

Abstract
The preferred pathway for prothrombin activation by prothrombinase involves initial cleavage at Arg320 to produce meizothrombin, which is then cleaved at Arg271 to liberate thrombin. Exosite binding drives substrate affinity and is independent of the bond being cleaved. The pathway for cleavage is determined by large differences in Vmax for cleavage at the two sites within intact prothrombin. By fluorescence binding studies in the absence of catalysis, we have assessed the ability of the individual cleavage sites to engage the active site of Xa within prothrombinase at equilibrium. Using a panel of recombinant cleavage site mutants, we show that in intact prothrombin, the Arg320 site effectively engages the active site in a 1:1 interaction between substrate and enzyme. In contrast, the Arg271 site binds to the active site poorly in an interaction that is ~600-fold weaker. Perceived substrate affinity is independent of active site engagement by either cleavage site. We further show that prior cleavage at the 320 site or the stabilization of the uncleaved zymogen in a proteinase-like state facilitates efficient docking of Arg271 at the active site of prothrombinase. Therefore, we establish direct relationships between docking of either cleavage site at the active site of the catalyst, the Vmax for cleavage at that site, substrate conformation, and the resulting pathway for prothrombin cleavage. Exosite tethering of the substrate in either the zymogen or proteinase conformation dictates which cleavage site can engage the active site of the catalyst and enforces the sequential cleavage of prothrombin by prothrombinase.
Thrombin, the key effector product of the coagulation cascade, is produced by specific proteolysis of the zymogen precursor, prothrombin (1). The physiologically relevant catalyst for this reaction is considered to be prothrombinase, a complex that assembles through reversible interactions between the serine proteinase, factor Xa, and the cofactor protein, factor Va on membranes containing acidic phospholipids (2, 3). The fundamental role of this reaction system in normal hemostasis is evident from profound bleeding associated with deleterious mutations in any one of its components (4, 5).
Prothrombinase acts specifically on prothrombin and is not known to catalyze cleavage of other coagulation zymogens at an appreciable rate. A series of studies have established a predominant role for interactions between extended surfaces on prothrombinase (exosites) and its protein substrate, independent of the active site of the catalyst, in determining function (6). Exosite binding drives substrate affinity and confers specificity by restricting the action of factor Xa within prothrombinase to protein substrate species that can engage the enzyme complex in this way (6). Docking of residues flanking the cleavage site in the substrate to the active site of factor Xa within prothrombinase occurs in a second, intramolecular binding step that contributes little to substrate affinity but has instead been proposed to affect the Vmax for bond cleavage (7).
Thrombin formation requires proteolytic cleavage of prothrombin following Arg271 and Arg320 (Scheme I, reaction 1).2 When catalyzed by prothrombinase, the reaction occurs through two sequential enzyme-catalyzed reactions that proceeds largely via initial cleavage of prothrombin at Arg320 followed by subsequent cleavage at Arg271 (8). Meizothrombin (mIIa),3 produced as the sole detectable intermediate, accumulates transiently at concentrations in vast excess over the concentration of enzyme and cleavage of the two bonds in the opposite order is below experimental limits of detection (9, 10). Although recent studies indicate that some intermediate produced via the alternate pathway can be detected (11), it is clear that both cleavage sites in prothrombin are accessible to externally added proteinase, yet prothrombinase preferentially recognizes and cleaves Arg320 in the intact zymogen (12). Mechanisms that enforce enzymic specificity and restrict the action of prothrombinase to prothrombin must impact the ability of the enzyme to discriminate between the two bonds within the substrate itself.
SCHEME I. Cleavage of prothrombin and variants.

Prothrombinase converts prothrombin (IIWT) to thrombin (IIa) and the activation peptide F1.2 by catalyzing cleavage at Arg271 and Arg320 (reaction 1). The individual reactions have been studied using recombinant derivatives (IIQ271, IIQ320) or the proteolytic intermediate mIIa with the indicated representative steady state kinetic constants (12). The overall reaction can be accounted for by the essentially ordered cleavage of prothrombin at Arg320 to form mIIa (reaction 2) followed by cleavage at Arg271 (reaction 3). Cleavage of bonds in the opposite order is not readily detected because of the low Vmax for cleavage at Arg271 in IIQ320 to yield prethrombin 2 (P2) and F1.2 (reaction 4). Although IIQQ is not cleaved by prothrombinase (reaction 5), it acts as a competitive inhibitor for the cleavage of the other substrates.
Studies with a series of activation site mutants as well as the two possible intermediates of prothrombin activation have only recently provided a potential quantitative explanation for the long standing problem of the essentially ordered action of prothrombinase on prothrombin (12). In otherwise intact prothrombin, Arg320 is cleaved with a ~30-fold greater Vmax than Arg271 and prior cleavage at Arg320 to produce mIIa corrects the Vmax for subsequent cleavage at Arg271 (Scheme I, reactions 2–4). Whereas this provides a kinetic explanation for the largely ordered cleavage of prothrombin, its molecular basis has been shown to likely relate to the conformational transition associated with the conversion of the zymogen to proteinase that accompanies cleavage at Arg320 (13). All possible substrate species regardless of the availability of a cleavage site bind with equivalent affinity and in a mutually exclusive manner to prothrombinase (Scheme I) (12). This is in line with the conclusion that all substrate species bind to the enzyme in a comparable way through exosite interactions that are independent of the cleavage site or the active site of the enzyme (14). It follows that the observed differences in Vmax must arise from differences in the ability of the two sites to engage the active site of the catalyst that is somehow influenced by whether the substrate is a zymogen or a proteinase (15).
The unexpected and untested relationship between substrate binding at the active site and the perceived rate constant for catalysis is a key feature of the current model proposed for the ordered action of prothrombinase on prothrombin. The Circe Effect provides a formal consideration of the strategies by which enzymes may utilize the intrinsic binding energy of the substrate to affect the rate constant for catalysis (16). However, this relationship is typically inferred through negative findings and has proved difficult to establish experimentally with an unambiguous mechanistic basis (16, 17). Furthermore, kinetic approaches, such as those that have been used to frame our ideas, are notorious for yielding interpretations that can be greatly affected by the model and assumptions chosen for analysis. We now directly examine the previously proposed relationship between binding and catalysis. Our approach employs equilibrium binding measurements in the absence of catalysis to provide a model-independent assessment of the ability of the individual cleavage site prothrombin derivatives to dock at the active site of factor Xa within prothrombinase.
EXPERIMENTAL PROCEDURES
Reagents
Small unilamellar phospholipid vesicles (PCPS) composed of 75% (w/w) hen egg L-α-phosphatidylcholine and 25% (w/w) porcine brain L-α-phosphatidylserine (Avanti Polar Lipids) were prepared and characterized as described (9). Large unilamellar vesicles of the same composition (PCPSLUV) used for initial velocity studies were prepared and characterized as before (12). D-Phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (FPR-CH2Cl, Calbiochem), 4-aminobenzamidine (pAB, Sigma), and H-D-phenylalanyl-L-pipecolyl-L-arginine-p-nitroanilide (Chromogenix) were purchased from the indicated suppliers. The concentration of pAB was determined using E293 = 15,000 M−1 cm−1 (18). Crude lyophilized venoms from Echis carinatus pyramidum and Daboia russelli russelli (Russell’s viper) were obtained from Latoxan (Valence, France). Human plasma used for protein isolation was a generous gift of the Plasmapheresis Unit of the Hospital of the University of Pennsylvania. All fluorescence and activity measurements were performed in 20 mM Hepes, 150 mM NaCl, 5 mM CaCl2, 0.1% (w/v) polyethylene glycol 8000, pH 7.5 (Assay Buffer), at 25 °C.
Proteins
Ecarin and the factor X activator (RVVXCP) were purified from the appropriate crude venom using established procedures (14, 19). A fragment of staphylocoagulase containing an additional Met residue at the N terminus and a His6 extension at the COOH terminus (Met-SC1–325-His6) was expressed in Escherichia coli and purified as described (13, 20). Procedures for the purification of factors V and X from human plasma have been described (7, 21). Factors Va and Xa were produced by preparative activation of factor V by thrombin or factor X by RVVXCP followed by re-purification and quality control as previously detailed (7, 21). Recombinant wild type human prothrombin (IIWT), prothrombin containing Gln in place of Arg at position 320 (IIQ320), prothrombin containing Gln in place of Arg at position 271 (IIQ271), and prothrombin containing Gln in place of Arg at both positions 271 and 320 (IIQQ) (Scheme I) were expressed in HEK293 cells on a large scale, purified, and characterized as described (12). Recombinant factor X containing Ala in place of Ser at position 1954 was prepared by site-directed mutagenesis of the cDNA encoding wild type factor X, expressed in HEK293 cells, and purified as described for other recombinant factor X mutants (22, 23). This factor X derivative was activated with RVVXCP and the resulting XaS195A was purified by affinity chromatography using soybean trypsin inhibitor Sepharose (Sigma) (24). N-terminal sequencing and quantitative chemical analysis of 4-carboxyglutamic acid content established that XaS195A and all recombinant derivatives of prothrombin possessed a correctly processed N terminus with 4-carboxyglutamic acid content that was indistinguishable from that of their counterparts purified from plasma. IIQ320 was converted to mIIa and catalytically inactivated by preparative cleavage using ecarin in the presence of FPR-CH2Cl followed by purification as described (12). IIQ320 was conformationally activated using Met-SC1–325-His6 and stabilized in a proteinase-like state by covalent inactivation with FPR-CH2Cl followed by dissociation of Met-SC1–325-His6 and purification of the conformationally stabilized FPR-IIQ320 adduct as described (13). All prothrombin derivatives and XaS195A were exchanged into Assay Buffer by centrifugal gelfiltration before use. Protein concentrations were determined by using the following molecular weights and extinction coefficients (E280, mg−1)cm2 : Xa and XaS195A, 45,300, 1.16 (25); Va, 173,000, 1.78 (26); all recombinant II variants and mIIa, 72,000, 1.47 (27).
Steady State Fluorescence Measurements
All fluorescence measurements were performed using a PTI QuantaMaster fluorescence spectrophotometer (Photon Technology International) using 0.5 × 0.5-cm quartz cells maintained at 25 °C. Samples (0.5 ml) were mixed manually with a polystyrene paddle following additions to the cuvette and before each measurement. Ratiometric emission spectra were collected by scanning the emission monochromator between 350 and 450 nm using λEX = 320 nm with long pass filters (LP 345, CVI Laser) in the emission beam. Equivalence between multiple emission scans was used to rule out trivial effects due to photobleaching during the measurement period. The spectra presented are averages of three emission scans. Ratiometric measurements of fluorescence intensity following incremental additions of titrant were conducted using λEX = 320 nm and λEM = 374 nm with a long pass filter (LP 345, CVI Laser) in the emission beam. Intensities were obtained from the average signal calculated by integration for 30 s.
Binding of pAB to Xa and Prothrombinase
Fluorescence titrations were conducted using four reaction mixtures (0.5 ml each): A, buffer alone; B, 1 μM Xa; C, 1.2 μM Va, 200 μM PCPS; and D, 1 μM Xa, 1.2 μM Va, 200 μM PCPS. Equal microliter additions were made to each sample using a repeating microsyringe (Hamilton) to achieve the indicated concentration of pAB and fluorescence intensity was recorded. All intensities were corrected for dilution (maximum of 7.5%) and the signals from A and C were used to calculate correction factors for the inner filter effect (28, 29). The molar extinction coefficient for pAB at the exciting wavelength (6320 M−1 cm−1) and the fitted effective pathlength were applied to all four sets of measurements to derive the corrected fluorescence signal (FAcorr, FBcorrr, FCcorr, and FDcorr). The signal arising from the interaction between Xa and pAB was obtained from FBcorr − FAcorr and that for the interaction between prothrombinase and pAB was obtained from FDcorr − FCcorr. Each value was then divided by the corrected fluorescence signal from 1 μM pAB to aid the direct comparison of fluorescence intensity observed with different enzyme species. Equivalent experiments were performed with XaS195A.
Displacement of pAB from Prothrombinase by Prothrombin Variants
Three reaction mixtures (0.5 ml each) were used for these measurements: A (no probe), 1 μM XaS195A, 1.2 μM Va, 200 μM PCPS, and 10 μM FPR-CH2Cl; B (no enzyme), 1.2 μM Va, 200 μM PCPS, 10 μM FPR-CH2Cl, and 25 μM pAB; C (experimental), 1 μM XaS195A, 1.2 μM Va, 200 μM PCPS, 10 μM FPR-CH2Cl, and 25 μM pAB. Emission spectra were collected before and after the addition of the indicated concentrations of the prothrombin variant. For titrations, steady state intensity was measured 5 and 10 min after incremental additions of the indicated prothrombin variant. All emission spectra were corrected for dilution (maximum 6%) and spectra from samples B and C were corrected for scatter using reaction A. The resulting spectra from B and C were normalized by setting the peak of spectrum B (free probe) to 1. In intensity titrations, fluorescence intensity was corrected for scatter and expressed in terms of the fluorescence of free pAB using,
| (Eq. 1) |
where the subscripted terms for F indicate the fluorescence intensities measured for reactions A, B, and C. To accommodate minor variations in FOBS/FP,Free from systematic error in experiments conducted over several months, all titration curves were normalized to a reference data set with FOBS/FP,Free = 1.371 in the absence of added prothrombin. Control experiments documented that the inclusion of 10 μM FPR-CH2Cl was without affect on the binding of pAB to XaS195A but was sufficient to abrogate any detectable cleavage of the prothrombin variants by trace contaminating active proteinase over the extended period of the experiment. Titrations were discontinued if the scattering signal was unexpectedly high or if the samples turned visibly turbid. Problems associated with turbidity and high scattering were minimized by the use of PCPS within 2 days of preparation.
Kinetic Studies of IIQ271 Cleavage
Initial velocity studies of IIQ271 cleavage were conducted as previously detailed (12). Reaction mixtures in assay buffer and maintained at 25 °C, containing 0.3 μM IIQ271, 27.5 μM PCPSLUV, 31 nM Va, and increasing concentrations of either IIQQ or IIQ320 were initiated with 0.02 nM Xa. Samples were quenched at 0, 0.5, 1, 1.5, 2, and 3 min and product formation was assessed discontinuously as described (15), to yield the initial, steady state rate of IIQ271 cleavage.
Data Analysis
Representative results from two or more experiments, typically done with different protein preparations, are presented throughout. Data were fitted by the indicated equations using the Levenberg-Marquardt algorithm (30). Fitted constants are presented ±95% confidence limits. Analysis according to Scheme II was performed by combining the numerical solution of multiple equilibria with non-linear error minimization using Dynafit (31), obtained as a generous gift from Petr Kuzmic (BioKin, Pullman, WA). In this case, errors in the parameters reflect linear approximations of 95% confidence limits.
SCHEME II. Equilibria for the binding of active site-directed probe and substrate to prothrombinase.
The protein substrate (S) binds to prothrombinase (E) through an initial exosite binding step determined by KE,S followed by active site engagement to yield ES*. The equilibrium constant for active site docking is given by Ks*. This dimensionless unimolecular equilibrium constant is defined as Ks* = [ES]/[ES*]. The binding of pAB (P) to the active site is determined by KE,P. Based on previous kinetic studies, α is assumed to be indistinguishable from 1 (15).
In all experiments with prothrombinase, the concentrations of Va and PCPS were chosen to saturably incorporate all Xa (or XaS195A) into prothrombinase based on measured equilibrium constants and stoichiometries (32). Therefore, the concentration of prothrombinase was considered equal to the limiting concentration of factor Xa. The rationale for using PCPSLUV in activity measurements at subnanomolar concentrations of enzyme has been described (12). This approach permits the use of saturating concentrations of phospholipid yet minimizes kinetic complexity associated with non-productive binding of substrate to vesicles lacking enzyme (12).
Fluorescence titrations to assess pAB binding assumed a stoichiometry of 1 mol of pAB bound per mol of enzyme at saturation. Analysis as previously described (29) yielded fitted values for Kd and the maximal fluorescence change at saturation (ΔFMAX/FFree), which reflects the -fold increase in fluorescence that accompanies the binding of pAB to enzyme.
Analysis according to Scheme II assumed KE,S = Km (or Ki), with KE,S fixed at 260 nM, which represents an average of the affinity terms listed in Scheme I. KE,P was fixed at the measured value of 50 μM, 1 mol of P was assumed to be bound per mol of E at saturation and α was assumed to be equal to 1 based on previous kinetic studies (15). In addition, the fluorescence yields of P bound to either E or ES were assumed to be equal. Fitting according to Scheme II with these constraints provided estimates for Ks* and the fluorescence signals for free and enzyme-bound P normalized on a molar basis. Division of these two fluorescence yields along with the propagation of the individual errors provided a fitted value for ΔFMAX/FFree equivalent to the term derived from the pAB binding studies. In titrations with a saturable decrease in normalized fluorescence to 1, uncertainty in the stoichiometry for the binding of S to E was accommodated by also fitting the concentration of E. This approach is valid because the fixed concentration of E was substantially greater than KE,S. The stoichiometry determined in this way was fixed in titrations that produced only a small change in fluorescence. Although Ks* was robustly determined within these constraints, the fitted value of Ks* was found to be highly correlated with the assumption that EP and EPS exhibit identical fluorescence properties. For this reason, we have deliberately qualified fitted values of Ks* as upper or lower limit estimates without ascribing significance to fitted confidence limits in this parameter.
RESULTS
Experimental Strategy
We pursued studies with XaS195A and pAB to develop a strategy assessing the ability of the substrate to engage the active site but in the absence of catalysis. This approach is based on the extensive literature in the serine proteinase field establishing that mutation of the catalytic serine (Ser195) or its chemical conversion to dehydroalanine abolishes catalysis without impacting, in a major way, binding interactions at the active site (33–36). The use of pAB as a reversible fluorescent probe for the S1 pocket5 of arginine-specific serine proteinases, including Xa, has been established in numerous studies (28). Because pAB and other ligands cannot simultaneously be bound at the S1 site, probe binding and dissociation, inferred from fluorescence, has been used extensively to characterize reaction pathways between inhibitors and proteinases (28).
The binding of pAB to the active site of Xa in solution or to the active site of factor Xa within prothrombinase was accompanied by a large and saturable increase in probe fluorescence intensity (Fig. 1). Addition of 100 μM FPR-CH2Cl to covalently modify the active site led to a rapid decay in fluorescence denoting the displacement of pAB from the active site of the catalyst (not shown). The assembly of factor Xa into prothrombinase produced a further enhancement in probe fluorescence, suggesting some detectable perturbation in the active site of Xa when it assembles into prothrombinase. The equilibrium dissociation constant inferred for the binding of pAB to Xa (Table 1), was in excellent agreement with published results (28). The assembly of factor Xa into prothrombinase was found to produce a minor but detectable enhancement in pAB binding (Table 1). Equivalent results were obtained with XaS195A either in solution or upon its assembly into prothrombinase with saturating concentrations of Va and PCPS although the enhancement in probe fluorescence was considerably lower (Table 1). These results were unrelated to the inefficient incorporation of XaS195A into prothrombinase as binding studies established that XaS195A was indistinguishable from Xa in its ability to assemble into prothrombinase (not shown). Lower probe fluorescence observed with XaS195A could reflect the contribution of hydrogen bonding between the amino group of the probe and Ser195 absent in this Xa variant (37, 38). These data establish the ability of pAB to bind reversibly to the S1 site of XaS195A within prothrombinase, albeit with lower fluorescence intensity. Substrates or ligands that engage the active site are expected to displace the probe and upon saturation, reduce fluorescence intensity to that of pAB in solution.
FIGURE 1. Binding of pAB to Xa and prothrombinase.

Fluorescence titrations were performed as described under “Experimental Procedures” by the addition of increasing concentrations of pAB to 1 μM Xa (○) or to 1 μM prothrombinase (1 μM Xa, 1.2 μM Va, 200 μM PCPS) (●). Fluorescence was corrected, normalized, and analyzed as described. The lines are drawn using the fitted parameters listed in Table 1.
TABLE 1. Binding of 4-aminobenzamidine to Xa and prothrombinase.
Equilibrium dissociation constants for pAB binding were inferred from fluorescence titrations as illustrated in Fig. 1.
| Enzyme speciesa | Kd ± S.D.b | ΔFMax/FFree ± S.D.c |
|---|---|---|
| μM | ||
| Xa | 85 ± 7.5 | 114 ±3 4 |
| Xa, Va, PCPS | 31 ± 0.8 | 189 ± 1 |
| XaS195A | 117 ± 14 | 23 ± 1 |
| XaS195A, Va, PCPS | 50 ± 5.5 | 30 ± 1 |
Fluorescence titrations were performed using 1.0 μM Xa or XaS195A with no other additions or in the presence of saturating concentrations of Va (1.2 μM) and PCPS (200 μM).
Fitted parameters are listed ±95% confidence limits.
The fitted amplitude of the fluorescence change at saturation reflects the fold increase in fluorescence of free PAB upon binding the enzyme assuming 1 mol of pAB bound per mol of enzyme.
Probe Displacement by Prothrombin Variants
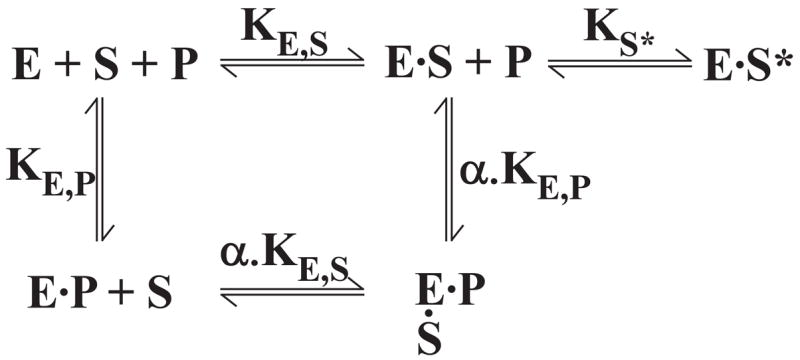
Because prothrombin is cleaved by prothrombinase, at least one of the two cleavage sites in the zymogen must engage the active site of the catalyst. The ability of prothrombin to displace pAB from XaS195A within prothrombinase was assessed using fluorescence emission spectra. Addition of XaS195A to a reaction mixture containing a non-saturating concentration of pAB (0.5 × Kd) and saturating concentrations of Va and PCPS yielded the expected increase in fluorescence intensity and a small blue shift in the emission spectrum (Fig. 2A). Subsequent addition of 1.4 μM IIWT decreased emission intensity to that observed in the absence of XaS195A, implying that IIWT could engage the active site of the catalyst and completely displace pAB (Fig. 2A).The addition of higher concentrations of IIWT did not further decrease probe fluorescence (below). In parallel experiments, addition of the same concentration of IIQQ, which is not cleaved by prothrombinase, produced no obvious evidence for displacement for pAB from the active site of prothrombinase (Fig. 2B). Because substitution of the two P1 Arg residues with Gln in IIQQ is expected to abrogate docking at the active site, these findings lend support to the initial conclusion that the decrease in pAB fluorescence to the value expected for free probe following addition of IIWT (Fig. 2A) reflects the displacement of pAB resulting from the engagement of at least one of the two possible cleavage sites in IIWT with the active site of XaS195A within prothrombinase.
FIGURE 2. Displacement of pAB from prothrombinase.
Technical fluorescence emission spectra (λEX = 320 nm) were obtained using reaction mixtures containing 25 μM pAB, 200 μM PCPS, 1.2 μM Va (P), the reaction mixture in P but also containing 1 μM XaS195A (P + E), and the reaction mixture in P + E also containing 1.4 μM IIWT (panel A, P + E + IIWT), 1.4 μM IIQQ (panel B, p + E + IIQQ), 1.4 μM IIQ271 (panel C, P + E + IIQ271) or 1.4 μM IIQ320 (panel D, P + E + IIQ320). Each spectrum, collected by averaging three scans, was corrected for scattering and normalized by setting the peak fluorescence of reaction P to 1.
This idea was further investigated using prothrombin variants (IIQ271 and IIQ320) in which the two cleavage sites were singly rendered uncleavable (12). Emission spectra obtained following the addition of IIQ271, which retains a cleavable site at Arg320, paralleled those obtained with IIWT (Fig. 2C). In contrast, spectra obtained following the addition of IIQ320, with a cleavable site at Arg271, mirrored those obtained with IIQQ (Fig. 2D). It follows from studies with these variants that in intact prothrombin, the Arg320 site can engage the active site of the catalyst, whereas the Arg271 site cannot do so as readily. It also follows that probe displacement by IIWT can be accounted for by active site docking by the Arg320 site. These conclusions are in agreement with the ability of cleavage at Arg320 and not at Arg271, to quantitatively explain the initial action of prothrombinase on prothrombin (Scheme I, reaction 2 versus 4) (12).
Contribution of Active Site Docking to Substrate Affinity
The inability of IIQQ and IIQ320 to displace pAB from the active site of XaS195A within prothrombinase could arise from a compromised affinity for the enzyme. This possibility was assessed by initial velocity studies of IIQ271 cleavage by prothrombinase in the presence of IIQQ as an inhibitor or IIQ320 as an alternate substrate (Fig. 3). Near equivalent curves were obtained with either prothrombin variant. The data could be adequately described by the rate expression for complete competitive inhibition to yield approximately equal affinities for IIQ271, IIQ320, and IIQQ (Fig. 3). The findings indicate that the affinity of these variants for functional prothrombinase is independent of their ability to engage the active site of prothrombinase as inferred from the displacement of pAB.
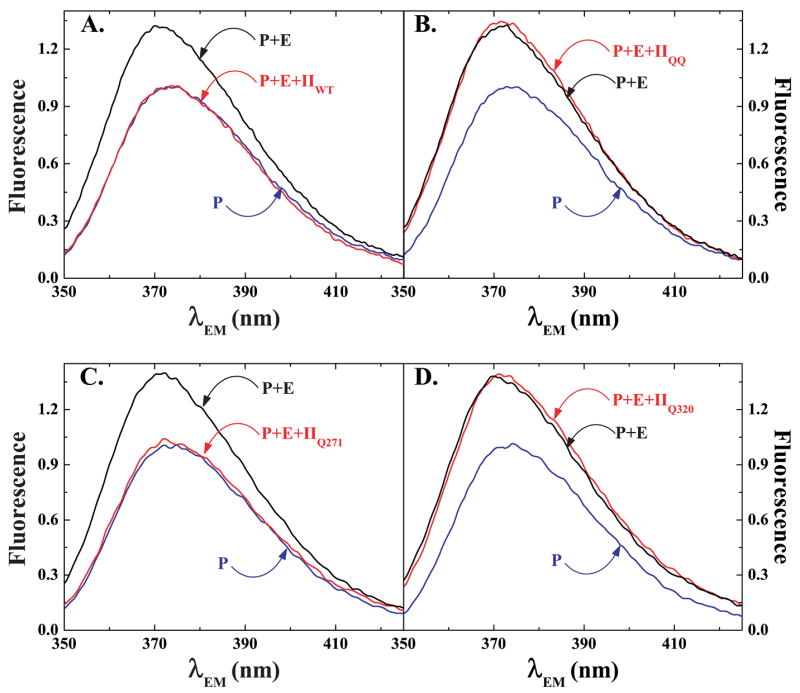
FIGURE 3. Inhibition of IIQ271 cleavage.
Initial velocities for the cleavage of 300 nM IIQ271 by 0.02 nM prothrombinase (0.02 nM Xa, 31 nM Va, 27.5 μM PCPSLUV) were determined in the presence of increasing concentrations of IIQ320 (●) or IIQQ (○). The line is drawn following simultaneous analysis of both data sets with the rate expression for classical competitive inhibition using an assumed value of V/E = 94 s−1 (Scheme I) and fitted values for Km = 231 ± 11 nM and Ki = 336 ± 23 nM.
In classical competitive inhibition, the initial rate for the indicator reaction tends to zero at infinite concentrations of inhibitor or alternate substrate (39). Adequate description of the data by the rate expression for this form of inhibition over a relatively wide range of concentrations of IIQ320 and IIQQ (as high as ~22 × Ki with S = 1.5 × Km) obviates the need to invoke partial inhibition by alternate substrates in this system as has previously been suggested (10).
Analysis of Active Site Docking by the Individual Sites in Intact Prothrombin
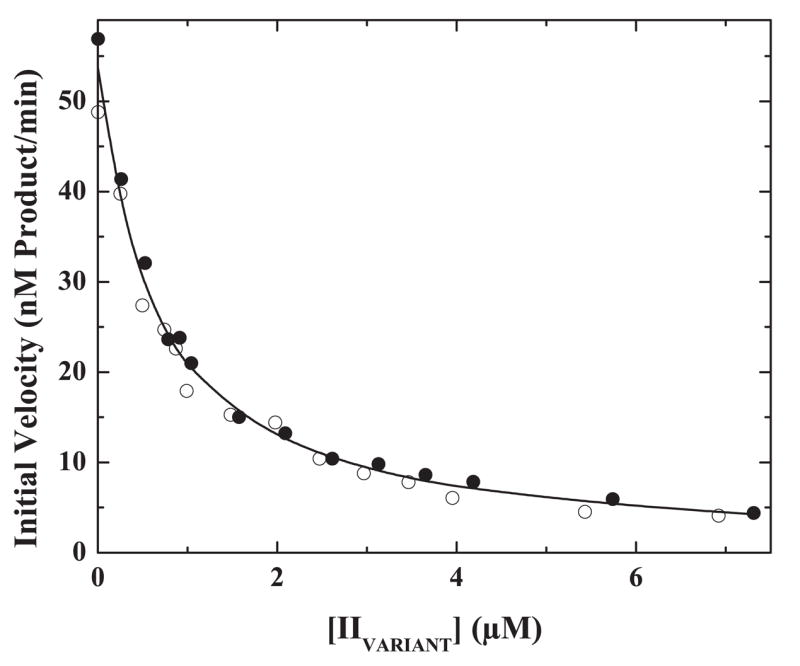
The displacement of pAB from the active site of XaS195A within prothrombinase was studied further with more comprehensive fluorescence titrations (Fig. 4). Increasing concentrations of IIWT and IIQ271 yielded sharp breaking displacement curves that were indistinguishable from each other (Fig. 4). In either case, fluorescence intensity saturated at the signal for free pAB at approximately 1 eq of added substrate (Fig. 4). The data imply that active site docking by either IIWT or IIQ271 and the associated displacement of pAB, results from a tight binding interaction, wherein, 1 mol of either prothrombin variant is bound per mol of prothrombinase.
FIGURE 4. Active site docking by prothrombin and its variants.
Fluorescence intensity normalized to that of free pAB was measured in reaction mixtures containing 25 μM pAB, 1 μM prothrombinase (1 μM XaS195A, 1.2 μM Va, 200 μM PCPS), and 10 μM FPR-CH2Cl with increasing concentrations of IIWT (●), IIQ271 (○), IIQ320 (▲), or IIQQ (△). The line drawn through the IIWT and IIQ271 data sets was obtained by analysis according to Scheme II with the indicated assumptions and the fitted terms ΔFMAX/ΔFFREE = 31.3 ± 1.4, 0.88 ± 0.03 mol of S/mol of E at saturation and Ks* ≤ 0.017. The line drawn through the IIQQ and IIQ320 data sets corresponds to the fitted terms ΔFMAX/ΔFFREE = 34.1 ± 2.3 and Ks* ≥12.3 assuming 0.88 mol of S/mol of E at saturation.
Even though IIQ320 and IIQQ bind to prothrombinase with the same affinity as IIQ271 (above), increasing concentrations of IIQ320 or IIQQ produced only a minor change in fluorescence (Fig. 4). These findings again suggest the inability of these variants to effectively engage the active site of the catalyst (Fig. 4). Whereas IIQQ is not cleaved by prothrombinase and is unlikely to engage the S1 pocket in the enzyme, the Arg271 site in IIQ320 is indeed cleaved by prothrombinase (Scheme I, reaction 4) and therefore must, however poorly, engage the active site of the catalyst. Comparable findings with IIQ320 and IIQQ suggest that any possible active site docking by Arg271 in IIQ320 occurs so weakly so as to be within the precision of the measurement and cannot be distinguished from a completely negative result.
A series of previous studies have established that pAB acts as a classical noncompetitive inhibitor of protein substrate cleavage by prothrombinase. This is because it has no effect on exosite-dependent substrate binding but interferes with the subsequent docking of the substrate with the active site of the enzyme (15). These ideas, illustrated in Scheme II, provide a formal framework for further consideration of the behavior of the prothrombin variants in the pAB displacement studies. Analysis according to Scheme II, using the constraints and assumptions outlined under “Data Analysis, ” provided an adequate description of the data (Fig. 4). Fitting of displacement curves obtained with increasing concentrations of either IIWT or IIQ271 yielded a stoichiometry of near 1 mol of S bound/mol of E at saturation, a ratio of free and bound fluorescence yields that was consistent with the directly measured value and a fitted upper limit estimate of Ks* = 0.02 (Fig. 4). For data obtained with IIQ320 and IIQQ, analysis yielded a lower limit estimate for Ks* = 12 (Fig. 4). These estimates for Ks* provide the quantitative basis for the suggestion that Arg320 in intact prothrombin readily engages the active site of prothrombinase, whereas Arg271 does so poorly, with an estimated 600-fold larger equilibrium constant.
Rescue of the Defective Ability of IIQ320 to Engage the Active Site of Prothrombinase
Prior cleavage of prothrombin at Arg320 enhances the action of prothrombinase on the Arg271 site (Scheme I) (12). The action of prothrombinase on Arg271 can also be enhanced without prior substrate cleavage but by stabilizing the uncleaved zymogen in a proteinase-like state (13). Both approaches were applied to IIQ320 so as to rescue its defective ability to engage the active site of prothrombinase. Ecarin can cleave the 320 site in IIQ320 to form mIIa despite the absence of Arg at P1 (12). IIQ320 was converted to mIIa with ecarin, covalently inactivated with FPR-CH2Cl, and purified (Fig. 5, inset). IIQ320 was also conformationally activated using Met-SC1–325-His6, covalently modified with FPR-CH2Cl, and re-purified following dissociation of the staphylocoagulase fragment to yield an uncleaved IIQ320 species (FPR-IIQ320) (Fig. 5, inset), stabilized in a proteinase-like state by covalently bound inhibitor (13). Fluorescence titrations revealed that mIIa produced from IIQ320 could robustly displace pAB from the active site of XaS195A within prothrombinase (Fig. 5). Analysis according to Scheme II yielded an adequate description of the data and fitted parameters that were similar to those obtained with IIWT and IIQ271 with an upper limit estimate for Ks* = 0.02 (Figs. 4 and 5). The weak ability of the Arg271 site in IIQ320 to engage the active site of the catalyst was also enhanced in the uncleaved FPR-IIQ320 derivative, although perhaps not to the same extent as seen following prior cleavage at the 320 site (Fig. 5). Thus, cleavage at the 320 site in IIQ320 increases the affinity of the Arg271 site for active site docking by an estimated 600-fold. Enhanced active site binding by the Arg271 site can be recapitulated by the uncleaved derivative stabilized in a proteinase-like state. It therefore follows that appropriate presentation of the Arg271 site for active site docking requires substrate in the proteinase state or in a proteinase-like configuration.
FIGURE 5.
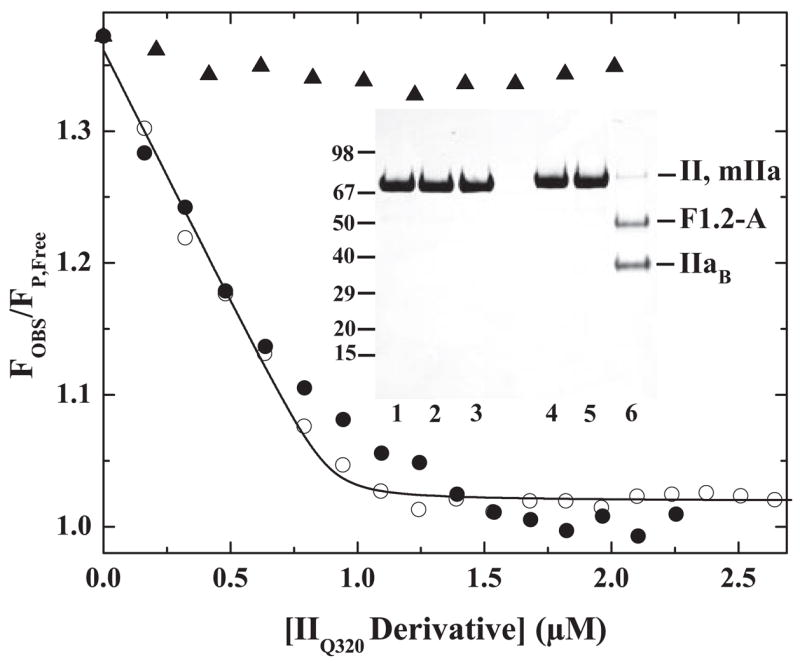
Active site engagement by Arg271 upon proteinase formation. Normalized fluorescence intensity of pAB was measured in reaction mixtures containing 25 μM pAB, 1 μM prothrombinase (1 μM XaS195A, 1.2 μM Va, 200 μM PCPS), and 10 μM FPR-CH2Cl with increasing concentrations of FPR-II320 (●) or mIIa produced from IIQ320 (○). Data obtained with increasing concentrations of IIQ320 (▲) are provided for reference. The line drawn by analysis of data obtained with mIIa according to Scheme II and the fitted terms ΔFMAX/ΔFFREE = 30.8 ± 1.1, 0.89 ± 0.02 mol of S/mol of E at saturation, and Ks* ≤ 0.018. Inset, SDS-PAGE analysis of IIQ320 (lanes 1 and 4), FPR-IIQ320 (lanes 2 and 5), and mIIa produced from IIQ320 (lanes 3 and 6) either before (lanes 1–3) or after (lanes 4 – 6) disulfide bond reduction. Approximately 5 μg of protein was loaded per lane.
DISCUSSION
The use of XaS195A and fluorescent S1 site probe, pAB, has permitted the investigation of the ability of prothrombin and its derivatives to engage the active site of factor Xa within prothrombinase but in the absence of catalysis. In intact prothrombin, structures flanking the Arg320 cleavage site can effectively bind the active site of prothrombinase and displace pAB. In contrast, structures flanking the Arg271 cleavage site do not readily engage the active site of the catalyst. Prior cleavage of the zymogen at Arg320 or the stabilization of otherwise uncleaved prothrombin in the proteinase-like state rescues the defective ability of the Arg271 site to dock at the active site of prothrombinase. Qualitative interpretation of these findings provides the model-independent conclusion that the ability of the individual cleavage sites to dock at the active site of the enzyme, a prerequisite for catalysis, is dictated by whether the substrate is in the zymogen or in the proteinase configuration. This conclusion now establishes a direct relationship between substrate binding at the active site and the perceived rate constant for catalysis (Scheme I). It also provides a physical explanation for the largely ordered action of prothrombinase on prothrombin.
The variants IIQQ, IIQ271, and IIQ320 all bind in a mutually exclusive manner to prothrombinase with equal affinity despite their differential abilities to engage the active site of the enzyme. Thus, the energetics of active site docking contributes undetectably to substrate affinity. This conclusion verifies a key, yet previously untested, prediction that arises from the multistep pathway for substrate recognition by prothrombinase initially proposed from kinetic studies (6). Accordingly, active site engagement inferred from pAB displacement can adequately be accounted for by the reactions illustrated in Scheme II assuming directly measured values of KE,S and KE,P. This quantitative analysis establishes that approximately 1 mol of substrate is bound per mol of prothrombinase at saturation and provides measured estimates for Ks*, the unimolecular equilibrium constant for active site engagement by the individual cleavage sites for the substrate in either the zymogen or proteinase configuration.
Limitations arising from the small fluorescence change exhibited upon the binding of pAB to XaS195A and the need for large amounts of recombinant protein precluded more comprehensive studies to establish Ks* reliably. Nevertheless, the present estimates differ considerably from those provided by initial velocity studies with bovine prothrombinase using proteolytic derivatives of prothrombin lacking the membrane binding domain of the substrate (14, 15). Protein-membrane and possibly protein-protein interactions mediated by the fragment 1 domain of the substrate, by definition, provide additional exosite interactions beyond those observed in derivatives lacking fragment 1. Such interactions likely require the inclusion of additional unimolecular binding steps in Scheme II to explain the behavior of intact prothrombin. Thus, although the simplified Scheme II, developed from previous studies, can adequately describe the data, fitted values of Ks* are probably complex functions of two or more unimolecular binding steps. This possibility represents one likely explanation for the discrepancy in values of Ks* in this and previous studies (14, 15). Other possible contributing factors to this discrepancy include differences in the human versus bovine systems or a failure of the rapid equilibrium assumption used to interpret constants derived from kinetic studies.
In contrast to the interpretations outlined here, it has alternately been proposed that the recognition and cleavage of the two bonds in prothrombin is accomplished by two different isoforms of prothrombinase that are differentially specific for the two sites (10, 11). This alternate interpretation, which requires that the affinity of prothrombinase for the substrate is cleavage site-dependent, is inconsistent with a series of kinetic findings and with the fact that IIQQ is a classical competitive inhibitor of all possible half-reactions of prothrombin activation (12). Nevertheless, the approaches used in the present work permit us to differentiate between these two opposing ideas in the absence of uncertainties in kinetic interpretation or concern regarding slowly equilibrating forms of the enzyme. In the limit that recognition of the two cleavage sites is accomplished by two distinct and equal populations of enzyme that do not interconvert, either cleavage site mutant (IIQ271 or IIQ320) would be expected to displace only 50% of the enzyme-bound pAB. Maximum displacement would be expected to occur at 0.5 mol of variant/mol of prothrombinase. If the two enzymic isoforms were to readily interconvert with an equilibrium constant near 1, as has been suggested (10, 11), either cleavage site mutant (IIQ271 or IIQ320) would be expected to displace all enzyme-bound pAB. These and other related models requiring two enzyme forms are inconsistent with our observations (Figs. 4 and 5), and thus can be excluded by the findings of the present work.
The enzymology literature is replete with examples in which knowledge-based structural alterations in either the enzyme or the substrate expected to perturb substrate binding, yield disproportionately large changes in the rate constant for catalysis relative to substrate affinity. Such observations usually implicate the Circe Effect, proposed by Jencks (16), which encompasses a series of mechanisms by which intrinsic substrate binding energy may be coupled to drive ground state destabilization, decrease ΔG‡, and improve catalysis. However, experimental approaches to establish the quantitative contributions of such mechanisms remain a challenge and computational approaches have yielded controversial solutions (40, 41).
When taken together, the kinetic and binding approaches with prothrombinase establish a direct relationship between substrate binding at the active site and the perceived rate constant for catalysis. The kinetic explanation for this relationship lies in the ordered multistep pathway for protein substrate recognition that results from exosite binding followed by active site engagement, which occurs in a unimolecular step prior to catalysis (6). This result is generalizable to any reaction in which substrate binds to the enzyme through stepwise interactions although the precise contribution of any unimolecular binding step to rate at saturating substrate would depend on the magnitude of the rate and equilibrium dissociation constants for the individual steps. Such a mechanism relating substrate binding energies to catalysis could be considered potentially trivial because it does not arise from ground state destabilization and instead from interpretation of kinetics with incomplete consideration of the full mechanism for substrate binding. It may also be expected to be most relevant to reaction systems in which the substrate is a macromolecule and can engage the enzyme in an extended way. Nevertheless, because substrate affinity is typically determined by multiple bonds between the substrate and enzyme even in small substrates, it is unlikely that all such contacts occur simultaneously or that their stepwise formation is accounted for in most interpretations. Thus, this relationship between substrate binding and the perceived rate constant for catalysis may apply widely to interpretations of structure-function relationships in enzyme-catalyzed reactions.
For enzymes that act on macromolecular substrates, the relationship between binding and the perceived rate constant for catalysis is likely to greatly impact interpretations when the likelihood of substrate engagement through extended interactions is not fully accounted for. An illustration of such problems is provided by the fact that the Arg271 site in intact prothrombin is cleaved poorly because the substrate is technically non-productively bound to the enzyme. Yet, comparison of the steady state kinetic constants for cleavage at the individual sites provides no indication of this fact based on the definition of non-productive binding developed to explain the behavior of simpler enzyme systems (42).
Numerous studies have established the predominant role for exosite binding in determining the affinity of prothrombinase for its protein substrate (6). This strategy plays a major role in restricting the action of prothrombinase to prothrombin or its derivatives despite the fact that the active site of factor Xa, within the enzyme complex, can accommodate a wide variety of peptidyl sequences including those found at the activation sites of the other coagulation zymogens (7). By equilibrium binding studies, we now directly show that active site docking by the substrate that follows exosite binding enforces an additional level of specificity evident in the ability of prothrombinase to discriminate between the two cleavage sites in the substrate. Consequently, the largely ordered action of prothrombinase on prothrombin arises from differences in Vmax and not Km for the individual cleavage reactions related to the differential ability of the individual cleavage sites to engage the active site of the catalyst depending on whether the substrate is a zymogen or a proteinase. This guides the activation pathway via the formation of mIIa, a proteinase with a different spectrum of biological activities than thrombin (5). Geometric constraints imposed by exosite-dependent substrate tethering likely play a significant role in affecting active site docking by the two cleavage sites that are expected to be ~36 Å apart in the zymogen and that probably reposition following initial cleavage at the Arg320 site. This idea highlights a testable physical correlate of the findings in this and in our previous work (13).
Exosite-dependent substrate recognition is increasingly evident in the action of the other enzymes of coagulation on their protein substrates (6). Furthermore, many reactions of coagulation require multiple cleavage events in the substrate that frequently occur in a seemingly ordered fashion (2). Thus, the two step “dock and lock” strategy for substrate recognition and associated consequences delineated for prothrombin cleavage by prothrombinase may also apply to explain biological specificity and function in the other coagulation reactions.
Footnotes
This work was supported by National Institutes of Health Grants HL-74124 (to S. K., and R. M. C.), HL-47465 (to S. K.), HL-038779 (to P. E. B.), and HL-071544 (to P. E. B.).
Residue numbers in prothrombin represent those obtained by consecutive numbering of the 579 residues in the mature protein.
The abbreviations used are: mIIa, meizothrombin; FPR-CH2Cl, D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone; FPR-IIQ320, IIQ320 stabilized in the proteinase-like state with FPR-CH2Cl following conformational activation by staphylocoagulase; IIQ271, prothrombin with Arg271 replaced with Gln; IIQ320, prothrombin with Arg320 replaced with Gln; IIQQ, prothrombin containing Gln replacing both Arg271 and Arg320; IIWT, recombinant wild type human prothrombin; Met-SC1–325His6, recombinant fragment containing an N-terminal methionine, residues 1–325 of staphylocoagulase followed by a His tag; pAB, 4-aminobenzamidine; PC, L-α-phosphatidylcholine; PS, L-α-phosphatidylserine; PCPS, small unilamellar vesicles composed of 75% (w/w) PC and 25% (w/w) PS; PCPSLUV, large unilamellar vesicles containing 75% (w/w) PC and 25% (w/w) PS; RVVXCP, factor X activating proteinase from Russell’s viper venom; XaS195A, recombinant human factor Xa containing Ala in place of the catalytic Ser195; HEK, human embryonic kidney.
Residue 195 in factor Xa is identified based on chymotrypsinogen numbering of the catalytic domain sequence (43).
Nomenclature of Schechter and Berger (44).
References
- 1.Jackson CM, Nemerson Y. Annu Rev Biochem. 1980;49:765–811. doi: 10.1146/annurev.bi.49.070180.004001. [DOI] [PubMed] [Google Scholar]
- 2.Mann KG, Jenny RJ, Krishnaswamy S. Annu Rev Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 3.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 4.Camire RM, Pollack ES. In: Hemostasis and Thrombosis, Basic Principles and Clinical Practice. Colman RW, Marder VJ, Clowes AJ, George JN, Goldhaber SZ, editors. Lippincott Williams & Wilkins; Philadelphia: 2006. pp. 59–89. [Google Scholar]
- 5.Jenny NS, Lundblad RL, Mann KG. In: Hemostasis and Thrombosis, Basic Principles and Clinical Practice. Colman RW, Marder VJ, Clowes AJ, George JN, Goldhaber SZ, editors. Lippincott Williams & Wilkins; Philadelphia: 2006. pp. 193–213. [Google Scholar]
- 6.Krishnaswamy S. J Thromb Haemostasis. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 7.Orcutt SJ, Pietropaolo C, Krishnaswamy S. J Biol Chem. 2002;277:46191–46196. doi: 10.1074/jbc.M208677200. [DOI] [PubMed] [Google Scholar]
- 8.Krishnaswamy S, Church WR, Nesheim ME, Mann KG. J Biol Chem. 1987;262:3291–3299. [PubMed] [Google Scholar]
- 9.Walker RK, Krishnaswamy S. J Biol Chem. 1994;269:27441–27450. [PubMed] [Google Scholar]
- 10.Brufatto N, Nesheim ME. J Biol Chem. 2003;278:6755–6764. doi: 10.1074/jbc.M206413200. [DOI] [PubMed] [Google Scholar]
- 11.Kim PY, Nesheim ME. J Biol Chem. 2007 August 28; doi: 10.1074/jbc.M701781200. [DOI] [PubMed] [Google Scholar]
- 12.Orcutt SJ, Krishnaswamy S. J Biol Chem. 2004;279:54927–54936. doi: 10.1074/jbc.M410866200. [DOI] [PubMed] [Google Scholar]
- 13.Bianchini EP, Orcutt SJ, Panizzi P, Bock PE, Krishnaswamy S. Proc Natl Acad Sci U S A. 2005;102:10099–10104. doi: 10.1073/pnas.0504704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boskovic DS, Krishnaswamy S. J Biol Chem. 2000;275:38561–38570. doi: 10.1074/jbc.M006637200. [DOI] [PubMed] [Google Scholar]
- 15.Krishnaswamy S, Betz A. Biochemistry. 1997;36:12080–12086. doi: 10.1021/bi970979+. [DOI] [PubMed] [Google Scholar]
- 16.Jencks WP. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 17.Jencks WP. Methods Enzymol. 1989;171:145–163. doi: 10.1016/s0076-6879(89)71010-7. [DOI] [PubMed] [Google Scholar]
- 18.Evans SA, Olson ST, Shore JD. J Biol Chem. 1982;257:3014–3017. [PubMed] [Google Scholar]
- 19.Furie BC, Furie B. Methods Enzymol. 1976;45:191–205. doi: 10.1016/s0076-6879(76)45019-x. [DOI] [PubMed] [Google Scholar]
- 20.Panizzi P, Friedrich R, Fuentes-Prior P, Kroh HK, Briggs J, Tans G, Bode W, Bock PE. J Biol Chem. 2006;281:1169–1178. doi: 10.1074/jbc.M507955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baugh RJ, Krishnaswamy S. J Biol Chem. 1996;271:16126–16134. doi: 10.1074/jbc.271.27.16126. [DOI] [PubMed] [Google Scholar]
- 22.Camire RM. J Biol Chem. 2002;277:37863–37870. doi: 10.1074/jbc.M203692200. [DOI] [PubMed] [Google Scholar]
- 23.Camire RM, Larson PJ, Stafford DW, High KA. Biochemistry. 2000;39:14322–14329. doi: 10.1021/bi001074q. [DOI] [PubMed] [Google Scholar]
- 24.Bock PE, Craig PA, Olson ST, Singh P. Arch Biochem Biophys. 1989;273:375–388. doi: 10.1016/0003-9861(89)90496-7. [DOI] [PubMed] [Google Scholar]
- 25.Di Scipio RG, Hermodson MA, Davie EW. Biochemistry. 1977;16:5253–5260. doi: 10.1021/bi00643a015. [DOI] [PubMed] [Google Scholar]
- 26.Toso R, Camire RM. J Biol Chem. 2004;279:21643–21650. doi: 10.1074/jbc.M402107200. [DOI] [PubMed] [Google Scholar]
- 27.Mann KG, Elion J, Butkowski RJ, Downing M, Nesheim ME. Methods Enzymol. 1981;80:286–302. doi: 10.1016/s0076-6879(81)80025-0. [DOI] [PubMed] [Google Scholar]
- 28.Craig PA, Olson ST, Shore JD. J Biol Chem. 1989;264:5452–5461. [PubMed] [Google Scholar]
- 29.Betz A, Vlasuk GP, Bergum PW, Krishnaswamy S. Biochemistry. 1997;36:181–191. doi: 10.1021/bi962060g. [DOI] [PubMed] [Google Scholar]
- 30.Bevington PR, Robinson KD. Data Reduction and Error Analysis for the Physical Sciences. 2. McGraw-Hill; New York: 1992. pp. 161–166. [Google Scholar]
- 31.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 32.Buddai SK, Toulokhonova L, Bergum PW, Vlasuk GP, Krishnaswamy S. J Biol Chem. 2002;277:26689–26698. doi: 10.1074/jbc.M202507200. [DOI] [PubMed] [Google Scholar]
- 33.Ako H, Foster RJ, Ryan CA. Biochemistry. 1974;13:132–139. doi: 10.1021/bi00698a021. [DOI] [PubMed] [Google Scholar]
- 34.Vincent JP, Peron-Renner M, Pudles J, Lazdunski M. Biochemistry. 1974;13:4205–4211. doi: 10.1021/bi00717a023. [DOI] [PubMed] [Google Scholar]
- 35.Hosokawa K, Ohnishi T, Shima M, Nagata M, Koide T. Biochem J. 2001;354:309–313. doi: 10.1042/0264-6021:3540309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krem MM, Di Cera E. Biophys Chem. 2003;100:315–323. doi: 10.1016/s0301-4622(02)00289-2. [DOI] [PubMed] [Google Scholar]
- 37.Bode W, Schwager P. J Mol Biol. 1975;98:693–717. doi: 10.1016/s0022-2836(75)80005-2. [DOI] [PubMed] [Google Scholar]
- 38.Bajaj SP, Schmidt AE, Agah S, Bajaj MS, Padmanabhan K. J Biol Chem. 2006;281:24873–24888. doi: 10.1074/jbc.M509971200. [DOI] [PubMed] [Google Scholar]
- 39.Segel IH. Enzyme Kinetics, Behaviour and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. 1. John Wiley & Sons; New York: 1975. pp. 100–125. [Google Scholar]
- 40.Warshel A, Florian J. Proc Natl Acad Sci U S A. 1998;95:5950–5955. doi: 10.1073/pnas.95.11.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warshel A, Florian J, Strajbl M, Villa J. Chembiochem. 2001;2:109–111. doi: 10.1002/1439-7633(20010202)2:2<109::AID-CBIC109>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 42.Fersht A. Enzyme Structure and Mechanism. 1. W. H. Freeman and Co; San Franciso: 1977. pp. 109–111. [Google Scholar]
- 43.Padmanabhan K, Padmanabhan KP, Tulinsky A, Park CH, Bode W, Huber R, Blankenship DT, Cardin AD, Kisiel W. J Mol Biol. 1993;232:947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 44.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]