

Abstract
Bioinformatic tools are widely utilized to predict functional single nucleotide polymorphisms (SNPs) for genotyping in molecular epidemiological studies. However, the extent to which these approaches are mirrored by epidemiological findings has not been fully explored. In this study, we first surveyed SNPs examined in case-control studies of lung cancer, the most extensively-studied cancer type. We then computed SNP functional scores using four popular bioinformatics tools: SIFT, PolyPhen, SNPs3D, and PMut, and determined their predictive potential using the odds ratios (ORs) reported. Spearman’s correlation coefficient (r) for the association with SNP score from SIFT, PolyPhen, SNPs3D, and PMut, and the summary ORs were r = −0.36 (p = 0.007), r = 0.25 (p = 0.068), r = −0.20 (p = 0.205), and r = −0.12 (p = 0.370) respectively. By creating a combined score using information from all four tools we were able to achieve a correlation coefficient of r = 0.51 (p < 0.001). These results indicate that scores of predicted functionality could explain a certain fraction of the lung cancer risk detected in genetic association studies and more accurate predictions may be obtained by combining information from a variety of tools. Our findings suggest that bioinformatic tools are useful in predicting SNP functionality and may facilitate future genetic epidemiological studies.
Keywords: SNP, SIFT, PolyPhen, SNPs3D, PMut and Lung Cancer
1. Introduction
SNP-disease association research in the emerging field of genetic and molecular epidemiology has been driven by the candidate gene and genome-wide approaches. In the candidate gene approach, an interesting SNP will typically be investigated in a population-based study if a plausible biological mechanism, which relates the gene harboring the SNP to disease etiology or progression, has been proposed. Our understanding of disease etiology is continuing to improve and with it a greater number of disease candidate genes are being discovered. Concurrent with this are numerous SNP-finding efforts that are generating millions of putative SNPs for potential association studies. The entirety of this multitude cannot be immediately assessed and therefore, the current obstacle lies in extracting useful and informative SNPs from the public databases [1]. Approaches to prioritizing these functional SNPs will tremendously enhance SNP-based association research in molecular epidemiology.
Combining modern molecular phenotypic assays with polymorphisms (i.e. SNP data) will provide an ultimate assessment of the effect that genetic variations have on the network of interacting molecular and physiologic systems under normal and pathological human conditions. This, however, is not an easy task and it currently poses one of the greatest challenges to modern medical scientists. In addition to this, functional assays are currently not available for most candidate SNPs [2].
Taking advantage of recent developments in evolutionary biology, protein structural genomics, and transcriptomics, several computational methods may be utilized to discriminate between neutral SNPs, which constitute the majority of genetic variations, and the small portion of SNPs of likely functional importance. The most straightforward approach to predicting SNP functionality involves determining the importance of the SNP’s location. For example, exonic SNPs may alter amino acid sequences and consequently influence the kinetic parameters of enzymes, the DNA-binding properties of proteins that regulate transcription, the signal transduction activities of transmembrane receptors, and the architectural roles of structural proteins [3]. SNPs may also change the sequence of DNA splicing sites in both exons and introns, and in turn, affect post-transcriptional processes [4]. Moreover, from an evolutionary perspective, SNPs altering a conserved amino acid site are more likely to have functional importance [5].
Many SNPs in candidate disease-related genes have been genotyped in molecular epidemiological studies, especially in the field of cancer research. This provides a great opportunity to validate these bioinformatic tools by correlating predicted SNP functional scores to findings from case-control studies. In this study, we first surveyed previous publications which genotyped SNPs in case-control studies of lung cancer, the most extensively examined cancer type. We then used four major bioinformatic tools, SIFT [5], PolyPhen [3], SNPs3D [6], and PMut [7] to predict the functional impact of these SNPs. Finally, we tested the hypothesis that SNPs predicted to have a significant impact on protein function are more likely to be associated with cancer risk in terms of odds ratios by correlating the functional scores to ORs obtained from actual published case-control studies.
2. Methods
2.1. Literature search
We performed a PubMed search using the keywords “lung cancer polymorphism case control,” while setting the limits to case-control studies examining SNPs and lung cancer risk, published in English from January, 1994 to December, 2006. The time restriction was imposed as few studies were identified prior to 1994, and these studies might be of questionable methodological quality. The bioinformatic tools we used make predictions about the impact of a given SNP based on the resulting amino acid substitution. As such, we imposed a further restriction on the studies to include only those which investigated non-synonymous SNPs (nsSNPs), i.e. those resulting in an amino acid change. Studies matching this additional criteria were manually identified from the larger pool of studies returned from the initial keyword search. We chose the overall odds ratio (OR) based on the dominant disease model, comparing carriers of combined heterozygous and homozygous variant allele with the homozygous wild type allele carriers.
2.2 Assessment of nsSNP functionality
The impact of nsSNPs can be assessed by evaluating the importance of the amino acids they affect. Four widely-used computational tools for determining the functional significance of nsSNPs were employed for this study. The SIFT software (blocks.fhcrc.org/~pauline/SIFT.html) was used to determine the conservation level of a particular amino acid position in a protein, which leads to a tolerance index (from 0 to 1) for SNP functionality [5]. The higher a tolerance index, the less functional impact a particular amino acid substitution is likely to have, as a higher tolerance index indicates that the position is less conserved across species. We also used the PolyPhen tool (http://coot.embl.de/PolyPhen/) to estimate the structural and functional impact of an amino acid substitution, which returns a “Position-Specific Independent Count” (PSIC) score [3]. Large values of this PSIC score indicate that the substitution is rarely or never observed in the protein family, suggesting likelihood that the amino acid replacement will be deleterious. SNPs3D (http://www.snps3d.org) uses two methods for determining whether a SNP will be deleterious to protein function[6]. The first makes predictions based on the estimated impact of the nsSNP on protein stability[8], while the second considers conservation of the given amino acid within a protein family[9]. In both cases, a negative score indicates a deleterious substitution, while a positive number corresponds to a neutral change, with greater absolute values indicating stronger confidence in the prediction. In cases where both scores were available for a given SNP, we used the more confident prediction. No score was available from either model for 12 of the 54 SNPs under study. The final bioinformatic tool we employed was PMut (http://mmb2.pcb.ub.es:8080/PMut/). This tool uses neural networks trained using a large database of disease-associated and neutral SNPs to predict the impact of a given amino acid substitution[7,10]. We used the prediction tool which returns a neural network output value from 0 to 1, with 0 corresponding to a “highly reliable” neutral prediction and 1 corresponding to a “highly reliable” deleterious prediction. As values approach 0.5 they become less robust. All searches performed on each of the four tools were performed using the respective default parameters.
2.3 Calculation of summary score
In order to incorporate data from all four tools, a summary score was created which combined the scores returned from each individual algorithm. In order to provide a meaningful summary value, an equation had to be generated which took into consideration the direction corresponding to a deleterious SNP, along with the range of possible output values for a particular tool. Since larger values for the scores generated by Polyphen and PMut indicate deleterious SNPs, these were included together in the numerator. Furthermore, since PMut scores take on a value between 0 and 1, while PolyPhen scores can be greater than 2, Polyphen scores were divided by the PMut score, to create one value in the numerator which would increase as confidence in a deleterious prediction increased. Since larger SIFT and SNPs3D values correspond to neutral SNPs, these were placed in the denominator of the final equation and multiplied by one another. In order to generate comparable values from the numerator and denominator portions of the summary equation, we took the square root of the numerator and the square of the denominator. The resulting range of values for the numerator piece of the equation was 0.39 to 6.46, while the range for the denominator was 0 to 5.85, indicating fairly equal contributions from the two halves of the equation. The net effect of this final equation was to create one summary value which drew on information from each of the 4 tools, and would take on larger values with increasing confidence in a deleterious prediction for each SNP. The final equation was as follows:
| (1) |
2.4 Statistical analysis
All statistical analyses were performed using the STATA statistical software (StataCorp.; College Station, TX) unless otherwise specified. Meta-analysis was first performed to estimate summary ORs for SNPs examined in multiple studies in order to generate one data point for each SNP in the subsequent correlation analysis. Overall ORs were calculated using a random-effects model [11] which includes both within- and between-study variations. Briefly, ORs, standard errors, and 95% Confidence Intervals (CIs) were calculated for all studies using published frequencies for cases and controls of the genotypes of interest. Weighting was applied in the calculation by using the inverse of an individual ORs’ variance in order to take into account the quality of information available (e.g. the sample size of the study and the precision of the point estimate). If a SNP had been found to be protective (OR < 1) in a study, we re-expressed the odds ratio in terms of the risk genotypes (OR > 1) in order to facilitate the comparisons, which were based on the strength of the association, rather than the direction. Neither the odds ratios nor their natural logarithms followed a Gaussian distribution, so correlations were determined by nonparametric methods (Spearman’s rank correlation) using the re-expressed ORs. In addition to correlating the scores from each bioinformatic tool to the observed odds ratios for all SNPs, Spearman’s rank correlation coefficients were also calculated to assess the correlation among the various functional scores obtained for each SNP.
Results
Our PubMed search identified 51 case-control studies examining a total of 54 nsSNPs in 37 different genes for risk estimates of lung cancer (Table 1). All of these SNPs are located in the coding regions of cancer related-genes, such as those involved in DNA repair, metabolism, and cell cycle checkpoints.
Table 1.
List of SNPs investigated in lung cancer case-control studies.
| Gene | Amino Acid change | SNP ID | OR* | SIFT Score | PSIC Score | SNPs3D Score | PMut Score | Reference |
|---|---|---|---|---|---|---|---|---|
| ADRB2 | Gly16Arg | rs1042713 | 1.01 | 0.40 | 1.353 | 1.96 | 0.44 | [16] |
| APE1/APEX1 | Gln51His | rs1048945 | 1.04 | 0.12 | 0.093 | 0.53 | 0.0341 | [17] |
| APE1/APEX1 | Ile64Val | rs2307486 | 0.60 | 0.33 | 0.911 | 0.82 | 0.0226 | [17] |
| APE1/APEX1 | Asp148Glu | rs1130409 | 0.92 | 1 | 0.467 | 1.42 | 0.0876 | [17–19] |
| ATR | Thr211Met | rs2227928 | 0.64 | 0.21 | 0.980 | - | 0.3969 | [17] |
| CDKN1A | Ser31Arg | rs1801270 | 0.92 | 0.80 | 0.488 | 2.58 | 0.7492 | [20] |
| CYP1A1 | Ile462Val | rs1048943 | 1.35 | 0.05 | 0.785 | −0.64 | 0.0485 | [21–28] |
| CYP1B1 | Leu432Val | rs1056836 | 1.78 | 0.13 | 0.277 | −0.25 | 0.2893 | [27,29] |
| EPHX1 | His113Tyr | rs1051740 | 0.69 | 0.01 | 2.261 | −0.78 | 0.2091 | [30,31] |
| EPHX1 | His139Arg | rs2234922 | 0.74 | 0.49 | 0.141 | 0.07 | 0.166 | [30,31] |
| ERCC2/XPD | His201Tyr | rs1799792 | 1.05 | 1 | 0.350 | 1.95 | 0.1941 | [17] |
| ERCC2/XPD | Asp312Asn | rs1799793 | 1.03 | 0.58 | 0.333 | 2.03 | 0.345 | [17,19,32,33] |
| ERCC2/XPD | Lys751Gln | rs13181 | 1.29 | 0.84 | 0.472 | 2.88 | 0.697 | [17,19,32–34] |
| ERCC4/XPF | Arg415Gln | rs1800067 | 1.20 | 0.28 | 1.892 | −0.91 | 0.6268 | [17] |
| ERCC5/XPG | Cys529Ser | rs2227869 | 1.32 | 0.80 | 0.660 | 1.63 | 0.2999 | [32] |
| ERCC5/XPG | His1104Asp | rs17655 | 1.11 | 0 | 1.628 | −0.98 | 0.6505 | [32,35,36] |
| EXO1 | Glu589Lys | rs1047840 | 0.81 | 0.72 | 0.252 | - | 0.5013 | [17] |
| GSTP1 | Ile105Val | rs1695 | 1.07 | 1 | 0.166 | 1.36 | 0.0276 | [29,37–41] |
| GSTP1 | Ala114Val | rs1138272 | 1.40 | 0.12 | 1.455 | 1.22 | 0.498 | [40] |
| HDAC5 | Asp593Glu | rs228757 | 1.19 | 1 | 0.114 | 1.04 | 0.0274 | [42] |
| LIG4 | Ile658Val | rs2232641 | 1.20 | 0.60 | 0.432 | 1.13 | 0.0402 | [35] |
| MYCL1 | Ser362Thr | rs3134614 | 0.68 | 0.04 | 1.235 | - | 0.0296 | [43] |
| MBD1 | Pro345Ala | rs125555 | 1.29 | 0.40 | 0.866 | −0.7 | 0.8374 | [44] |
| MGMT | Leu84Phe | rs12917 | 1.12 | 0.71 | 0.024 | −1.16 | 0.1301 | [17,45] |
| MGMT | Ile143Val | rs2308321 | 1.35 | 0.70 | 0.193 | 0.78 | 0.0332 | [17,46] |
| MGMT | Lys178Arg | rs2308327 | 1.24 | 0.52 | 0.112 | 0.64 | 0.0701 | [17,47] [47] |
| MMP9 | Arg279Gln | rs17576 | 1.29 | 0.35 | 1.537 | 2.28 | 0.2577 | [48] |
| MMP9 | Pro574Arg | rs2250889 | 1.61 | 0.50 | 0.764 | 2.4 | 0.8465 | [48] |
| MMP9 | Arg668Gln | rs2274756 | 1.04 | 1 | 0.084 | 1.9 | 0.5389 | [48] |
| MTHFR | Ala222Val | rs1801133 | 0.91 | 0.04 | 1.266 | −2.01 | 0.3939 | [49] |
| MTHFR | Ala429Glu | rs1801131 | 1.12 | 0.28 | 0.786 | - | 0.6345 | [49] |
| MTHFR | Arg594Gln | rs2274976 | 1.23 | 0.06 | 1.729 | 0.3811 | [49] | |
| MTR | Asp919Gly | rs1805087 | 0.93 | 0.33 | 0.390 | 1.29 | 0.7541 | [49] |
| MTRR | Leu22Met | rs1801394 | 1.36 | 0.03 | 1.267 | − | 0.1041 | [49] |
| NBS1 | Glu185Gln | rs1805794 | 1.27 | 0.64 | 0.624 | 1.78 | 0.0689 | [17,50,51] |
| NQO1 | Pro187Ser | rs1800566 | 1.1 | 0.40 | 1.499 | −1.29 | 0.1403 | [52–55] |
| OGG1 | Ser326Cys | rs1052133 | 1.19 | 0.22 | 0.397 | −0.46 | 0.707 | [17,29,56] |
| p53 | Arg72Pro | rs1042522 | 1.20 | 0.54 | 0.521 | 2.63 | 0.7672 | [20,35,57–59] |
| PARP1 | Val762Ala | rs1136410 | 1.26 | 0.66 | 1.357 | 0.54 | 0.2393 | [60] |
| PARP1 | Lys940Arg | rs3219145 | 1.30 | 0.21 | 1.678 | − | 0.0431 | [35] |
| PDZ-RhoGEF | Ser1416Gly | rs868188 | 0.90 | 0.12 | 1.323 | - | 0.2736 | [61] |
| POLB | Pro242Arg | rs3136797 | 2.31 | 0.07 | 2.587 | 0.97 | 0.4716 | [17] |
| POLI | Thr706Ala | rs8305 | 1.17 | 0.67 | 0.227 | - | 0.3617 | [17,35] |
| RAD23B | Ala249Val | rs1805329 | 1.81 | 0.29 | 1.060 | 0.11 | 0.6915 | [32] |
| REV1 | Phe257Ser | rs3087386 | 1.00 | 0.90 | 0.342 | 2.25 | 0.0736 | [35] |
| SOD2 | Ala16Val | rs4880 | 1.24 | 0.53 | 0.173 | - | 0.5608 | [58] |
| SULT1A1 | Arg213His | rs9282861 | 1.36 | 0.07 | 1.725 | 0.71 | 0.465 | [62,63] |
| XPC | Ala499Val | rs2228000 | 1.11 | 0.73 | 0.346 | - | 0.3404 | [32,64,65] |
| XPC | Lys939Gln | rs2228001 | 0.99 | 0 | 2.068 | - | 0.2929 | [32,64,65] |
| XRCC1 | Arg194Trp | rs1799782 | 0.99 | 0.11 | 2.495 | 1.47 | 0.7963 | [17,66] |
| XRCC1 | Arg280His | rs25489 | 1.11 | 0.14 | 1.595 | 2.1 | 0.5637 | [17,66] |
| XRCC1 | Arg399Gln | rs25487 | 0.97 | 0.80 | 0.201 | 1.25 | 0.8085 | [17–19,51,58,60,66] |
| XRCC2 | Arg188His | rs3218536 | 2.82 | 0.20 | 0.631 | 2.3 | 0.1764 | [17] |
| XRCC3 | Thr241Met | rs861539 | 0.86 | 0.61 | 1.068 | 1.79 | 0.3058 | [17,19,51] |
Only summary OR is listed for a SNP investigated in multiple studies.
Correlations between each of the SNP functional scores obtained for each nsSNP were assessed by calculating Spearman’s rank correlation coefficients. Significant correlations were found between the SIFT tolerance index and PSIC score (r = −0.607, p < 0.001), as well as SIFT and SNPs3D scores (r = 0.348, p = 0.024). PMut scores did not significantly correlate with any other scores (Table 2).
Table 2.
Correlations among predicted SNP functionality scores and observed odds ratios.
| Sift | PolyPhen | SNPs3D | Pmut | Combined | ||
|---|---|---|---|---|---|---|
| OR | −0.361 | 0.250 | −0.200 | −0.124 | 0.513 | Rho* |
| 0.007 | 0.068 | 0.205 | 0.370 | <0.001 | p-value* | |
| 54 | 54 | 42 | 54 | 42 | N | |
| Sift | −0.607 | 0.348 | −0.013 | −0.828 | Rho* | |
| <.001 | 0.024 | 0.923 | <.001 | p-value* | ||
| 54 | 42 | 54 | 42 | N | ||
| PolyPhen | −0.154 | 0.181 | 0.488 | Rho* | ||
| 0.330 | 0.189 | 0.001 | p-value* | |||
| 42 | 54 | 42 | N | |||
| SNPs3d | 0.191 | −0.636 | Rho* | |||
| 0.225 | <.001 | p-value* | ||||
| 42 | 42 | N | ||||
| PMut | −0.150 | Rho* | ||||
| 0.344 | p-value* | |||||
| 42 | N |
Rhos and p-values are for Spearman’s rank correlation coefficient.
SIFT, PSIC, SNPs3D, and PMut scores were also correlated to the observed ORs associated with the corresponding nsSNPs detected in molecular epidemiologic studies of lung cancer. Spearman’s rank correlation showed that of the four tools, SIFT scores were most strongly correlated with observed ORs (r = −0.361, p = 0.007). PolyPhen scores were modestly correlated (r = 0.250, p = 0.068), while SNPs3d and PMut demonstrated very weak associations (r = −0.200, p = 0.205, and r = −0.124, p = 0.370), respectively (Figure 1). The summary score, which considers the magnitude of the scores returned from each tool, as well as which direction corresponds to a deleterious substitution, was computed for each of the 42 nsSNPs for which scores were available from all four tools (N = 42). Spearman’s rank correlation coefficient for this summary score and the observed ORs was r = 0.513 (p < 0.001) (Figure 2).
Fig. 1.

Correlation between log of observed ORs and SNP scores from each of the four tools.
Fig. 2.
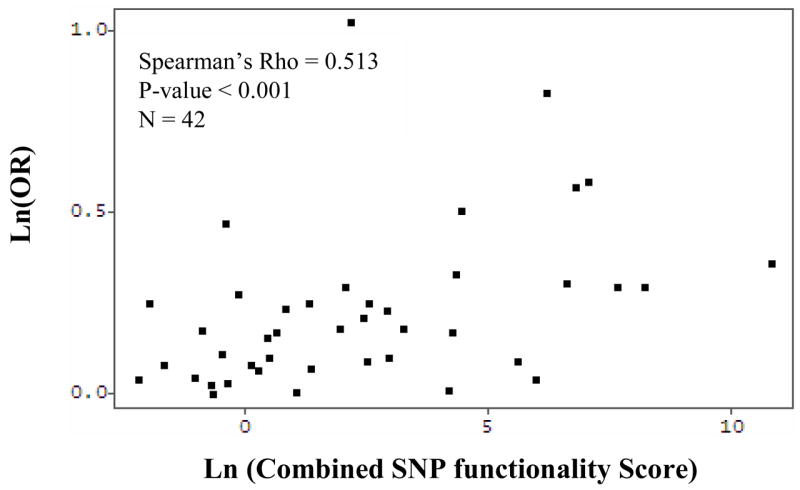
Correlation between log of observed ORs and log of combined SNP scores
Discussion
One unsolved issue of SNP genotyping in molecular epidemiological studies is how to choose target SNPs for investigation, since functionalities of most SNPs are unknown. Though haplotype tagging SNPs (htSNPs) generated from the HapMap project tremendously facilitates genotyping for genetic association studies, the ultimate goal of such studies is still to locate functional genetic changes linked to htSNPs in candidate genes. Given the lack of phenotypic data for most of the SNPs identified, results from our analyses suggest that bioinformatic tools based on recent findings from evolutionary biology, protein structure research, and computational biology may provide useful information in assessing the functional importance of SNPs.
Our findings are congruent with our previous observations, which demonstrated that SNPs altering conserved amino acids assessed by the SIFT tool are more likely to be associated with cancer risk [12]. The current study expanded on the previous analysis in three important ways, resulting in more meaningful and interpretable results. First, we included functional scores from SIFT, PolyPhen, SNPs3D, and PMut tools, each of which employs fundamentally different algorithms that can be used to assess the functionality of the same nsSNPs. Second, we restricted the analyses to case-control studies investigating lung cancer only, as the importance of a given gene in carcinogenesis may vary significantly across cancer types. Moreover, lung cancer is the most extensively studied cancer type, which allowed us to collect a reasonable number of studies for our analysis. Third, meta-analysis was performed to obtain summary ORs for SNPs examined in multiple studies in order to generate one data point for each SNP in the analysis.
While previous studies have investigated the predictive power and accuracy of computational approaches[13,14], this analysis is unique in that it draws on risk estimates derived directly from human epidemiological studies. While correlations based on predicted protein functionality or mutagenesis studies are important, we believe that those obtained by observational studies of human risk are even more relevant. Our data suggests that individual tools correlate modestly with observed odds ratio, and that combining information from a variety of tools may significantly increase the predictive power for determining the functional impact of a given nsSNP.
Our results demonstrate a significant agreement between the SIFT and PolyPhen tools, as well as SIFT and SNPs3D in the prediction of SNP functionality. This finding supports results from a previous analysis, which showed that the predicted scores of SNP functionality from the two algorithms are highly associated, with concordance in the predicted impact observed for approximately 62% of the variants [15].
However, while the combined scores yielded a respectable correlation coefficient (r) with observed ORs, the correlations for the individual tools detected in our analyses were fairly modest. This implies that the functional impact of a SNP on a gene may not be predicted 100% correctly by computational tools. It might also indicate that molecular phenotype (e.g. functional SNPs) may not always penetrate through to clinical phenotype (e.g. ORs). Furthermore, lung cancer is a complex environment-related cancer type, and disease-associated SNPs may only trigger tumorigenesis in the presence of certain environmental exposures such as tobacco carcinogens.
Although the correlation we observed is readily apparent, the current study has several limitations. First, there is the concern that many of the SNPs under study may not in fact be singly causal, but may associate with disease in more complicated ways. While linkage disequilibrium between the SNPs under study and other causative SNPs cannot be ruled out, each of these tools make predictions for missense mutations only, and base their predictions on the impact of the amino acid change on protein function. In addition, the ORs reported for each of the SNPs included in the analysis were based on studies designed using a candidate gene approach, rather than an array-based method. As such, the SNPs under study lie within genes that have established relevance to carcinogenesis such as DNA repair and metabolic genes, and are therefore likely to affect cancer risk directly. Couple this with the fact that the tools make predictions based on the specific SNP’s effect on protein function, and the result should be a fairly specific relationship between the bioinformatic prediction and the epidemiologic finding. Furthermore, the utility of these tools and others like them, lies in their ability to make predictions on the functional impact of an amino acid change for which little or no direct experimental evidence is available, and then to use this information in further studies to relate genetic variations to disease etiology. It is possible that a SNP predicted harmful does not show an association with cancer because multiple factors, such as gene-gene and/or gene-environment interaction are involved in tumorigenesis. It is also possible that a SNP predicted benign is actually associated with cancer, if the SNP is genetically linked to adjacent causal variations. Second, the accuracy with which a SNP predicts cancer risk depends on the alignment obtained in some bioinformatic tools. For example, the SIFT method depends on homologous sequence alignments among different species. The number of sequences available from different species may be different from gene to gene, which in turn may affect the accuracy of the prediction. The PolyPhen and SNPs3D predictions depend on protein structure information available for a given gene product, which may be of varying quality across the human proteome.
Nevertheless, our data indicates that bioinformatic tools are indeed useful in predicting the functional impact of SNPs, as our findings could explain a respectable fraction of the lung cancer risk that has been detected. Although these algorithms have been developed based on empirical data, correlations between predictive scores and findings from human studies have not been explored, with the exception of SIFT. The results of our study have therefore provided novel evidence of the correlation using human data, which in turn facilitate genotyping efforts in future molecular epidemiological studies and provide targets for phenotypic analysis of genetic variants. These results can also be used to refine the bioinformatic algorithms. These findings warrant a more comprehensive approach that includes other cancer types and more available bioinformatic tools in future analyses.
Acknowledgments
This study was supported by NIH grants CA122676, CA110937, and CA108369.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schork NJ, Fallin D, Lanchbury JS. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet. 2000;58:250–264. doi: 10.1034/j.1399-0004.2000.580402.x. [DOI] [PubMed] [Google Scholar]
- 2.Imyanitov EN, Togo AV, Hanson KP. Searching for cancer-associated gene polymorphisms: promises and obstacles. Cancer Lett. 2004;204:3–14. doi: 10.1016/j.canlet.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 3.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 4.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 8.Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Yue P, Moult J. Identification and analysis of deleterious human SNPs. J Mol Biol. 2006;356:1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 10.Ferrer-Costa C, Orozco M, de la Cruz X. Sequence-based prediction of pathological mutations. Proteins. 2004;57:811–819. doi: 10.1002/prot.20252. [DOI] [PubMed] [Google Scholar]
- 11.Laird NM, Mosteller F. Some statistical methods for combining experimental results. Int J Technol Assess Health Care. 1990;6:5–30. doi: 10.1017/s0266462300008916. [DOI] [PubMed] [Google Scholar]
- 12.Zhu Y, Spitz MR, Amos CI, Lin J, Schabath MB, Wu X. An evolutionary perspective on single-nucleotide polymorphism screening in molecular cancer epidemiology. Cancer Res. 2004;64:2251–2257. doi: 10.1158/0008-5472.can-03-2800. [DOI] [PubMed] [Google Scholar]
- 13.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–1325. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xi T, Jones IM, Mohrenweiser HW. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics. 2004;83:970–979. doi: 10.1016/j.ygeno.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Hao B, Chen X, Zhao N, Cheng G, Jiang Y, Liu Y, Lin C, Tan W, Lu D, Wei Q, Jin L, Lin D, He F. Beta-2 adrenergic receptor gene (ADRB2) polymorphism and risk for lung adenocarcinoma: A case-control study in a Chinese population. Cancer Lett. 2005 doi: 10.1016/j.canlet.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 17.Zienolddiny S, Campa D, Lind H, Ryberg D, Skaug V, Stangeland L, Phillips DH, Canzian F, Haugen A. Polymorphisms of DNA repair genes and risk of non-small cell lung cancer. Carcinogenesis. 2006;27:560–567. doi: 10.1093/carcin/bgi232. [DOI] [PubMed] [Google Scholar]
- 18.Ito H, Matsuo K, Hamajima N, Mitsudomi T, Sugiura T, Saito T, Yasue T, Lee KM, Kang D, Yoo KY, Sato S, Ueda R, Tajima K. Gene-environment interactions between the smoking habit and polymorphisms in the DNA repair genes, APE1 Asp148Glu and XRCC1 Arg399Gln, in Japanese lung cancer risk. Carcinogenesis. 2004;25:1395–1401. doi: 10.1093/carcin/bgh153. Epub 2004 Mar 1325. [DOI] [PubMed] [Google Scholar]
- 19.Popanda O, Schattenberg T, Phong CT, Butkiewicz D, Risch A, Edler L, Kayser K, Dienemann H, Schulz V, Drings P, Bartsch H, Schmezer P. Specific combinations of DNA repair gene variants and increased risk for non-small cell lung cancer. Carcinogenesis. 2004;25:2433–2441. doi: 10.1093/carcin/bgh264. [DOI] [PubMed] [Google Scholar]
- 20.Popanda O, Edler L, Waas P, Schattenberg T, Butkiewicz D, Muley T, Dienemann H, Risch A, Bartsch H, Schmezer P. Elevated risk of squamous-cell carcinoma of the lung in heavy smokers carrying the variant alleles of the TP53 Arg72Pro and p21 Ser31Arg polymorphisms. Lung Cancer. 2007;55:25–34. doi: 10.1016/j.lungcan.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Hamada GS, Sugimura H, Suzuki I, Nagura K, Kiyokawa E, Iwase T, Tanaka M, Takahashi T, Watanabe S, Kino I, et al. The heme-binding region polymorphism of cytochrome P450IA1 (CypIA1), rather than the RsaI polymorphism of IIE1 (CypIIE1), is associated with lung cancer in Rio de Janeiro. Cancer Epidemiol Biomarkers Prev. 1995;4:63–67. [PubMed] [Google Scholar]
- 22.Larsen JE, Colosimo ML, Yang IA, Bowman R, Zimmerman PV, Fong KM. Risk of non-small cell lung cancer and the cytochrome P4501A1 Ile462Val polymorphism. Cancer Causes Control. 2005;16:579–585. doi: 10.1007/s10552-004-7842-3. [DOI] [PubMed] [Google Scholar]
- 23.London SJ, Yuan JM, Coetzee GA, Gao YT, Ross RK, Yu MC. CYP1A1 I462V genetic polymorphism and lung cancer risk in a cohort of men in Shanghai, China. Cancer Epidemiol Biomarkers Prev. 2000;9:987–991. [PubMed] [Google Scholar]
- 24.Ng DP, Tan KW, Zhao B, Seow A. CYP1A1 polymorphisms and risk of lung cancer in non-smoking Chinese women: influence of environmental tobacco smoke exposure and GSTM1/T1 genetic variation. Cancer Causes Control. 2005;16:399–405. doi: 10.1007/s10552-004-5476-0. [DOI] [PubMed] [Google Scholar]
- 25.Song N, Tan W, Xing D, Lin D. CYP 1A1 polymorphism and risk of lung cancer in relation to tobacco smoking: a case-control study in China. Carcinogenesis. 2001;22:11–16. doi: 10.1093/carcin/22.1.11. [DOI] [PubMed] [Google Scholar]
- 26.Sugimura H, Wakai K, Genka K, Nagura K, Igarashi H, Nagayama K, Ohkawa A, Baba S, Morris BJ, Tsugane S, Ohno Y, Gao C, Li Z, Takezaki T, Tajima K, Iwamasa T. Association of Ile462Val (Exon 7) polymorphism of cytochrome P450 IA1 with lung cancer in the Asian population: further evidence from a case-control study in Okinawa. Cancer Epidemiol Biomarkers Prev. 1998;7:413–417. [PubMed] [Google Scholar]
- 27.Wenzlaff AS, Cote ML, Bock CH, Land SJ, Santer SK, Schwartz DR, Schwartz AG. CYP1A1 and CYP1B1 polymorphisms and risk of lung cancer among never smokers: a population-based study. Carcinogenesis. 2005;26:2207–2212. doi: 10.1093/carcin/bgi191. [DOI] [PubMed] [Google Scholar]
- 28.Yang XR, Wacholder S, Xu Z, Dean M, Clark V, Gold B, Brown LM, Stone BJ, Fraumeni JF, Jr, Caporaso NE. CYP1A1 and GSTM1 polymorphisms in relation to lung cancer risk in Chinese women. Cancer Lett. 2004;214:197–204. doi: 10.1016/j.canlet.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 29.Liang G, Pu Y, Yin L. Rapid detection of single nucleotide polymorphisms related with lung cancer susceptibility of Chinese population. Cancer Lett. 2005;223:265–274. doi: 10.1016/j.canlet.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 30.To-Figueras J, Gene M, Gomez-Catalan J, Pique E, Borrego N, Corbella J. Lung cancer susceptibility in relation to combined polymorphisms of microsomal epoxide hydrolase and glutathione S-transferase P1. Cancer Lett. 2001;173:155–162. doi: 10.1016/s0304-3835(01)00626-7. [DOI] [PubMed] [Google Scholar]
- 31.Voho A, Metsola K, Anttila S, Impivaara O, Jarvisalo J, Vainio H, Husgafvel-Pursiainen K, Hirvonen A. EPHX1 gene polymorphisms and individual susceptibility to lung cancer. Cancer Lett. 2006;237:102–108. doi: 10.1016/j.canlet.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 32.Shen M, Berndt SI, Rothman N, Demarini DM, Mumford JL, He X, Bonner MR, Tian L, Yeager M, Welch R, Chanock S, Zheng T, Caporaso N, Lan Q. Polymorphisms in the DNA nucleotide excision repair genes and lung cancer risk in Xuan Wei, China. Int J Cancer. 2005;116:768–773. doi: 10.1002/ijc.21117. [DOI] [PubMed] [Google Scholar]
- 33.Hu Z, Xu L, Shao M, Yuan J, Wang Y, Wang F, Yuan W, Qian J, Ma H, Liu H, Chen W, Yang L, Jing G, Huo X, Chen F, Jin L, Wei Q, Wu T, Lu D, Huang W, Shen H. Polymorphisms in the two helicases ERCC2/XPD and ERCC3/XPB of the transcription factor IIH complex and risk of lung cancer: a case-control analysis in a Chinese population. Cancer Epidemiol Biomarkers Prev. 2006;15:1336–1340. doi: 10.1158/1055-9965.EPI-06-0194. [DOI] [PubMed] [Google Scholar]
- 34.Yin J, Vogel U, Ma Y, Guo L, Wang H, Qi R. Polymorphism of the DNA repair gene ERCC2 Lys751Gln and risk of lung cancer in a northeastern Chinese population. Cancer Genet Cytogenet. 2006;169:27–32. doi: 10.1016/j.cancergencyto.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Sakiyama T, Kohno T, Mimaki S, Ohta T, Yanagitani N, Sobue T, Kunitoh H, Saito R, Shimizu K, Hirama C, Kimura J, Maeno G, Hirose H, Eguchi T, Saito D, Ohki M, Yokota J. Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int J Cancer. 2005;114:730–737. doi: 10.1002/ijc.20790. [DOI] [PubMed] [Google Scholar]
- 36.Cui Y, Morgenstern H, Greenland S, Tashkin DP, Mao J, Cao W, Cozen W, Mack TM, Zhang ZF. Polymorphism of Xeroderma Pigmentosum group G and the risk of lung cancer and squamous cell carcinomas of the oropharynx, larynx and esophagus. Int J Cancer. 2006;118:714–720. doi: 10.1002/ijc.21413. [DOI] [PubMed] [Google Scholar]
- 37.Larsen JE, Colosimo ML, Yang IA, Bowman R, Zimmerman PV, Fong KM. CYP1A1 Ile462Val and MPO G-463A interact to increase risk of adenocarcinoma but not squamous cell carcinoma of the lung. Carcinogenesis. 2006;27:525–532. doi: 10.1093/carcin/bgi227. [DOI] [PubMed] [Google Scholar]
- 38.Lewis SJ, Cherry NM, Niven RM, Barber PV, Povey AC. GSTM1, GSTT1 and GSTP1 polymorphisms and lung cancer risk. Cancer Lett. 2002;180:165–171. doi: 10.1016/s0304-3835(02)00028-9. [DOI] [PubMed] [Google Scholar]
- 39.Schneider J, Bernges U, Philipp M, Woitowitz HJ. GSTM1, GSTT1, and GSTP1 polymorphism and lung cancer risk in relation to tobacco smoking. Cancer Lett. 2004;208:65–74. doi: 10.1016/j.canlet.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Spitz MR, Schabath MB, Ali-Osman F, Mata H, Wu X. Association between glutathione S-transferase p1 polymorphisms and lung cancer risk in Caucasians: a case-control study. Lung Cancer. 2003;40:25–32. doi: 10.1016/s0169-5002(02)00537-8. [DOI] [PubMed] [Google Scholar]
- 41.Wenzlaff AS, Cote ML, Bock CH, Land SJ, Schwartz AG. GSTM1, GSTT1 and GSTP1 polymorphisms, environmental tobacco smoke exposure and risk of lung cancer among never smokers: a population-based study. Carcinogenesis. 2005;26:395–401. doi: 10.1093/carcin/bgh326. [DOI] [PubMed] [Google Scholar]
- 42.Park JM, Lee GY, Choi JE, Kang HG, Jang JS, Cha SI, Lee EB, Kim SG, Kim CH, Lee WK, Kam S, Kim DS, Jung TH, Park JY. No association between polymorphisms in the histone deacetylase genes and the risk of lung cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1841–1843. doi: 10.1158/1055-9965.EPI-05-0200. [DOI] [PubMed] [Google Scholar]
- 43.Spinola M, Conti B, Ravagnani F, Fabbri A, Incarbone M, Cataldo I, Pira E, Pelucchi C, La Vecchia C, Dragani TA. A new polymorphism (Ser362Thr) of the L-myc gene is not associated with lung adenocarcinoma risk and prognosis. Eur J Cancer Prev. 2004;13:87–89. doi: 10.1097/00008469-200402000-00014. [DOI] [PubMed] [Google Scholar]
- 44.Jang JS, Lee SJ, Choi JE, Cha SI, Lee EB, Park TI, Kim CH, Lee WK, Kam S, Choi JY, Kang YM, Park RW, Kim IS, Cho YL, Jung TH, Han SB, Park JY. Methyl-CpG binding domain 1 gene polymorphisms and risk of primary lung cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:2474–2480. doi: 10.1158/1055-9965.EPI-05-0423. [DOI] [PubMed] [Google Scholar]
- 45.Chae MH, Jang JS, Kang HG, Park JH, Park JM, Lee WK, Kam S, Lee EB, Son JW, Park JY. O6-alkylguanine-DNA alkyltransferase gene polymorphisms and the risk of primary lung cancer. Mol Carcinog. 2006;45:239–249. doi: 10.1002/mc.20171. [DOI] [PubMed] [Google Scholar]
- 46.Cohet C, Borel S, Nyberg F, Mukeria A, Bruske-Hohlfeld I, Constantinescu V, Benhamou S, Brennan P, Hall J, Boffetta P. Exon 5 polymorphisms in the O6-alkylguanine DNA alkyltransferase gene and lung cancer risk in non-smokers exposed to second-hand smoke. Cancer Epidemiol Biomarkers Prev. 2004;13:320–323. doi: 10.1158/1055-9965.epi-03-0120. [DOI] [PubMed] [Google Scholar]
- 47.Jang JS, Lee SJ, Choi JE, Cha SI, Lee EB, Park TI, Kim CH, Lee WK, Kam S, Choi JY, Kang YM, Park RW, Kim IS, Cho YL, Jung TH, Han SB, Park JY. Methyl-CpG Binding Domain 1 Gene Polymorphisms and Risk of Primary Lung Cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:2474–2480. doi: 10.1158/1055–9965.EPI-05-0423. [DOI] [PubMed] [Google Scholar]
- 48.Hu Z, Huo X, Lu D, Qian J, Zhou J, Chen Y, Xu L, Ma H, Zhu J, Wei Q, Shen H. Functional polymorphisms of matrix metalloproteinase-9 are associated with risk of occurrence and metastasis of lung cancer. Clin Cancer Res. 2005;11:5433–5439. doi: 10.1158/1078-0432.CCR-05-0311. [DOI] [PubMed] [Google Scholar]
- 49.Shi Q, Zhang Z, Li G, Pillow PC, Hernandez LM, Spitz MR, Wei Q. Polymorphisms of methionine synthase and methionine synthase reductase and risk of lung cancer: a case-control analysis. Pharmacogenet Genomics. 2005;15:547–555. doi: 10.1097/01.fpc.0000170916.96650.70. [DOI] [PubMed] [Google Scholar]
- 50.Lan Q, Shen M, Berndt SI, Bonner MR, He X, Yeager M, Welch R, Keohavong P, Donahue M, Hainaut P, Chanock S. Smoky coal exposure, NBS1 polymorphisms, p53 protein accumulation, and lung cancer risk in Xuan Wei, China. Lung Cancer. 2005;49:317–323. doi: 10.1016/j.lungcan.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Ryk C, Kumar R, Sanyal S, de Verdier PJ, Hemminki K, Larsson P, Steineck G, Hou SM. Influence of polymorphism in DNA repair and defence genes on p53 mutations in bladder tumours. Cancer Lett. 2006;241:142–149. doi: 10.1016/j.canlet.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 52.Alexandrie AK, Nyberg F, Warholm M, Rannug A. Influence of CYP1A1, GSTM1, GSTT1, and NQO1 genotypes and cumulative smoking dose on lung cancer risk in a Swedish population. Cancer Epidemiol Biomarkers Prev. 2004;13:908–914. [PubMed] [Google Scholar]
- 53.Bock CH, Wenzlaff AS, Cote ML, Land SJ, Schwartz AG. NQO1 T allele associated with decreased risk of later age at diagnosis lung cancer among never smokers: results from a population-based study. Carcinogenesis. 2005;26:381–386. doi: 10.1093/carcin/bgh314. Epub 2004 Oct 2021. [DOI] [PubMed] [Google Scholar]
- 54.Lawson KA, Woodson K, Virtamo J, Albanes D. Association of the NAD(P)H:quinone oxidoreductase (NQO1) 609C->T polymorphism with lung cancer risk among male smokers. Cancer Epidemiol Biomarkers Prev. 2005;14:2275–2276. doi: 10.1158/1055-9965.EPI-05-0375. [DOI] [PubMed] [Google Scholar]
- 55.Saldivar SJ, Wang Y, Zhao H, Shao L, Lin J, Spitz MR, Wu X. An association between a NQO1 genetic polymorphism and risk of lung cancer. Mutat Res. 2005;582:71–78. doi: 10.1016/j.mrgentox.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 56.Park J, Chen L, Tockman MS, Elahi A, Lazarus P. The human 8-oxoguanine DNA N-glycosylase 1 (hOGG1) DNA repair enzyme and its association with lung cancer risk. Pharmacogenetics. 2004;14:103–109. doi: 10.1097/00008571-200402000-00004. [DOI] [PubMed] [Google Scholar]
- 57.Jain N, Singh V, Hedau S, Kumar S, Daga MK, Dewan R, Murthy NS, Husain SA, Das BC. Infection of human papillomavirus type 18 and p53 codon 72 polymorphism in lung cancer patients from India. Chest. 2005;128:3999–4007. doi: 10.1378/chest.128.6.3999. [DOI] [PubMed] [Google Scholar]
- 58.Liu G, Zhou W, Park S, Wang LI, Miller DP, Wain JC, Lynch TJ, Su L, Christiani DC. The SOD2 Val/Val genotype enhances the risk of nonsmall cell lung carcinoma by p53 and XRCC1 polymorphisms. Cancer. 2004;101:2802–2808. doi: 10.1002/cncr.20716. [DOI] [PubMed] [Google Scholar]
- 59.Schabath MB, Wu X, Wei Q, Li G, Gu J, Spitz MR. Combined effects of the p53 and p73 polymorphisms on lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2006;15:158–161. doi: 10.1158/1055-9965.EPI-05-0622. [DOI] [PubMed] [Google Scholar]
- 60.Zhang X, Miao X, Liang G, Hao B, Wang Y, Tan W, Li Y, Guo Y, He F, Wei Q, Lin D. Polymorphisms in DNA base excision repair genes ADPRT and XRCC1 and risk of lung cancer. Cancer Res. 2005;65:722–726. [PubMed] [Google Scholar]
- 61.Gu J, Wu X, Dong Q, Romeo MJ, Lin X, Gutkind JS, Berman DM. A nonsynonymous single-nucleotide polymorphism in the PDZ-Rho guanine nucleotide exchange factor (Ser1416Gly) modulates the risk of lung cancer in Mexican Americans. Cancer. 2006;106:2707–2715. doi: 10.1002/cncr.21944. [DOI] [PubMed] [Google Scholar]
- 62.Liang G, Miao X, Zhou Y, Tan W, Lin D. A functional polymorphism in the SULT1A1 gene (G638A) is associated with risk of lung cancer in relation to tobacco smoking. Carcinogenesis. 2004;25:773–778. doi: 10.1093/carcin/bgh053. [DOI] [PubMed] [Google Scholar]
- 63.Pachouri SS, Sobti RC, Kaur P, Singh J, Gupta SK. Impact of polymorphism in sulfotransferase gene on the risk of lung cancer. Cancer Genet Cytogenet. 2006;171:39–43. doi: 10.1016/j.cancergencyto.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 64.Hu Z, Wang Y, Wang X, Liang G, Miao X, Xu Y, Tan W, Wei Q, Lin D, Shen H. DNA repair gene XPC genotypes/haplotypes and risk of lung cancer in a Chinese population. Int J Cancer. 2005;115:478–483. doi: 10.1002/ijc.20911. [DOI] [PubMed] [Google Scholar]
- 65.Lee GY, Jang JS, Lee SY, Jeon HS, Kim KM, Choi JE, Park JM, Chae MH, Lee WK, Kam S, Kim IS, Lee JT, Jung TH, Park JY. XPC polymorphisms and lung cancer risk. Int J Cancer. 2005;115:807–813. doi: 10.1002/ijc.20900. [DOI] [PubMed] [Google Scholar]
- 66.Hao B, Miao X, Li Y, Zhang X, Sun T, Liang G, Zhao Y, Zhou Y, Wang H, Chen X, Zhang L, Tan W, Wei Q, Lin D, He F. A novel T-77C polymorphism in DNA repair gene XRCC1 contributes to diminished promoter activity and increased risk of non-small cell lung cancer. Oncogene. 2006;25:3613–3620. doi: 10.1038/sj.onc.1209355. [DOI] [PubMed] [Google Scholar]