

Abstract
Checkpoint with forkhead-associated and RING (Chfr) is a ubiquitin ligase (E3) that establishes an antephase or prometaphase checkpoint in response to mitotic stress. Though ubiquitination is essential for checkpoint function, the sites, linkages and ubiquitin conjugating enzyme (E2) specificity are controversial. Here we dissect the function of the two Chfr homologs in S. cerevisiae, Chf1 and Chf2, overexpression of which retard cell cycle at both G1 and G2. Using a genetic assay, we establish that Ubc4 is required for Chf2-dependent G1 cell cycle delay and Chf protein turnover. In contrast, Ubc13/Mms2 is required for G2 delay and does not contribute to Chf protein turnover. By reconstituting cis and trans-ubiquitination activities of Chf proteins in purified systems and characterizing sites modified and linkages formed by tandem mass spectrometry, we discovered that Ubc13/Mms2-dependent modifications are a distinct subset of those catalyzed by Ubc4. Mutagenesis of Lys residues identified in vitro indicates that site-specific Ubc4-dependent Chf protein autoubiquitination is responsible for Chf protein turnover. Thus, combined genetic and biochemical analyses indicate that Chf proteins have dual E2 specificity accounting for different functions in the cell cycle.
Keywords: Chfr, E3 ubiquitin ligase, E2 ubiquitin conjugating enzyme, Ubc4, Ubc13/Mms2, yeast genetics, in vitro reconstitution, tandem mass spectrometry
INTRODUCTION
Post-translational modification by ubiquitination alters the cellular lifetime, localization and functions of target proteins. Ubiquitination occurs through an ATP-dependent enzyme cascade requiring ubiquitin (Ub), a Ub-activating enzyme or E1 (Uba1 in yeast), a Ub-conjugating enzyme or E2 (11 in yeast), and a Ub ligase or E3 (dozens in yeast). Reaction of Ub-activated E2 with an E3 and a protein target results in formation of an isopeptide linkage between the C-terminal Gly of Ub and the ε-amino group of a Lys in the target.1 Proteins can be monoubiquitinated, multiply monoubiquitinated on different Lys residues, or polyubiquitinated, in which Ub chains are polymerized by formation of an isopeptide linkage between the attacking carboxy-terminus of Ub and an internal Lys of Ub. Indeed, because there are many Lys residues on target proteins and 7 Lys residues on Ub, the number of ways in which a single protein can be modified is massively combinatorial.
Target and linkage specificity are determined by the E2/E3 combination and interactions between the E2/E3 machinery with substrates. A major class of E3 ligases is defined by the presence of a really interesting new gene (RING) domain, a ~70 amino acid motif featuring conserved Cys and His residues that coordinate two structurally and enzymatically important Zn2+ ions.2 RING E3s act as scaffolds and specificity factors to promote association of a target with Ub-activated E2 to catalyze ubiquitination.3
Depending on the type of ubiquitination, targets are directed to highly distinct outcomes. Many membrane receptors are regulated by mono- or di-ubiquitination which establishes a signal for internalization and lysosomal degradation.4–7 Polyubiquitination can occur at multiple primary sites on target proteins. There are seven Lys residues in Ub—all seven have been observed to be modified by polyubiquitination in yeast.8 The most abundantly formed poly-Ub linkages are generated through Lys48 and Lys63 Ub modification. While Lys48-linked Ub chains of 4 or more units usually comprise a signal for degradation by the 26S proteasome,1 Lys63-linked chains have a multitude of signaling functions in the regulation of transcription,9, 10 translation,11, 12 kinase activation,9 endocytosis13, 14 and DNA repair.15, 16
The Ubc13/Mms2 heterodimer is a specialized E2 that forms Lys63-linked polyubiquitin15 whereas Ubc4 and Ubc5 are considered prototypical E2 enzymes that form Lys48-linked polyubiquitin to target short-lived proteins to the proteasome.17, 18 However, despite this generalization, not all effects of Lys63-linked polyubiquitin conjugation are due to Ubc13/Mms2 activity. A substantial amount of Lys63-linked polyubiquitin depends on presence of Ubc4 and Ubc5.19 In addition, yeast strains expressing Ub-K63R as the sole source of Ub are sensitive to inhibitors of translation, which ubc13 and mms2 mutants are not; and Lys63-linked polyubiquitination of ribosomal protein L28 requires Ubc4 but not Ubc13/Mms2.11, 12 Adding complexity to the E2-dependence of the Ub system, in some cases, endocytosis of plasma membrane receptors is mediated by Ubc4 and Ubc5 monoubiquitination.4, 5, 20
Biochemical analysis indicates that individual E2 enzymes can functionally pair with multiple E3 ligases21 while several purified E3 ligases function in vitro with multiple E2 conjugating enzymes.22 However, the apparent promiscuity of in vitro reactions casts doubt on reactions that have not been validated by genetic analysis.
Chfr and Rnf8 are human RING E3 ubiquitin ligases that each consist of an N-terminal forkhead-associated (FHA) domain, a RING domain and a distinct protein-protein interaction domain (a C-terminal Cys-rich region in Chfr and a central coiled-coil domain in Rnf8). FHA domains are typically modules found in DNA repair and checkpoint proteins that bind phosphoThr.23 Chfr was identified as a key component of an early mitotic, prometaphase checkpoint because chfr deficiency causes cells to fail to delay chromosome condensation and nuclear envelope breakdown in response to taxol or nocodazole.24 However, video micrographic assays indicate that the point at which Chfr acts is in late G2 or “antephase”.25 Chfr levels are down-regulated in human cancers originating in colon,26–29 gastric,30–32 esophageal33 and lung34 epithelia by hypermethylation of the Chfr promoter.
Though Chfr-deficient tumors are characterized by sensitivity to microtubule poisons and loss of in vitro ubiquitination activity is associated with loss of checkpoint function,24, 25, 35–38 the sites, linkages and E2-dependence of Chfr functions remain controversial. One body of work reports that the Chfr RING domain can auto-ubiquitinate when Ubc4, UbcH5A or UbcH5B is supplied as the E2.35, 39 Proposed proteolytic targets of Chfr include Polo-like kinase 136, 38 and Aurora A kinase.40 Though Ubc4 and Ubc5 clearly work with Chfr in vitro, another body of work argues that such reactions are artefactual and that the Ubc13/Mms2 heterodimer is the E2 that authentically works with Chfr to form Lys63-linked polyubiquitin chains that perform a signaling function essential for the mitotic checkpoint.25, 37
Rnf8 is a nuclear protein that interacts with retinoid X receptor α(RXRα) and functions to enhance RXRα transcriptional activation activity in a RING dependent manner.41 In vitro assays with Rnf8 have demonstrated autoubiquitination when UbcH6, UBE2E2, UBE2E3, UbcH542 or Ubc13/Mms243 is supplied as the E2. There have been no genetic studies to determine the E2-dependence of Chfr or Rnf8 in vivo.
Chf1 and Chf2 from S. cerevisiae,44 alternately termed Dma1 and Dma2,45 possess Chfr and Rnf8-like domain structures consisting of variable N-terminal regions, central FHA domains and C-terminal RING domains. Consistent with the requirement of Chfr to impose a checkpoint late in the cell cycle,24 S. pombe dma1 plays an important role in cytokinesis.46, 47 Chf1 and Chf2 interact with the positive cell cycle regulator Cdc123 and are required for both the G1 and G2 cell cycle phenotypes of cdc123-4.44 At the permissive temperature of cdc123-4, most cells are delayed in G2/M and the mitotic accumulation of cdc123-4 cells is lost in cdc123-4 chf1 chf2 mutants.44 Additionally, chf1 chf2 mutants fail to assemble a septin ring properly.45 However, two lines of evidence indicate that Chf proteins function in G1 in addition to G2/M. First, at the restrictive temperature, cdc123-4 cells arrest at G1 in a manner that depends on Chf proteins. Second, overexpression of Chf1 or Chf2 retards the cell cycle in a RING and FHA-dependent manner and produces an excess of cells in G1 phase.44
Here we report a detailed biochemical and genetic analysis of the functions of the Chfr and Rnf8 related proteins, Chf1 and Chf2. We provide genetic evidence that Ubc4 and Ubc13/Mms2 are required for distinct cell cycle-delaying activities of Chf2, early and late in the cell cycle respectively. Moreover, we describe site-specific and linkage-specific differences in the biochemical activities of Chf1 and Chf2 when paired with Ubc4 and Ubc13/Mms2. We show that Ubc4 but not Ubc13/Mms2 downregulates Chf protein levels and establish that residues identified as Ub targets in vitro play important roles in Chf protein stability in vivo. The combined analysis indicates that Chfr homologs define a distinct class of E3 ligases capable of targeting proteins with Lys48-linked and Lys63-linked polyubiquitin via specific pairing with Ubc4 and Ubc13/Mms2. Dual E2 specificity of Chf1 and Chf2 allows these enzymes to fulfill different functions early and late in the cell cycle.
MATERIALS AND METHODS
Yeast strains
BY4742 was used as the wild-type S. cerevisiae strain and parent for all experiments in this study. Nine deletion mutants in the BY4742 background, chf1, ubc5, ubc7, ubc8, ubc11, ubc13, mms2, rad6 and pex4 were from the deletion consortium.48 The UBC1 gene was replaced with the natMX resistance cassette and the UBC4 gene was replaced with HIS3 by PCR mediated gene disruption.49 UBC13 and MMS2 were both replaced with kanMX. Strain names and genotypes are provided in Supplementary Table 1.
Plasmid constructions
Plasmids containing the constructs for His-tagged Ubc4 and Ubc11 were kindly provided by Aled Edwards.22 Plasmid pC105 was constructed by amplifying wild-type CHF2 with added PstI and XhoI sites for insertion into pRS425GAL1. Plasmid pC110 was generated by amplifying the yeast MMS2 gene from a yeast cDNA library with NdeI and XhoI sites for insertion into plasmid pSGA04,50 which provides an N-terminal His tag. UBC13 was amplified from yeast cDNA with flanking EcoRI and XhoI sites and cloned into pGEX4T-2 to generate the glutathione S-transferase (GST)-Ubc13 expression vector, pC111. Expression vectors for GST-Chf1 (wild-type, RING mutant Chf1-C345S, H350A and FHA mutants Chf1-G192E and Chf1-S220A, H223L) were constructed by amplifying each gene with flanking BamHI and XhoI sites for insertion into pGEX4T-2 to generate pC103, pC114, pC115 and pC116, respectively. Plasmids for GST-Chf2 (wild-type, RING mutant Chf2-C451S, H456A and FHA mutants Chf2-S326A, H329L and Chf2-G298E) expression were generated by PCR amplification of each gene to add EcoRI and XhoI sites followed by insertion into pGEX4T-2 to yield pB308, pC117, pC118 and pC119, respectively. To generate constructs for N-terminally tagged Chf proteins, a “Z” tag (sequence of an IgG binding domain of Protein A) was inserted into the NdeI site of pB299 (pGAL-CHF1)44 and into the PstI site of pC105 (pGAL-CHF2) yielding pC145 and pC146, respectively. Chf1 Lys to Arg mutations were made in pC145 using QuikChange Site-directed mutagenesis (Stratagene) to generate plasmids pC153, pC155, pC156, pC157, pC158, pC159 and pC160. All plasmids were confirmed by DNA sequencing (Norris Cotton Cancer Center Molecular Biology Core Facility). A plasmid summary is provided in Supplementary Table 2.
Suppression of Chf2 growth inhibition
Wild-type and E2 null mutants were transformed with plasmids pRS425GAL1 and pC105 (GAL1-CHF2). These strains were grown to log phase and then stamped in 5-fold serial dilutions on SC -leu 2% glucose and SC- leu 2% galactose media. Galactose plates were incubated for seven days at 28°C.
Measurement of doubling time and G1 phase
Yeast strains (BY4742, ubc4, ubc13, mms2 and ubc11) containing pRS425GAL1 or pC105 (pGAL-CHF2) were grown overnight in liquid SC -leu 2% glucose media at 28°C. These cultures were used to inoculate fresh media to an OD600 nm of 0.05 the next morning. The transformants were monitored by OD600 nm to measure the doubling times for each strain in SC -leu containing 2% glucose or 2% galactose as the carbon source. Doubling time = ln2 / ((ln (A/Ao)) / t) where A = cell density at time t; Ao = initial cell density. The length of G1 phase for each strain was calculated by counting the number of unbudded and budded cells in log phase culture.51 Length of G1 phase = T[1 − log(2 − F ·unbud/log2)], in which T is the doubling time and F · unbud is the fraction of unbudded cells.
Enzyme expression and purification
All GST fusion proteins (GST-Ubc13, GST-Chf1 and GST-Chf2 as well as Chf1/Chf2 FHA and RING mutants) were expressed in E. coli strain BL21 Star (DE3). Starter cultures were grown in LB + 100 μg/mL ampicillin overnight at 37 °C and used to inoculate 1 liter LB + ampicillin. The large cultures were grown to an OD600 nm of ~0.6 at which time the temperature was reduced to room temperature and protein expression was induced for ~18 hours by the addition of 0.4 mM IPTG. Cells were harvested by centrifugation, washed with water and stored at −20 °C.
Purification of the GST-tagged proteins was carried out essentially as described52 with some modifications. Briefly, cell pellets were resuspended in chilled 50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 4 mM MgCl2, 5 mM DTT, 10% glycerol, 1 M NaCl, 1 mM PMSF and complete mini EDTA-free protease inhibitors (Roche) and lysed by sonication. The cell suspension was clarified by centrifugation at 4°C. Following centrifugation, an equal volume of buffer without NaCl was added to the clarified lysate. GST-fusion proteins were purified using glutathione sepharose 4B (Amersham Biosciences) by a sequential batch and column procedure at 4°C. The beads were washed extensively with 50 mM Tris-HCl, pH 7.5, 4 mM MgCl2, 1 mM DTT, 10% glycerol and 0.5 M NaCl. The GST-fusion proteins were eluted in wash buffer containing 20mM NaOH and 25 mM reduced glutathione (Sigma). Purified proteins were dialyzed overnight against storage buffer (50mM Tris-HCl, pH 7.5, 5 mM MgCl2, 2 mM NaF, 0.6 mM DTT, 10 μM ZnSO4, 10% glycerol) and stored at −80 °C.
E2 enzymes Ubc4, Ubc11 and E2 variant Mms2, were expressed as described above for the GST fusion proteins. The enzymes were purified as described22 with minor modifications. Cell pellets were resuspended in 20 mM HEPES, pH 8.0, 500 mM NaCl, 10% glycerol, 10 μM ZnCl2, 5 mM imidazole, 0.5 mM Tris (2-carboxyethyl) phosphine hydrochloride, and protease inhibitor tablets (Roche), and then lysed by sonication. The lysate was clarified by centrifugation at 4 °C. Clarified lysate was applied to TALON Superflow Metal Affinity Resin (Clontech) equilibrated with lysis buffer and the tagged proteins were purified using a tandem batch/column procedure. After binding, the resin was washed extensively first with lysis buffer, followed by the same buffer containing 30 mM imidazole. The enzymes were eluted in the same buffer containing 500 mM imidazole, then dialyzed against 50 mM HEPES pH 7.5, 100 mM NaCl, 5 mM MgOAc, 1 mM DTT and 10% glycerol and stored at −80°C.
Ubiquitination assays
Chf protein Ub ligase activity assays were carried out essentially as described53 using purified reaction components including ~500 ng (~1.5 μg in reactions for mass spec analysis) GST-Chf1 or GST-Chf2, ~300 ng of an E2, 100 ng yeast E1 (Boston Biochem), 1 μg N-terminally FLAG-tagged Ub (Sigma), and 2 mM ATP. Ubiquitinated species were separated on 10% SDS-PAGE gels under non-reducing conditions (Cambrex). Reactions were visualized with GelCode Blue stain reagent (Pierce) and by immunoblotting with Anti-FLAG M2 monoclonal antibody-peroxidase conjugate (Sigma).
LC-MS/MS analysis
Ubiquitination reactions were performed as described above and reaction products were separated by SDS-PAGE on a 10% gel. Gel regions corresponding to mono-, di- and tri-ubiquitinated Chf1 or Chf2 were excised from the gel, trypsinized and analyzed on a hybrid linear ion trap-Fourier transform mass spectrometer (Thermo Electron) as described.54 Resulting tandem mass-spectra were data-searched8, 55 to identify sites and linkages of ubiquitination (Table 2, Supplementary Table 3).
TABLE 2.
Chf1 and Chf2 ubiquitination sites and linkages
| Chf1 ubiquitinated peptides | sites | Ubc4+Chf1 | Ubc13/Mms2+Chf1 | Ubc4+Chf2 |
|---|---|---|---|---|
| M* DK# HGLFSIR | K150 | X | ||
| EAISK# IPDQYHPVVFK | K204 | X | X | |
| THGCFK# VDDQGNWFLK | K217 | X | ||
| VDDQGNWFLK# DVK | K237 | X | X | |
| DVK# SSSGTFLNHQR | K240 | X | ||
| LSSASTTSK# DYLLHDGDIIQLGMDFR | K260 | X | ||
| LK# ANAFNK# EALSR | K300, K306 | X | ||
| ANAFNK# EALSR | K306 | X | X | X |
| IK# NLQK# LTTGLEQEDCSICLNK | K313, K317 | X
X |
||
| NLQK# LTTGLEQEDCSICLNK | K317 | X | ||
|
| ||||
| LIFAGK# QLEDGR | K48 | X | X | |
| TLSDYNIQK# ESTLHLVLR | K63 | X | X | |
|
| ||||
| Chf2 ubiquitinated peptides | Sites | Ubc4+Chf2 | Ubc13/Mms2+Chf2 | Ubc4+Chf1 |
|
| ||||
| NIVGGADGSTIVNNSQEMYK# NLR | K211 | X | X | |
| K# DKHGLFSIR | K256 | X | ||
| KDK# HGLFSIR | K258 | X | X | |
| K# AGPGSQLVIGR | K288 | X | X | |
| DAISK# IPEQYHPVVFK1 | K310 | X | X | X |
| THGCFK# VDSQGNWYIK | K333 | X | ||
| VDSQGNWYIK# DVK | K343 | X | X | |
| DVK# SSSGTFLNHQR | K346 | X | X | |
| LSPASSLSK# DTPLR1 | K366 | X | X | X |
| LK# ANSFNK# EALQR | K406, K412 | X
X |
||
| ANSFNK# EALQR | K412 | X | X | |
| LQNLQK# LTTGIEEEDCSICLCK | K423 | X | ||
| Ubiquitin peptides | ||||
|
| ||||
| LIFAGK# QLEDGR | K48 | X | X | |
| TLSDYNIQK# ESTLHLVLR | K63 | X | X | |
Oxidation (+/− 15.99644 Da)
GlyGly (114.05421 Da)
Same K# detected on peptide with M* and/or longer peptide.
RESULTS
Cell Cycle Delay of Chf2 Depends on Ubc4 and Ubc13/Mms2
To address E2 utilization by Chf proteins in vivo, we exploited the slow cell division phenotype of Chf2 overexpression44 to identify genes encoding factors essential for Chf2-dependent cell cycle delays. Because the slow cell division phenotypes of GAL1-driven Chf proteins depend on intact RING domains, we hypothesized that overexpression of the E3 ligases causes depletion or dysregulation of one or more cell cycle regulators by ubiquitination in a manner that depends on a specific E2 Ub conjugating enzyme.
A panel of 11 yeast strains, consisting of a wild-type strain, nine haploviable E2 null mutant strains (ubc1, ubc4, ubc5, ubc7, ubc8, ubc11, ubc13, pex4, rad6), and mms2, which is deleted for the yeast ubiquitin E2 variant gene, were transformed with a control vector and a plasmid encoding GAL1-driven Chf2. As shown in Fig. 1A, the toxicity of Chf2 over-expression was largely rescued by ubc4 and rescued to a lesser degree by ubc13 or mms2 deletion. None of the other E2 mutants tested suppressed the Chf2 expression phenotype, though the ubc1 deletion was not informative because this mutant has a gal-phenotype. Surprisingly, deletion of ubc5 did not suppress Chf2 over-expression even though Ubc4 and Ubc5 are >90% identical in sequence and are reportedly redundant in multiple functions.5, 17, 20, 56 As Ubc13 and Mms2 function as a heterodimer,15 the similar phenotypes of ubc13 and mms2 can be easily rationalized.
Figure 1.
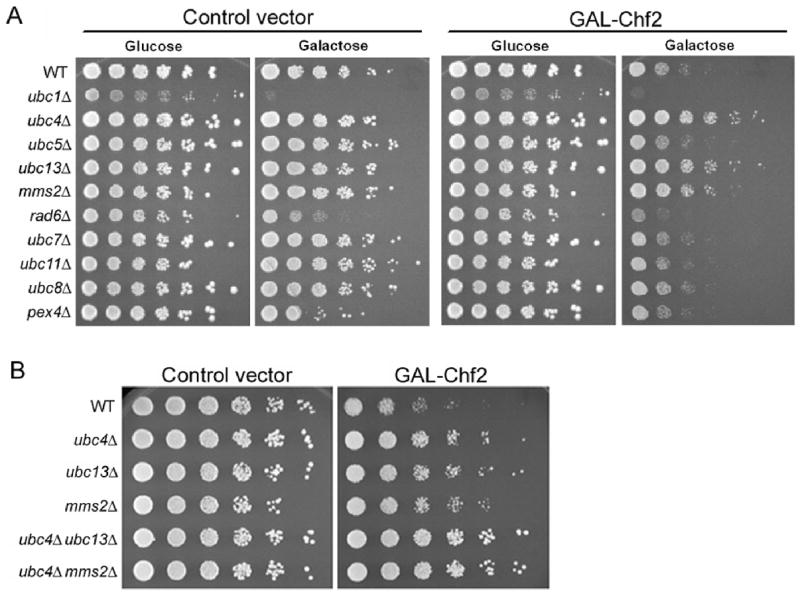
Chf2-mediated growth inhibition depends on Ubc4, Ubc13 and Mms2. (A) In the left panel, growth of a set of isogenic strains is examined as a function of E2 gene deletions and carbon source. In the right panel, the galactose-specific growth-inhibitory effect of GAL1-driven Chf2 expression is demonstrated and is shown to be suppressed by ubc4, ubc13 and mms2 deletions. (B) On galactose media, the suppressor activity of ubc4 deletion is shown to be additive with ubc13 or mms2 deletion such that ubc4 ubc13 and ubc4 mms2 are fully epistatic to the growth-inhibitory effect of Chf2 overexpression.
To test whether the residual cell division disadvantage of ubc4, ubc13 and mms2 cells overexpressing Chf2 might be due to expression of the other genetically interacting E2, we constructed ubc4 ubc13 and ubc4 mms2 double mutants. As shown in Fig. 1B, these double mutants were completely resistant to the effects of Chf2 overexpression.
To quantify Chf2 overexpression phenotypes in wild-type, ubc4, ubc13, mms2 and ubc11 (negative control) strains, we grew control and GAL1-CHF2 transformants in liquid glucose and galactose media and determined doubling times. As shown in Table 1, all transformants grew similarly in glucose media with doubling times between 1.9 and 2.3 hours. Strains containing the control vector exhibit moderately slower cell division on galactose media, ranging from 2.5 to 2.7 hours. Upon galactose induction of Chf2 expression, doubling times were increased in wild-type and ubc11 mutants to 4 and 4.2 hours respectively. The 1.3 hr increase in the doubling time for wild-type was reduced by deletion of ubc4 to 0.7 hrs. Upon deletion of ubc13 or mms2, Chf2 overexpression extended doubling time by 1 hr and 0.9 hrs, respectively. As was observed in colony forming assays (Fig. 1), these data establish that Chf2 dually depends on Ubc4 and Ubc13/Mms2 functions in vivo.
TABLE 1.
Length of cell division cycle and G1 phase (hrs)
| Control | GAL-Chf2 | |||||
|---|---|---|---|---|---|---|
| Glucose Cell cycle | Galactose Cell cycle (G1) | Glucose Cell cycle | Galactose Cell cycle(G1) | Chf2 delay | G1 delay | |
| WT | 2.0 | 2.7 (1.5) | 1.9 | 4.0 (1.9) | 1.3 | 0.4 |
| ubc4 | 2.3 | 2.7 (1.8) | 2.2 | 3.4 (1.7) | 0.7 | −0.1 |
| ubc13 | 2.0 | 2.5 (1.3) | 1.9 | 3.5 (1.8) | 1.0 | 0.5 |
| mms2 | 2.0 | 2.6 (1.3) | 2.0 | 3.5 (1.6) | 0.9 | 0.3 |
| ubc11 | 1.9 | 2.5 (1.3) | 1.9 | 4.2 (1.8) | 1.7 | 0.5 |
Ubc4 is Required for the G1 Delay Phenotype of Chf2
Whereas a substantial body of evidence indicates that Chfr24, 25, 35–38, 40 and fungal Chf proteins44–47 function late in the cell cycle, we stand alone in observing a role in G1.44 To determine whether Ubc4 or Ubc13/Mms2 might mediate a Chf2-dependent delay early in the cell cycle, we determined the length of G151 in Chf2-overexpressing strains as a function of E2 gene deletion. As shown in Table 1, the wild-type and ubc11 strains exhibit a 0.4 to 0.5 hr extension in the length of G1 upon Chf2 overexpression. Though ubc13 and mms2 deletions reduce the cell cycle delay by Chf2, the G1 phase remains extended by 0.5 hr and 0.3 hr, respectively. However, ubc4 was completely epistatic to the ability of Chf2 to extend G1 phase. These data indicate that the G1-delaying activity of Chf2 depends on Ubc4 while the major mediator of delay later in the cell cycle is Ubc13/Mms2.
Chf Autoubiquitination Activity with Ubc4 and Ubc13/Mms2
Because genetic analysis indicates that the cell cycle delaying activity of Chf2 depends on the E2 functions of Ubc4, Ubc13 and Mms2, we attempted to reconstitute purified ubiquitination reactions consisting of yeast Uba1, Flag-tagged Ub, His-tagged E2 enzymes purified from E. coli, and full-length Chf1 or Chf2 as GST fusions purified from E. coli. As shown in Fig. 2A, whereas both Ubc4 and Ubc11 formed FLAG-tagged Ub adducts when incubated with Uba1, ATP and FLAG-Ub, Ubc4 but not Ubc11 was capable of conjugating long-chain Ub adducts to Chf1 and Chf2. As shown in Fig. 2B, Ubc13 but not Mms2 formed an activated Ub species upon incubation with Uba1, ATP and Ub. Upon incubation with Uba1, ATP, Ub and a reconstituted Ubc13/Mms2 heterodimer, we observed formation of polyubiquitinated species, consistent with assembly on an activated E2 complex.15 Addition of Chf1 and Chf2 resulted in significant autoubiquitination dependent upon the presence of both Ubc13 and Mms2. Thus, Chf1 and Chf2 possess potent Ubc4 and Ubc13/Mms2-dependent autoubiquitination activities in genetically validated in vitro reactions that require no yeast cytosol.
Figure 2.
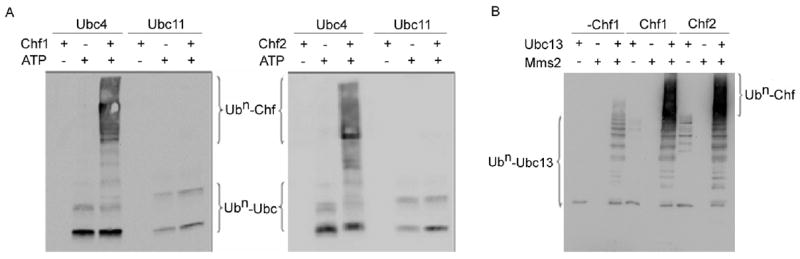
In vitro reconstitution of Ubc4-dependent and Ubc13/Mms2-dependent Chf protein autoubiquitination activities. (A) In side-by-side reactions in which Ubc4 and Ubc11 both form ATP and E1-dependent Ub-activated species, Ubc4 but not Ubc11 allows conjugation of Ub to Chf1 and to Chf2. (B) Whereas Ubc13 is sufficient, with ATP and E1, to form a Ub-activated E2 species, a reconstituted Ubc13/Mms2 heterodimer is required for Chf1 and Chf2 autoubiquitination.
Chf ubiquitination activity is necessary but not sufficient for cell cycle delay
Previous studies demonstrated that Chf1 and Chf2 mutants with amino acid substitution in the FHA and RING domains are highly stabilized in vivo.44 Despite stabilization of the mutant proteins, FHA and RING mutations greatly reduce the cell cycle delays induced by overexpression of wild-type Chf1 and Chf2.44 To identify the biochemical correlates of these mutants and clarify the nature of Chf-imposed cell division arrest, we purified Chf1 and Chf2 mutants and assayed each with respect to autoubiquitination in assays with ATP, Ub, Uba1 and either Ubc4 or Ubc13/Mms2 as the E2. As shown in Fig. 3A, RING mutants of Chf1 and Chf2 (Chf1-Cys345Ser, His350Ala and Chf2-Cys451Ser, His456Ala) were completely devoid of autoubiquitination activity with Ubc4 as E2. Two different FHA mutant alleles of each Chf protein (Chf1-Gly192Glu, Chf1-Ser220Ala, His223Leu, Chf2-Gly298Glu and Chf2-Ser326Ala, His329Leu) were assayed for autoubiquitination activity. Whereas a report indicated that FHA deletion forms of Chfr were activated with respect to autoubiquitination,36 our data indicate that Chf1 and Chf2 are neither activated nor inhibited by FHA domain point mutations. As shown in Fig. 3B, Ubc13/Mms2-dependent reactions were equally RING-dependent and FHA-independent. Combined with our earlier in vivo analysis44 these data suggest that, while autoubiquitination activity and an intact FHA domain are required for the normal instability of Chf proteins, substrate targeting by the FHA domain is critical for cell cycle delaying activity.
Figure 3.
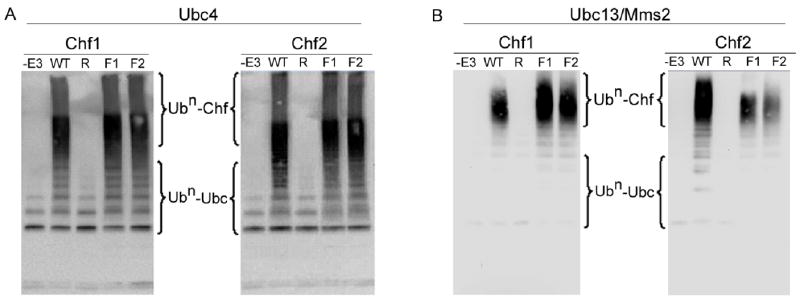
Chf protein autoubiquitination is RING-dependent and FHA-independent. Chf RING mutants “R” (Chf1-C345S, H350A and Chf2-C451S, H456A) and FHA mutants “F1” (Chf1-G192E and Chf2-G298E) and “F2” (Chf1-S220A, H223L and Chf2- S326A, H329L) were tested for autoubiquitination activity with Ubc4 (A) or Ubc13/Mms2 (B) supplied as the E2. In these assays, RING domain mutants are undistinguishable from omitting the E3 ligase, whereas FHA domain mutants are undistinguishable from wild-type.
Ubc4 down-regulates Chf protein levels in vivo
In Fig. 3, we demonstrated that RING mutants are unable to auto-ubiquitinate in vitro. It has also been established that mutations in the Chf RING domains lead to dramatic Chf protein stabilization.44 The intrinsic instability of Chf proteins suggests that Chf protein turnover may depend on a specific E2 conjugating enzyme. We expressed an epitope-tagged Chf2 under the control of the GAL1 promoter as a function of deletions of genes encoding E2 enzymes. As shown in Fig. 4, Chf2 levels are not altered by mms2 and ubc13 deletion. In contrast, deletion of ubc4 leads to a ~3-fold accumulation of the Chf2 protein. These data suggest that the Ubc4-dependent autoubiquitination activities reconstituted in Fig. 2A function to promote Chf protein turnover. However, because Chf2 protein is more abundant but not G1-delaying in the ubc4 mutant, we argue that Ubc4 functions with Chf proteins both for autoubiquitination and for substrate ubiquitination with substrate ubiquitination being critical for G1 delay.
Figure 4.
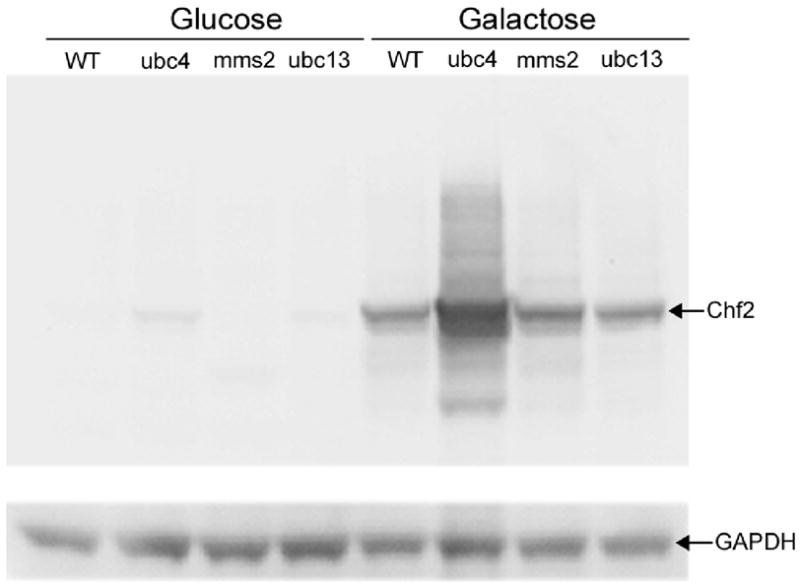
Ubc4 regulates Chf protein levels. To determine whether a specific E2 promotes the intrinsic instability of Chf proteins, the levels of Chf2 protein in E2 deletion strains were examined in cells expressing GAL1-driven Chf2. Whereas mms2 deletion and ubc13 deletion did not stabilize Chf2, ubc4 deletion increased Chf2 protein level by ~3-fold with respect to a GAPDH loading control, suggesting that Ubc4-dependent Chf protein ubiquitination drives Chf protein turnover.
Mass spectrometric analysis of Chf1/2 autoubiquitination sites and linkages
Because Chf1 and Chf2 depend on an intact RING domain for cellular instability, Lys48-linked autoubiquitination would be expected to be an important mediator of Chf protein signal termination.44 Additionally, Lys63-linked autoubiquitination was proposed as the key mechanism of Chfr cellular signaling.37 Despite the existence of these models, the sites and linkage-specificity of Ub modification of Chfr-homologous ligases have never been determined. We considered it particularly important to determine if the sites and linkages of Chf1 and Chf2 autoubuiquitination might be altered by use of Ubc4 versus Ubc13/Mms2 as the E2. As shown in Fig. 5, we scaled up purified, reconstituted reaction systems with either Ubc4 or Ubc13/Mms2 supporting Chf1 and Chf2 autoubiquitination. Coomassie-stained bands corresponding to Chf1 or Chf2 with one to three Ub modifications were excised from SDS-PAGE gels and digested with trypsin. Peptides were separated by microcapillary reversed phase liquid chromatography (LC) and analyzed by high performance shotgun tandem mass spectrometry (MS/MS). The resulting fragmentation spectra were correlated to peptide sequences by database-matching in which Lys residues were allowed to be modified by the addition of 114.0429 Da.8 Because the carboxy-terminus of Ub is Arg↓Gly-Gly, complete tryptic cleavage leaves a 114.0429 Da Gly-Gly isopeptidic remnant on Lys residues modified by mono- or polyubiquitination. All peptides were positively identified with mass accuracies of 13 parts per million or better (Supplementary Data).
Figure 5.

Identification of Ubc4 and Ubc13/Mms2-dependent Chf protein sites of autoubiquitination. Coomassie-stained gels of purified Ubc4-dependent autoubiquitination reactions are shown above the sequences of Chf1 (A) and Chf2 (B). As described in Materials and Methods and as shown in Table 2, sites of ubiquitination were identified by tandem mass spectrometry. Lys residues are shown in magenta, with sites ubiquitinated by Ubc4 in gray and sites ubiquitinated by both Ubc4 and Ubc13/Mms2 in yellow. Sites ubiquitinated in trans reactions are boxed. FHA domains are in green and RING domains are in blue.
As shown in Fig. 5 and Table 2, when Ubc4 was provided as the E2, Chf1 modified at least 10 of 21 Lys residues and Chf2 modified at least 11 of 19 Lys residues. The majority of Lys residues that were not detected as Ub-modified by LC/MS-MS would have been identified on very small or large peptides, which are typically under-represented in this type of analysis. As shown in Fig. 5, the N-terminal regions of Chf1 and Chf2 are relatively Lys-poor such that the majority of Lys residues and modified Lys residues occur in the FHA domains, the RING domains and in the 53 amino acid segments between the FHA and RING domains. Ubc4-Chf1 and Ubc4-Chf2 reactions formed two polyubiquitin species detected by LC-MS/MS, namely Lys48-linked and Lys63-linked polyubiquitin.
Reactions catalyzed by Ubc13/Mms2 exhibited greater specificity in the sites and linkages of autoubiquitination. Scoring the peptides recovered by LC-MS/MS, Chf1 modified only Lys residues 237 and 306 while Chf2 modified Lys residues 258, 310, 346 and 366. These six sites were among the 21 sites modified in Ubc4 reactions. Whereas Ubc4 generated both Lys48 and Lys63-linked chains, the Ubc13/Mms2 heterodimer produced exclusively Lys63-linked Ub chains on the Chf proteins. Thus, in terms of autoubiquitination, the Ubc13/Mms2 reactions produced a subset of the reactions catalyzed by Ubc4.
Chf1 and Chf2 ubiquitination in trans
In protein kinase signaling, it has long been established that a great deal of auto-phosphorylation is mediated by trans-autophosphorylation mechanisms mediated by kinase dimerization.57 Similar questions in autoubiquitination remain largely unexplored. Indeed, if Chf proteins were to function entirely by autoubiquitination, one might expect ubiquitination to be an obligate intramolecular reaction. To test this hypothesis, we performed in vitro Ubc4-catalyzed reactions utilizing the inactive RING mutant Chf1 protein as a substrate of wild-type Chf2 protein and the inactive RING mutant Chf2 as a substrate of wild-type Chf1. After separating reaction components by SDS-PAGE, we excised bands corresponding to modified mutant E3 proteins. After tryptic digestion and LC-MS/MS, we identified two Lys residues on Chf1 and seven Lys residues on Chf2 that were subject to trans-autoubiquitination. In both cases, the sites of trans-autoubiquitination were subsets of the sites modified in reactions in which the E3 served as both substrate and enzyme. The fact that subsets of the same Lys residues modified by Ubc4-Chf1 and Ubc4-Chf2 are modified by Ubc13/Mms2-Chf1, Ubc13/Mms2-Chf2 and in Ubc4 trans-autoubiquitination reactions suggests that particular Chf1 and Chf2 Lys residues are preferred Ub acceptors independent of the E2-Ub-E3 complex transferring the modification.
Mutation of identified ubiquitination sites leads to stabilization of Chf1 in vivo
Analysis of Chf autoubiquitination sites by LC/MS-MS identified numerous sites of ubiquitination with a few sites modified in multiple different reactions. To test whether these were key sites that play regulatory roles in vivo, we mutated various Lys residues alone and in combination (Fig. 6A). The stability of mutants was compared with that of wild-type by immunoblotting (Fig. 6B and C). We observed moderate stabilization when any of these Lys residues was substituted by Arg. These data suggest that Chf1 protein is functionally modified at multiple sites.
Figure 6.
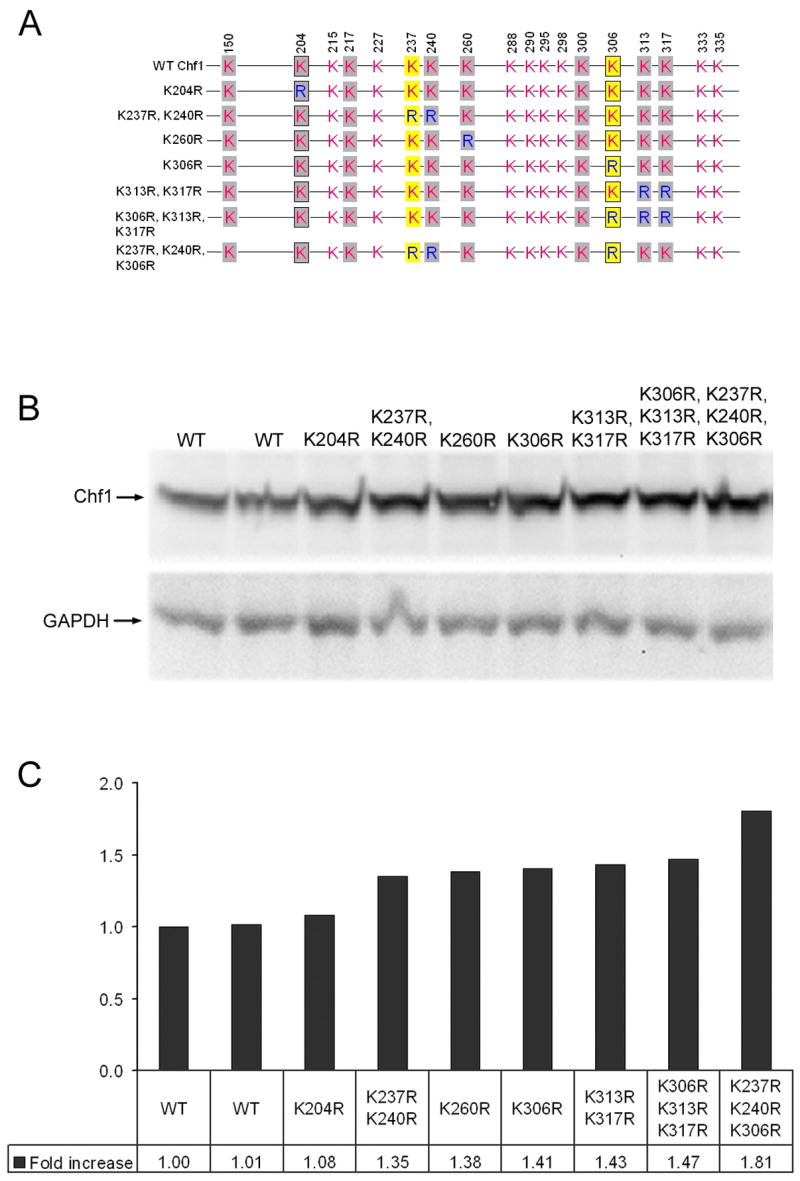
Stabilization of Chf1 by mutation of ubiquitination sites. (A) A schematic diagram of Chf1 Lys to Arg mutants. Sites auto-ubiquitinated with Ubc4 are in gray. Sites additionally auto-ubiquitinated with Ubc13/Mms2 are in yellow. Sites that are trans-ubiquitinated by Chf2 are boxed. As shown by immunoblot (B) and relative quantitation versus GAPDH (C), individual Lys mutants such as Lys260Arg and Lys306Arg stabilize Chf2 protein alone and in combination with other Lys to Arg mutations, such as Lys237Arg, Lys240Arg, validating these residues as sites of biological regulation.
Chf1 has only two sites that are modified in vitro by Ubc4-dependent autoubiquitination and by Ubc4-dependent Chf2 trans-ubiquitination. Though the Lys204Arg substitution, which would preclude modification at one such in vitro defined site, produced only an ~8% relative increase in Chf1 accumulation, the Lys306Arg mutation produced a ~41% increase in Chf1 protein. Combination of Lys306Arg with a ~35% stabilizing Lys237Arg, Lys240Arg mutation produced a form of Chf1 with an additive (~81%) effect on Chf1 accumulation. The data indicate that this is a degenerate system in which Ubc4-dependent modification of multiple Lys sites contributes to Chf protein turnover.
DISCUSSION
Proteolytic and nonproteolytic models have been proposed to connect Chfr protein function in the cell cycle with RING-dependent ubiquitination activity. According to the proteolytic models, Chfr ligates Lys48-linked polyubiquitin to cell division-promoting proteins to target them for proteolysis.35, 38–40 According to the nonproteolytic models, Ubc13/Mms2 is the biologically relevant E2, which works with Chfr to conjugate Lys63-linked ubiquitination as a G2/M arrest signal.25, 37 We turned to yeast homologs of Chfr to address these issues. As first steps to solving the problems of Chf protein function, we determined the E2-dependence of Chf protein function at two cell cycle stages, examined which E2 is responsible for Chf protein downregulation, identified sites and linkages catalyzes by genetically validated E2s, and demonstrated the biological relevance of modification sites identified by mass spectrometry.
Whereas Chfr has only been reported to act late in the cell cycle,24, 25 our work on yeast Chf proteins suggested that Chf1 and Chf2 act at both major cell cycle transitions.44 We find that the ability of Chf2 expression to retard the cell cycle depends jointly on Ubc4 and Ubc13/Mms2. Ubc4 is the mediator of the G1 delay, whereas Ubc13/Mms2 is the mediator of late cell cycle delay. Thus, our work indicates Chf proteins are authentically dual specificity ligases with different E2 partners early and late in the cell cycle.
Considering the high sequence similarity of Ubc4 and Ubc5 and the frequently observed overlapping functions of Ubc4 and Ubc5,5, 17, 20, 56 assignment of Ubc4 to the G1 function of Chf2 and genetic exclusion of Ubc5 was surprising. However, the expression of UBC4 mRNA is highest in exponentially growing cells, while UBC5 mRNA is primarily expressed in stationary phase.17
Genetic (Figs. 4 and 6) and biochemical (Fig. 5 and Table 2) data establish Ubc4 as the E2 that catalyzes Chf protein autoubiquitination in order to promote in vivo degradation. The sites of autoubiquitination identified by mass spectrometry appear to be authentic because alteration of these Lys residues to Arg is stabilizing. However, because ubc4 deletion stabilizes Chf2 yet eliminates Chf2-dependent G1 cell cycle delay, we argue that an additional protein must be targeted by Ubc4-Chf2 for the G1 cell cycle delay.
Much of the debate on Chfr function has centered on what type of polyubiquitin chains are formed by this E3 ligase. Our genetic validation of Ubc4 and Ubc13/Mms2 allowed us to characterize Chf-dependent reactions in vitro. In reactions with Ubc4 as the E2, Chf1 and Chf2 formed Lys48-linked and Lys63-linked polyubiquitin. As expected, with Ubc13/Mms2 as the E2, Chf1 and Chf2 formed Lys63-linked polyubiquitin and no other Ub-Ub linkage.
The targets of Ubc13/Mms2-Chf protein modification would not be expected to be proteolyzed as a consequence of Lys63-linked polyubiquitination.15, 58 Moreover, we have excluded Ubc4 from the late cycle cycle function of Chf2 (Table 1), and shown that Chf2 is capable of ubiquitinating an external substrate (Table 2). Our analysis of FHA domain mutants strongly suggests the existence of G1 targets, potentially including eIF2γ, 44 and G2 targets in Chf protein function. Because the FHA domain mutant, chf1-G192A is highly stabilized by this mutation but extremely deficient in cell cycle arresting function,44 yet is eminently capable of autoubiquitination (Fig. 3), we suggest that substrate-targeted Lys48-linked and Lys63-linked polyubiquitination mediated by Ubc4 and by Ubc13/Mms2 are required for Chf protein function at both cell cycle stages. Because temperature inactivation of Cdc123 also leads to Chf protein accumulation and Thr274 of Cdc123 was genetically mapped as a Chf1 FHA domain interacting residue,44 Cdc123 association may be competitive with external substrates and drive Ubc4-dependent Chf protein turnover. Genetic and biochemical systems developed herein, as applied to Chf1 and Chf2-specific interactors, are expected to clarify the cycle cycle-specific direct targets of the four E2-E3 combinations.
Supplementary Material
Acknowledgments
Work was supported by NIGMS grant R01-081665, awarded to CB. GLL and SAG were additionally supported by NCI training grant T32-CA09658 and NCRR COBRE P20-018787, respectively.
Abbreviations
- Chfr
checkpoint with forkhead associated and RING
- E3
ubiquitin ligase
- E2
ubiquitin conjugating enzyme
- Ub
ubiquitin
- E1
ubiquitin activating enzyme
- RING
really interesting new gene domain
- FHA
forkhead associated domain
- GST
glutathione S-transferase
References
- 1.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Borden KL. RING domains: master builders of molecular scaffolds? J Mol Biol. 2000;295:1103–12. doi: 10.1006/jmbi.1999.3429. [DOI] [PubMed] [Google Scholar]
- 3.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–33. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 4.Hicke L. Gettin’ down with ubiquitin: turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 1999;9:107–12. doi: 10.1016/s0962-8924(98)01491-3. [DOI] [PubMed] [Google Scholar]
- 5.Horak J, Wolf DH. Glucose-induced monoubiquitination of the Saccharomyces cerevisiae galactose transporter is sufficient to signal its internalization. J Bacteriol. 2001;183:3083–8. doi: 10.1128/JB.183.10.3083-3088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haglund K, Di Fiore PP, Dikic I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci. 2003;28:598–603. doi: 10.1016/j.tibs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Haglund K, Dikic I. Ubiquitylation and cell signaling. Embo J. 2005;24:3353–9. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–6. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 9.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–61. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 10.Laine A, Topisirovic I, Zhai D, Reed JC, Borden KL, Ronai Z. Regulation of p53 localization and activity by Ubc13. Mol Cell Biol. 2006 doi: 10.1128/MCB.01156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spence J, Sadis S, Haas AL, Finley D. A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol. 1995;15:1265–73. doi: 10.1128/mcb.15.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spence J, Gali RR, Dittmar G, Sherman F, Karin M, Finley D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell. 2000;102:67–76. doi: 10.1016/s0092-8674(00)00011-8. [DOI] [PubMed] [Google Scholar]
- 13.Kee Y, Lyon N, Huibregtse JM. The Rsp5 ubiquitin ligase is coupled to and antagonized by the Ubp2 deubiquitinating enzyme. Embo J. 2005;24:2414–24. doi: 10.1038/sj.emboj.7600710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kee Y, Munoz W, Lyon N, Huibregtse JM. The Ubp2 deubiquitinating enzyme modulates Rsp5-dependent K63-linked polyubiquitin conjugates in Saccharomyces cerevisiae. J Biol Chem. 2006 doi: 10.1074/jbc.M608756200. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–53. doi: 10.1016/s0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- 16.Brusky J, Zhu Y, Xiao W. UBC13, a DNA-damage-inducible gene, is a member of the error-free postreplication repair pathway in Saccharomyces cerevisiae. Curr Genet. 2000;37:168–74. doi: 10.1007/s002940050515. [DOI] [PubMed] [Google Scholar]
- 17.Seufert W, Jentsch S. Ubiquitin-conjugating enzymes UBC4 and UBC5 mediate selective degradation of short-lived and abnormal proteins. Embo J. 1990;9:543–50. doi: 10.1002/j.1460-2075.1990.tb08141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chuang SM, Madura K. Saccharomyces cerevisiae Ub-conjugating enzyme Ubc4 binds the proteasome in the presence of translationally damaged proteins. Genetics. 2005;171:1477–84. doi: 10.1534/genetics.105.046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnason T, Ellison MJ. Stress resistance in Saccharomyces cerevisiae is strongly correlated with assembly of a novel type of multiubiquitin chain. Mol Cell Biol. 1994;14:7876–83. doi: 10.1128/mcb.14.12.7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth AF, Davis NG. Ubiquitination of the yeast a-factor receptor. J Cell Biol. 1996;134:661–74. doi: 10.1083/jcb.134.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kraft E, Stone SL, Ma L, Su N, Gao Y, Lau OS, Deng XW, Callis J. Genome analysis and functional characterization of the E2 and RING-type E3 ligase ubiquitination enzymes of Arabidopsis. Plant Physiol. 2005;139:1597–611. doi: 10.1104/pp.105.067983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kus BM, Caldon CE, Andorn-Broza R, Edwards AM. Functional interaction of 13 yeast SCF complexes with a set of yeast E2 enzymes in vitro. Proteins. 2004;54:455–67. doi: 10.1002/prot.10620. [DOI] [PubMed] [Google Scholar]
- 23.Durocher D, Smerdon SJ, Yaffe MB, Jackson SP. The FHA domain in DNA repair and checkpoint signaling. Cold Spring Harb Symp Quant Biol. 2000;65:423–31. doi: 10.1101/sqb.2000.65.423. [DOI] [PubMed] [Google Scholar]
- 24.Scolnick DM, Halazonetis TD. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 2000;406:430–5. doi: 10.1038/35019108. [DOI] [PubMed] [Google Scholar]
- 25.Matsusaka T, Pines J. Chfr acts with the p38 stress kinases to block entry to mitosis in mammalian cells. J Cell Biol. 2004;166:507–16. doi: 10.1083/jcb.200401139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toyota M, Sasaki Y, Satoh A, Ogi K, Kikuchi T, Suzuki H, Mita H, Tanaka N, Itoh F, Issa JP, Jair KW, Schuebel KE, Imai K, Tokino T. Epigenetic inactivation of CHFR in human tumors. Proc Natl Acad Sci U S A. 2003;100:7818–23. doi: 10.1073/pnas.1337066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertholon J, Wang Q, Falette N, Verny C, Auclair J, Chassot C, Navarro C, Saurin JC, Puisieux A. Chfr inactivation is not associated to chromosomal instability in colon cancers. Oncogene. 2003;22:8956–60. doi: 10.1038/sj.onc.1207078. [DOI] [PubMed] [Google Scholar]
- 28.Corn PG, Summers MK, Fogt F, Virmani AK, Gazdar AF, Halazonetis TD, El-Deiry WS. Frequent hypermethylation of the 5′ CpG island of the mitotic stress checkpoint gene Chfr in colorectal and non-small cell lung cancer. Carcinogenesis. 2003;24:47–51. doi: 10.1093/carcin/24.1.47. [DOI] [PubMed] [Google Scholar]
- 29.Brandes JC, van Engeland M, Wouters KA, Weijenberg MP, Herman JG. CHFR promoter hypermethylation in colon cancer correlates with the microsatellite instability phenotype. Carcinogenesis. 2005;26:1152–6. doi: 10.1093/carcin/bgi058. [DOI] [PubMed] [Google Scholar]
- 30.Honda T, Tamura G, Waki T, Kawata S, Nishizuka S, Motoyama T. Promoter hypermethylation of the Chfr gene in neoplastic and non-neoplastic gastric epithelia. Br J Cancer. 2004;90:2013–6. doi: 10.1038/sj.bjc.6601849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang HC, Kim IJ, Park JH, Shin Y, Park HW, Ku JL, Yang HK, Lee KU, Choe KJ, Park JG. Promoter hypermethylation and silencing of CHFR mitotic stress checkpoint gene in human gastric cancers. Oncol Rep. 2004;12:129–33. [PubMed] [Google Scholar]
- 32.Koga Y, Kitajima Y, Miyoshi A, Sato K, Sato S, Miyazaki K. The significance of aberrant CHFR methylation for clinical response to microtubule inhibitors in gastric cancer. J Gastroenterol. 2006;41:133–9. doi: 10.1007/s00535-005-1732-7. [DOI] [PubMed] [Google Scholar]
- 33.Shibata Y, Haruki N, Kuwabara Y, Ishiguro H, Shinoda N, Sato A, Kimura M, Koyama H, Toyama T, Nishiwaki T, Kudo J, Terashita Y, Konishi S, Sugiura H, Fujii Y. Chfr expression is downregulated by CpG island hypermethylation in esophageal cancer. Carcinogenesis. 2002;23:1695–9. doi: 10.1093/carcin/23.10.1695. [DOI] [PubMed] [Google Scholar]
- 34.Mizuno K, Osada H, Konishi H, Tatematsu Y, Yatabe Y, Mitsudomi T, Fujii Y, Takahashi T. Aberrant hypermethylation of the CHFR prophase checkpoint gene in human lung cancers. Oncogene. 2002;21:2328–33. doi: 10.1038/sj.onc.1205402. [DOI] [PubMed] [Google Scholar]
- 35.Chaturvedi P, Sudakin V, Bobiak ML, Fisher PW, Mattern MR, Jablonski SA, Hurle MR, Zhu Y, Yen TJ, Zhou BB. Chfr regulates a mitotic stress pathway through its RING-finger domain with ubiquitin ligase activity. Cancer Res. 2002;62:1797–801. [PubMed] [Google Scholar]
- 36.Kang D, Chen J, Wong J, Fang G. The checkpoint protein Chfr is a ligase that ubiquitinates Plk1 and inhibits Cdc2 at the G2 to M transition. J Cell Biol. 2002;156:249–59. doi: 10.1083/jcb.200108016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bothos J, Summers MK, Venere M, Scolnick DM, Halazonetis TD. The Chfr mitotic checkpoint protein functions with Ubc13-Mms2 to form Lys63-linked polyubiquitin chains. Oncogene. 2003;22:7101–7. doi: 10.1038/sj.onc.1206831. [DOI] [PubMed] [Google Scholar]
- 38.Shtivelman E. Promotion of mitosis by activated protein kinase B after DNA damage involves polo-like kinase 1 and checkpoint protein CHFR. Mol Cancer Res. 2003;1:959–69. [PubMed] [Google Scholar]
- 39.Kang D, Wong J, Fang G. A Xenopus cell-free system for analysis of the Chfr ubiquitin ligase involved in control of mitotic entry. Methods Mol Biol. 2004;280:229–43. doi: 10.1385/1-59259-788-2:229. [DOI] [PubMed] [Google Scholar]
- 40.Yu X, Minter-Dykhouse K, Malureanu L, Zhao WM, Zhang D, Merkle CJ, Ward IM, Saya H, Fang G, van Deursen J, Chen J. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet. 2005;37:401–6. doi: 10.1038/ng1538. [DOI] [PubMed] [Google Scholar]
- 41.Takano Y, Adachi S, Okuno M, Muto Y, Yoshioka T, Matsushima-Nishiwaki R, Tsurumi H, Ito K, Friedman SL, Moriwaki H, Kojima S, Okano Y. The RING finger protein, RNF8, interacts with retinoid X receptor alpha and enhances its transcription-stimulating activity. J Biol Chem. 2004;279:18926–34. doi: 10.1074/jbc.M309148200. [DOI] [PubMed] [Google Scholar]
- 42.Ito K, Adachi S, Iwakami R, Yasuda H, Muto Y, Seki N, Okano Y. N-Terminally extended human ubiquitin-conjugating enzymes (E2s) mediate the ubiquitination of RING-finger proteins, ARA54 and RNF8. Eur J Biochem. 2001;268:2725–32. doi: 10.1046/j.1432-1327.2001.02169.x. [DOI] [PubMed] [Google Scholar]
- 43.Plans V, Scheper J, Soler M, Loukili N, Okano Y, Thomson TM. The RING finger protein RNF8 recruits UBC13 for lysine 63-based self polyubiquitylation. J Cell Biochem. 2006;97:572–82. doi: 10.1002/jcb.20587. [DOI] [PubMed] [Google Scholar]
- 44.Bieganowski P, Shilinski K, Tsichlis PN, Brenner C. Cdc123 and checkpoint forkhead associated with RING proteins control the cell cycle by controlling eIF2gamma abundance. J Biol Chem. 2004;279:44656–66. doi: 10.1074/jbc.M406151200. [DOI] [PubMed] [Google Scholar]
- 45.Fraschini R, Bilotta D, Lucchini G, Piatti S. Functional characterization of Dma1 and Dma2, the budding yeast homologues of Schizosaccharomyces pombe Dma1 and human Chfr. Mol Biol Cell. 2004;15:3796–810. doi: 10.1091/mbc.E04-02-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murone M, Simanis V. The fission yeast dma1 gene is a component of the spindle assembly checkpoint, required to prevent septum formation and premature exit from mitosis if spindle function is compromised. Embo J. 1996;15:6605–16. [PMC free article] [PubMed] [Google Scholar]
- 47.Guertin DA, Venkatram S, Gould KL, McCollum D. Dma1 prevents mitotic exit and cytokinesis by inhibiting the septation initiation network (SIN) Dev Cell. 2002;3:779–90. doi: 10.1016/s1534-5807(02)00367-2. [DOI] [PubMed] [Google Scholar]
- 48.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–6. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 49.Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh S, Lowenstein JM. A multifunctional vector system for heterologous expression of proteins in Escherichia coli. Expression of native and hexahistidyl fusion proteins, rapid purification of the fusion proteins, and removal of fusion peptide by Kex2 protease. Gene. 1996;176:249–55. doi: 10.1016/0378-1119(96)00260-0. [DOI] [PubMed] [Google Scholar]
- 51.Rivin CJ, Fangman WL. Cell cycle phase expansion in nitrogen-limited cultures of Saccharomyces cerevisiae. J Cell Biol. 1980;85:96–107. doi: 10.1083/jcb.85.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martzen MR, McCraith SM, Spinelli SL, Torres FM, Fields S, Grayhack EJ, Phizicky EM. A biochemical genomics approach for identifying genes by the activity of their products. Science. 1999;286:1153–5. doi: 10.1126/science.286.5442.1153. [DOI] [PubMed] [Google Scholar]
- 53.Furukawa M, Andrews PS, Xiong Y. Assays for RING family ubiquitin ligases. Methods Mol Biol. 2005;301:37–46. doi: 10.1385/1-59259-895-1:037. [DOI] [PubMed] [Google Scholar]
- 54.Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villen J, Gygi SP. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–37. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- 55.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 56.Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT alpha 2 repressor. Cell. 1993;74:357–69. doi: 10.1016/0092-8674(93)90426-q. [DOI] [PubMed] [Google Scholar]
- 57.Frattali AL, Treadway JL, Pessin JE. Transmembrane signaling by the human insulin receptor kinase. Relationship between intramolecular beta subunit trans- and cis-autophosphorylation and substrate kinase activation. J Biol Chem. 1992;267:19521–8. [PubMed] [Google Scholar]
- 58.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.