

Abstract
The mechanism by which a single mutant cell clonally expands is usually assumed to involve an additional mutation in a cell cycle regulatory gene. An alternative mechanism for driving clonal expansion is apoptosis, which might create vacant stem cell compartments that can be repopulated by mutant cells. This model predicts that in a mouse with reduced apoptotic capacity (i) more mutated cells will appear initially but (ii) these cells will expand into clones more slowly than in wild-type animals. To test this hypothesis for ultraviolet B (UVB)-induced skin carcinogenesis, we examined UVB-induced p53 mutant clones and tumors in a transgenic (Tg) mouse (K14-Survivin) with skin-specific expression of the apoptosis inhibitor Survivin. To limit the effects of Survivin on apoptosis, without affecting epidermal proliferation or differentiation, we used Survivin expression levels and UVB doses that resulted in a 2-fold reductioninkeratinocyte apoptosis. After 5 weeks of chronic UVB irradiation, newly created p53 mutant keratinocyte clones (indicative of initial mutation frequency) were 1.4-fold more frequent in K14-Survivin mice (P = 4 ×10−6). As predicted, this effect was reversed for clones growing by clonal expansion, which were rarer in Tg skin by 1.7-fold (P = 0.047). At 10 weeks large expanding Tg clones were rarer by a magnitude approaching the apoptosis differential (~2-fold, P = 4 ×10−5). Survivin expression also retarded clonal expansion at later stages of tumor development. By 20 weeks 95% of animals carried tumors (primarily papillomas), which were 1.6-fold rarer in apoptosis-defective Tg mice (P = 0.03). In contrast, the rate of tumors attaining large size (≥3 mm, P = 0.048) and converting to carcinoma was increased ~2-fold in Tg mice. Thus, Survivin-regulated apoptosis appears to suppress two stages that involve new mutations, initiation and malignant conversion, yet drives clonal expansion of existing p53 mutant cells.
Introduction
Apoptosis is conventionally viewed as a mechanism of cancer defense. It acts by opposing the first of two steps by which mutant cells emerge during tumor development—creation of a single mutant cell and expansion of this cell into a clone. In tissues exposed to chemical or physical carcinogens, apoptotic cell death removes cells with DNA damage that might have led to point mutations or chromosomal abnormalities (1–3). Once mutations arise, apoptosis also removes tumorigenic cells that are aberrantly proliferating due to overexpressed c-Myc or defective Rb (4). Tumors can evade apoptosis by acquiring suppressive mutations in oncogenes such as Ras or inactivating mutations in proapoptotic genes such as p53 (4) or by expressing inhibitor of apoptosis (IAP) proteins such as Survivin (5). When such suppression is removed, some tumors regress (6). Yet, once an apoptosis-resistant mutant has arisen, protective apoptosis could backfire by enhancing the clonal expansion step. Apoptosis will preferentially delete cells that retain normal apoptosis sensitivity while sparing apoptosis-resistant mutants, thereby acting as a selection pressure favoring clonal expansion of mutant cells (1,7). In this way, tumor generation could actually be increased by apoptosis if greater clonal expansion offsets the reduced mutation frequency.
Non-melanoma skin cancers, such as squamous cell carcinoma (SCC), are the most common of human cancers and are caused primarily by chronic exposure to ultraviolet radiation in the 290–320 nm range (UVB) (8). The sequence of carcinogenic events includes UVB-induced p53 mutation, formation and expansion of p53 mutant clones, creation of precancers and malignant conversion of precancers to carcinoma in situ and SCC (1,8). It is known that UVB-induced epidermal apoptosis minimizes the accumulation of mutant epidermal keratinocytes (2,3) and involves p53 (1). Sunlight-induced p53 mutations occur homogeneously throughout both cancers and precancers (1,9–11) and are frequent in normal sun-exposed skin (1,12), suggesting that mutations arise early and apoptosis may be a critical determinant of early events in skin cancer. Clones of p53 mutant keratinocytes are largest in UV-exposed skin (13,14) and provide a molecular indicator to correlate tumor development with formation or expansion of premalignant clones. Similar clones arise in chronically UVB-irradiated murine skin and are tightly correlated with subsequent development of papillomas and carcinomas (15). If apoptosis reduces survival of mutant cells yet drives clonal expansion in this system, one might predict that suppression of apoptosis would increase the number of UVB-induced p53 mutant clones but these clones would be constrained by their surviving normal neighbors and thus not attain their usual size. Such a scenario could result in a paradoxical reduction in tumor formation, which relies heavily on clonal expansion.
We therefore sought to manipulate epidermal apoptosis without compromising the in vivo pathophysiology of the events involved. Dissecting long-term physiological events requires care because small changes can have large cumulative effects and many proteins have pleiotropic effects or back-up pathways. As an example of the first, a mere 5% difference in the rate of cell death over proliferation can determine whether a skin tumor regresses or grows (16). Yet, because most apoptotic pathways are highly buffered by interconnection and overlap, profound changes typically require disruptive and embryoniclethal manipulations such as inactivating both Jnk1 and Jnk2 (17) or Bax and Bak (18). Here we have employed a transgenic mouse with epidermis-specific expression of Survivin that exhibits a 2-fold reduction in UVB-induced keratinocyte apoptosis. This reduced susceptibility to apoptosis is similar to the apoptosis differential created by a p53+/− mutation (1) and does not disturb epidermal proliferation or differentiation. We then examined the consequences of this 2-fold apoptotic differential for formation of UVB-induced p53 mutant clones, clonal expansion, creation and growth of papillomas and conversion to SCC. We find that apoptosis inhibition resulting from Survivin expression exerts two opposing activities in skin carcinogenesis: enhancing mutant clone creation and malignant progression, but restricting clone expansion.
Materials and methods
Mice
Generation of keratin-14 (K14)-Survivin transgenic (Tg) mice is described elsewhere (19). Animals on a hairless background were derived from fourth or fifth generation backcrosses of K14-Survivin mice with SKH1 hairless mice (Charles River Laboratories, Wilmington, MA). All littermates were genotyped for the presence of the Survivin transgene by PCR as described (19) and used at 7–9 weeks of age. Animal procedures were approved by the Institutional Animal Care and Use Committees at the University of Utah and Yale University.
UVB-induced sunburn cells
The UVB irradiation chamber contained a bank of four UVB lamps (FS20T12-UVB; National Biological Corp., Twinsburg, OH) that emit wavelengths between 250 and 420 nm (72.6% UVB, 27.4% UVA, 0.01% UVC), with peak emission at 313 nm, according to the manufacturer. A filter (Kodacel TA422; Eastman Kodak, Rochester, NY) was placed over the cages to block residual UVC. Dosimetry was determined using a calibrated UVB-500C meter (National Biological Corp.). The irradiation rate was ~2 J/m2/s. Mice were exposed unrestrained to a single dose of UVB and 24 h later dorsal skin was harvested and sunburn cells were visualized in situ and quantitated as described previously (19).
UVB carcinogenesis
Mice were UVB irradiated unrestrained three times weekly (Monday, Wednesday and Friday) for 20 weeks using a modified regimen from that (20) described previously. Individual treatment doses began at ~700 J/m2 the first week and were increased by 25% each week until a maximum dose of ~2100 J/m2 was attained. This gradual increase in exposure produced minimal erythema and was not associated with scaling or ulceration. Tumor number and size (diameter) were documented weekly. The large number of tumors produced on many of the animals precluded reliable assessment of growth or regression of individual tumors.
Histological analysis of normal mouse skin
Mouse dorsal skin was excised and stained with hematoxylin and eosin as described previously (19). For assessment of proliferating cells in situ, mice were injected i.p. with 50 mg/kg bromodeoxyuridine (BrdU) (Sigma Chemical Co., St Louis, MO) in phosphate-buffered saline and skin was harvested 2 h later. Staining for BrdU was performed using a kit (Zymed Laboratories, San Francisco, CA) as described previously (19).
Histological analysis of tumors
Tumors were excised, fixed, paraffin embedded and stained with hematoxylin and eosin as described previously (21). All slides were reviewed twice in blind fashion by a dermatopathologist (S.R.F.) and assessed for tumor architecture, keratinocyte differentiation, cytololgical atypia and inflammation. Tumors were classified as SCC, atypical papillomas or typical papillomas based on architecture and cytological atypia as described previously (21). Some UVB-induced tumors on the face proved to be abscesses and these were not included in the final tumor analysis. Tumors <2 mm in diameter proved too small for reliable histological analysis and were assumed to be papillomas.
Tumors with sufficient residual tissue remaining in the paraffin block were subjected to further analysis. Five micrometer sections were cut and the slides processed as described previously (21). Mitotic index was assessed using anti-proliferating cell nuclear antigen (PCNA) antibody (0.5 μg/ml; BD Transduction Laboratories, Palo Alto, CA) and a HistoMouse AEC kit (Zymed Laboratories) as described previously (22). Apoptotic index was assessed by TUNEL staining using the ApopTag kit (Intergen, Purchase, NY) as described previously (22). Survivin expression was confirmed by in situ hybridization using a mouse Survivin riboprobe (23).
p53 mutant clone detection
K14-Survivin mice and non-Tg littermates were treated with escalating doses of UVB three times weekly as above for a total of 5, 7, 10 or 13 weeks. These experiments used a similar UVB source (24). Three days after the last irradiation (sufficient time for wild-type P53 up-regulated by UVB to return to baseline levels), epidermal sheets were prepared from multiple strips of dorsal skin as described (24). Most p53 mutations increase P53 protein stability and allow antibody detection (13,24). Clones of p53 mutant keratinocytes were detected by immunohistochemistry using NCL-p53-CM5p antibody (Vector Laboratories, Burlingame, CA) (24).
Statistics
For sunburn cell and tumor formation, data derived from multiple animals was subjected to survival analysis and unpaired t-tests with Welch’s correction using Prism (Graphpad, San Diego, CA). Analysis of p53 mutant clones was performed using StatXact 5.0.3. software (StatXact, Cambridge, MA). For analyses of fractional clone size distribution between groups of mice, a Pearson’s χ2 test was applied. For analyses of differences in clone density within predetermined size ranges (bins), the assumption was made that the random observed number of clones of a given size should follow a Poisson distribution, with mean proportional to the total area. P values of ≤0.05 were considered statistically significant.
Results
Survivin expression affects keratinocyte apoptosis but not proliferation or differentiation
To investigate the potential role of Survivin and apoptosis in UVB-induced skin cancer, we placed the K14-Survivin transgene (19) on a SKH1-hairless background, shown previously to be susceptible to UVB-induced cutaneous carcinogenesis (20). To confirm transgene function, mice were exposed to single doses of UVB and after 24 h the skin was excised and examined histologically for the presence of apoptotic keratinocytes (sunburn cells). Compared with non-Tg littermates, K14-Survivin mice were 2- to 5-fold (P < 0.01, 0.05) more resistant to UVB-induced sunburn cell formation depending on dose (Figure 1a). The Tg skin (in the absence of UVB) did not demonstrate epidermal hyperplasia, hyperkeratosis or any other histological variation compared with non-Tg skin (Figure 1b), suggesting that keratinocyte differentiation and proliferation were not affected. The lack of effect on basal proliferation was directly confirmed by BrdU incorporation, which was comparable in Tg and non-Tg epidermis (Figure 1c). Thus modest levels of constitutive Survivin expression (19) confers an anti-apoptotic phenotype in keratinocytes without affecting epidermal differentiation or proliferation.
Fig. 1.
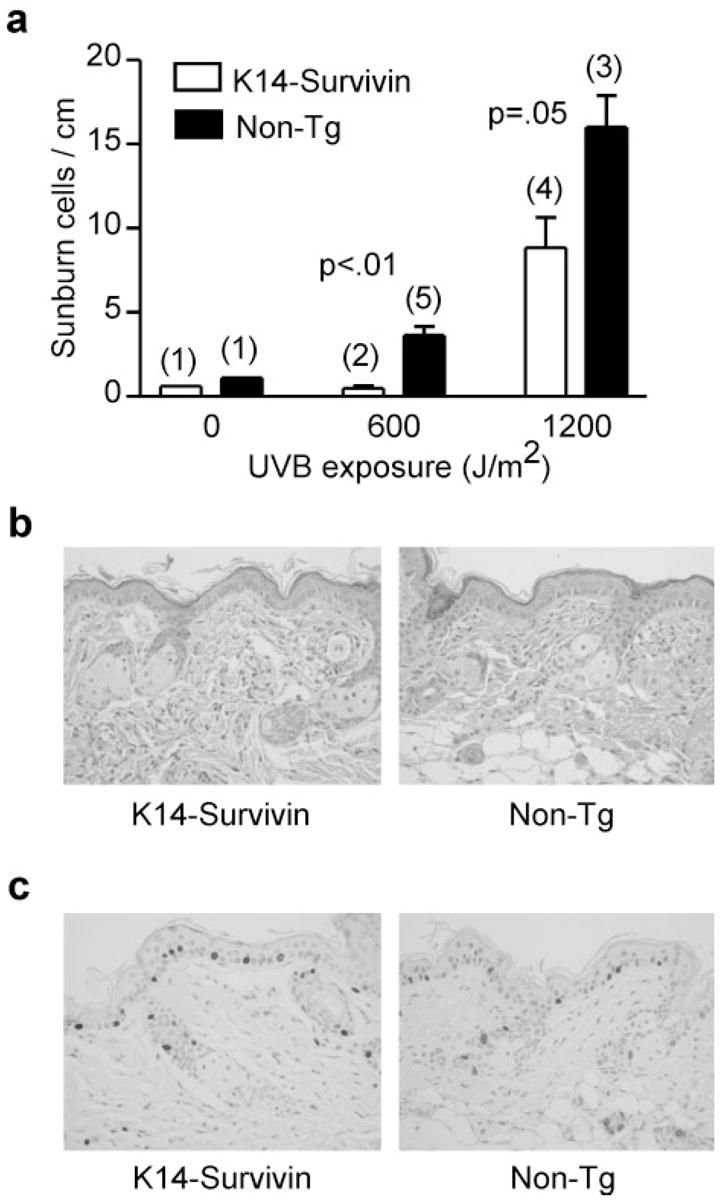
Survivin expression suppresses UVB-induced apoptosis but does not perturb epidermal differentiation or proliferation. (a) K14-Survivin (open bars) and non-Tg (filled bars) littermates were exposed to the indicated dose of UVB. The figure shows sunburn cell formation as detected histologically in skin sections harvested 24 h following UVB exposure. Data are expressed as sunburn cell number per linear cm of skin. Error bars denote SEM. Actual numbers of mice in each group are indicated in parentheses above the bars. P values for comparisons between groups of K14-Survivin and non-Tg mice are indicated. (b) Histological sections of Survivin Tg and non-Tg skin stained with hematoxylin and eosin, demonstrating similar epidermal thickness and absence of hyperkeratosis in Tg skin. (c) Staining of a skin section from Tg and non-Tg mice for BrdU, indicating comparable rates of BrdU incorporation. Dark staining cells have incorporated BrdU.
Effect of Survivin expression on p53 mutant clones
To investigate early events prior to tumor formation, we examined the induction and expansion of p53 mutant clones in UVB-irradiated animals. K14-Survivin and non-Tg littermates were exposed to UVB for 5, 7, 10 and 13 weeks using a previously described escalating dose schedule that ultimately leads to tumors (20). These exposures mainly lie in the dose range at which a 2-fold differential in apoptosis was observed. Clones were visualized by immunohistochemistry using an antibody to P53 protein (Figure 2a and b) and the number of cells constituting each clone was recorded. Clone number was normalized for total skin area. At 5 weeks total clone density was mildly increased (15.2 versus 13.3 clones/cm2; P = 0.032) in K14-Survivin mice compared with non-Tg littermates (Table I). By 7 weeks increased clone density was more apparent and highly significant (46.0 versus 31.1 clones/cm2; P < 10−8) in Tg mice (Table I). This difference in mutation induction likely reflects the anti-mutagenic effect of apoptosis.
Fig. 2.

Representative UVB-induced tumors and visualization of Survivin staining and p53 mutant clones. Immunostaining of (a) small (arrow) and (b) large p53 mutant clones at 13 weeks in K14-Survivin mouse. Clinical and histological photographs of (c) isolated papilloma on K14-Survivin mouse, (d) atypical papillomas (arrows) on non-Tg mouse, (e) SCC on K14-Survivin mouse and (f) cyst on non-Tg mouse. Atypical papilloma from K14-Survivin mouse demonstrating (g) in situ staining of Survivin and (h) control staining in the absence of the riboprobe. Original magnifications of histological photographs: (a)–(e), ×200; (f), ×100; (g)–(h), ×400.
Table I.
Time-dependent induction of p53 mutant clone densitya
| Exposure(weeks) | K14-Survivin(clones/cm2) | Non-Tg | P valueb |
|---|---|---|---|
| 5c | 15.2 | 13.3 | 0.032 |
| 7d | 46.0 | 31.1 | <10−8 |
| 10e | 76.6 | 102 | <10−8 |
| 13f | 209 | 241 | <10−8 |
K14-Survivin and non-Tg littermates were exposed to UVB for the indicated times and the total number of p53 mutant clones determined. Densities were normalized by dividing total clone number by total skin area. Data for all mice in each group were pooled.
For analyses of fractional clone size distribution between groups of mice at each time point a Pearson’s χ2 test was applied.
Six Tg (1217 clones, 44.9 cm2) and five non-Tg mice (1011 clones, 35.0 cm2).
Three Tg (465 clones, 10.1 cm2) and three non-Tg mice (794 clones, 25.5 cm2).
Three Tg (1003 clones, 13.1 cm2) and three non-Tg mice (1153 clones, 11.3 cm2).
Three Tg (3833 clones, 18.3 cm2) and three non-Tg mice (4313 clones, 17.9 cm2).
We categorized the clone data by sorting clones into bins based on multiples of the previously determined size of murine epidermal stem cell compartments (~16 cells) (24,25). As shown in Figure 3a, most of the clones at 5 weeks were small, with 60–80% falling into one of the first three size bins, and less than 10% of clones were large. Application of Pearson’s χ2 test revealed a highly significant difference (P = 4 ×10−5) in the fractional size distribution of Survivin Tg compared with non-Tg skin. We next tested whether the differences in the density of clones within each bin was statistically significant. The assumption was made that the random observed number of clones of a given size should follow a Poisson distribution, with mean proportional to the total area. At 5 weeks very small p53 mutant keratinocyte clones (1–16 cells, ≤1 stem cell compartment), which reflect initial mutation frequency, were 1.4-fold more frequent (P = 4 ×10−6) in Survivin Tg mice (Figure 3a). This experiment was repeated, with similar results (not shown). At 7 weeks the ratio of Tg to non-Tg small clone densities was 1.6-fold and highly significant (P < 10−8) (Figure 3b). Survivin expression thus promotes the creation of new mutant clones.
Fig. 3.
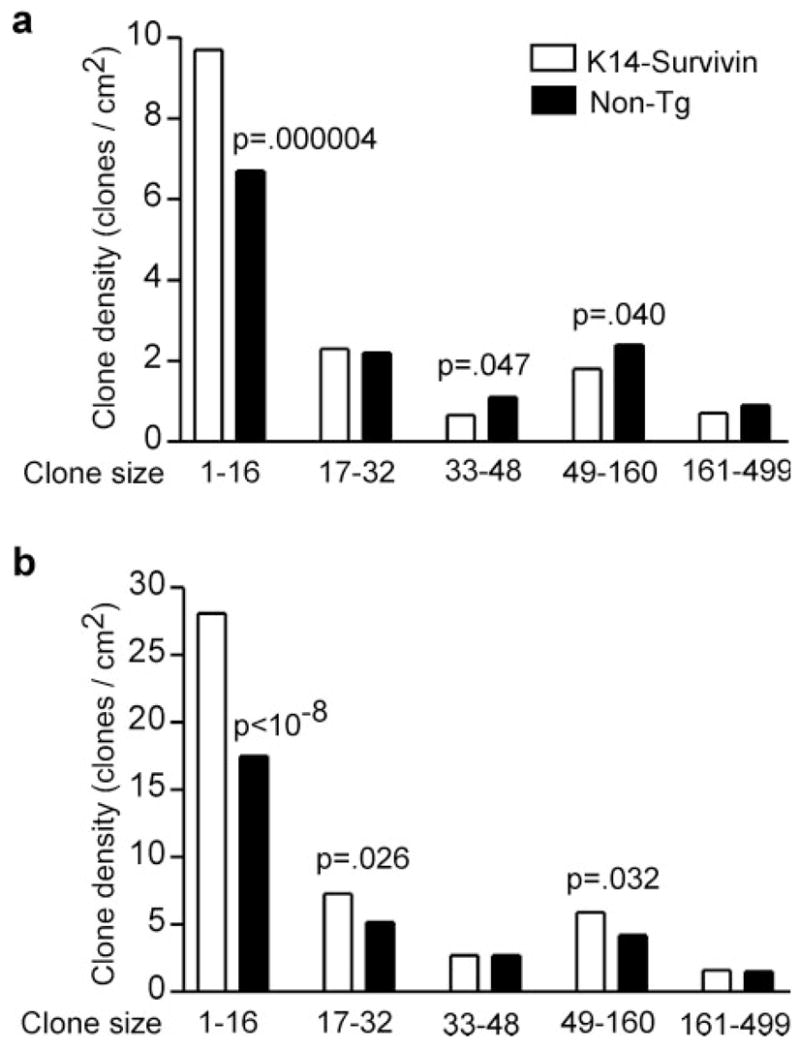
Survivin expression enhances creation of new p53 mutations. K14-Survivin (open bars) and non-Tg littermates (filled bars) were UVB-irradiated for (a) 5 or (b) 7 weeks. Skin was harvested and analyzed for p53 mutant clones by immunohistochemistry. Data are expressed as number of clones per cm2 of skin for various clone size bins. Bins represent ≤1 stem cell compartment (1–16 cells), and multiples of 2, 3, 4–10 and 11–31 compartments. The P values for comparisons of clonal densities from Tg and non-Tg mice are two-sided and significant values are indicated above the bars. For statistical analysis the assumption was made that the random observed number of clones of a given size follows a Poisson distribution, with mean proportional to the total area.
The effect of Survivin expression was quite different for larger clones, which was most evident at 10 and 13 weeks. Although the small size bins at 10 and 13 weeks still contained a greater fraction of Tg compared with non-Tg clones, this ratio was reversed in larger bins representing existing clones growing by clonal expansion. Intermediate and large clones (2–10 stem cell compartments) in Tg skin were 1.7-fold rarer at 10 weeks (Figure 4a) and 1.4-fold rarer at 13 weeks (Figure 4b). These differences were highly significant (Figure 4a and b). By 13 weeks some clones had reached over 2000 cells in size (>125 stem cell compartments) and these were 1.5-fold rarer (P = 0.019) in Survivin Tg mice (Figure 4b). These results indicate that Survivin expression enhances the formation of small clones, whereas clonal expansion (the tendency of small clones to become large clones) is impaired in apoptosis-defective K14-Survivin mice. At these later time points newly created clones are outnumbered by accumulating growing clones and the increased capacity for new clone formation in Tg skin makes a smaller contribution to the total clone density (Table I).
Fig. 4.
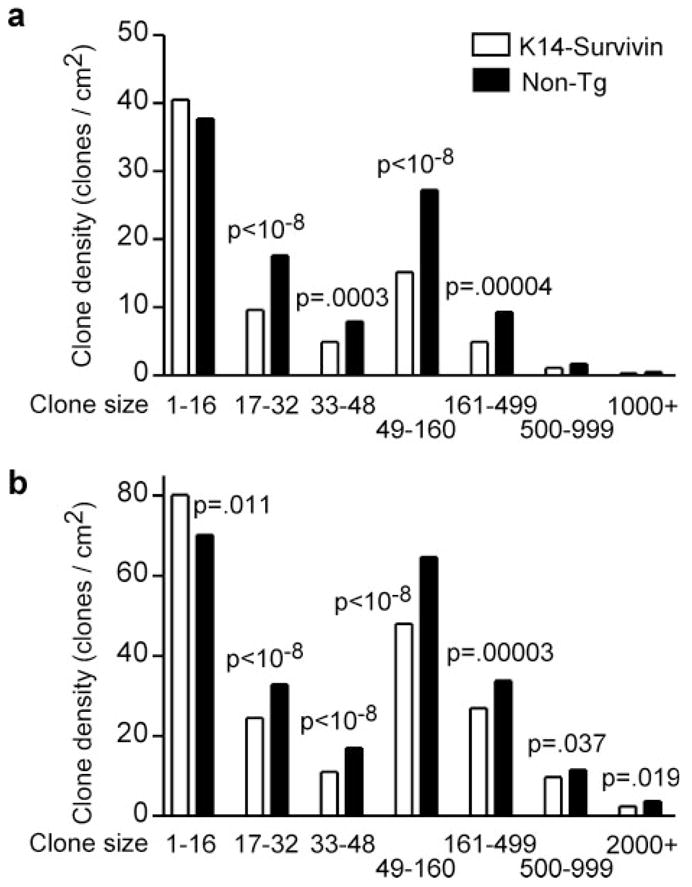
Survivin expression impedes expansion of existing p53 mutnt clones. K14-Survivin (open bars) and non-Tg littermates (filled bars) were UVB irradiated for (a) 10 or (b) 13 weeks. Skin was harvested, and p53 mutant clones were analyzed as in Figure 3. Fewer intermediate and large clones are found in Tg skin.
UVB-induced tumor formation and progression
K14-Survivin mice and non-Tg littermates were also examined for tumor induction in response to chronic UVB irradiation. UVB was administered using the same escalating regimen (20) and continued for 20 weeks. Treated mice were examined weekly for tumor formation and total tumor number and size were recorded. The large number of tumors forming on some animals (39 tumors in one case) precluded reliable tracking of growth or regression of individual tumors. Twenty weeks of treatment was sufficient to induce tumors in 95% of animals and served as the experimental end-point. All tumors arose on dorsal skin that had been exposed to UVB and untreated animals did not spontaneously develop tumors.
Tumor burden was reduced 1.5-fold (P = 0.03) in K14-Survivin mice, which formed 222 tumors with a mean of 5.97 tumors/animal, compared with non-Tg mice which formed 328 tumors with a mean of 8.71 tumors/animal (Table II). An increase in tumors would have been expected if the major effect of Survivin-mediated inhibition of apoptosis was to increase survival of precancerous cells. In contrast, twice as many tumors in Survivin Tg animals attained large size (≥3 mm) (7.7 versus 4.0%; P = 0.048) (Table II). As shown in Figure 5a, the median time to UVB-induced tumor formation was slightly less in K14-Survivin mice compared with non-Tg littermates (16 versus 17 weeks), although this difference was not statistically significant (P = 0.18).
Table II.
Summary of UVB-induced tumorsa
| K14-Survivin | Non-Tg | P value | |
|---|---|---|---|
| Mice with tumorsb (%) | 36/38 (95%) | 38/40 (95%) | |
| Total tumor numberc | 222 | 328 | |
| Tumor densityd | 5.97 (±0.86) | 8.71 (±1.18) | 0.03 |
| Tumor sizee | 1.36 (±0.05) | 1.26 (±0.05) | 0.18 |
| Papillomas (% of tumors) | 192 (86%) | 302 (92%) | 0.11f |
| Tumors ≥3 mm (% of tumors) | 17 (7.7%) | 13 (4.0%) | 0.048f |
| SCC (% of tumors) | 12 (5.4%) | 10 (3.0%) | 0.11f |
Seventy-nine mice (39 K14-Survivin and 40 non-Tg littermates) were exposed to UVB for 20 weeks. One Tg animal died at 6 weeks (without tumors).
Percentage of mice with tumors at the 20 weeks end-point.
Total number of tumors formed at the end-point.
Mean number of tumors per animal with tumors at the end-point. SEM in parentheses. The P value is two-sided.
Mean tumor size in mm. SEM in parentheses. The P value is two-sided.
The P values for percent tumors is one-sided.
Fig. 5.
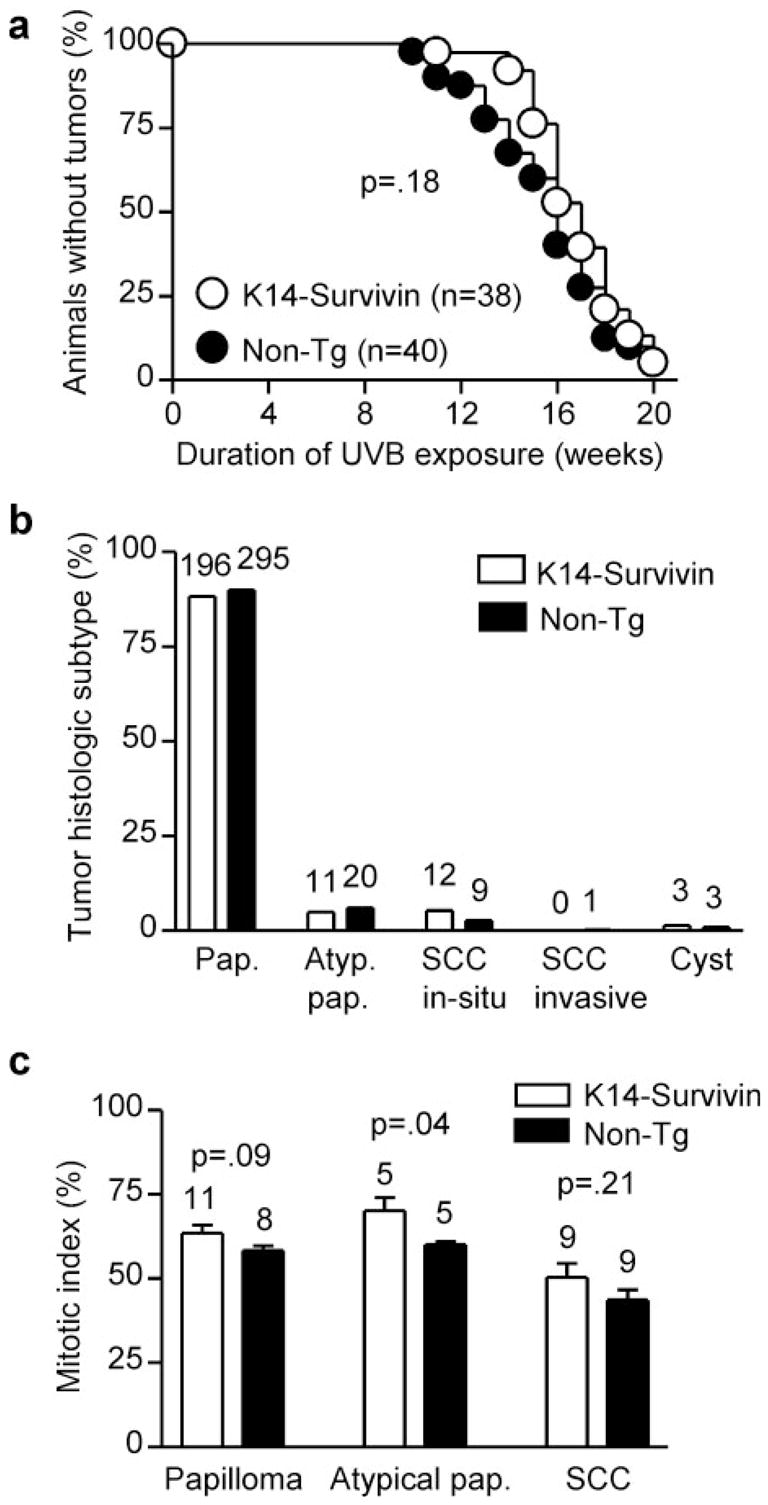
Tumor development and histological type induced by chronic UVB treatment. K14-Survivin (open circles and bars) and non-Tg (filled circles and bars) littermates were irradiated with UVB as described for 20 weeks. (a) Percent animals that did not form tumors. The number of animals in each group is indicated in parentheses. The P value for comparison of survival curves is 0.18. (b) Histological analysis of UVB-induced tumors. Tumors <2 mm in diameter were assumed to be papillomas. Percentages of total tumors formed at the end-point for each group are shown. The actual numbers of tumors are indicated above bars. (c) Mitotic index in UVB-induced tumors. Representative papillomas, atypical papillomas and SCC were stained for PCNA and the mitotic index was determined by calculating percent positive cells in multiple fields. The actual numbers of tumors analyzed in each group and P values are indicated above bars.
Tumors <2 mm in size were assumed to be papillomas, while the remaining tumors were subjected to histological examination and classified as typical papillomas, atypical papillomas, SCC in situ or with focal invasion or frankly invasive SCC as described previously (21). Representative examples of the clinical and histological spectrum of UVB-induced tumors are depicted in Figure 2. The majority of tumors (>90%) in both K14-Survivin and non-Tg animals were papillomas, with similar percentages of typical and atypical papillomas in each group (Figure 5b). Notably, however, the rate of SCC conversion (from papillomas) in K14-Survivin mice was almost twice that (5.4 versus 3.0%) observed in non-Tg littermates (Table II), although the small number of lesions reaching this stage resulted in borderline statistical significance (P = 0.11). Most of the SCC found in both groups were carcinoma in situ or demonstrated only focal invasion (Figure 5b) and were not further classified with respect to grade of differentiation. Finally, a few animals in each group developed large yellow subcutaneous masses that proved to be epidermoid cysts (Figure 2f ). Some tumors were not associated with inflammation, while others were characterized by a dense inflammatory infiltrate, but generally there were no trends noted between tumors in the two groups with respect to host inflammatory response (not shown). Thus, although Survivin expression was associated with decreased papilloma formation, there was an enhanced rate of malignant conversion from papilloma to SCC in Tg animals.
A subset of UVB-induced tumors were also analyzed for Survivin expression and for markers of proliferation and apoptosis. Survivin expression was detected in situ (Figure 2g) in 100% of tumors examined from both K14-Survivin mice (2 of 2 SCC and 5 of 5 atypical/typical papillomas) and non-Tg mice (2 of 2 SCC and 7 of 7 atypical/typical papillomas). There was no consistent pattern related to tumor subtype or mouse genotype. Mitotic index in representative tumors from both groups of mice was determined by PCNA staining. As shown in Figure 5c, all tumor types from K14-Survivin mice were associated with higher rates of proliferation, although the difference was significant (P = 0.04) only for the atypical papillomas. Apoptotic index was determined in representative tumors (6 SCC and 7 atypical/typical papillomas) from each group of mice by TUNEL staining. Sections revealed only 0–2 TUNEL-positive cells, with no apparent differences in spontaneous apoptosis between tumor subtypes or mouse genotypes (not shown).
Discussion
Opposing effects of apoptosis on tumor development
Despite the widespread supposition that apoptosis is anti-tumorigenic, direct tests of the role of apoptosis in skin tumor models have given paradoxical results. In our previous studies on chemical carcinogenesis in K14-Survivin mice the transgene exerted opposing effects on tumor formation and progression (21). A similar paradox has been reported in mice transgenic for bcl-2 (26) or defective in p53 (27,28). Although the tumor-initiating event in chemical carcinogenesis is known, namely the induction of Ha-Ras mutant keratinocytes (29), other early events in this pathway are a ‘black box’, since tumor precursors cannot be visualized. In contrast, detection of p53 mutant clones provides a window on the early steps in UVB carcinogenesis.
Survivin, like other IAP proteins and indeed many cellular proteins, can have multiple effects on cellular physiology (5). We sought to limit the study to apoptotic roles by using moderate overexpression of Survivin. Use of the Survivin Tg mouse allowed us to study the effects of manipulating apoptosis in keratinocytes without the major known confounding effects, on cell proliferation or differentiation, although it is always possible that additional cellular functions of Survivin will be discovered.
Evidence suggests that Survivin and P53 represent opposing signals acting on a common apoptosis pathway. Although certain IAP molecules function by binding to caspases and blocking their proteolytic activity (30), and interactions of Survivin with caspase-9 have been demonstrated (31,32), recent studies suggest that Survivin mediates its anti-apoptotic activity by restraining mitochondrial events (33,34). In contrast, P53 promotes the mitochondrial apoptotic pathway through up-regulation of the caspase-9 cofactor Apaf-1 (35,36) and may counteract Survivin by silencing its transcription (37,38). We previously demonstrated that apoptosis resistance in K14-Survivin mice was further enhanced when the transgene was placed on a p53+/− background (19); this differential makes feasible the present studies of p53+/− cells present in a field of cells with constitutive Survivin expression.
The early stage of clonal growth can be divided into two phases: UVB-induced p53 mutant clone formation, in which the first mutant cell is created and proliferates to fill its stem cell compartment; clonal expansion, in which the mutant cells extend beyond the borders of their original stem cell compartment. We were able to discern differential control of these early phases by Survivin and gain insight into the role of apoptosis at each step in the UVB carcinogenesis pathway.
Mutational steps: tumor initiation and malignant progression
The absence of Survivin in normal skin and its presence in actinic keratoses (23,39) suggest that constitutive Survivin expression promotes early steps in the generation of UVB-induced skin tumors. Survivin expression did enhance tumor initiation, as reflected by increased numbers of p53 mutant clones. This association was most significant for formation of very small (1–16 cell) clones at 5 and 7 weeks of UVB exposure. These clones reflect creation of the mutant cell and its limited proliferation within a stem cell compartment (Figure 6, panel 1). This difference in mutation induction is expected from the anti-mutagenic effect of apoptosis, which has been shown by others to reduce mutations by several fold at early times after UVB irradiation (2,40). The ability of Survivin to block apoptosis of DNA-damaged keratinocytes would similarly lead to an increase in cells mutated in other genes. Thereafter, apoptosis inhibition and the potential activity of Survivin as a promoter of cell cycle progression (41) would promote both survival and mitosis of the newly generated mutants.
Fig. 6.

Schematic model for the role of apoptosis in multi-step UVB carcinogenesis, showing the effects of apoptosis suppression resulting from transgenic expression of Survivin. Under normal conditions (top) UVB induces a p53 mutation in a keratinocyte stem cell, which then populates its own stem cell compartment (black hexagon). Further UVB exposures delete neighboring compartments by apoptosis (white hexagons) and these compartments can become repopulated by mutant cells (third panel). Continued UVB promotes expansion of the premalignant clone. Large clones will become papillomas, and in some papillomas cells acquire additional mutations (stippled black hexagons) that result in conversion to SCC. Under conditions of apoptosis suppression (bottom) resulting from Survivin expression, the creation of initial p53 mutations is enhanced (first panel). However, diminished apoptosis (second panel) impedes clonal expansion resulting in fewer large clones (third panel) and ultimately fewer papillomas. Reduced apoptosis promotes mutations in additional genes, as with p53, and there is enhanced malignant conversion (double arrows) of papillomas to SCC.
Although fewer total tumors were induced in K14-Survivin mice, due to the reduction in their papilloma precursors, tumor progression, as evidenced by the fraction of large tumors, was increased. This suggests that the barrier to clonal expansion imposed by apoptosis inhibition reduced tumor formation, but once tumors had formed reducing apoptosis in adjacent keratinocytes did not have a significant impact on tumor growth for most tumors. This may also be reflected in the lack of difference between Tg and non-Tg mice in onset of first tumor formation. Only a fraction of these tumors attained large size and there was a greater proportion of these in the Tg mice, perhaps reflecting the effect of enhanced Survivin expression in the tumors. Similarly, the rate of malignant conversion, the fraction of papillomas that evolved to SCC, was also increased. Because malignant conversion of papillomas is thought to be due to new mutations (42), Survivin expression was again associated with the failure of mutant cells to be eliminated (Figure 6, panel 5). The association of Survivin expression with heightened tumor progression is consistent with its being a predictor of poor prognosis in many cancers (43), including SCC (44).
Clonal expansion steps: p53 mutant clones and papillomas
For intermediate sized and large clones which are growing by clonal expansion, clone density was less in K14-Survivin mice than in non-Tg mice. This result implies that although Survivin expression promotes clone formation, it impedes clone expansion. Apoptosis therefore may act to facilitate clonal expansion of apoptosis-resistant mutant clones beyond the initial stem compartment. Whereas the development of a small clone occurs within a single epidermal compartment, the transition from small clones to large clones necessarily involves breaching the barrier presented by adjacent keratinocytes (45) in order to extend into neighboring compartments (24; Figure 6, panels 2–3). We previously demonstrated that p53 mutant clones cease expanding and regress if UVB exposure is discontinued (24), indicating that UVB is driving clonal expansion by a physiological mechanism rather than by creating additional mutations. Possible mechanisms for the UVB dependence of clonal expansion include stimulation of keratinocyte growth factors, impaired immunosurveillance or apoptotic removal of neighboring keratinocytes. While sustained UVB exposure stimulates keratinocyte proliferation, resulting in epidermal hyperplasia (19), this would not be selective for keratinocytes harboring p53 mutations. We recently found that clonal expansion is not altered in immunodeficient mice (46). The present results support the remaining explanation, with apoptosis induction in adjacent keratinocytes providing space to accommodate an expanding clone (Figure 6, panels 2–3). This side-effect of the UVB-induced apoptosis mechanism relies on the apoptosis-resistance phenotype of p53+/− mutant cells. It is intriguing to consider the suggestion that dying apoptotic cells may play an active, rather than passive, role in driving clonal expansion. We do not know if the phenomena of compensatory proliferation (47) and super-competition (48,49) described in Drosophila are occurring in our system and have no way to test it directly.
This behavior is consistent with observations in organotypic cultures. Normal human keratinocytes mixed at a ratio of 4:1 (but not 1:1) with cells carrying two mutant p53 alleles and mutant HRAS eliminate the transformed cells by inducing cell cycle arrest and differentiation (50). Low doses of UVB, however, induce apoptosis and differentiation only in the normal cells, allowing the transformed cells to overtake the culture (50). In the core of advanced tumors hypoxia induces apoptosis of normal cells but allows p53 mutant cells to increase in number (7). By comparing several stages of skin tumor development, we were able to test the possibility that, by creating larger clonal targets, such numerical increases can also accelerate the rate at which multistage carcinogenesis transits from one stage to the next.
Papillomas, which result from cycles of clonal expansion and mutation creation, will reflect the sum of Survivin’s opposing effects on these processes. The reduced frequency of papillomas in Survivin Tg animals suggests that the negative effect of Survivin on clonal expansion predominated over its promutational activity. The net effect of apoptosis was to favor papilloma growth (Figure 6, top panel 4). We found relatively linear relationships between apoptosis, clones and papillomas: a 2-fold reduction in apoptosis induction was associated with 1.5- to 1.8-fold changes in clone density, ~2-fold changes in papilloma number and ~2-fold changes in rate of SCC conversion. Similar phenomena may account for the reduced tumor yield reported in bcl-2 Tg mice (26) and mice deficient in p53 (27,28). Those Tg mice would also be expected to have reduced apoptosis in neighboring keratinocytes that serve as a barrier to clonal expansion. The balance between greater mutant formation and reduced clonal expansion expression could, however, depend on the expression level or timing associated with a particular promoter. For example, expression of Bcl-2 using a different promoter resulted in increased susceptibility to tumorigenesis (51). Varying results could also reflect differences in the epidermal compartment targeted (stem cell versus transient amplifying cell). Paradoxical roles of apoptosis have also been observed in other tumor models and clinical studies in patients with cancer (52). In conclusion, our findings of the opposing effects of Survivin expression on tumor formation and malignant conversion suggest that apoptosis functions in these pathways as a double-edged blade: thwarting clone formation and malignant progression but facilitating clonal expansion.
Acknowledgments
We thank Cathy Adrada for technical assistance. This work was supported by NIH grants KO8AR48618 and RO3AR48953 (to D.G.) and RO1CA78735 and RO1CA55737 (to D.E.B). D.G. was also supported by the Huntsman Cancer Foundation and a Fellowship-to-Faculty Transition Award from the University of Utah funded in part by the Howard Hughes Medical Institute.
Abbreviations
- BrdU
bromodeoxyuridine
- IAP
inhibitor of apoptosis
- K14
keratin-14
- SCC
squamous cell carcinoma
- Tg
transgenic
- UVB
ultraviolet B (290–320 nm)
References
- 1.Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 2.Hill LL, Ouhtit A, Loughlin SM, Kripke ML, Ananthaswamy HN, Owen-Schaub LB. Fas ligand: a sensor for DNA damage critical in skin cancer etiology. Science. 1999;285:898–900. doi: 10.1126/science.285.5429.898. [DOI] [PubMed] [Google Scholar]
- 3.Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature. 2000;407:777–783. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- 4.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 5.Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 6.Felsher DW. Cancer revoked: oncogenes as therapeutic targets. Nature Rev Cancer. 2003;3:375–380. doi: 10.1038/nrc1070. [DOI] [PubMed] [Google Scholar]
- 7.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 8.Grossman D, Leffell DJ. The molecular basis of nonmelanoma skin cancer: new understanding. Arch Dermatol. 1997;133:1263–1270. [PubMed] [Google Scholar]
- 9.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taguchi M, Watanabe S, Yashima K, Murakami Y, Sekiya T, Ikeda S. Aberrations of the tumor suppressor p53 gene and p53 protein in solar keratosis in human skin. J Invest Dermatol. 1994;103:500–503. doi: 10.1111/1523-1747.ep12395643. [DOI] [PubMed] [Google Scholar]
- 11.Nataraj AJ, Trent JC, Ananthaswamy HN. p53 gene mutations and photocarcinogenesis. Photochem Photobiol. 1995;62:218–230. doi: 10.1111/j.1751-1097.1995.tb05262.x. [DOI] [PubMed] [Google Scholar]
- 12.Nakazawa H, English D, Randell PL, Nakazawa K, Martel N, Armstrong BK, Yamasaki H. UV and skin cancer: specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc Natl Acad Sci USA. 1994;91:360–364. doi: 10.1073/pnas.91.1.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE, Brash DE. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci USA. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren ZP, Ponten F, Nister M, Ponten J. Two distinct p53 immunohistochemical patterns in human squamous cell skin cancer, precursors and normal epidermis. Int J Cancer. 1996;69:174–179. doi: 10.1002/(SICI)1097-0215(19960621)69:3<174::AID-IJC4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 15.Rebel H, Mosnier LO, Berg RJ, Westermande Vries A, van Steeg H, van Kranen HJ, de Gruijl FR. Early p53-positive foci as indicators of tumor risk in ultraviolet-exposed hairless mice: kinetics of induction, effects of DNA repair deficiency and p53 heterozygosity. Cancer Res. 2001;61:977–983. [PubMed] [Google Scholar]
- 16.Burns FJ, Vanderlaan M, Sivak A, Albert RE. Regression kinetics of mouse skin papillomas. Cancer Res. 1976;36:1422–1427. [PubMed] [Google Scholar]
- 17.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 18.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:624–626. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grossman D, Kim PJ, Blanc-Brude OP, Brash DE, Tognin S, Marchisio PC, Altieri DC. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J Clin Invest. 2001;108:991–999. doi: 10.1172/JCI13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Gruijl FR, Forbes PD. UV-induced skin cancer in a hairless mouse model. Bioessays. 1995;17:651–660. doi: 10.1002/bies.950170711. [DOI] [PubMed] [Google Scholar]
- 21.Allen SM, Florell SR, Hanks AN, Alexander A, Diedrich MJ, Altieri DC, Grossman D. Survivin expression in mouse skin prevents papilloma regression and promotes chemical-induced tumor progression. Cancer Res. 2003;63:567–572. [PubMed] [Google Scholar]
- 22.Grossman D, McNiff JM, Li F, Altieri DC. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J Invest Dermatol. 1999;113:1076–1081. doi: 10.1046/j.1523-1747.1999.00776.x. [DOI] [PubMed] [Google Scholar]
- 23.Bowen AR, Hanks AN, Murphy KJ, Florell SR, Grossman D. Proliferation, apoptosis and survivin expression in keratinocytic neoplasms and hyperplasias. Am J Dermatopathol. 2003;26:177–181. doi: 10.1097/00000372-200406000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang W, Remenyik E, Zelterman D, Brash DE, Wikonkal NM. Escaping the stem cell compartment: sustained UVB exposure allows p53-mutant keratinocytes to colonize adjacent epidermal proliferating units without incurring additional mutations. Proc Natl Acad Sci USA. 2001;98:13948–13953. doi: 10.1073/pnas.241353198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potten CS, Morris RJ. Epithelial stem cells in vivo. In: Lord BI, Dexter TM, editors. Stem Cells. Vol. 10. Company of Biologists Ltd; Cambridge: 1988. pp. 45–62. [Google Scholar]
- 26.Rossiter H, Beissert S, Mayer C, Schon MP, Wienrich BG, Tschachler E, Kupper TS. Targeted expression of bcl-2 to murine basal epidermal keratinocytes results in paradoxical retardation of ultraviolet- and chemical-induced tumorigenesis. Cancer Res. 2001;61:3619–3626. [PubMed] [Google Scholar]
- 27.Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74:813–822. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- 28.Greenhalgh DA, Wang XJ, Donehower LA, Roop DR. Paradoxical tumor inhibitory effect of p53 loss in transgenic mice expressing epidermal-targeted v-rasHa, v-fos, or human transforming growth factor alpha. Cancer Res. 1996;56:4413–4423. [PubMed] [Google Scholar]
- 29.Brown K, Buchmann A, Balmain A. Carcinogen-induced mutations in the mouse c-Haras gene provide evidence of multiple pathways for tumor progression. Proc Natl Acad Sci USA. 1990;87:538–542. doi: 10.1073/pnas.87.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 31.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci USA. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, Reed JC. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729–2740. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanc-Brude OP, Mesri M, Wall NR, Plescia J, Dohi T, Altieri DC. Therapeutic targeting of the survivin pathway in cancer: initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin Cancer Res. 2003;9:2683–2692. [PubMed] [Google Scholar]
- 34.Liu T, Brouha B, Grossman D. Rapid induction of mitochondrial events and caspase-independent apoptosis in Survivin-targeted melanoma cells. Oncogene. 2003;23:39–48. doi: 10.1038/sj.onc.1206978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- 36.Robles AI, Bemmels NA, Foraker AB, Harris CC. APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res. 2001;61:6660–6664. [PubMed] [Google Scholar]
- 37.Mirza A, McGuirk M, Hockenberry TN, et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene. 2002;21:2613–2622. doi: 10.1038/sj.onc.1205353. [DOI] [PubMed] [Google Scholar]
- 38.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 39.Grossman D, McNiff JM, Li F, Altieri DC. Expression of the apoptosis inhibitor, survivin, in nonmelanoma skin cancer and gene targeting in a keratinocyte cell line. Lab Invest. 1999;79:1121–1126. [PubMed] [Google Scholar]
- 40.Ouhtit A, Muller HK, Davis DW, Ullrich SE, McConkey D, Ananthaswamy HN. Temporal events in skin injury and the early adaptive responses in ultraviolet-irradiated mouse skin. Am J Pathol. 2000;156:201–207. doi: 10.1016/S0002-9440(10)64720-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 42.Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. J Dermatol Sci. 1998;17:1–7. doi: 10.1016/s0923-1811(97)00071-6. [DOI] [PubMed] [Google Scholar]
- 43.Altieri DC. Validating survivin as a cancer therapeutic target. Nature Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 44.Lo Muzio L, Staibano S, Pannone G, et al. Expression of the apoptosis inhibitor survivin in aggressive squamous cell carcinoma. Exp Mol Pathol. 2001;70:249–254. doi: 10.1006/exmp.2001.2367. [DOI] [PubMed] [Google Scholar]
- 45.Hennings H, Robinson VA, Michael DM, Pettit GR, Jung R, Yuspa SH. Development of an in vitro analogue of initiated mouse epidermis to study tumor promoters and antipromoters. Cancer Res. 1990;50:4794–4800. [PubMed] [Google Scholar]
- 46.Remenyik E, Wikonkal NM, Zhang W, Paliwal V, Brash DE. Antigen-specific immunity does not mediate acute regression of UVB-induced p53-mutant clones. Oncogene. 2003;22:6369–6376. doi: 10.1038/sj.onc.1206657. [DOI] [PubMed] [Google Scholar]
- 47.Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–1266. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 48.de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- 49.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 50.Mudgil AV, Segal N, Andriani F, Wang Y, Fusenig NE, Garlick JA. Ultraviolet-B irradiation induces expansion of intraepithelial tumor cells in a tissue model of early cancer progression. J Invest Dermatol. 2003;121:191–197. doi: 10.1046/j.1523-1747.2003.12320.x. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez-Villanueva J, Greenhalgh D, Wang XJ, Bundman D, Cho S, Delehedde M, Roop D, McDonnell TJ. Human keratin-1. bcl-2 transgenic mice aberrantly express keratin 6, exhibit reduced sensitivity to keratinocyte cell death induction and are susceptible to skin tumor formation. Oncogene. 1998;16:853–863. doi: 10.1038/sj.onc.1201610. [DOI] [PubMed] [Google Scholar]
- 52.Gurova KV, Gudkov AV. Paradoxical role of apoptosis in tumor progression. J Cell Biochem. 2003;88:128–137. doi: 10.1002/jcb.10382. [DOI] [PubMed] [Google Scholar]