Nicolaides, et al. 2001. Interleukin 9 promotes influx and local maturation of eosinophils. Blood. 97:1035–1042. [DOI] [PubMed] [Google Scholar]
- 13.Patkai, J., B. Mesples, M.A. Dommergues, G. Fromont, E.M. Thornton, J. Renauld, P. Evrard, and P. Gressens. 2001. Deleterious effects of IL-9-activated mast cells and neuroprotection by antihistamine drugs in the developing mouse brain. Pediatr. Res. 50:222–230. [DOI] [PubMed] [Google Scholar]
- 14.Ventura, M.T., L. Polimeno, A.C. Amoruso, F. Gatti, E. Annoscia, M. Marinaro, E. Di Leo, M.G. Matino, R. Buquicchio, S. Bonini, et al. 2006. Intestinal permeability in patients with adverse reactions to food. Dig. Dis. Sci. 38:732–736. [DOI] [PubMed] [Google Scholar]
- 15.Heyman, M. 2005. Gut barrier dysfunction in food allergy. Eur. J. Gastroenterol. Hepatol. 17:1279–1285. [DOI] [PubMed] [Google Scholar]
- 16.Hollander, D. 1993. Permeability in Crohn's disease: altered barrier functions in health relatives? Gastroenterology. 104:1848–1851. [DOI] [PubMed] [Google Scholar]
- 17.Yacyshyn, B.R., and J.B. Meddings. 1995. CD45RO expression on circulating CD19+ B cells in crohns disease correlates with intestinal permeability. Gastroenterology. 108:132–137. [DOI] [PubMed] [Google Scholar]
- 18.May, G.R., L.R. Sutherland, and J.B. Meddings. 1993. Is small intestine permeability really increased in relatives of patients with Crohns disease? Gastroenterology. 104:1627–1632. [DOI] [PubMed] [Google Scholar]
- 19.Peeters, M., B. Geypens, D. Claus, H. Nevens, Y. Ghoos, G. Verbeke, F. Baert, S. Vermeire, R. Vlietinck, and P. Rutgeerts. 1997. Clustering of increased small intestinal permeability in families with Crohn's disease. Gastroenterology. 113:802–807. [DOI] [PubMed] [Google Scholar]
- 20.Perdue, M.H., S. Masson, B.K. Wershil, and S.J. Galli. 1991. Role of mast cells in ion transport abnormalities associated with intestinal anaphylaxis. Correction of the diminished secretory response in genetically mast cell-deficient W/Wv mice by bone marrow transplantation. J. Clin. Invest. 87:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perdue, M., R. D'Inca, S. Crowe, P. Sestini, and J. Marshall. 1989. Effects of Mast Cells on Epithelial Function. Raven Press Ltd., New York. 295–305 pp.
- 22.Sweetser, D.A., E.H. Birkenmeier, P.C. Hoppe, D.W. McKeel, and J.I. Gordon. 1988. Mechanisms underlying generation of gradients in gene expression within the intestine: an analysis using transgenic mice containing fatty acid binding protein-human growth hormone fusion genes. Genes Dev. 2:1318–1332. [DOI] [PubMed] [Google Scholar]
- 23.Cohn, S.M., T.C. Simon, K.A. Roth, E.H. Birkenmeier, and J.I. Gordon. 1992. Use of transgenic mice to map cis-acting elements in the intestinal fatty acid binding protein gene (Fabpi) that control its cell lineage–specific and regional patterns of expression along the duodenal–colonic and crypt-villus axes of the gut epithelium. J. Cell Biol. 119:27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandt, E.B., R.T. Strait, D. Hershko, Q. Wang, E.E. Muntel, T.A. Scribner, N. Zimmermann, F.D. Finkelman, and M.E. Rothenberg. 2003. Mast cells are required for experimental oral allergen-induced diarrhea. J. Clin. Invest. 112:1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDermott, J.R., R.E. Bartram, P.A. Knight, H.R. Miller, D.R. Garrod, and R.K. Grencis. 2003. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc. Natl. Acad. Sci. USA. 100:7761–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madden, K.B., L. Whitman, C. Sullivan, W.C. Gause, J.F. Urban Jr., I.M. Katona, F.D. Finkelman, and T. Shea-Donohue. 2002. Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. J. Immunol. 169:4417–4422. [DOI] [PubMed] [Google Scholar]
- 27.Grencis, R.K., K.J. Else, J.F. Huntley, and S.I. Nishikawa. 1993. The in vivo role of stem cell factor (c-kit ligand) on mastocytosis and host protective immunity to the intestinal nematode Trichinella spiralis in mice. Parasite Immunol. 15:55–59. [DOI] [PubMed] [Google Scholar]
- 28.Romert, P., and H.B. Mikkelsen. 1998. c-kit immunoreactive interstitial cells of Cajal in the human small and large intestine. Histochem. Cell Biol. 109:195–202. [DOI] [PubMed] [Google Scholar]
- 29.Young, H.M., D. Ciampoli, B.R. Southwell, and D.F. Newgreen. 1996. Origin of interstitial cells of Cajal in the mouse intestine. Dev. Biol. 180:97–107. [DOI] [PubMed] [Google Scholar]
- 30.Sampson, H.A., S.H. Sicherer, and A.H. Birnbaum. 2001. AGA technical review on the evaluation of food allergy in gastrointestinal disorders. American Gastroenterological Association. Gastroenterology. 120:1026–1040. [DOI] [PubMed] [Google Scholar]
- 31.Sicherer, S.H., and H.A. Sampson. 2006. 9. Food allergy. J. Allergy Clin. Immunol. 117:S470–S475. [DOI] [PubMed] [Google Scholar]
- 32.Bischoff, S., and S.E. Crowe. 2005. Gastrointestinal food allergy: new insights into pathophysiology and clinical perspectives. Gastroenterology. 128:1089–1113. [DOI] [PubMed] [Google Scholar]
- 33.Mitsuhata, H., H. Takeuchi, J. Saitoh, N. Hasome, Y. Horiguchi, and R. Shimizu. 1995. An inhibitor of nitric oxide synthase, N omega-nitro-L-arginine-methyl ester, attenuates hypotension but does not improve cardiac depression in anaphylaxis in dogs. Shock. 3:447–453. [PubMed] [Google Scholar]
- 34.Strait, R.T., S.C. Morris, K. Smiley, J.F. Urban Jr., and F.D. Finkelman. 2003. IL-4 exacerbates anaphylaxis. J. Immunol. 170:3835–3842. [DOI] [PubMed] [Google Scholar]
- 35.Leng, W., K. Chang, J.R. Williamson, and B.A. Jakschik. 1989. Increased regional vascular albumin permeation in the rat during anaphylaxis. J. Immunol. 142:1982–1985. [PubMed] [Google Scholar]
- 36.Castro-Faria-Neto, H.C., P.T. Bozza, A.R. Silva, P.M. Silva, M.C. Lima, M.A. Martins, R.S. Cordeiro, and B.B. Vargaftig. 1992. Interference of azelastine with anaphylaxis induced by ovalbumin challenge in actively sensitized rats. Eur. J. Pharmacol. 213:183–188. [DOI] [PubMed] [Google Scholar]
- 37.Finkelman, F.D., M.E. Rothenberg, E.B. Brandt, S.C. Morris, and R.T. Strait. 2005. Molecular mechanisms of anaphylaxis: lessons from studies with murine models. J. Allergy Clin. Immunol. 115:449–457. [DOI] [PubMed] [Google Scholar]
- 38.Townsend, J.M., G.P. Fallon, J.D. Matthews, P. Smith, E.H. Jolin, and N.A. McKenzie. 2000. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity. 13:573–583. [DOI] [PubMed] [Google Scholar]
- 39.Matsuzawa, S., K. Sakashita, T. Kinoshita, S. Ito, T. Yamashita, and K. Koike. 2003. IL-9 enhances the growth of human mast cell progenitors under stimulation with stem cell factor. J. Immunol. 170:3461–3467. [DOI] [PubMed] [Google Scholar]
- 40.Fallon, P.G., H.E. Jolin, P. Smith, C.L. Emson, M.J. Townsend, R. Fallon, and A.N. McKenzie. 2002. IL-4 induces characteristic Th2 responses even in the combined absence of IL-5, IL-9, and IL-13. Immunity. 17:7–17. [DOI] [PubMed] [Google Scholar]
- 41.Bischoff, S.C., G. Sellge, A. Lorentz, W. Sebald, R. Raab, and M.P. Manns. 1999. IL-4 enhances proliferation and mediator release in mature human mast cells. Proc. Natl. Acad. Sci. USA. 96:8080–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ochi, H., N.H. De Jesus, F.H. Hsieh, F.K. Austen, and J.A. Boyce. 2000. IL-4 and IL-5 prime human mast cells for different profiles of IgE-dependent cytokine production. Proc. Natl. Acad. Sci. USA. 97:10509–10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lorentz, A., M. Wilke, G. Sellge, H. Worthmann, J. Klempnauer, M.P. Manns, and S.C. Bischoff. 2005. IL-4-induced priming of human intestinal mast cells for enhanced survival and Th2 cytokine generation is reversible and associated with increased activity of ERK1/2 and c-Fos. J. Immunol. 174:6751–6756. [DOI] [PubMed] [Google Scholar]
- 44.Matsuda, K., A.M. Piliponsky, M. Likura, S. Nakae, E.W. Wang, S.M. Dutta, T. Kawakami, M. Tsai, and S.J. Galli. 2005. Monomeric IgE enhances human mast cell chemokine production: IL-4 augments and dexamethasone suppresses the response. J. Allergy Clin. Immunol. 116:1357–1363. [DOI] [PubMed] [Google Scholar]
- 45.Van der Kleij, H.P.M., D. Ma, F.A.M. Redegeld, A.D. Kraneveld, F.P. Nijkamp, and J. Bienenstock. 2003. Functional expression of neurokinin 1 receptors on mast cells induced by IL-4 and stem cell factor. J. Immunol. 171:2074–2079. [DOI] [PubMed] [Google Scholar]
- 46.Madden, K.B., J.F. Urban Jr., H.J. Ziltener, J.W. Schrader, F.D. Finkelman, and I.M. Katona. 1991. Antibodies to IL-3 and IL-4 suppress helminth-induced intestinal mastocytosis. J. Immunol. 147:1387–1391. [PubMed] [Google Scholar]
- 47.Urban, J.F., N. Noben-Trauth, D.D. Donaldson, K.B. Madden, S.C. Morris, M. Collins, and F.D. Finkelman. 1998. IL-13, IL-4Ralpha, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity. 8:255–264. [DOI] [PubMed] [Google Scholar]
- 48.Urban, J.F., L. Schopf, S.C. Morris, T. Orekhova, K.B. Madden, C.J. Betts, H.R. Gamble, C. Byrd, D. Donaldson, K. Else, and F.D. Finkelman. 2000. Stat6 signaling promotes protective immunity against Trichinella spiralis through a mast cell- and T cell-dependent mechanism. J. Immunol. 164:2046–2052. [DOI] [PubMed] [Google Scholar]
- 49.Godfraind, C., J. Louahed, H. Faulkner, A. Vink, G. Warnier, R. Grencis, and J.C. Renauld. 1998. Intraepithelial infiltration by mast cells with both connective tissue-type and mucosal-type characteristics in gut, trachea, and kidneys of IL-9 transgenic mice. J. Immunol. 160:3989–3996. [PubMed] [Google Scholar]
- 50.Eklund, K.K., N. Ghildyal, K.F. Austen, and R.L. Stevens. 1993. Induction by IL-9 and suppression by IL-3 and IL-4 of the levels of chromosome-14-derived transcripts that encode late-expressed mouse mast cell proteases. J. Immunol. 151:4266–4273. [PubMed] [Google Scholar]
- 51.Ghildyal, N., H.P. McNeil, S. Stechschulte, K.F. Austen, D. Silberstein, M.F. Gurish, L.L. Somerville, and R.L. Stevens. 1992. IL-10 induced transcription of the gene for mouse mast cell protease-1, a serine protease preferentially expressed in mucosal mast cells of Trichinella spiralis-infected mice. J. Immunol. 149:2123–2129. [PubMed] [Google Scholar]
- 52.Demoulin, J.B., J. Louahed, L. Dumoutier, M. Stevens, and J.C. Renauld. 2003. MAP kinase activation by interleukin-9 in lymphoid and mast cell lines. Oncogene. 22:1763–1770. [DOI] [PubMed] [Google Scholar]
- 53.Miyajima, I., D. Dombrowicz, T.R. Martin, J.V. Ravetch, J.P. Kinet, and S.J. Galli. 1997. Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J. Clin. Invest. 99:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oettgen, H.C., T.R. Martin, A. Wynshaw-Boris, C. Deng, J.M. Drazen, and P. Leder. 1994. Active anaphylaxis in IgE-deficient mice. Nature. 370:367–370. [DOI] [PubMed] [Google Scholar]
- 55.Strait, R.T., S.C. Morris, M. Yang, X.W. Qu, and F.D. Finkelman. 2002. Pathways of anaphylaxis in the mouse. J. Allergy Clin. Immunol. 109:658–668. [DOI] [PubMed] [Google Scholar]
- 56.Renauld, J.C., N. van der Lugt, A. Vink, M. van Roon, C. Godfraind, G. Warnier, H. Merz, A. Feller, A. Berns, and J. Van Snick. 1994. Thymic lymphomas in interleukin 9 transgenic mice. Oncogene. 9:1327–1332. [PubMed] [Google Scholar]
- 57.Knoops, L., J. Louahed, J. Van Snick, and J.C. Renauld. 2005. IL-9 promotes but is not necessary for systemic anaphylaxis. J. Immunol. 175:335–341. [DOI] [PubMed] [Google Scholar]
- 58.Kweon, M.N., M. Yamamoto, M. Kajiki, I. Takahashi, and H. Kiyono. 2000. Systemically derived large intestinal CD4+ Th2 cells play a central role in STAT-6-mediated allergic diarrhea. J. Clin. Invest. 106:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li, X.M., B.H. Schofield, C.K. Huang, G.I. Kleiner, and H.A. Sampson. 1999. A murine model of IgE-mediated cow's milk hypersensitivity. J. Allergy Clin. Immunol. 103:206–214. [DOI] [PubMed] [Google Scholar]
- 60.Arendse, B., J. Van Snick, and F. Brombacher. 2005. IL-9 is a susceptibility factor in Leishmania major infection by promoting detrimental Th2/type 2 responses. J. Immunol. 174:2205–2211. [DOI] [PubMed] [Google Scholar]
- 61.Shea-Donohue, T., C. Sullivan, F.D. Finkelman, K.B. Madden, S.C. Morris, J. Goldhill, V. Pineiro-Carrero, and J.F. Urban Jr. 2001. The role of IL-4 in Heligmosomoides polygyrus-induced alterations in murine intestinal epithelial cell function. J. Immunol. 167:2234–2239. [DOI] [PubMed] [Google Scholar]
- 62.Brandt, E.B., R.T. Strait, Q. Wang, D. Hersko, E. Muntel, F.D. Finkelman, and M.E. Rothenberg. 2003. Oral antigen-induced intestinal anaphylaxis requires IgE-dependent mast cell degranulation. J. Allergy Clin. Immunol. 111:S339. [Google Scholar]
- 63.Buhner, S., C. Buning, J. Genschel, K. Kling, D. Herrmann, A. Dignass, I. Kuechler, S. Krueger, H.H. Schmidt, and H. Lochs. 2006. Genetic basis for increased intestinal permeability in families with Crohn's disease: role of CARD15 3020insC mutation? Gut. 55:342–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takeuchi, K., L. Maiden, and I. Bjarnason. 2004. Genetic aspects of intestinal permeability in inflammatory bowel disease. In Inflammatory Bowel Disease—Crossroads of Microbes, Epithelium and Immune Systems. D. Chadwick, and J. Goode, editors. Wiley, Chichester. pp. 151–163. [PubMed]
- 65.Van Elburg, R.M., H.S. Heymans, and J.G. De Monchy. 1993. Effect of disodiumcromoglycate on intestinal permeability changes and clinical response during cow's milk challenge. Pediatr. Allergy Immunol. 4:79–85. [DOI] [PubMed] [Google Scholar]
- 66.Schrander, J.J., R.W. Unsalan-Hooyan, P.P. Forget, and J. Jansen. 1990. [51Cr]EDTA intestinal permeability in children with cow's milk tolerance. J. Pediatr. Gastroenterol. Nutr. 10:189–192. [DOI] [PubMed] [Google Scholar]
- 67.Postma, D.S., E.R. Bleecker, P.J. Amelung, K.J. Holroyd, J. Xu, C.I. Panhuysen, D.A. Meyers, and R.C. Levitt. 1995. Genetic susceptibility to asthma–bronchial hyperresponsiveness coinherited with a major gene for atopy. N. Engl. J. Med. 333:894–900. [DOI] [PubMed] [Google Scholar]
- 68.Melen, E., S. Umerkajeff, F. Nyberg, M. Zucchelli, A. Lindstedt, H. Gullsten, M. Wickman, G. Pershagen, and J. Kere. 2006. Interactions between variants in the interleukin-4 receptor a and interleukin-9 receptor genes in childhood wheezing: evidence from a birth cohort study. Clin. Exp. Allergy. 36:1391–1398. [DOI] [PubMed] [Google Scholar]
- 69.Blanchard, S.S., M. Gerrek, S. Czinn, G. Chelimsky, D. Seaman, C. Segal, and J. Splawski. 2006. Food protein sensitivity with partial villous atrophy after pediatric liver transplantation with tacrolimus immunosuppression. Pediatr. Transplant. 10:529–532. [DOI] [PubMed] [Google Scholar]
- 70.Ozdemir, O., A. Arrey-Mensah, and R.U. Sorensen. 2006. Development of multiple food allergies in children taking tacrolimus after heart and liver transplantation. Pediatr. Transplant. 10:380–383. [DOI] [PubMed] [Google Scholar]
- 71.Madsen, K.L., N.L. Yanchar, D.L. Sigalet, T. Reigel, and R.N. Fedorak. 1995. FK 506 increases permeability in rat intestine by inhibiting mitochondrial function. Gastroenterology. 109:107–114. [DOI] [PubMed] [Google Scholar]
- 72.Yanchar, N.L., T.M. Riegel, G. Martin, R.N. Fedorak, N.M. Kneteman, and D.L. Sigalet. 1996. Tacrolimus (FK506)–its effects on intestinal glucose transport. Transplantation. 61:630–634. [DOI] [PubMed] [Google Scholar]
- 73.Asante-Korang, A., G.J. Boyle, S.A. Webber, S.A. Miller, and F.J. Fricker. 1996. Experience of FK506 immune suppression in pediatric heart transplantation: a study of long-term adverse effects. J. Heart Lung Transplant. 15:415–422. [PubMed] [Google Scholar]
- 74.Prabhakaran, K., H.T. Lau, B. Wise, K. Schwarz, and P.M. Colombani. 1994. Incidence of allergic symptoms in pediatric liver transplant recipients treated with tacrolimus based immunosuppression. Pediatrics. 104:786–787. [Google Scholar]
- 75.Darmon, N., M. Heyman, C. Candalh, M.A. Blaton, and J.F. Desjeux. 1996. Anaphylactic intestinal response to milk proteins during malnutrition in guinea pigs. Am. J. Physiol. 270:G442–G448. [DOI] [PubMed] [Google Scholar]
- 76.Darmon, N., M.A. Pelissier, C. Candalh, P. Chappuis, M.A. Blaton, R. Albrecht, J.F. Desjeux, and M. Heyman. 1997. Zinc and intestinal anaphylaxis to cow's milk proteins in malnourished guinea pigs. Pediatr. Res. 42:208–213. [DOI] [PubMed] [Google Scholar]
- 77.Ellmark, P., J. Ingvarsson, A. Carlsson, B.S. Lundin, C. Wingren, and C.A. Borrebaeck. 2006. Identification of protein expression signatures associated with Helicobacter pylori infection and gastric adenocarcinoma using recombinant antibody microarrays. Mol. Cell. Proteomics. 5:1638–1646. [DOI] [PubMed] [Google Scholar]
- 78.Gessner, A., H. Blum, and M. Rollinghoff. 1993. Differential regulation of IL-9 expression after infection with Leishmania major in susceptible and resistant mice. Immunobiology. 189:419–435. [DOI] [PubMed] [Google Scholar]
- 79.Nashed, B.F., Y. Maekawa, M. Takashima, T. Zhang, K. Ishii, T. Dainichi, H. Ishikawa, T. Sakai, H. Hisaeda, and K. Himeno. 2000. Different cytokines are required for induction and maintenance of the Th2 type response in DBA/2 mice resistant to infection with Leishmania major. Microbes Infect. 2:1435–1443. [DOI] [PubMed] [Google Scholar]
- 80.Matsuo, T., Y. Ikura, M. Ohsawa, M. Ogami, S. Kayo, N. Yoshimi, E. Hai, T. Naruko, M. Ohishi, K. Higuchi, et al. 2003. Mast cell chymase expression in helicobacter pylori-associated gastritis. Histopathology. 43:538–549. [DOI] [PubMed] [Google Scholar]
- 81.Corrado, G., I. Luzzi, S. Lucarelli, T. Frediani, C. Pacchiarotti, M. Cavaliere, P. Rea, and E. Cardi. 1998. Positive association between Helicobacter pylori infection and food allergy in children. Scand. J. Gastroenterol. 33:1135–1139. [DOI] [PubMed] [Google Scholar]
- 82.Cherner, J.A., R.T. Jensen, A. Dubois, T.M. O'Dorisio, J.D. Gardner, and D.D. Metcalfe. 1988. Gastrointestinal dysfunction in systemic mastocytosis. A prospective study. Gastroenterology. 95:657–667. [DOI] [PubMed] [Google Scholar]
- 83.Horan, R.F., A.L. Sheffer, and K.F. Austen. 1990. Cromolyn sodium in the management of systemic mastocytosis. J. Allergy Clin. Immunol. 85:852–855. [DOI] [PubMed] [Google Scholar]
- 84.Horan, R.F., and K.F. Austen. 1991. Systemic mastocytosis: retrospective review of a decades clinical experience at the Brigham and Women's Hospital. J. Invest. Dermatol. 96:5S–14S. [PubMed] [Google Scholar]
- 85.Webb, D.C., K.I. Matthaei, Y. Cai, A.N. McKenzie, and P.S. Foster. 2004. Polymorphisms in IL-4R alpha correlate with airways hyperreactivity, eosinophilia, and Ym protein expression in allergic IL-13−/− mice. J. Immunol. 172:1092–1098. [DOI] [PubMed] [Google Scholar]
- 86.Webb, D.C., S. Mahalingam, Y. Cai, K.I. Matthaei, D.D. Donaldson, and P.S. Foster. 2003. Antigen-specific production of interleukin (IL)-13 and IL-5 cooperate to mediate IL-4Ralpha-independent airway hyperreactivity. Eur. J. Immunol. 33:3377–3385. [DOI] [PubMed] [Google Scholar]
- 87.Foster, P.S., D.C. Webb, M. Yang, C. Herbert, and R.K. Kumar. 2003. Dissociation of T helper type 2 cytokine-dependent airway lesions from signal transducer and activator of transcription 6 signalling in experimental chronic asthma. Clin. Exp. Allergy. 33:688–695. [DOI] [PubMed] [Google Scholar]
- 88.Rothenberg, M.E., A.D. Luster, C.M. Lilly, J.M. Drazen, and P. Leder. 1995. Constitutive and allergen-induced expression of eotaxin mRNA in the guinea pig lung. J. Exp. Med. 181:1211–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Forbes, E., V.E. Smart, A. D'Aprile, P. Henry, M. Yang, K.I. Matthaei, M.E. Rothenberg, P.S. Foster, and S.P. Hogan. 2004. T helper-2 immunity regulates bronchial hyperresponsiveness in eosinophil-associated gastrointestinal disease in mice. Gastroenterology. 127:105–118. [DOI] [PubMed] [Google Scholar]
- 90.Finkelman, F.D., and S.C. Morris. 1999. Development of an assay to measure in vivo cytokine production in the mouse. Int. Immunol. 11:1811–1818. [DOI] [PubMed] [Google Scholar]
- 91.Green, T.P., D.E. Johnson, R.P. Marchessault, and C.W. Gatto. 1988. Transvascular flux and tissue accrual of Evans blue: effects of endotoxin and histamine. J. Lab. Clin. Med. 111:173–183. [PubMed] [Google Scholar]
- 92.Strait, R.T., S.C. Morris, and F.D. Finkelman. 2006. IgG-blocking antibodies inhibit IgE-mediated anaphylaxis in vivo through both antigen interception and Fc gamma RIIb cross-linking. J. Clin. Invest. 116:833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
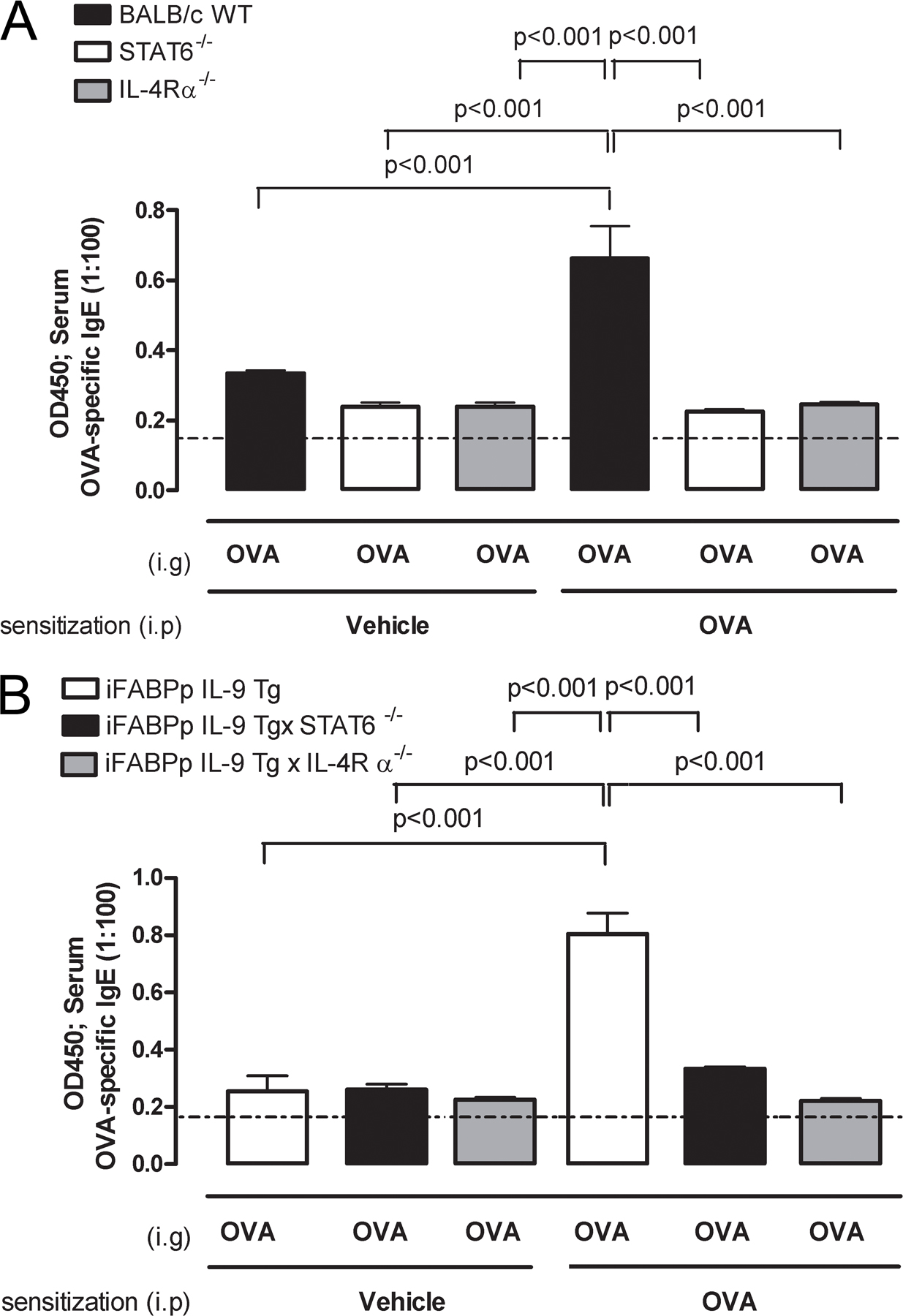{kind=link}
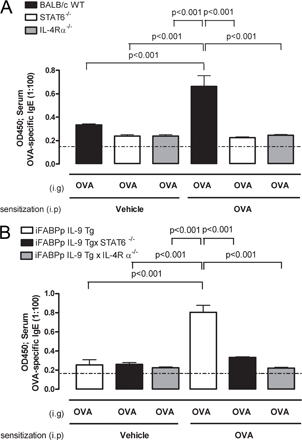{kind=link}
{kind=link}
{kind=link}