

Abstract
Oxidative stress, a hallmark of Alzheimer disease (AD), has been shown to induce lipid peroxidation and apoptosis disrupting cellular homeostasis. Normally, the aminophospholipid phosphatidylserine (PtdSer) is asymmetrically distributed on the cytosolic leaflet of the lipid bilayer. Under oxidative stress conditions, asymmetry is altered, characterized by the appearance of PtdSer on the outer leaflet, to initiate the first stages of an apoptotic process. PtdSer asymmetry is actively maintained by the ATP-dependent translocase flippase, whose function is inhibited if covalently bound by lipid peroxidation products, 4-hydroxynonenal (HNE) and acrolein, within the membrane bilayer in which they are produced. Additionally, pro-apoptotic proteins Bax and caspase-3 have been implemented in the oxidative modification of PtdSer resulting in subsequent asymmetric collapse, while anti-apoptotic protein Bcl-2 has been found to prevent this process.
The current investigation focused on detection of PtdSer on the outer leaflet of the bilayer in synaptosomes from brain of subjects with AD and amnestic mild cognitive impairment (MCI), as well as expression levels of apoptosis-related proteins Bcl-2, Bax, and caspase-3. Fluorescence and Western blot analysis suggest PtdSer exposure on the outer leaflet is significantly increased in brain from subjects with MCI and AD contributing to early apoptotic elevation of pro- and anti- apoptotic proteins and finally neuronal loss. MCI is considered a possible transition point between normal cognitive aging and probable AD. Brain from subjects with MCI is reported to have increased levels of tissue oxidation; therefore, the results of this study could mark the progression of patients with MCI into AD. This study contributes to a model of apoptosis-specific oxidation of phospholipids consistent with the notion that PtdSer exposure is required for apoptotic-cell death.
Keywords: Mild Cognitive Impairment (MCI), Alzheimer disease (AD), phosphatidylserine (PtdSer), phospholipid asymmetry, apoptosis, Bax, Bcl-2, caspase-3, oxidative stress
Introduction
The asymmetric distribution and localization of the aminophospholipid phosphatidylserine (PtdSer) to the cytosolic leaflet of the cellular membrane is actively regulated by the ATP-dependent membrane bound aminophospholipid translocase flippase, a P-type ATPase known to transport phospholipids across the bilayer (Daleke, 2003; Paulusma and Oude Elferink, 2005; Tang et al., 1996). In the event asymmetry is altered, PtdSer is exposed to the cell surface, initiating early stages of apoptosis considered crucial to selective recognition and mononuclear phagocytosis of target cells by macrophages and fibroblasts (Castegna et al., 2004; Fadok et al., 2001; Tyurina et al., 2004b). Exposure of PtdSer to the outer leaflet has been noted to affect protein function involving platelet aggregation, activity of membrane receptors and transport proteins, signal transduction pathways, and cellular morphology via interference with lipid-protein and protein-protein interactions (Balasubramanian and Schroit, 2003; Paulusma and Oude Elferink, 2005; Verkleij and Post, 2000).
Previous studies have shown that lipid peroxidation, leading to loss of phospholipid asymmetry (Castegna et al., 2004; Kagan et al., 2002; Mohmmad Abdul and Butterfield, 2005), ultimately leads to apoptosis (Fadok et al., 1992; Kagan et al., 2003; Shimohama, 2000). Oxidative stress and oxidative damage, hallmarks of Alzheimer disease (AD) pathology, can lead to increased production of lipid peroxidation products, 4-hydroxynonenal (HNE) and acrolein (2-propenal), in the membrane bilayer (Butterfield et al., 2001; 2002; Butterfield and Lauderback, 2002; Lovell et al., 2001; Markesbery and Lovell, 1998). These reactive alkenals have been found to interfere with PtdSer asymmetry via redox dependent flippase (Castegna et al., 2004; Daleke, 2003; Tyurina et al., 2004b). Furthermore, much evidence indicates that oxidative modification of flippase by reactive alkenals and/or apoptotic proteins, resulting in asymmetric collapse (Castegna et al., 2004; Kagan et al., 2002; Mandal et al., 2005; Mohmmad Abdul and Butterfield, 2005; Tyurina et al., 2004a), is greatly elevated in the early apoptotic phenotype of AD models (Herrmann and Devaux, 1990; Kagan et al., 2000).
Oxidative stress conditions in AD can also result in cytochrome c release from mitochondria and disruption of electron transport to initiate apoptosis (Ott et al., 2007), known to play an important role in neuronal loss in vitro and in vivo in AD (Cras et al., 1995; Honig and Rosenberg, 2000). Apoptosis is modulated by the B-cell lymphoma-2 (Bcl-2) family, that involve both pro- and anti- apoptotic members (Pellegrini and Strasser, 1999). Among them, the Bcl-2 protein can block apoptotic cell death and promote survival of neurons. Bcl-2 expression has been found significantly increased in AD temporal and frontal cortex, but not significantly increased in AD cerebellum (Engidawork et al., 2001; Kitamura et al., 1998). Bcl-2 has also been found to block oxidation of PtdSer (Fabisiak et al., 1997), implying oxidative modification of PtdSer or flippase is an essential ingredient to the apoptotic process. Another Bcl-2 family member, the Bcl-2-associated X protein (Bax), is believed to promote apoptosis. Both Bcl-2 and Bax have been found constitutively expressed in neurons of the central and peripheral nervous systems (Castren et al., 1994; Oltvai et al., 1993).
In addition, aspartate-specific cysteine proteases (caspases) function in both cell disassembly (effectors) and in initiating this disassembly in response to pro-apoptotic signals (initiators) (Wolf and Green, 1999). Activated caspase-3 immunoreactivity has been detected in both neurons and astrocytes and found elevated in frontotemporal dementia (Su et al., 2000) and AD (Su et al., 2001). PtdSer exposure due to apoptosis has been found to occur downstream of caspase activation in some cell types (Mandal et al., 2002; Martin et al., 1996; Vanags et al., 1996); in non-neuronal cells like human erythrocytes, caspase activation is associated with loss of aminophospholipid translocase activity and subsequent PtdSer externalization (Mandal et al., 2005).
Amnestic mild cognitive impairment (MCI) is considered a transition point between normal cognitive aging and dementia or probable AD (Petersen et al., 1999; Winblad et al., 2004). Brain samples from MCI subjects are reported to have increased oxidative stress (Butterfield et al., 2006b; 2007; Keller et al., 2005; Markesbery et al., 2005; Petersen et al., 1999) that can be associated with increased levels of reactive alkenals (Butterfield et al., 2006a). Unfortunately, not much is known about apoptotic processes with respect to MCI to date. However, one recent study ascertained that the effector caspase-6 was intimately linked to pathological hallmarks of AD in the hippocampus of persons with all stages of AD, including MCI and NCI (severely aged cognitively normal) (Albrecht et al., 2007). The present study investigated the hypothesis that PtdSer asymmetry is significantly altered in the inferior parietal lobule (IPL) of subjects with MCI and AD, which suggests that increased production of lipid peroxidation products (Butterfield et al., 2006a) and levels of apoptotic proteins may work together to affect PtdSer asymmetry even in MCI, arguably the earliest form of AD.
Materials and Methods
All chemicals used in lipid asymmetry studies were purchased from Sigma-Aldrich (St. Louis, MO, USA) with the exception of NBD-PS [1-palmitoyl-2-[6(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]caproyl-sn-glycero-3-phosphoserine], obtained from Avanti Polar Lipids (Alabaster, AL, USA). All apoptotic expression chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA), with exceptions of nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), electrophoretic transfer system (Trans-blot semi-dry Transfer Cell; Bio-Rad), anti-Bcl-2 monoclonal antibody (Stressgen Biotech, Ann Arbor, MI, USA), anti-Bax polyclonal antibody (Calbiochem, LA Jolla, CA, USA), anti-caspase-3 polyclonal antibody (Calbiochem, La Jolla, CA, USA), and anti-mouse IgG alkaline phosphatase secondary antibody (Chemicon International, Temulca, CA, USA).
Phospholipid Asymmetry Studies
Control, MCI, and AD brain
Frozen IPL samples from MCI, AD, and age-matched controls were obtained from the University of Kentucky Rapid Autopsy Program of the Alzheimer Disease Clinical Center (UK ADC). All AD patients met NINCDS-ARDA Workgroup criteria for clinical diagnosis of probable AD (McKhann et al., 1984) and also displayed progressive intellectual decline. Control subjects were without history of dementia or other neurological disorders and underwent annual mental status testing and semi-annual physical and neurological exams as part of the UK ADC normal volunteer longitudinal aging study, and had test scores in the normal range (Table II). Samples and demographics used for the AD study were described previously (Sultana et al., 2006). Additional demographic parameters of controls and subjects with MCI and AD without personal identifiers were made available by the UK ADC and are provided in Table I and Table II.
Table II.
Characteristics of control and AD patients available from medical records.
| Parameters: Demographic Variables | Control Subjects | AD Subjects |
|---|---|---|
| Number of subjects | 5 | 5 |
| Gender (male/female) | 3/2 | 3/2 |
| Age at death (yrs)a | 87.0 ± 3.94 | 85.8 ± 6.02 |
| Postmortem interval (h)a | 2.9 ± 0.70 | 3.4 ± 1.4 |
| MMSE; number of months prior to death test takena | 28 ± 0.8; 6.6 ± 1.4 | 28 ± 0.8; 6.6 ± 1.4 |
| APOE genotype, if known (N) | 3/3 (3) 3/4 (2) | ND |
| Cause of death | surgery complications, cardiac failure; COPD | surgery complications, cardiac failure; COPD |
(mean ± SD); AD, Alzheimer Disease; MMSE, Mini-Mental State Examination; APOE, apolipoprotein E; ND, not determined; N, number of individuals; SD, standard deviation; COPD, chronic obstructive pulmonary disease [adapted from (Sultana et al., 2006)].
Table I.
Characteristics of control and MCI patients where brain was obtained.
| Parameters: Demographic Variables | Control Subjects | MCI Subjects |
|---|---|---|
| Number of subjects | 5 | 5 |
| Gender (male/female) | 2/3 | 2/3 |
| Age at death (yrs)a | 82 ± 2.6 | 88 ± 1.5 |
| Postmortem Interval (h)a | 2.87 ± 1.14 | 3.125 ± 1.033 |
| Brain Weight (g)a | 1260 ± 120 | 1120 ± 61 |
| Braak Stage | I–II | III–V |
(mean ± SD)
Synaptosome Preparation
Synaptosome fractions from MCI and AD IPL were isolated as described previously (Keller et al., 2000; Mohmmad Abdul and Butterfield, 2005) with modifications. In brief, fractions were homogenized in an isolation buffer (0.32 M sucrose, 0.2 mM PMSF, 2 mM EDTA, 2 mM EGTA, 20 mM HEPES, 4 µg/ml leupeptin, 4 µg/ml pepstatin, 5 µg/ml aprotinin, and 20 µg/ml trypsin inhibitor). The homogenate was then centrifuged at 4000 rpm (1940 × g) for 10 min at 0°C and supernatant (S1) decanted. The pellet was rehomogenized in isolation buffer and centrifuged again at 4000 rpm (1940 × g) for 10 min at 0°C. The second pellet was discarded, while the supernatant (S2) was mixed with S1. The combined supernatants (S1+S2) were centrifuged at 14,800 rpm (25,400 × g) for 12 min at 0°C. The resulting pellet was mixed with a small volume of cold isolation buffer and layered onto cold discontinuous sucrose gradients containing 10 ml each of 1.18 M (pH 8.5), 1.0 M (pH 8.0), and 0.85 M (pH 8.0) sucrose, as well as 2 mM EDTA, 2 mM EGTA, and 10 mM HEPES. Gradients were centrifuged in a Beckman-Coulter Optima L-90K ultracentrifuge at 22,000 rpm (82,500 × g) for 1 h at 4°C. Resulting purified synaptosomes were removed from the 1.18/1.0 M interface and washed three times in Locke’s buffer (pH 7.4) at 15,500 rpm (29,100 × g) for 15 min at 0°C. Protein concentrations were determined according to the Pierce BCA method (Pierce, Rockford, IL, USA).
NBD-PS assay
This assay was completed as previously described (Comfurius et al., 1996; Mohmmad Abdul and Butterfield, 2005) to study the externalization of PtdSer. Synaptosome samples (200 µg) were covered and incubated for 1 h with NBD-PS, then washed twice in Locke’s buffer (pH 7.4). After resuspension in 200 µl Locke’s buffer (pH 7.4), samples were loaded onto a fluorescence plate and treated with 7.5 mM sodium dithionite (Na2S2O4) to quench fluorescence by reduction of the exposed, NBD labeled PtdSer residues. Loss of residual fluorescence was measured in a Molecular Devices SpectraMAX Gemini fluorescence plate reader (wavelengths of Ex/Em: 460/514 nm).
Mouse Brain Tissue
Post-Mortem Intervals
All animals were euthanized with CO2 in a chamber. At death, six FVB mice were placed in a Styrofoam box containing two 710 ml bottles of water at ~37°C. This delayed the postmortem cooling of the mouse brain in order to more closely approximate the human brain cooling curve (Geddes et al., 1986; Schwab et al., 1994). Postmortem intervals (PMIs) used were chosen based on the minimum and maximum human PMIs obtained from the UK ADC found in Table I and Table II. After PMIs of 2.8 h and 3.4 h, mouse brains were removed and frozen at −80°C. Three additional mouse brains were rapidly removed after death to represent a 0 h PMI (i.e., fresh samples), and synaptosomes processed directly alongside thawed 2.8 h (N=3) and 3.4 h (N=3) brain samples, according to the synaptosome preparation procedure described above.
Mg2+ ATPase activity
This assay was completed according to previous procedures using FVB mice (Castegna et al., 2004; Sadrzadeh et al., 1993). Synaptosome aliquots (7.5 µg) were suspended in ATPase assay buffer (18 mM histidine, 18 mM imidazole, 80 mM NaCl, 15 mM KCl, 3 mM MgCl2, 0.1 mM EGTA, pH 7.1) and treated with 0.1 mM of the Na+K+ATPase inhibitor ouabain to a final volume of 100 µl in a microtiter plate. After a 10 min incubation at 37°C, 3 mM ATP was added, and the reaction allowed to proceed for 60 min at 37°C. Then, 5 µl of 5% SDS and 125 µL of color reagent (0.36 g ascorbic acid mixed with 15 ml of molybdate acid solution) were added to each well. The activity of Mg2+ ATPase was measured with a UV plate reader as the amount of phosphate produced by the enzyme (Sadrzadeh et al., 1993).
Apoptotic Protein Expression Studies
Sample preparation
Samples were prepared as described previously (Butterfield et al., 2006b), with exceptions. These samples from control, MCI, and AD IPL were sonicated in lysis buffer (pH 7.4) containing 10 mM HEPES buffer, 137 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4, and 0.6 mM MgSO4, as well as proteinase inhibitors leupeptin (0.5 mg/mL), pepstatin (0.7 mg/mL), type II S soybean trypsin inhibitor (0.5 mg/mL), and PMSF (40 mg/mL). Homogenates were centrifuged at 14,000 × g for 10 min to remove debris. Protein concentration in the supernatant was determined by the Pierce BCA method (Pierce, Rockford, IL, USA).
Western Blotting
For immunoblot analysis, samples were separated by 12.5% SDS-polyacrylamide gel electrophoresis (PAGE). Equal amounts of protein (75 µg) were loaded in each lane with loading buffer containing 0.5 M Tris (pH 6.8), 40% glycerol, 8% SDS, 20% β-mercaptoethanol, and 0.01% bromophenol blue. Samples were heated at 95 °C for 10 min prior to gel loading. Proteins separated by gel electrophoresis were transferred to nitrocellulose membranes using an electrophoretic transfer system (Bio-Rad Trans-blot semi-dry Transfer Cell) at 45 mA for 2 h. After, the membranes were blocked with 5% non-fat dried milk in phosphate-buffered saline containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 (PBST) at 4°C for 1 h.
Following this procedure, membranes were incubated for 2 h at room temperature with primary antibodies: anti-Bcl-2 monoclonal antibody (1:1000), anti-Bax polyclonal antibody (1:1000), and anti-caspase-3 polyclonal antibody (1:2000). After washing the blots three times in PBST for 5 min each, the blots were incubated with anti-rabbit or anti-mouse IgG alkaline phosphatase secondary antibody (1:3000) in PBST for 1 h at room temperature. Membranes were then washed in PBST three times for 5 min and developed using 5-Bromo-4-chloro-3-indolyl-phosphate/Nitroblue tetrazolium (BCIP/NBT) color developing reagent. Blots were dried and scanned with Adobe Photoshop and quantitated with Scion Image (PC version of Macintosh-compatible NIH Image) software.
Statistics
All statistical analysis were performed using a two-tailed Student’s t-test. P<0.05 was considered significantly different from control.
Results
Detection of PtdSer asymmetry in MCI and AD IPL synaptosomes by NBD-PS assay
Loss of lipid asymmetry is marked by the exposure of PtdSer to the plasma membrane surface following lipid peroxidation (Mohmmad Abdul and Butterfield, 2005). After pretreatment of synaptosomes with NBD-PS, a PtdSer with a fluorescent tag, exposed fluorescence was quenched by chemical modification of the fluorophore employing sodium dithionite (Na2S2O4), resulting in a decrease in fluorescence for samples compared to control. This method had a value-added characteristic of addressing potential concerns with probe access to the inner leaflet of an unstable system creating incorrect interpretation of asymmetric collapse, as Na2S2O4 does not readily diffuse through the plasma membrane (Castegna et al., 2004; Mohmmad Abdul and Butterfield, 2005). Pretreatment with NBD-PS suggested significant exposure of PtdSer in brain from subjects with MCI (Fig. 1; **P<0.00001) and AD (Fig. 1; *P<0.0001) compared to that of controls.
Figure 1. NBD-PS assay in synaptosomes from brain of subjects with MCI and AD.

HumanMCI, AD, and control synaptosomes were treated with the fluorescent phospholipid NBD-PS. PtdSer exposure to the outer leaflet of the membrane bilayer is measured as the percent decrease in fluorescence signal after Na2S2O4 quenching. The control value was set to 100%, to which experimental values were compared. These data, in arbitrary units on the ordinate axis, are presented as mean ±S.D. AD, N=5, *P<0.0001; MCI, N=5, **P<0.00001.
Detection of PtdSer asymmetry as a function of PMI in mouse synaptosomes by NBD-PS assay
For these assays, exposed NBD-PS fluorescence was quenched by chemical modification of the fluorophore by Na2S2O4. However, as this study was focused on the effects of postmortem intervals, in addition to freeze-thawing, 2.8 h and 3.4 h samples obtained from mice held at ~37°C were compared to fresh (0 h) samples. Therefore, data were not analyzed as a percent decrease from control rather the average fluorescence signal of each sample set was measured, in arbitrary units, and compared to the average 0 h signal. Post-mortem autopsy of patients from the UK ADC usually occurs just hours after death in order to preserve brain tissue from further oxidation and degradation; however, during the time between death and tissue extraction, it is possible that significant alteration of tissue could occur influencing results. In addition, during the freeze-thawing process of samples post-mortem, freeze-fracturing of the cell membrane conceivably can occur, potentially causing a significant increase of PtdSer exposure to the outer leaflet that could artifactually alter results.
In order to address these potential difficulties, synaptosomes pretreated with the fluorescent phospholipid NBD-PS suggested no significant PtdSer exposure difference between 0 h, 2.8 h, and 3.4 h samples (Fig. 2; 2.8 h, P<0.65; 3.4 h, P<0.80). During synaptosome preparation, one sample of the 2.8 h PMI was irretrievably lost, reducing the sample number to N=2. The results of this investigation suggest that significant results observed in MCI and AD samples were presumably from oxidative processes brought about by development of MCI or AD, and not artifactual in nature.
Figure 2. PMI NBD-PS assay.

FVB mouse synaptosomes were treated with the fluorescent phospholipid NBD-PS. PtdSer exposure to the outer leaflet of the plasma membrane is measured as a decrease in fluorescence signal, compared to 0 h, after quenching with Na2S2O4. Since samples were not compared as a percentage of control, negative values are not seen, but graphical interpretation is relatively the same. A fluorescence decrease, compared to 0 h, denotes an increased amount of PtdSer exposed to the lipid bilayer outer leaflet. Data shown in arbitrary fluorescence units on the ordinate axis as mean ±S.D. 0 h, N=3; 2.8 h, N=2, P<0.65; 3.4 h, N=3, P<0.80.
Measurement of Mg2+ ATPase activity in mouse synaptosomes
Determination of Mg2+ ATPase activity in sample sets of the PMI study was completed as a representative of the membrane-bound ATP-requiring enzyme flippase, as highly specific antibodies to flippase have not yet been obtained (Auland et al., 1994). This indirect measure of activity was performed by measurement of the net amount of inorganic phosphate (Pi) formed (Sadrzadeh et al., 1993). Decreased UV-absorbance (810 nm) of 2.8 h and 3.4 h PMI sample sets compared to 0 h PMI samples denotes decreased Mg2+ATPase activity (Fig. 3; 2.8 h, *P<0.04; 3.5 h, P<0.26).
Figure 3. Mg2+ATPase activity assay.

FVB mouse synaptosome Mg2+ATPase activity was measured to represent flippase activity, an ATP-dependent membrane bound translocase. Decreased UV-absorbance (810 nm) compared to 0 h denotes decreased Mg2+ATPase activity. Data shown, in arbitrary absorbance units, as mean ±S.D. 0 h, N=3; 2.8 h, N=2, *P<0.04; 3.5 h, N=3, P<0.26.
Bcl-2 levels in MCI and AD IPL
The expression of anti-apoptotic protein Bcl-2 was measured by Western blot analysis in MCI and AD brain compared with controls. Equal amounts of protein extracts from IPL were separated by SDS-PAGE and transferred to a nitrocellulose membrane. In both MCI and AD samples, a 26 kDa Bcl-2 band was probed with a monoclonal anti-Bcl-2 antibody (Fig. 4a). Equivalent levels of β-tubulin were detected to normalize the amount of protein loaded in each well of the gel (Fig. 4a). Quantification was completed by densitometric scanning with a phosphoimager (Molecular Dynamics, Sunnyvale, CA, USA), and analysis showed that the level of Bcl-2 was significantly increased in both MCI (Fig. 4b; *P<0.006) and AD (Fig. 4c; *P<0.04).
Figure 4. Bcl-2 levels in brain from subjects with MCI and AD.


Bcl-2 levels were measured in IPL of MCI and AD patients by Western blot analysis. (a) MCI and AD gels were run with equal amounts of protein (75 µg/lane). Increased band intensity after normalization with β-tubulin represents an increase in the amount of Bcl-2 protein present. (b) & (c) Graphical analysis of MCI and AD band intensities, respectively. The control value was set to 100%, to which experimental values were compared. Data shown in arbitrary units on the ordinate axis as mean ±S.D. in (a) MCI, N=3, *P<0.006; in (b) AD N=3, *P<0.04.
Bax levels in MCI and AD IPL
The levels of the pro-apoptotic protein Bax in IPL were measured using Western blot analysis in brain from both MCI and AD compared to controls. After separation of proteins by SDS-PAGE and transfer to a nitrocellulose membrane, the polyclonal antibody anti-Bax was used to probe the immunoblots. The antibody recognized a band at 21 kDa for both MCI and AD samples (Fig. 5a). Again, β-tubulin was used to normalize the blot (Fig. 5a). Analysis shows Bax levels significantly increased in MCI samples compared to control (Fig. 5b; *P<0.03), but significantly decreased in AD (Fig. 5c; *P<0.005). The ratios of Bax to Bcl-2 in MCI and AD were 2.38 and 0.23, respectively.
Figure 5. Bax levels in brain from subjects with MCI and AD.


Bax levels were measured in IPL of MCI and AD patients by Western blot analysis. (a) MCI and AD gels were run with equal amounts of protein (75 µg/lane). Increased band intensity after normalization with β-tubulin represents an increase in the amount of Bax protein present. (b) & (c) Graphical analysis of MCI and AD band intensities, respectively. The control value was set to 100%, to which experimental values were compared. Data shown in arbitrary units on the ordinate axis as mean ±S.D. in (a) MCI, N=3, *P<0.03; in (b) AD N=3, *P<0.005.
Caspase-3 levels in MCI and AD IPL
Caspase-3 is synthesized as an inactive precursor (34 kDa), which upon receiving an apoptotic signal, is activated and subsequently cleaved into two subunits (20 kDa and 12 kDa). Equal amounts of protein from MCI, AD, and control were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. The antibody anti-caspase-3 was used to recognize and probe both precursor and activated caspase fragments on the immunoblots; β-tubulin was used to normalize (Fig. 6a). Only the band relative to the caspase-3 precursor was recognized, while bands relative to activated fragments were not detected (Fig. 6a). Quantification of immunoreactivity revealed a significant increase of caspase-3 precursor levels in both MCI (Fig 6b; *P<0.02) and AD (Fig. 6c; *P<0.04) compared to controls.
Figure 6. Caspase-3 levles in brain from subjects with MCI and AD.
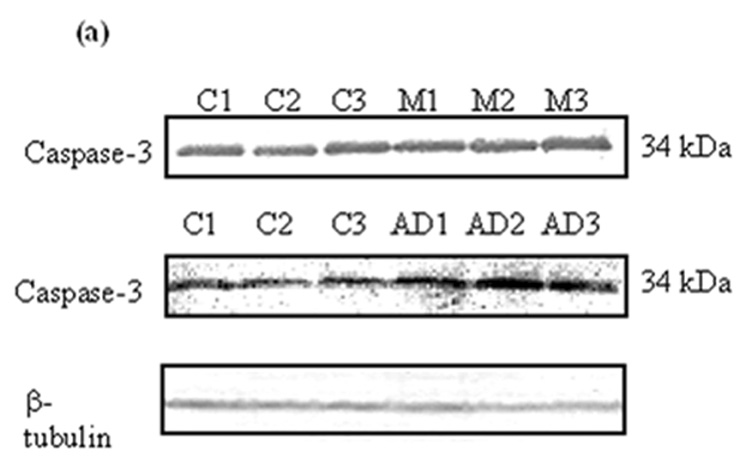
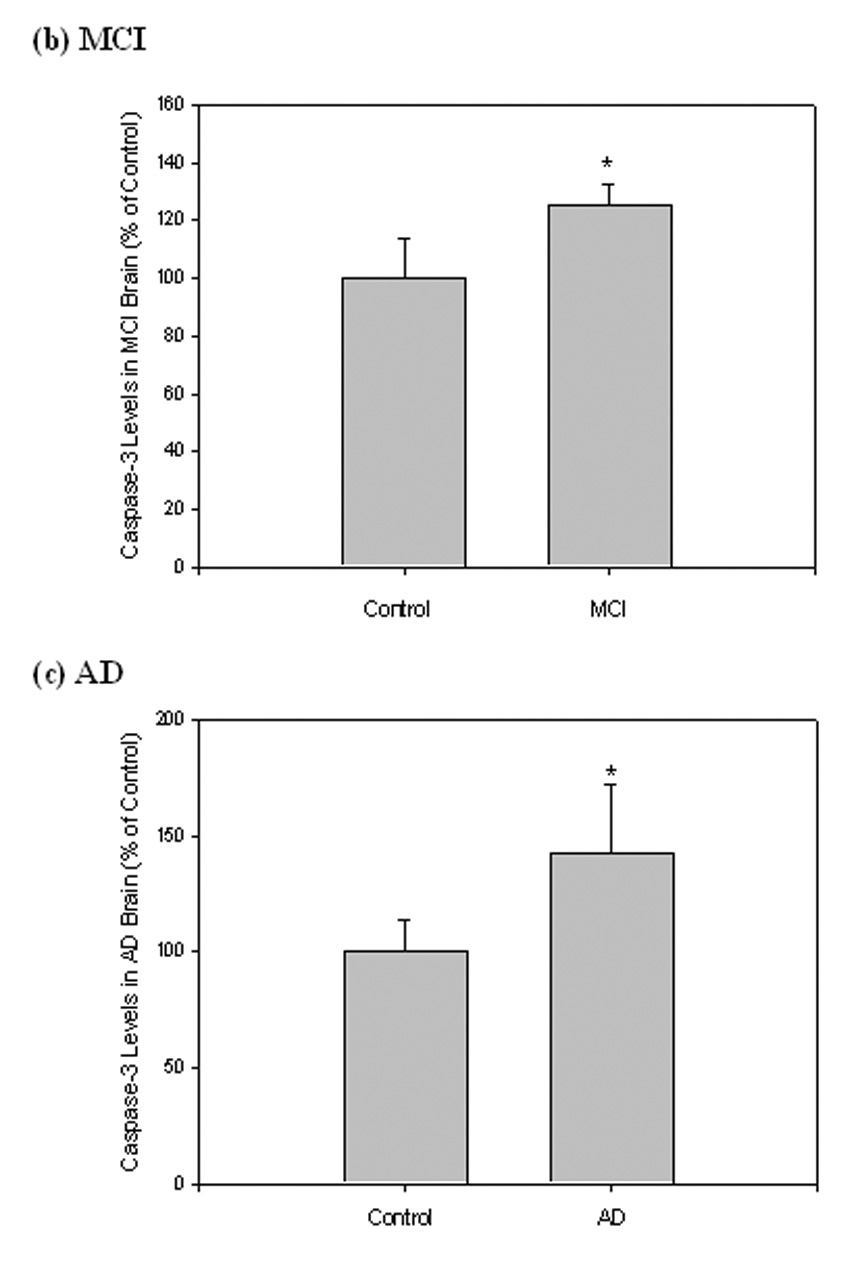
Caspase-3 levels were measured in IPL of MCI and AD patients by Western blot analysis. (a) MCI and AD gels were run with equal amounts of protein (75 µg/lane). Increased band intensity after normalization with β-tubulin represents an increase in the amount of caspase-3 present. (b) & (c) Graphical analysis of MCI and AD band intensities, respectively. The control value was set to 100%, to which experimental values were compared. Data shown in arbitrary units on the ordinate axis as mean ±S.D. in (a) MCI, N=3, *P<0.02; in (b) AD N=3, *P<0.04.
Discussion
The present study investigated synaptosomal PtdSer asymmetry by a fluorescent assay, and its relation to apoptotic protein levels, in order to determine the role of apoptosis-specific oxidation of PtdSer and/or flippase in the progression of MCI into AD. NBD-PS assay results demonstrate a significant fluorescence decrease in MCI and AD samples (Fig. 1) compared to control, suggesting that PtdSer externalization could be an important contributor to neurodegeneration found in AD brain and, notably, a potential link to those patients with MCI that may eventually develop AD.
A central path contributing to elevated exposure of PtdSer on the outer leaflet of synaptic membranes in MCI and AD begins with oxidative stress, predominant in both AD and MCI (Butterfield et al., 2001; 2002; Butterfield and Lauderback, 2002; Butterfield et al., 2006b; 2007; Lovell et al., 2001; Markesbery and Lovell, 1998; Markesbery et al., 2005). Oxidative conditions can contribute to lipid peroxidation products, HNE and acrolein, within the bilayer of apoptotic cells (Butterfield et al., 2006a; Lovell et al., 2001; Markesbery and Lovell, 1998) that could interfere with PtdSer asymmetry by initiating further lipid peroxidation via free radical mechanisms (Prasad et al., 1998). By diffusing from their formation sites, these neurotoxic alkenals could react via Michael addition with flippase by covalently binding a critical cysteine residue of this transporter (Daleke and Lyles, 2000; Daleke, 2003; Paulusma and Oude Elferink, 2005; Tyurina et al., 2004a), suggesting a definite relation between PtdSer asymmetry and lipid peroxidation.
Likewise, oxidative stress conditions can also initiate early apoptosis, known to play an important role in neuronal loss in vitro and in vivo in AD (Cotman and Anderson, 1995; Cras et al., 1995; Honig and Rosenberg, 2000; MacGibbon et al., 1997; Smale et al., 1995); however, not much is known about apoptotic pathways in MCI. Several studies have found increased Bcl-2 expression in the hippocampus and entorhinal cortex of AD brains (Satou et al., 1995; Su et al., 1996). Western blot analysis conducted in the IPL shows Bcl-2 basal levels were significantly increased in AD brain (Fig. 5a,c), congruent with previous studies, and for the first time in MCI brain (Fig. 5a,b). The up-regulation of anti-apoptotic Bcl-2 levels can be rationalized as a cellular compensatory response in an attempt to protect the remaining neurons from cell death. Since PtdSer externalization during apoptosis has been previously reported to be blocked by Bcl-2 (Fabisiak et al., 1997), it is likely such a response is an attempt to block oxidative modification of PtdSer and/or flippase, reinforcing the notion that PtdSer exposure is required for apoptotic-cell phagocytosis. Indeed, earlier studies showed that cells with Bcl-2 overexpressed were refractive to Aβ-induced lipid peroxidation (Bruce-Keller et al., 1998).
The pro-apoptotic protein Bax is known to compete with Bcl-2 as increased levels of Bax can induce cell death, while increased levels of Bcl-2 prevent it (Oltvai et al., 1993). In many regions of AD brain such as the hippocampus, entorhinal cortex, frontal and temporal cortices, Bax immunoreactivity has been found to be increased, with the exception of dentate granule hippocampal cells (Giannakopoulos et al., 1999; Nagy and Esiri, 1997; Tortosa et al., 1998). In the current study, MCI samples showed Bax levels significantly increased compared to control, as expected (Fig. 5a,b). By contrast, however, we found a significant decrease in Bax levels in AD IPL compared to control (Fig. 5a,c). These results suggest Bax is involved in neurodegeneration during the earliest stages of AD, while later in the disease progression, attenuation of Bax may be related to the relative resistance of neurons against apoptosis, as evidenced by higher Bcl-2 levels in AD samples compared to MCI. This is consistent with the notion that in AD IPL the pro-apoptotic system is progressively more neutralized by the anti-apoptotic system causing a substantial reduction of Bax.
Additionally, caspases are critically involved in PtdSer externalization and apoptosis (Mandal et al., 2002; Nicholson, 1999). Activated caspase-3 is found only in cells undergoing apoptosis, and consists of p18 (amino acids 29–175; 20 kDa) and p12 subunits (amino acids 176–277; 12 kDa) (Nicholson et al., 1995), which are derived from the 32 kDa pro-enzyme (pro-caspase-3). The antibody used in this study should have recognized both precursor and active fragments if present; however, results only show an increase in precursor caspase-3 (Fig. 6a), while no signal correlated to either activated fragment in MCI or AD samples. This is most likely an indicator that levels of key components of the apoptotic system are altered and may be the initial cause of neuronal loss. Unfortunately, the relative contribution of apoptosis to neuronal loss in AD is difficult to assess due to the chronic nature of the process. Some neurons exhibit morphological features of apoptosis, but many degenerating neurons do not show evidence of apoptosis, indicating apoptosis is not the only mechanism of neurodegeneration in AD (Su et al., 1994), or even MCI.
The process of PtdSer externalization leading to asymmetric collapse and apoptosis under oxidative stress conditions is relatively dependent on the activation of caspase-3, which is also associated with impairment of flippase activity (Mandal et al., 2002; Martin et al., 1996; Vanags et al., 1996). Previous studies have shown that excessive outer leaflet exposure of PtdSer on erythrocytes can be blocked comparatively by pretreatment with a caspase-3 inhibitor (Mandal et al., 2002). Additionally, loss of flippase activity was blocked after pretreatment with the same caspase-3 inhibitor prior to oxidation (Mandal et al., 2002). Such findings imply PtdSer oxidation and subsequent exposure is an intrinsic feature of the apoptotic process due to flippase inactivation (Kagan et al., 2002).
Other considerations could conceivably contribute to the observed loss of phospholipid asymmetry in MCI and AD brain as in the current study. Flippase is known to be sensitive towards the cellular influx of Ca2+ in such a way that its translocating activity is significantly affected at [Ca2+]i of 0.2 µM or higher (Bitbol et al., 1987; Daleke, 2003). HNE and acrolein have also been found to disrupt Ca2+ homeostasis, again interfering with PtdSer asymmetry (Castegna et al., 2004; Mohmmad Abdul and Butterfield, 2005). Even though elevated Ca2+ levels may contribute to asymmetric collapse, it is doubtful elevated Ca2+ is the chief contributor (Castegna et al., 2004; Mohmmad Abdul and Butterfield, 2005). Furthermore, changes in membrane cholesterol also are observed in response to oxidative stress, elevated levels of amyloid-beta protein (Aβ), and aging (Lopez-Revuelta et al., 2007; Wood et al., 2002; Wood et al., 2003).
As a physiochemical modulator of the plasma membrane, cholesterol has been found to affect many membrane functions, including susceptibility to oxidative stress (Lopez-Revuelta et al., 2007), control of Aβ synthesis, and interactions between Aβ and neuronal membranes (Eckert et al., 2003). One study suggests cholesterol depletion promotes PtdSer externalization induced by oxidative stress associated with significant inhibition of flippase activity and an increased uptake of altered cells by macrophages (Lopez-Revuelta et al., 2007). Moreover, oxidative stress and neurodegeneration are known to be induced in vivo by Aβ (Mohmmad Abdul and Butterfield, 2005; Mohmmad Abdul et al., 2006). Proteolytic cleavage of the amyloid precursor protein (APP) at the amino- and carboxy- termini by β-secretase and γ-secretase, respectively, produces Aβ(1–42) peptide within the lipid membrane. Aβ(1–42) is known to elicit neurotoxic effects by inserting into the lipid bilayer as small, soluble aggregates (Drake et al., 2003; Klein, 2006).
Loss of membrane phospholipid asymmetry is also reported to be caused by Aβ, a process that can be inhibited by antioxidants (Mohmmad Abdul and Butterfield, 2005). Thus, cholesterol asymmetry and Aβ-induced oxidative stress conceivably could contribute to the loss of PtdSer asymmetry observed in MCI and AD brain in the present study. However, it is important to note that although there are many parameters contributing to asymmetric collapse, all studies converge on the premise that inactivation of flippase via oxidative processes is a central cause.
Abnormal distribution of aminophospholipids has been reported in AD brain (Prasad et al., 1998). Our results are consistent with the notion that oxidative modification of PtdSer and/or flippase occurred not only in human AD brain, but also MCI brain, causing a significant increase in outer leaflet exposure of PtdSer and cell death. Findings suggest oxidative stress increases production of lipid peroxidation products (Butterfield et al., 2006a) and levels of apoptosis-related proteins that affect PtdSer asymmetry thereby causing neuronal cell death as early as MCI.
This is the first investigation of MCI phospholipid asymmetry and its relation to apoptotic factor expression. By looking at both disease stages, the model of apoptosis-specific oxidation of phospholipids confirms that PtdSer exposure is required for apoptotic-cell phagocytosis. Furthermore, it is considered essential that flippase activity be highly maintained in order to prevent premature PtdSer exposure, early apoptosis, and subsequent excessive neurodegeneration. Once specific antibodies to flippase become available, it will be more feasible to directly examine this aminophospholipid translocase and its response to oxidation phenomena apart from PtdSer.
Acknowledgements
This work was supported in part by NIH grants [AG-05119; AG-10836].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am. J. Pathol. 2007;170:1200–1209. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auland ME, Roufogalis BD, Devaux PF, Zachowski A. Reconstitution of ATP-dependent aminophospholipid translocation in proteoliposomes. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10938–10942. doi: 10.1073/pnas.91.23.10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian K, Schroit AJ. Aminophospholipid asymmetry: A matter of life and death. Annu. Rev. Physiol. 2003;65:701–734. doi: 10.1146/annurev.physiol.65.092101.142459. [DOI] [PubMed] [Google Scholar]
- Bitbol M, Fellmann P, Zachowski A, Devaux PF. Ion regulation of phosphatidylserine and phosphatidylethanolamine outside-inside translocation in human erythrocytes. Biochim. Biophys. Acta. 1987;904:268–282. doi: 10.1016/0005-2736(87)90376-2. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Begley JG, Fu W, Butterfield DA, Bredesen DE, Hutchins JB, Hensley K, Mattson MP. Bcl-2 protects isolated plasma and mitochondrial membranes against lipid peroxidation induced by hydrogen peroxide and amyloid beta-peptide. J Neurochem. 1998;70:31–39. doi: 10.1046/j.1471-4159.1998.70010031.x. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid β-peptide. Trends Mol. Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid β-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol. Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006a;397:170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol. Dis. 2006b;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer's disease. Brain Res. 2007;1148:243–248. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castegna A, Lauderback CM, Mohmmad-Abdul H, Butterfield DA. Modulation of phospholipid asymmetry in synaptosomal membranes by the lipid peroxidation products, 4-hydroxynonenal and acrolein: implications for Alzheimer's disease. Brain Res. 2004;1004:193–197. doi: 10.1016/j.brainres.2004.01.036. [DOI] [PubMed] [Google Scholar]
- Castren E, Ohga Y, Berzaghi MP, Tzimagiorgis G, Thoenen H, Lindholm D. Bcl-2 messenger RNA is localized in neurons of the developing and adult rat brain. Neuroscience. 1994;61:165–177. doi: 10.1016/0306-4522(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Comfurius P, Williamson P, Smeets EF, Schlegel RA, Bevers EM, Zwaal RF. Reconstitution of phospholipid scramblase activity from human blood platelets. Biochemistry. 1996;35:7631–7634. doi: 10.1021/bi9606859. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Anderson AJ. A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol. Neurobiol. 1995;10:19–45. doi: 10.1007/BF02740836. [DOI] [PubMed] [Google Scholar]
- Cras P, Smith MA, Richey PL, Siedlak SL, Mulvihill P, Perry G. Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer disease. Acta Neuropathol. (Berl.) 1995;89:291–295. doi: 10.1007/BF00309621. [DOI] [PubMed] [Google Scholar]
- Daleke DL, Lyles JV. Identification and purification of aminophospholipid flippases. Biochim. Biophys. Acta. 2000;1486:108–127. doi: 10.1016/s1388-1981(00)00052-4. [DOI] [PubMed] [Google Scholar]
- Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J. Lipid Res. 2003;44:233–242. doi: 10.1194/jlr.R200019-JLR200. [DOI] [PubMed] [Google Scholar]
- Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid β-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol. Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- Eckert GP, Kirsch C, Leutz S, Wood WG, Muller WE. Cholesterol modulates amyloid β-peptide's membrane interactions. Pharmacopsychiatry. 2003;36 Suppl 2:S136–S143. doi: 10.1055/s-2003-43059. [DOI] [PubMed] [Google Scholar]
- Engidawork E, Gulesserian T, Seidl R, Cairns N, Lubec G. Expression of apoptosis related proteins in brains of patients with Alzheimer's disease. Neurosci. Lett. 2001;303:79–82. doi: 10.1016/s0304-3940(01)01618-4. [DOI] [PubMed] [Google Scholar]
- Fabisiak JP, Kagan VE, Ritov VB, Johnson DE, Lazo JS. Bcl-2 inhibits selective oxidation and externalization of phosphatidylserine during paraquat-induced apoptosis. Am. J. Physiol. 1997;272:C675–C684. doi: 10.1152/ajpcell.1997.272.2.C675. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 2001;276:1071–1077. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Chang-Chui H, Cooper SM, Lott IT, Cotman CW. Density and distribution of NMDA receptors in the human hippocampus in Alzheimer's disease. Brain Res. 1986;399:156–161. doi: 10.1016/0006-8993(86)90611-6. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Kovari E, Savioz A, de Bilbao F, Dubois-Dauphin M, Hof PR, Bouras C. Differential distribution of presenilin-1, Bax, and Bcl-X(L) in Alzheimer's disease and frontotemporal dementia. Acta Neuropathol. (Berl.) 1999;98:141–149. doi: 10.1007/s004010051062. [DOI] [PubMed] [Google Scholar]
- Herrmann A, Devaux PF. Alteration of the aminophospholipid translocase activity during in vivo and artificial aging of human erythrocytes. Biochim. Biophys. Acta. 1990;1027:41–46. doi: 10.1016/0005-2736(90)90045-p. [DOI] [PubMed] [Google Scholar]
- Honig LS, Rosenberg RN. Apoptosis and neurologic disease. Am. J. Med. 2000;108:317–330. doi: 10.1016/s0002-9343(00)00291-6. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Fabisiak JP, Shvedova AA, Tyurina YY, Tyurin VA, Schor NF, Kawai K. Oxidative signaling pathway for externalization of plasma membrane phosphatidylserine during apoptosis. FEBS Lett. 2000;477:1–7. doi: 10.1016/s0014-5793(00)01707-5. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Gleiss B, Tyurina YY, Tyurin VA, Elenstrom-Magnusson C, Liu SX, Serinkan FB, Arroyo A, Chandra J, Orrenius S, Fadeel B. A role for oxidative stress in apoptosis: oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing Fas-mediated apoptosis. J. Immunol. 2002;169:487–499. doi: 10.4049/jimmunol.169.1.487. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Borisenko GG, Serinkan BF, Tyurina YY, Tyurin VA, Jiang J, Liu SX, Shvedova AA, Fabisiak JP, Uthaisang W, Fadeel B. Appetizing rancidity of apoptotic cells for macrophages: oxidation, externalization, and recognition of phosphatidylserine. Am. J. Physiol. Lung Cell Mol. Physiol. 2003;285:L1–L17. doi: 10.1152/ajplung.00365.2002. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J. Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Kamoshima W, Ota T, Matsuoka Y, Nomura Y, Smith MA, Perry G, Whitehouse PJ, Taniguchi T. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer's disease. Brain Res. 1998;780:260–269. doi: 10.1016/s0006-8993(97)01202-x. [DOI] [PubMed] [Google Scholar]
- Klein WL. Synaptic targeting by Aβ oligomers (ADDLS) as a basis for memory loss in early Alzheimer’s disease. Alzheimer's & Dementia. 2006;2:43–55. doi: 10.1016/j.jalz.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Lopez-Revuelta A, Sanchez-Gallego JI, Garcia-Montero AC, Hernandez-Hernandez A, Sanchez-Yague J, Llanillo M. Membrane cholesterol in the regulation of aminophospholipid asymmetry and phagocytosis in oxidized erythrocytes. Free Radic. Biol. Med. 2007;42:1106–1118. doi: 10.1016/j.freeradbiomed.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures. Neurobiol. Aging. 2001;22:187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- MacGibbon GA, Lawlor PA, Sirimanne ES, Walton MR, Connor B, Young D, Williams C, Gluckman P, Faull RL, Hughes P, Dragunow M. Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimer's disease hippocampus. Brain Res. 1997;750:223–234. doi: 10.1016/s0006-8993(96)01351-0. [DOI] [PubMed] [Google Scholar]
- Mandal D, Moitra PK, Saha S, Basu J. Caspase 3 regulates phosphatidylserine externalization and phagocytosis of oxidatively stressed erythrocytes. FEBS Lett. 2002;513:184–188. doi: 10.1016/s0014-5793(02)02294-9. [DOI] [PubMed] [Google Scholar]
- Mandal D, Mazumder A, Das P, Kundu M, Basu J. Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J. Biol. Chem. 2005;280:39460–39467. doi: 10.1074/jbc.M506928200. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Lovell MA. 4-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol. Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann. Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Finucane DM, Amarante-Mendes GP, O'Brien GA, Green DR. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J. Biol. Chem. 1996;271:28753–28756. doi: 10.1074/jbc.271.46.28753. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Butterfield DA. Protection against amyloid β-peptide (1–42)-induced loss of phospholipid asymmetry in synaptosomal membranes by tricyclodecan-9-xanthogenate (D609) and ferulic acid ethyl ester (FAEE): implications for Alzheimer's disease. Biochim. Biophys. Acta. 2005;1741:140–148. doi: 10.1016/j.bbadis.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid β-peptide (1–42), H2O2 and kainic acid: implications for Alzheimer's disease. J. Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- Nagy ZS, Esiri MM. Apoptosis-related protein expression in the hippocampus in Alzheimer's disease. Neurobiol. Aging. 1997;18:565–571. doi: 10.1016/s0197-4580(97)00157-7. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- Paulusma CC, Oude Elferink RP. The type 4 subfamily of P-type ATPases, putative aminophospholipid translocases with a role in human disease. Biochim. Biophys. Acta. 2005;1741:11–24. doi: 10.1016/j.bbadis.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Strasser A. A portrait of the Bcl-2 protein family: life, death, and the whole picture. J. Clin. Immunol. 1999;19:365–377. doi: 10.1023/a:1020598632068. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch. Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer's disease. Neurochem. Res. 1998;23:81–88. doi: 10.1023/a:1022457605436. [DOI] [PubMed] [Google Scholar]
- Sadrzadeh SM, Vincenzi FF, Hinds TR. Simultaneous measurement of multiple membrane ATPases in microtiter plates. J. Pharmacol. Toxicol. Methods. 1993;30:103–110. doi: 10.1016/1056-8719(93)90013-5. [DOI] [PubMed] [Google Scholar]
- Satou T, Cummings BJ, Cotman CW. Immunoreactivity for Bcl-2 protein within neurons in the Alzheimer's disease brain increases with disease severity. Brain Res. 1995;697:35–43. doi: 10.1016/0006-8993(95)00748-f. [DOI] [PubMed] [Google Scholar]
- Schwab C, Bondada V, Sparks DL, Cahan LD, Geddes JW. Postmortem changes in the levels and localization of microtubule-associated proteins (tau, MAP2 and MAP1B) in the rat and human hippocampus. Hippocampus. 1994;4:210–225. doi: 10.1002/hipo.450040212. [DOI] [PubMed] [Google Scholar]
- Shimohama S. Apoptosis in Alzheimer's disease--an update. Apoptosis. 2000;5:9–16. doi: 10.1023/a:1009625323388. [DOI] [PubMed] [Google Scholar]
- Smale G, Nichols NR, Brady DR, Finch CE, Horton WE., Jr Evidence for apoptotic cell death in Alzheimer's disease. Exp. Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer's disease. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- Su JH, Satou T, Anderson AJ, Cotman CW. Up-regulation of Bcl-2 is associated with neuronal DNA damage in Alzheimer's disease. NeuroReport. 1996;7:437–440. doi: 10.1097/00001756-199601310-00015. [DOI] [PubMed] [Google Scholar]
- Su JH, Nichol KE, Sitch T, Sheu P, Chubb C, Miller BL, Tomaselli KJ, Kim RC, Cotman CW. DNA damage and activated caspase-3 expression in neurons and astrocytes: evidence for apoptosis in frontotemporal dementia. Exp. Neurol. 2000;163:9–19. doi: 10.1006/exnr.2000.7340. [DOI] [PubMed] [Google Scholar]
- Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW. Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res. 2001;898:350–357. doi: 10.1016/s0006-8993(01)02018-2. [DOI] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Tang X, Halleck MS, Schlegel RA, Williamson P. A subfamily of P-type ATPases with aminophospholipid transporting activity. Science. 1996;272:1495–1497. doi: 10.1126/science.272.5267.1495. [DOI] [PubMed] [Google Scholar]
- Tortosa A, Lopez E, Ferrer I. Bcl-2 and Bax protein expression in Alzheimer's disease. Acta Neuropathol. (Berl.) 1998;95:407–412. doi: 10.1007/s004010050817. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Serinkan FB, Tyurin VA, Kini V, Yalowich JC, Schroit AJ, Fadeel B, Kagan VE. Lipid antioxidant, etoposide, inhibits phosphatidylserine externalization and macrophage clearance of apoptotic cells by preventing phosphatidylserine oxidation. J. Biol. Chem. 2004a;279:6056–6064. doi: 10.1074/jbc.M309929200. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Tyurin VA, Zhao Q, Djukic M, Quinn PJ, Pitt BR, Kagan VE. Oxidation of phosphatidylserine: a mechanism for plasma membrane phospholipid scrambling during apoptosis? Biochem. Biophys. Res. Commun. 2004b;324:1059–1064. doi: 10.1016/j.bbrc.2004.09.102. [DOI] [PubMed] [Google Scholar]
- Vanags DM, Porn-Ares MI, Coppola S, Burgess DH, Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J. Biol. Chem. 1996;271:31075–31085. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- Verkleij AJ, Post JA. Membrane phospholipid asymmetry and signal transduction. J. Membr. Biol. 2000;178:1–10. doi: 10.1007/s002320010009. [DOI] [PubMed] [Google Scholar]
- Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, Nordberg A, Backman L, Albert M, Almkvist O, Arai H, Basun H, Blennow K, de Leon M, DeCarli C, Erkinjuntti T, Giacobini E, Graff C, Hardy J, Jack C, Jorm A, Ritchie K, van Duijn C, Visser P, Petersen RC. Mild cognitive impairment--beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J. Intern. Med. 2004;256:240–246. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J. Biol. Chem. 1999;274:20049–20052. doi: 10.1074/jbc.274.29.20049. [DOI] [PubMed] [Google Scholar]
- Wood WG, Schroeder F, Igbavboa U, Avdulov NA, Chochina SV. Brain membrane cholesterol domains, aging and amyloid β-peptides. Neurobiol. Aging. 2002;23:685–694. doi: 10.1016/s0197-4580(02)00018-0. [DOI] [PubMed] [Google Scholar]
- Wood WG, Eckert GP, Igbavboa U, Muller WE. Amyloid β-protein interactions with membranes and cholesterol: causes or casualties of Alzheimer's disease. Biochim. Biophys. Acta. 2003;1610:281–290. doi: 10.1016/s0005-2736(03)00025-7. [DOI] [PubMed] [Google Scholar]