

Abstract
Bioelectronic coupling of multi-redox-site membrane proteins was accomplished with cytochrome c oxidase (CcO) as an example. A biomimetic membrane system was used for the oriented immobilization of the CcO oxidase on a metal electrode. When the protein is immobilized with the CcO binding side directed toward the electrode and reconstituted in situ into a lipid bilayer, it is addressable by direct electron transfer to the redox centers. Electron transfer to the enzyme via the spacer, referred to as electronic wiring, shows an exceptionally high rate constant. This allows a kinetic analysis of all four consecutive electron transfer steps within the enzyme to be carried out. Electron transfer followed by rapid scan cyclic voltametry in combination with surface-enhanced resonance Raman spectroscopy provides mechanistic and structural information about the heme centers. Probing the enzyme under turnover conditions showed mechanistic insights into proton translocation coupled to electron transfer. This bioelectronic approach opens a new field of activity to investigate complex processes in a wide variety of membrane proteins.
INTRODUCTION
Membrane proteins represent a crucial class of biological agents. They are the key factors in many vital functions of the cell, including respiration. Typically, the proteins of the respiratory chain are multi-redox site membrane proteins. They use the energy released by electron transfer (ET) between the redox centers to actively transport protons across the lipid membrane. This process is also referred to as proton pumping. Cytochrome c oxidase (CcO) is the most prominent example of such a proton pump, illustrated schematically in Fig. 1. ET is shown to proceed stepwise along four redox centers, CuA, heme a, heme a3, and CuB and to be coupled to proton transfer (Fig. 1, inset) (1). Although x-ray crystallography (2) and genetic engineering (3,4) have shown a great deal of detail regarding amino acids involved in the proton pumping process, the precise mechanism is not elucidated completely (5,6).
FIGURE 1.

Schematic representation of CcO embedded into a protein-tethered bilayer lipid membrane. The protein is attached to the surface of a template stripped gold film by the his-tag attached to SU II, allowing for direct ET. The lipid bilayer is assembled around the protein. The box on the right shows the stepwise ET inside the CcO (44) and the box on the left shows the details of the surface modification (11).
For mechanistic studies, CcO must be functionally reconstituted in a lipid membrane. A majority of the previous investigations made use of proteoliposomes, with reconstituted enzymes (3,4). In these preparations the proteins are arranged in random orientations not allowing the application of defined electric fields, an essential parameter to control the charge transfer processes. Likewise, proteoliposomes do not allow for bioelectronic coupling, i.e., the direct ET between the protein and an electrode. Control of the oxidation state of redox centers in potentiometric titrations (3) or spectroscopic investigations (4) was accomplished indirectly by the use of soluble redox mediators that shuttle electrons to and away from redox centers.
Attempts have been made to pinpoint membrane proteins on surfaces. CcO for example has been incorporated previously into solid supported bilayer lipid membranes (7,8). With solid supported bilayer lipid membranes the application of defined electric fields was possible (8) and direct ET to the protein was achieved, albeit slow and without alluding to catalytic activity (7). These systems are limited by the absence of a hydrophilic layer adjacent to the electrode that separates the membrane from the surface thus providing a reservoir for ions. Aqueous submembrane layers have been provided through tethered bilayer lipid membranes (tBLMs) (9,10). tBLMs, however, do neither solve the problem of the random orientation nor do they offer the possibility of bioelectronic coupling. ET into the CcO was achieved, but only indirectly via cytochrome c, the natural substrate of the CcO (10).
To overcome all of these limitations, we developed a biomimetic membrane system, which tethers proteins to electrode surfaces through a histidine (his)-tag engineered onto the enzyme in a strict orientation (Fig. 1). A lipid bilayer is then reconstituted in situ around the bound proteins, forming a protein-tethered bilayer lipid membrane (ptBLM) (11,12). First attempts to address the protein by direct electronic coupling seemed promising indicating the direct electrochemical access to the ET pathway of the protein using electrochemically controlled surface-enhanced resonance Raman spectroscopy (SERRS) (13). Electrochemically-induced ET to multi-centered redox proteins has been the subject of research in recent years, providing a vast amount of kinetic information, but due to the lack of a biomimetic membrane system, mostly for soluble (14) or membrane-extrinsic proteins (15). The benefit of direct ET is the accessibility of the internal redox centers avoiding the diffusion limitation of mediators.
A further advantage inherent in the geometry of the ptBLM is the hydrophilic layer between the electrode and the lipid membrane provided by the tether molecules. The experimental benefit is the sensitivity of the small interstitial reservoir (∼0.2 nL cm−2) to changes in proton concentration as a result of enzyme activity. Thus this methodology presents a new approach to separately examine proton and electron transfer reactions in membrane proteins with significant consequences for the elucidation of the mechanism of proton pumping.
As a model of the 500 kDa large CcO complex located in the inner mitochondrial membrane of higher organisms (2), this study uses the smaller versions (125 kDa) from the cell membrane of bacteria, i.e., Rhodobacter sphaeroides (16) and Paracoccus denitrificans (17). X-ray crystallography suggests these versions are identical with respect to their electron and proton pathways (2,16,17). His-tags were attached to the C-terminus of SU II of R. sphaeroides and to C-terminus of SU I of P. denitrificans. The enzyme can thus be immobilized in different orientations, either with the cytochrome c binding side (Fig. 1) or, with the O2 reduction side, directed toward the electrode.
MATERIALS AND METHODS
If not otherwise stated, materials and methods were used as described previously (11). CcO from R. sphaeroides with a His-tag engineered to the C-terminus of subunit II was expressed and purified according to Mitchell and Gennis (18). Template stripped gold (19) electrodes, used for surface plasmon resonance and electrochemistry measurements, were immersed for 120 min in a solution of dithiobis (N-succinimidyl propionate) and dithiobis (propionic acid) in a 60:40 ratio, in dry DMSO (2 mg/ml). After rinsing with DMSO and ethanol, the slides were functionalized for 48 h in a 0.15 M solution of N-(5-amino-1-carboxypentyl) iminodiacetic acid buffered to pH 9.8 by adding 0.5 M KCO3. Finally, the glass slides were immersed for 30 min in 40 mM NiSO4 in acetate buffer (50 mM, pH 5.5) followed by thorough rinsing with the acetate buffer to remove the excess NiSO4. Immobilization of the protein was carried out as described previously (11). Dialysis was done by adding biobeads (Bio-Rad Laboratories GmbH, Munich, Germany) to the lipid-detergent-containing phosphate/dodecyl β-D-maltoside (DDM) buffer, DiPhyPC 0.05 mg/ml in phosphate/DDM buffer (K2HPO4 0.1 M, KCl 0.05 M, pH = 8, 0.1% DDM).
Electrochemistry
Electrochemical measurements were carried out using an Autolab instrument (PGSTAT302) equipped with an FRA2-module for impedance measurements, an ECD-module amplifier for low-currents, an ADC750 module for rapid scan measurements, and a SCAN-GEN module for analog potential scanning. Cyclic voltametry experiments were conducted with IR drop compensation, particularly at high scan rates. Measurements under anaerobic conditions were carried out in a buffer solution containing K2HPO4 0.1M, KCl 0.05M, pH = 8 and the oxygen trap consisting of glucose (0.3% w/w), glucose oxidase (75 μg/mL) and catalase (12.5 μg/mL) (20). This solution was flushed with Ar purged from oxygen by bubbling through the oxygen trap containing buffer solution for 1 or 2 h before the measurements to assure a completely deoxygenated solution. All electrochemical measurements were taken in a three-electrode configuration with template stripped gold as the working electrode, a Ag|AgCl,KClsat reference, and a platinum wire as the counter electrode. All electrode potentials are quoted versus normal hydrogen electrode (NHE).
SERRS
SERRS was carried out in a custom-made spectro electrochemical cell also connected to an Autolab instrument using an upside-down rotating disc electrode with a roughened silver surface (diameter, 10 mm) inserted into a Teflon mantle. An inner Teflon trough was fixed to the rotating disc electrode rotating in-line with the electrode. The cell was sealed to ambient air by a lid provided with inlets for the counter, reference (Ag|AgCl,KClsat) electrodes, an inlet for deoxygenation as well as for the microscope objective. The cell was flushed with Ar, purged from oxygen by washing through the oxygen trap containing buffer solution (see above). Surface enhanced resonance Raman (SERR) spectra were collected using a confocal Raman microscope (LabRam, HR800, HORIBA Jobin Yvon) equipped with a liquid nitrogen cooled back-illuminated CCD camera. The laser beam from a Kr+ laser (excitation wavelength, 413 nm) was focused on the surface of the above mentioned Ag-disc electrode by means of a water immersion objective. To prevent photoreduction of the enzyme, the electrode was rotated with 15 Hz or 900 rpm and the laser intensity at the sample surface was kept smaller than 100 μW. SERR spectra of several independent measurements and different samples were averaged to improve the signal/noise ratio.
RESULTS
Direct ET
CcO from R. sphaeroides engineered with a his-tag on subunit II (SU II) (16), was tethered to the surface of a gold electrode and reconstituted in situ into a ptBLM. Under these conditions, the cytochrome c binding side, and hence also the first electron acceptor, CuA, is oriented toward the electrode surface. The cyclic voltammogram (CV), taken in a strictly anaerobic bathing solution, shows a single reduction peak at −274 ± 7 mV and a corresponding oxidation peak at −209 ± 6 mV (Fig. 2 a) at low scan rates (<1 V s−1). As the scan rate is increased to ∼1 V s−1, the peak positions remain largely unchanged and the baseline-corrected current density increases linearly (Fig. 2 a, inset), which is consistent with surface confined ET (21). The constant peak separation at scan rates (<1 Vs−1), compatible with a fast ET (22) (for an explanation see below) is observed only with the lipid membrane formed around the protein. Membrane formation was checked by surface plasmon resonance and electrochemical impedance measurements (Supplementary Material, Figs. S1 and S2). Finally, we have estimated the apparent standard potential, 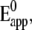 assuming symmetrical behavior of the redox system, to be −242 ± 7 mV. This
assuming symmetrical behavior of the redox system, to be −242 ± 7 mV. This 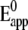 is shifted ∼ −450 mV from the standard potential of 230 mV determined for the CuA center in isolated CcO (23). This shift can be explained by the ET pathway to the enzyme via the Ni complex, taking into account the low standard potentials of chelated Ni2+/Ni+ couples (24). The multi-step ET to electron acceptors of very different standard potentials may result in a peak whose potential is located in between the single standard potentials, provided the first electron acceptor has a much lower standard potential compared to the following ones. To show this effect simulations were carried out (Figs. S5–S7) considering the Ni complex as a transient electron acceptor and taking into account the following standard redox potentials
is shifted ∼ −450 mV from the standard potential of 230 mV determined for the CuA center in isolated CcO (23). This shift can be explained by the ET pathway to the enzyme via the Ni complex, taking into account the low standard potentials of chelated Ni2+/Ni+ couples (24). The multi-step ET to electron acceptors of very different standard potentials may result in a peak whose potential is located in between the single standard potentials, provided the first electron acceptor has a much lower standard potential compared to the following ones. To show this effect simulations were carried out (Figs. S5–S7) considering the Ni complex as a transient electron acceptor and taking into account the following standard redox potentials 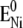 = −442 mV,
= −442 mV, 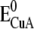 = 230 mV,
= 230 mV,  = 340 mV. The simulated CV displays a peak at −270 mV in reasonable agreement with our measurements. The simulations are based on concepts developed by Walz et al. (25) for analyzing bioelectrochemical processes across membranes. Details can be found in the supplementary information. The transient nature of ET to the Ni complex is shown by the probabilities of the conformations with Ni in the reduced state which do not exceed 6 × 10−8 (Fig. S7 b), in accordance with the absence of redox peaks when the Ni-NTA is chelated with imidazole instead of the his-tag (26).
= 340 mV. The simulated CV displays a peak at −270 mV in reasonable agreement with our measurements. The simulations are based on concepts developed by Walz et al. (25) for analyzing bioelectrochemical processes across membranes. Details can be found in the supplementary information. The transient nature of ET to the Ni complex is shown by the probabilities of the conformations with Ni in the reduced state which do not exceed 6 × 10−8 (Fig. S7 b), in accordance with the absence of redox peaks when the Ni-NTA is chelated with imidazole instead of the his-tag (26).
FIGURE 2.

Cyclic voltammograms of CcO with the his-tag attached to SU II under strictly anaerobic conditions. (a) Scan rates a between 0.1 and 1 Vs−1, 0.01 V s−1 (green), 0.02 V s−1 (orange), 0.03 V s−1 (dark blue), 0.05 V s−1 (light blue), 0.075 V s−1 (red), and 0.1 V s−1 (black). (Inset) Baseline-corrected current density plotted as a function of scan rate, reductive branch (red), oxidative branch (green). (b) Oxidative branches (baseline-corrected) of CVs at scan rates between 1 and 600 Vs−1 (current densities normalized by the scan rate, ν ≅ 1 Vs−1) 1 V s−1 (red), 3 V s−1 (black), 7.5 V s−1 (green), 20 V s−1 (orange), 40 V s−1 (pink), 300 V s−1 (blue), 400 V s−1 (violet), 500 V s−1 (gray), 600 V s−1 (light blue). (Inset) Example of a deconvolution into four Gaussian components.
Another reason for the peak shift such as a kinetic limitation of the direct ET must be excluded because the peak separation hardly changes at low scan rates (<1 V s−1). However, the peak separation of a surface-confined ET as indicated by the linear dependency of the current density from the scan rate, must be expected to be zero whereas an appreciable but unchanging peak separation accounts for an irreversible reaction after the reversible ET (21). Protonations of the heme centers of the CcO after reduction that are known from independent measurements (27) are considered most likely to be the reason for the observed peak separation.
A true change of the thermodynamic standard potential may also be ruled out considering this would mean the unfolding of the protein. This is highly unlikely taking into account the catalytic activity inhibited by cyanide observed by electrochemical measurements in the presence of oxygen as described below. Moreover, strong evidence for the integrity of the enzyme undergoing electrochemically induced ET was derived from Soret band excited SERRS taken as a function of potential, under strictly anaerobic conditions (Fig. 3 a). The maximum absorption feature of the heme a and a3 sites of the protein, the so-called Soret band, is located at 410 nm (1,28,29). Using an excitation wavelength of 413nm of the krypton laser, we take advantage of the selective resonant enhancement of the vibrational modes of the heme sites. The characteristic marker band region (1300–1750 cm−1) is not only very sensitive to the integrity of the enzyme, but it also provides information on the redox state, the ligation, spin and coordination state of the heme a/a3 structure. The bands at 1358 cm−1 and 1370 cm−1 originate from the ν4 modes of both hemes in the reduced (−350 mV) and oxidized state (−150 mV), respectively (13,29). Furthermore, the type-a hemes of CcO include conjugated vinyl and formyl substituents, which give rise to resonance Raman-active stretching modes between 1610 and 1680 cm−1 (28,29). Characteristic redox linked changes of the weakly bound formyl substituent of heme a3 cause a small change from 1663 cm−1 in the oxidized state to 1671 cm−1 in the reduced state, because the formyl group remains in a hydrophobic environment. A further characteristic marker for the reduced heme a is the intense and well-separated band at 1517 cm−1 (ν11) not present in the oxidized state. Redox induced changes of the C=O side group of heme a3 can be observed by a larger frequency downshift from 1646 to 1610 cm−1, which reflects a substantial redox-linked change of the stronger hydrogen bond interactions of this substituent with Gln471 and Arg52 (13,29). Both porphyrin modes (ν4 and ν11) and the formyl stretching modes allow monitoring of the oxidation states of the individual hemes of the immobilized CcO. The bands at 1500 (ν3) and 1585 cm−1 (ν2) result from a ferric hexacoordinated low-spin (6cLS) heme (13,28,29). These findings corroborate the results of earlier studies, showing the SERR spectra on the surface to be identical to resonance Raman spectra in solution (13). Furthermore, they show a gradual transition of the redox state as well as the coordination and spin state of both heme centers in the potential range between −150 mV and −350 mV, in agreement with the CV data (Fig. 2).
FIGURE 3.

SERR spectra of CcO at different potentials applied across the biomimetic protein-membrane architecture immersed in buffer solution (a) with the his-tag on SU II at −150 mV (1), −175 mV (2), −200 mV (3), −225 mV (4), −250 mV (5), −275 mV (6), −300 mV (7), −325 mV (8) and −350 mV (9). (b) With the his-tag on SU I, spectrum taken at +200 mV (1), −1100 mV (2) and after chemical reduction with dithionite (3).
Because the SERRS experiment is particularly designed for a selective resonance enhancement of the heme structure, the Cu centers are not visible. Evidence that ET to the hemes indeed occurs via CuA as described in previous studies (31,32) was derived from potential-controlled surface enhanced IR reflection absorption spectroscopy carried out on CcO immobilized in the same orientation in a ptBLM. These measurements make use of amide I bands of the protein environment assigned to specific redox states of CuA (3). Results will be presented in a forthcoming publication. Taken together with the Raman spectra, these results indicate that the electrochemical reduction peak is in fact due to direct ET to CuA and further on to the heme centers. This is further corroborated by the charge calculated from the integrated area of the reductive and oxidative peaks (Fig. S3). These results are consistent with a densely packed protein monolayer, assuming an ellipsoidal disk of 4.5 nm × 7.0 nm for the in-plane dimension of the protein deduced from the crystal structure of CcO from R. sphaeroides (33), but only if four electrons transferred per molecule are considered.
As a control, CcO from P. denitrificans with a his-tag engineered to the C-terminus of subunit I (SU I) (11,12,17) was immobilized in the same biomimetic system. The cytochrome c binding side, and hence CuA, is oriented toward the outside of the membrane opposite to that described above. This inverted orientation shows Raman spectra identical to those of CcO with the his-tag on SU II, thus indicating the same structural integrity of the protein. No indication of reduction can be seen in the potential range between +200 mV and −1100 mV (Fig. 3 b), suggesting that the enzyme remains in the oxidized state. Accordingly, no ET peaks can be observed with CV (see Fig. 5 a, blue curve). Nevertheless, this is not due to a functional impairment of the redox centers since they can be reduced chemically by adding dithionite (Fig. 3 b), and CcO may be activated by reduced cytochrome c (11). This experiment proves conclusively that ET between the electrode and CcO does not occur nonspecifically to any of the redox centers, but is possible only via CuA, with the cytochrome c binding side oriented toward the electrode. Thus, exquisite control of the orientation of the enzyme allows direct, verifiable ET reactions to and within CcO to be elucidated.
FIGURE 5.

Voltammograms of CcO with the his-tag attached to SU II under aerobic and anaerobic conditions. (a) Cyclic voltammograms at a scan rate of 0.01 Vs−1 anaerobic (red), aerobic (black), and CcO with the his-tag on SU I (blue). (b) Differential pulse voltammograms, anaerobic (red), aerobic (black), and aerobic in the presence of 40 μM cyanide (green). The dashed curves are enlarged for clarity, corresponding to the y axis on the right hand side.
Kinetics of ET
Although slow scan rate CV is sufficient to obtain basic ET mechanistic information, scan rates >1 V s−1 must be used to extract kinetic information (Fig. 2 b). The baseline-corrected current in the CV of a surface-confined redox couple can be represented by a Gaussian curve whose peak position and width are determined by the standard potential of the couple, the number of electrons, the rate constant of ET (k0) and the scan rate (21). At low scan rates the peak position is approximately constant and close to the standard potential. As the scan rate is increased to several Vs−1, kinetic constraints cause the peaks to separate, with reductive and oxidative peaks shifting to more negative and more positive potentials, respectively.
The four redox center CcO enzyme exhibits a more complicated behavior. Under anaerobic conditions the redox currents display single slightly asymmetric peaks up to a scan rate of ∼1 Vs−1 (cf. Fig. 2 a) indicating the contributions from multiple redox centers. At scan rates above 1 Vs−1 the baseline-corrected peaks become more asymmetric, a second maximum gradually appears as the scan rate exceeds 20 Vs−1 (Fig. 2 b). Complex voltammograms of multi-redox-site proteins were previously deconvoluted into a number of Gaussian components with the area of each peak corresponding to the number of electrons transferred in each step (34). Accordingly, the curves shown in Fig. 2 b were found to require four Gaussian components to adequately describe the system. However, due to the strong overlap, the peak areas are not evenly distributed, particularly regarding the last component. This is assumed to be caused by the strong coupling of the ET occurring sequentially through the four redox centers CuA, heme a, heme a3, and CuB in the enzyme (Fig. 2 b, inset), not observed in other enzymes (34).
Established procedures (34) were used to obtain kinetic rate constants by plotting the peak potentials versus scan rate in a logarithmic scale, to yield a “trumpet” plot (Fig. 4) (35). This plot was analyzed by means of the algorithm described by Jeuken et al. with the simplifying assumption of an individual one-step ET to each redox center (22,34). Values for the rate constant k0 and the apparent standard potential 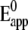 thus obtained are shown in Table 1. Because ET between redox centers in CcO occurs sequentially, the values of k0 for redox centers 2, 3, and 4 are approximations of lumped rate constants (37) to heme a, heme a3, and CuB through the ET chain. However, k0 for redox center 1 is attributed to ET to CuA. Hence, it is clear that ET to the subsequent redox centers is not restricted or rate-limited by the electrochemical ET to CuA. Therefore, the measurements in the ptBLM yield results of biological relevance.
thus obtained are shown in Table 1. Because ET between redox centers in CcO occurs sequentially, the values of k0 for redox centers 2, 3, and 4 are approximations of lumped rate constants (37) to heme a, heme a3, and CuB through the ET chain. However, k0 for redox center 1 is attributed to ET to CuA. Hence, it is clear that ET to the subsequent redox centers is not restricted or rate-limited by the electrochemical ET to CuA. Therefore, the measurements in the ptBLM yield results of biological relevance.
FIGURE 4.

Peak potentials as a function of scan rate (trumpet plots) of the four redox centers. Peak potentials are obtained by deconvolution of CVs into Gaussian components of oxidation and reduction branches of CcO with the his-tag attached to SU II under anaerobic conditions (cf. Fig. 2 b), redox center 1 (CuA) (black), 2 (red), 3 (green), 4 (blue). The error bars represent ranges resulting from the deconvolution procedure.
TABLE 1.
Apparent standard potentials 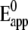 and rate constants k0 obtained from CV of CcO under anaerobic conditions
and rate constants k0 obtained from CV of CcO under anaerobic conditions
| Redox center |
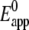 /mV /mV |
k0/s−1 |
|---|---|---|
| 1 (black) A CuA | −231 ± 8 | 4130 ± 390 |
| 2 (red) | −234 ± 7 | 1650 ± 170 |
| 3 (green) | −211 ± 6 | 68 ± 8 |
| 4 (blue) | −183 ± 7 | 11 ± 2 |
The errors represent ranges resulting from experimental errors, the deconvolution procedure, and the fit routine applied to the trumpet plot for four data sets of the same enzyme preparation.
Proton transport coupled to ET
To investigate the enzyme undergoing catalytic turnover, aerobic conditions were used. The CV of CcO with the cytochrome c binding side oriented toward the electrode showed two cathodic peaks (Fig. 5 a, black curve). The first peak at −202 ± 5mV, attributed to reduction of the four redox centers of CcO, showed a significant amplification of the current density compared to the peak under anaerobic conditions (Fig. 5 a, red curve). This is a clear indication of the catalytic turnover of the enzyme. Electrons transferred from the electrode to the redox centers of CcO are irreversibly transferred to oxygen, leading to a continuous ET (31,32). The current density (35 μA cm−2) of this peak is considerably larger than the one measured previously with the help of soluble substrates of membrane proteins immobilized in a tBLM (≤0.6 μA cm−2) (10), indicating a comparatively high activity of the CcO embedded in the ptBLM.
As a consequence of catalytic turnover of the CcO in the orientation with the his-tag attached to SU II, protons are pumped from the bulk solution into the interstitial space between the electrode and the ptBLM. Due to its small volume, this interstitial space is thereby highly acidified (as low as pH 2; see Fig. S4). These protons are electrochemically reduced to H2 giving rise to the second peak at −422 ± 6 mV (Fig. 5 a, black curve), as confirmed by experiments using the modified surface without the enzyme and lipid membrane being present (see Fig. S4). Moreover, a similar proton reduction peak has already been observed for H+/ATP-synthase reconstituted into a tBLM (38). The absence of direct ET and the H+ reduction peak in the inverted orientation (Fig. 5 a, blue curve) further confirms the orientation dependence of direct ET as well as transmembrane proton transport. The sloping baseline in the negative potential range of the control experiment (Fig. 5 a, blue curve) is explained in terms of the irreversible reduction of oxygen at the modified electrode. This holds also for the baseline in the CV of CcO subject to direct ET under aerobic (Fig. 5 a, black curve) as compared to anaerobic conditions (Fig. 5 a, red curve).
Improved sensitivity can be achieved by using differential pulse voltametry (DPV). Under aerobic conditions the amplified current density of the reductive peak (−190 mV) and the resultant proton reduction peak (−390 mV) can be observed (Fig. 5 b, black curve). More importantly, under anaerobic conditions no proton peak was recorded while a single peak is observed due to the reduction of the enzyme (Fig. 5 b, red curve). If only one proton was transferred across the membrane as all four redox centers are reduced, a proton reduction current of ∼0.8 μA cm−2 can be expected, which would be easily detectable by DPV. The same applies when the enzyme is reduced in the presence of the inhibitor cyanide under aerobic conditions; (Fig. 5 b, green curve) in this case no proton reduction peak is observed.
DISCUSSION
Bioelectronic coupling of a multi-redox-site membrane protein in a functionally active form was verified using CcO as an example. This is shown clearly by electrochemical and potential-controlled SERRS experiments. Direct ET to the enzyme is very specifically controlled by the orientation of the CcO immobilized on the surface. Electrons are transferred but only when the CcO is immobilized with the cytochrome c binding side directed toward the electrode, suggesting that the primary electron acceptor is CuA as in other biochemical assays. Electron transfer appears to be facilitated by the Ni1+/2+ complex, as deduced from the heterogeneous rate coefficient k0 > 4000 s−1, which is fast as compared to other (soluble) proteins. Previous attempts at coupling proteins to surfaces have yielded k0 values between 100 s−1 to 1600 s−1 (39,40). Electron injection into the CcO by other methods (41) is faster, e.g., by pulse radiolysis or photochemical excitation (42,43). In these methods, however, a substrate has to be bound first to the enzyme in a slow diffusion-controlled process before electron injection can be initiated. As a consequence, fast electron injection occurs only once, and a continuous or repetitive process is not possible. By contrast, electrochemical ET is not limited by diffusion. It can be repeatedly carried out many times in both the reductive and oxidative direction or alternatively it can be used to elicit a stationary state of ET. This makes electrochemical ET the method of choice for time-resolved measurements particularly for processes occurring in cycles, e.g., enzymatic cycles. The full enzyme cycle apparently occurs in CcO immobilized in the ptBLM as is evident from CV scans obtained under aerobic conditions.
ET is often rate-limited by protonation reactions; for example, proton uptake of the so-called charge compensating proton of reduced heme a/a3 occurs in the time scale of 200 μs (27) that would yield a first order rate constant of 5000 s−1. Protonation of the hemes is very likely to occur on electrochemical reduction of the CcO that would account for an overall k0 > 4000 s−1. Rates of transitions between other states in the enzyme cycle, e.g., transitions between the states labeled P and F, or F and O are considerably slower (in the ms time scale) (44). It is thus concluded that the bioelectronic approach is designed to carry out kinetic measurements of biological relevance.
Regarding mechanistic details, first insights into the performance of the electrochemical method have been gained by comparative measurements under aerobic and anaerobic conditions. Proton transfer into the interstitial space underneath the membrane only occurs in the presence of oxygen. This observation supports the proposed mechanism of proton pumping, in conjunction with the transition from state O to E, which requires a preceding full turnover (or an activated state OH) (45,46). Other proposals contradict this idea, claiming that the transfer of two electrons from state O to E is followed by a proton pumping step even with no preceding turnover of the catalytic cycle (27,44). The latter hypothesis seems questionable in the light of our findings which suggest that protons are not released to the submembrane space on a mere reduction of the hemes, i.e., proton pumping does not occur without oxygen present (45–47). It should be mentioned, however, that the uptake of protons in the O to E transition, [the so-called charge compensating protons (27)] is compatible with our experiments.
The results presented in this work show that the ptBLM is a promising new platform for kinetic and mechanistic studies of a large number of membrane proteins, in particular redox proteins. Studies of other membrane proteins would certainly also benefit from this strategy, considering that electric fields are often a controlling parameter, for example for ion transport through channel proteins. These options are particularly attractive with respect to the combined application of electrochemical and multiple surface analytical methods, including vibrational spectroscopy.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We are grateful to Shelagh Ferguson-Miller, Michigan State University, for providing us with CcO from R. sphaeroides engineered with the his-tag on SU II and to Bernd Ludwig, university of Frankfurt Germany, for providing us with CcO from P. denitrificans engineered with the his-tag on SU I.
Joseph W. F. Robertson's current address is Semiconductor Electronics Division, Electronics and Electrical Engineering Laboratory, National Institute of Standards and Technology, Gaithersburg, MD, 20899.
Dieter Walz's current address is Lerchenstr. 21, 4095 Basel, Switzerland.
Editor: Brian R. Dyer.
References
- 1.Richter, O.-M. H., and B. Ludwig. 2003. Cytochrome c oxidase structure, function and physiology of a redox-driven molecular machine. Rev. Physiol. Biochem. Pharmacol. 147:47–74. [DOI] [PubMed] [Google Scholar]
- 2.Tsukihara, T., H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, and Sh. Yoshikawa. 1995. Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 Å. Science. 269:1069–1074. [DOI] [PubMed] [Google Scholar]
- 3.Gorbikova, E. A., K. Vuorilehto, M. Wikström, and M. I. Verkhovsky. 2006. Redox titration of all electron carriers of cytochrome c oxidase by Fourier transform infrared spectroscopy. Biochemistry. 45:5641–5649. [DOI] [PubMed] [Google Scholar]
- 4.Hellwig, P., U. Pfitzner, J. Behr, B. Rost, R. P. Pesavento, W. von Donk, R. B. Gennis, H. Michel, B. Ludwig and W. Maentele. 2002. Vibrational modes of tyrosines in cytochrome c oxidase from Paracoccus denitrificans: FTIR and electrochemical studies on Tyr-D4-labeled and on Tyr280His and Tyr35Phe mutant enzymes. Biochemistry. 41:9116–9125. [DOI] [PubMed] [Google Scholar]
- 5.Zaslavsky, D., and R. B. Gennis. 2000. Proton pumping by cytochrome c oxidase: progress, problems and postulates. Biochim. Biophys. Acta. 1458:164–179. [DOI] [PubMed] [Google Scholar]
- 6.Wikström, M. 2004. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim. Biophys. Acta. 1655:241–247. [DOI] [PubMed] [Google Scholar]
- 7.Salamon, Z., J. Hazzard, and G. Tollin. 1993. Proc. Natl. Acad. Sci. USA. 90:6420–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cullison, J. K., F. M. Hawkridge, N. Nakashima, and Sh. Yoshikawa. 1994. A study of cytochrome c oxidase in lipid bilayer membranes on electrode surfaces. Langmuir. 10:877–882. [Google Scholar]
- 9.Naumann, R., E. K. Schmidt, A. Jonczyk, K. Fendler, B. Kadenbach, T. Liebermann, A. Offenhäusser, and W. Knoll. 1999. The peptide-tethered lipid membrane as a biomimetic system to incorporate cytochrome c oxidase in a functionally active form. Biosens. Bioelectron. 14:651–662. [Google Scholar]
- 10.Jeuken, L. J. C., S. D. Connell, P. J. F. Henderson, R. B. Gennis, St. D. Evans, and R. J. Bushby. 2006. Redox enzymes in tethered membranes. J. Am. Chem. Soc. 128:1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giess, F., M. G. Friedrich, J. Heberle, R. L. Naumann, and W. Knoll. 2004. The protein-tethered lipid bilayer: A novel mimic of the biological membrane. Biophys. J. 87:3213–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ataka, K., F. Giess, W. Knoll, R. Naumann, S. Haber-Pohlmeier, B. Richter, and J. Heberle. 2004. Oriented attachment and membrane reconstitution of his-tagged cytochrome c oxidase to a gold electrode: In situ monitoring by surface enhanced infrared absorption spectroscopy. J Am. Chem. Soc. 126:16199–16206. [DOI] [PubMed] [Google Scholar]
- 13.Friedrich, M. G., F. Giess, R. Naumann, W. Knoll, K. Ataka, J. Heberle, J. Hrabakova, D. H. Murgida, and P. Hildebrandt. 2004. Active site structure and redox processes of cytochrome c oxidase immobilised in a novel biomimetic lipid membrane. Chem. Commun. 2376–2377. [DOI] [PubMed]
- 14.Léger, Ch., S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones, and F. A. Armstrong. 2003. Enzyme electrokinetics: using protein film voltametry to investigate redox enzymes and their mechanisms. Biochemistry. 42:8653–8662. [DOI] [PubMed] [Google Scholar]
- 15.Hirst, J., A. Sucheta, B. A. C. Ackrell, and F. A. Armstrong. 1996. Electrocatalytic voltametry of succinate dehydrogenase: direct quantification of the catalytic properties of a complex electron-transport enzyme. J. Am. Chem. Soc. 118:5031–5038. [Google Scholar]
- 16.Hosler, J. P., J. Fetter, M. M. Tecklenburg, M. Espe, C. Lerma, and Sh. Ferguson-Miller. 1992. Cytochrome aa3 of Rhodobacter sphaeroides as a model for mitochondrial cytochrome c oxidase. Purification, kinetics, proton pumping, and spectral analysis. J. Biol. Chem. 267:24264–24273. [PubMed] [Google Scholar]
- 17.Pfitzner, U., A. Odenwal, Th. Ostermann, L. Weingard, B. Ludwig, and O.-M. H. Richter. 1998. Cytochrome c oxidase (Heme aa3) from Paracoccus denitrificans: analysis of mutations in putative proton channels of subunit I. J. Bioenerg. Biomembr. 30:3089–3097. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell, D. M., and R. B. Gennis. 1995. Rapid purification of wildtype and mutant cytochrome c oxidase from Rhodobacter sphaeroides by Ni2+-NTA affinity chromatography. FEBS Lett. 368:148–150. [DOI] [PubMed] [Google Scholar]
- 19.Naumann, R., St. M. Schiller, F. Giess, B. Grohe, K. B. Hartman, I. Kaercher, I. Koeper, J. Luebben, K. Vasilev, and W. Knoll. 2003. Tethered lipid bilayers on ultra flat gold surfaces. Langmuir. 19:5435–5443. [Google Scholar]
- 20.Vanderkooi, J. M., G. Maniar, T. J. Green, and D. Wilson. 1987. An optical method for measurement of dioxygen concentration based upon quenching of phosphorescence. J. Biol. Chem. 262:5476–5482. [PubMed] [Google Scholar]
- 21.Bard, A. J., and L. L. Faulkner. 2001. Electrochemical Methods: Fundamentals and Applications, 2nd Ed. John Wiley & Sons, New York.
- 22.Jeuken, L. J. C., J. P. McEvoy, and F. A. Armstrong. 2002. Insights into gated electron-transfer kinetics at the electrode-protein interface: a square wave voltammetry study of the blue copper protein azurin. J. Phys. Chem. B. 106:2304–2313. [Google Scholar]
- 23.Moody, A. J., U. Brandt, P. R. Rich. 1991. Single electron transfer of slow and fast cytochrome c oxidase. FEBS Lett. 293:101–105. [DOI] [PubMed] [Google Scholar]
- 24.Budnikova, Yu. G., O. E. Petrukhina, and Yu. M. Kargin. 1996. Metal-complex catalysis in organic electrochemical synthesis. The paramagnetic complexes [Ni(0)L2]-. in the reduction of organic halides. Zhurnal Obshchei Khimii. 66:1876–1880. [Google Scholar]
- 25.Walz, D., S. R. Caplan, D. R. L. Scriven, and D. C. Miculecky. 1995. Methods of mathematical modeling. In Bioelectrochemistry: Principles and Practice, Vol. 1 (Bioelectrochemistry: General Introduction). S. R. Caplan, I. R. Miller, and G. Milazzo, editors. Birkhäuser Verlag, Basel.
- 26.Blankespoor, R., B. Limoges, B. Schöllhorn, J.-L. Syssa-Magalé, and D. Yazidi. 2005. Dense monolayer of metal-chelating ligands covalently attached to carbon electrodes electrochemically and their useful application in affinity binding of histidine-tagged proteins. Langmuir. 21:3362–3375. [DOI] [PubMed] [Google Scholar]
- 27.Ruitenberg, M., A. Kannt, E. Bamberg, K. Fendler, and H. Michel. 2002. Reduction of cytochrome c oxidase by a second electron leads to proton translocation. Nature. 417:99–102. [DOI] [PubMed] [Google Scholar]
- 28.Döpner, S., J. Hudecek, B. Ludwig, H. Witt, and P. Hildebrandt. 2000. Structural changes in cytochrome c oxidase binding. A resonance Raman study. Biochim. Biophys. Acta. 1480:57–64. [DOI] [PubMed] [Google Scholar]
- 29.Heibel, G. E., P. Hildebrandt, B. Ludwig, P. Steinrücke, T. Soulimane, and G. Buse. 1993. Comparative resonance Raman study of cytochrome c oxidase from beef heart and Paracoccus denitrificans. Biochemistry. 32:10866–10877. [DOI] [PubMed] [Google Scholar]
- 30.Reference deleted in proof.
- 31.Verkhovsky, M. I., J. E. Morgan, and M. Wikström. 1995. Control of electron delivery to the oxygen reduction site of cytochrome c oxidase, a role for protons. Biochemistry. 34:7483–7491. [DOI] [PubMed] [Google Scholar]
- 32.Szundi, I., J. A. Cappucino, N. Borovok, B. Kotlyar, and O. Einarsdottir. 2001. Photo-induced electron transfer in the cytochrome c oxidase oxidase complex using thioredopyrenetrisulfonate-labeled cytochrome c. Optical multichannel detection. Biochemistry. 40:2186–2193. [DOI] [PubMed] [Google Scholar]
- 33.Svensson-Ek, M., J. Abramson, G. Larsson, S. Tornroth, P. Brzezinski, and S. Iwata. 2002. The x-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 321:329–339. [DOI] [PubMed] [Google Scholar]
- 34.Armstrong, F. A. 2002. Insights from protein film voltametry into mechanisms of complex biological electron-transfer reactions. J. Chem. Soc., Dalton Trans. 661–671.
- 35.Laviron, E. 1979. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 101:19–28. [Google Scholar]
- 36.Reference deleted in proof.
- 37.Hill, T. L. 1977. Free Energy Transduction in Biology. Academic Press, New York.
- 38.Naumann, R., T. Baumgart, P. Gräber, A. Jonczyk, A. Offenhäusser, and W. Knoll. 2002. Proton transport through a peptide-tethered bilayer lipid membrane by the H+-ATP synthase from chloroplasts measured by impedance spectroscopy. Biosens. Bioelectr. 17:25–34. [DOI] [PubMed] [Google Scholar]
- 39.Jeuken, L. J. C. 2003. Conformational reorganization in interfacial protein electron transfer. Biochim. Biophys. Acta. 1604:67–76. [DOI] [PubMed] [Google Scholar]
- 40.Hirst, J., J. L. C. Duff, G. N. L. Jameson, M. A. Kemper, B. K. Burgess, and F. A. Armstrong. 1998. Kinetics and mechanism of redox-coupled, long-range proton transfer in an iron-sulfur protein. Investigation by fast-scan protein-film voltammetry. J. Am. Chem. Soc. 120:7085–7094. [Google Scholar]
- 41.Winkler, J. R., B. G. Malmström, and H. B. Gray. 1995. Rapid electron injection into multisite metalloproteins: intramolecular electron transfer in cytochrome oxidase. Biophys. Chem. 54:199–205. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi, K., H. Une, and K. Hayashi. 1989. Electron transfer processes in cytochrome oxidase after pulse radiolysis. J. Biol. Chem. 264:7976–7980. [PubMed] [Google Scholar]
- 43.Nilsson, T. 1992. Photoinduced electron transfer from tris(2,2'-bipyridyl)ruthenium to cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 89:6497–6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michel, H. 1999. Cytochrome c oxidase: catalytic cycle and mechanisms of proton pumping—a discussion. Biochemistry. 38:15129–15140. [DOI] [PubMed] [Google Scholar]
- 45.Verkhovsky, M. I., A. Jasaitis, M. L. Verkhovskaya, J. E. Morgan, and M. Wikström. 1999. Proton translocation by cytochrome c oxidase. Nature. 400:480–483. [DOI] [PubMed] [Google Scholar]
- 46.Verkhovsky, M. I., I. Belevich, D. A. Bloch, and M. Wikström. 2006. Elementary steps of proton translocation in the catalytic cycle of cytochrome oxidase. Biochim. Biophys. Acta. 1757:401–407. [DOI] [PubMed] [Google Scholar]
- 47.Faxen, K., G. Gilderson, P. Adelroth, and P. Brzezinski. 2005. A mechanistic principle for proton pumping by cytochrome c oxidase. Nature. 437:286–289. [DOI] [PubMed] [Google Scholar]