

Abstract
The role of the streptokinase (SK) α-domain in plasminogen (Pg) and plasmin (Pm) interactions was investigated in quantitative binding studies employing active site fluorescein-labeled [Glu]Pg, [Lys]Pg, and [Lys]Pm, and the SK truncation mutants, SK-(55–414), SK-(70–414), and SK-(152–414). Lysine binding site (LBS)-dependent and -independent binding were resolved from the effects of the lysine analog, 6-aminohexanoic acid. The mutants bound indistinguishably, consistent with unfolding of the α-domain on deletion of SK-(1–54). The affinity of SK for [Glu]Pg was LBS-independent, and although [Lys]Pg affinity was enhanced 13-fold by LBS interactions, the LBS-independent free energy contributions were indistinguishable. α-Domain truncation reduced the affinity of SK for [Glu]Pg 2–7-fold and [Lys]Pg ≤2-fold, but surprisingly, rendered both interactions near totally LBS-dependent. The LBS-independent affinity of SK for [Lys]Pm, 3000-fold higher compared with [Lys]Pg, was reduced dramatically by α-domain truncation. Thermodynamic analysis demonstrates that the SK α-domain contributes substantially to affinity for all Pg/Pm species solely through LBS-independent interactions, and that the higher affinity of SK for [Lys]Pm compared with [Lys]Pg involves all three SK domains. The residual affinity of the SK βγ-fragment for all Pg/Pm species was increased by an enhanced contribution to complex stability from LBS-dependent interactions or free energy coupling between LBS-dependent and -independent interactions. Redistribution of the free energy contributions accompanying α-domain truncation demonstrates the interdependence of SK domains in stabilizing the SK-Pg/Pm complexes. The flexible segments connecting the SK α, β, and γ domains allow their rearrangement into a distinctly different bound conformation accompanying loss of the constraint imposed by interactions of the α-domain.
Streptokinase (SK)1 secreted by Streptococcus equisimilis activates the human fibrinolytic system by converting the zymogen, plasminogen (Pg) into the clot-dissolving proteinase, plasmin (Pm) (1). This activity is the basis for the use of SK as a thrombolytic drug for treatment of myocardial infarction. SK activates Pg by a unique mechanism initiated by rapid and reversible formation of an SK·Pg* complex in which the Pg catalytic site is activated conformationally (2–6). Recent equilibrium binding and kinetic studies of the SK-initiated pathway of Pm formation support a mechanism in which rapidly formed SK·Pg* binds Pg as a specific substrate, followed by intermolecular cleavage to form initial Pm (3, 4). SK has ~800- and 11,000-fold higher affinity for Pm compared with native [Lys]Pg and [Glu]Pg, respectively, which results in displacement of Pg from SK·Pg* and formation of SK·Pm, that then catalyzes complete conversion of free Pg to Pm (3, 4). The Pg activation mechanism is modulated by lysine-binding site (LBS) interactions between SK and kringle domains of Pg and Pm (3, 4, 7–13). LBS interactions are not absolutely required, but enhance the affinity of SK·Pg* and SK·Pm catalytic complex formation and facilitate binding of Pg as a substrate of these complexes (3, 4, 8, 11, 12, 14–16).
Many studies have sought to elucidate structure-function relationships involved in SK-initiated Pg activation. As revealed by the crystal structure of SK bound to the catalytic domain of Pm (micro-Pm), SK consists of three homologous, independently folded domains (α-(1–146), β147–290), and γ-(291–414)) connected by flexible linking segments (17). In solution, free SK is highly flexible, with the three domains able to assume many different conformations due to the linking sequences (18). SK assumes a stable, highly structured configuration on binding to micro-Pm, in which the domains surround the catalytic site (17), implying domain arrangement on binding. Several studies now indicate that the structural basis for conformational activation of Pg is insertion of the N-terminal Ile1 residue of SK into the N-terminal binding cleft of Pg, forming the critical salt bridge with Asp194 (chymotrypsinogen numbering) that triggers the activating conformational change (19–22). Structural proof of this “molecular sexuality” mechanism is lacking for SK but has been demonstrated recently for the conformational prothrombin activator, staphylocoagulase (23).
Various functions in Pg activation have been attributed to each of the SK domains based on studies of isolated proteolytic fragments, recombinant domains, two domain constructs, and point mutants. Collectively, the studies have yielded a complex mixture of functional effects and a variety of different interpretations. In addition to providing Ile1, the α-domain has been concluded to mediate Pg substrate binding (15, 24–27), LBS interactions with Pg kringles (25), and the SK-(1–59) segment produced by Pm cleavage at Lys59 has been reported to bind two Pg molecules (26). SK-(1–59) remains associated with the rest of the α-domain and the two-chain SK retains activity in Pg activation (10, 28–30) and affinity for Pm (16). When SK-(1–59) is removed, the α-domain unfolds and activity is lost (10, 26, 31). All three SK domains have been concluded to mediate LBS interactions (10, 12–15, 25, 32). The isolated α-domain has been reported to support weak activation of Pg (15, 32) and to bind Pg kringles (25). The crystal structure of the α-domain·micro-Pg complex shows that it binds in a different location than it does in the full-length SK·micro-Pm complex (24), indicating alternative binding modes for the isolated domain.
The function of the α-domain has become of greater interest based on the finding that an SK-(1–59) deletion mutant (SK-(60–414)) is a more fibrin specific Pg activator (25, 33, 34). SK-(60–414) exhibits greatly reduced activity in [Glu]Pg activation in the absence of fibrin fragments, but enhanced activity in their presence (25, 33, 34). Because of deletion of SK-(1–59), the mutant does not conformationally activate Pg and requires Pm to proteolytically activate Pg. Pm in the presence of SK-(60–414) or point mutants in SK-(1–59) has also been shown to be more susceptible to inhibition by antiplasmin compared with SK·Pm (25, 33). Further studies of the fibrinolytic activity of SK-(60–414) showed that it was greater than SK at high but not low SK concentrations because of the relatively higher activity of native SK in activating Pg in plasma, resulting in increased fibrinolysis by depletion of fibrinogen and Pg (33). The mechanisms underlying the functions of the SK α-domain and the behavior of SK-(60–414) are not understood clearly.
The contribution of the α-domain to the affinity of SK for Pg and Pm has not been characterized quantitatively. In studies with active-site fluorescein-labeled [Glu]Pg, [Lys]Pg, and [Lys]Pm analogs, SK binds weakly to [Glu]Pg, 13–16-fold tighter to [Lys]Pg, and 3300-fold tighter to [Lys]Pm (3, 9, 11, 16). [Glu]Pg binding is LBS-independent, whereas the affinities of SK for [Lys]Pg and [Lys]Pm are decreased 13–30-fold by saturating concentrations of the lysine analog, 6-aminohexanoic acid (6-AHA) (3, 9, 11, 16). The role of the α-domain in the intrinsic differences in affinity and the differential effects of 6-AHA is unknown. The present studies employing the fluorescein-labeled Pg and Pm analogs were undertaken to address the role of LBS-independent and -dependent interactions of the α-domain in the affinity for Pg and Pm species, and to provide information necessary to interpret more clearly the functions of the α-domain in Pg activation. The results demonstrate a major role for the α-domain in LBS-independent interactions with all Pg and Pm species and no evidence for direct participation of this domain in kringle-mediated LBS interactions. Thermodynamic analysis of the interactions of the SK α-domain truncation mutants, SK-(55–414), SK-(70–414), and the complete domain deletion mutant, SK-(152–414) demonstrates that loss of binding free energy from LBS-independent interactions of the α-domain is counteracted by expression of an enhanced contribution from LBS-dependent βγ-domain interactions or free energy coupling between LBS-independent and -dependent binding of the βγ-domain. The results demonstrate the interdependence of the interactions of the individual α-, β- and γ-domains of intact SK and the role of coupling of LBS-independent and -dependent interactions in determining the structure of SK bound in the SK·Pg* and SK·Pm complexes. In keeping with the flexibility between SK domains, deletion of the α-domain results in a different bound conformation of the βγ-domains. The results support the conclusion that loss of LBS-independent interactions mediated by the α-domain with the catalytic domain of Pg and Pm results in the βγ-fragment binding in a conformation that more effectively interacts with LBS of Pg and Pm kringles. The redistribution of LBS-independent and -dependent contributions to affinity and the resulting differences between the structures of SKα βγ·Pg*/Pm and SKβγ·Pg/Pm complexes provide new insight into the complex relationship between SK structure and function in Pg activation.
EXPERIMENTAL PROCEDURES
Protein Purification and Characterization
[Glu]Pg carbohydrate variant 2 was purified from human plasma (35, 36). [Lys]Pg was generated by incubation of 40 μM [Glu]Pg with 2 μM Pm in 50 mM Tris-Cl, 20 mM L-lysine, 0.1 M NaCl, pH 9.0, for 30 min at 25 °C, and isolated by affinity chromatography on soybean trypsin inhibitor-agarose and aminohexyl-agarose as described previously (3, 11). [Lys]Pm was prepared by activation of 10 μM [Glu]Pg with 90 units/ml urokinase (Calbiochem) in 10 mM MES, 10 mM Hepes, 0.15 M NaCl, 20 mM 6-AHA, 1 mg/ml polyethylene glycol 8000 (PEG), pH 7.4, at 25 °C, and purified by chromatography on soybean trypsin inhibitor-agarose (9, 37).
Recombinant wild-type SK (wtSK) and SK-(55–414) were prepared as described previously (19, 38) with minor modifications. Ammonium sulfate precipitation was omitted, and following affinity chromatography on Pm immobilized on Sulfolink (Pierce), SK was dialyzed into 50 mM Hepes, 50 mM NaCl, pH 7.4 and was further purified by Resource Q FPLC. SK was eluted with a salt gradient from 0.05 to 1 M, fractions containing the SK species were pooled and dialyzed against 50 mM Hepes, 0.125 M NaCl, pH 7.4. The SK mutants in which the first 69 and 151 N-terminal residues were deleted (SK-(70–414) and SK-(152–414)) were generated by PCR. SK-(70–414) and SK-(152–414) were amplified with sense primers CATATGTCACATAAACTTGAGAAAGCTGACTTACTAAGGC and CATATGCAAAACCAAGCGAAATCTGTTGATGTGG, respectively, and with the antisense primer, GGATCCTTATTTGTCGTTAGGGTTATCAGGTATAAGTGTCCC. Template DNA was pET3a/SKC (38). The resulting products were digested with NdeI and BamHI, and were ligated into similarly treated pET20b(+). Protein expression and purification for SK-(70–414) and SK-(152–414) were performed as described previously (19).
Protein concentrations were determined from the A280 nm absorbance with the following absorption coefficients ((mg/ml)−1 cm−1) and molecular weights: wtSK 0.95 and 47,000 (39, 40); [Glu]Pg 1.69 and 92,000; [Lys]Pg 1.69 and 84,000; [Lys]Pm 1.9 and 84,000 (35, 41, 42); SK-(55–414) 0.80 and 40,800; SK-(70–414) 0.80 and 38,000; SK-(152–414) 0.80 and 35,500. Absorption coefficients for the SK α-domain deletion mutants were determined as described by Gill and von Hippel (43).
Preparation of Active Site-labeled [Lys]Pm
ATA-FFR-CH2Cl was prepared as described previously (44, 45). ATA-FFR-[Lys]Pm was prepared by incubating 19 μM active site-titrated Pm with a 6-fold excess of inhibitor in 0.1 M Hepes, 0.3 M NaCl, 1 mM EDTA, 10 mM 6-AHA, 1 mg/ml PEG, pH 7.0, for 1 h at 25 °C, until activity was < 0.01% of the initial value. ATA-FFR-[Lys]Pm was dialyzed overnight against 500 volumes of 50 mM Hepes, 0.3 M NaCl, 1 mM EDTA, pH 7.0, at 4 °C. Incorporation of inhibitor measured by burst reaction with 5,5′-dithiobis(2-nitrobenzoic acid) (45, 46), gave a stoichiometry of 0.92 mol of thioester/mol of Pm active sites. For labeling with fluorescein, a 7-ml reaction of 18 μM ATA-FFR-Pm with a 5-fold excess of 5-(iodoacetamido)-fluorescein (Molecular Probes) was initiated by addition of 0.1 M NH2OH, incubated at 25 °C in the dark for 1 h, and excess fluorophore removed by chromatography on a 25 ml column of Sephadex G-25 (superfine). Purified [5F]FFR-[Lys]Pm was dialyzed against 500 volumes of 50 mM Hepes, 0.125 M NaCl, 1 mM EDTA, 1 mg/ml PEG, pH 7.4 buffer at 4 °C in the dark, overnight. Quantitation of fluorescein incorporation as described previously (46) gave stoichiometries of 0.88–0.99 mol fluorescein/mol Pm active sites.
Preparation of Catalytic Site-labeled Pg
[Glu]Pg and [Lys]Pg were labeled at the catalytic site, separated from free dye on Sephadex G-25, and purified by affinity chromatography on SK-Affi-Gel and Pm-Sulfolink as described previously, with the addition of 20 mM 6-AHA to all reaction buffers (9, 11). Stoichiometries of probe incorporation were 0.8–1.0 mol fluorescein/mol of Pg for [5F]FFR-[Glu]Pg, and [5F]FFR-[Lys]Pg.
Fluorescence Studies
Fluorescence titrations of [5F]FFR-[Glu]Pg, [5F]FFR-[Lys]Pg, and [5F]FFR-Pm with recombinant SK were done by serial addition of small volumes to labeled Pg or Pm in 50 mM Hepes, 0.125 M NaCl, 1 mM EDTA, 1 mg/ml PEG, 1 mg/ml bovine serum albumin, 1 μM FFR-CH2Cl, pH 7.4, with and without 10 mM 6-AHA. Titrations were performed with an SLM 8100 spectrofluorometer in the ratio mode, using polyethylene glycol 20,000-coated acrylic cuvettes. Excitation and emission wavelengths were 500 nm and 516 nm, respectively (8- or 16-nm bandpass). Changes in fluorescence were monitored after 7 min of equilibration, and expressed as the fractional change of the initial fluorescence (ΔF/Fo = (Fobs − Fo)/Fo). Titrations were analyzed by non-linear least-squares fitting of the quadratic binding equation, with the maximum fluorescence change (ΔFmax/Fo) and the dissociation constant (KD) as the fitted parameters. The stoichiometric factor (n) was fixed at 1 for Pg and 1.3 for Pm as determined previously (16). Titrations of [Glu]Pg in the extended conformation were performed similarly in buffer containing 1 mM NaCl and 124 mM sodium acetate (11, 47). Competitive binding of wtSK and SK-(152–414) to labeled [Glu]Pg and [Lys]Pg were analyzed with the cubic equation for tight competitive binding of two ligands to a common labeled acceptor (48). Fitting was done using Scientist (MicroMath), and errors in reported parameters are ± 2 S.D. The change in free energy of association for SK species binding to Pg or Pm was calculated from ΔG0 = RTln(KD).
RESULTS
The effect of SK α-domain truncation on the affinity of SK for [Glu]Pg, [Lys]Pg, and [Lys]Pm was quantitated in fluorescence-based binding studies with active site fluorescein-labeled Pg and Pm derivatives (3, 11, 16, 19). Binding studies were done in the absence and presence of near saturating (10 mM) 6-AHA to distinguish between LBS-independent and LBS-dependent contributions to binding. LBS-dependent interactions of SK occur through the kringle domains of Pg (8, 12, 14, 32), whereas interactions of SK with the catalytic domain are LBS-independent as shown by the crystal structure of the SK·micro-Pm complex (17). Interactions with both sites on Pg contribute to the total free energy of SK binding. The thermodynamics for the situation in which two distinct sites are involved in binding of a complex ligand has been described by Jencks (49) in terms of the total binding free energy involving both sites, the free energy of binding at each site, and a coupling free energy, as given by Equation 1,
| (Eq. 1) |
where represents LBS-dependent binding, is LBS-independent binding, and is the total binding free energy. The coupling free energy represents the free energy advantage (or disadvantage) of SK engaging both types of sites on Pg over the sum of the free energies of the independent interactions. Positive ΔGC values represent favorable coupling of the interactions. In the present studies, the affinities in the presence of 6-AHA were taken as measurements of and the affinities measured in the absence of 6-AHA represented the total free energy, . Because was not measured independently, the differences in the measured free energies represented (Equation 2).
| (Eq. 2) |
This limited the interpretation of the observed free energy differences to changes in either the free energy of LBS-dependent binding or changes in the coupling between LBS-dependent and -independent interactions. The differences in accompanying α-domain truncation were calculated as , where the values for wtSK and the mutants are represented as αβγ and βγ, respectively. In this format, a positive free energy difference represents more favorable coupling or LBS-dependent interactions of the truncation mutants.
Binding of SK α-domain Truncation Mutants to [Glu]Pg
wtSK bound [5F]FFR-[Glu]Pg with dissociation constants of 1000 ± 200 nM and 720 ± 170 nM in the absence and presence of 10 mM 6-AHA, respectively (Fig. 1, A and B). This was consistent with results obtained previously for native SK and no detectable contribution of LBS interactions to the overall affinity (9, 11). Deletion of 54 or 69 N-terminal residues of SK, and all 151 residues of the α-domain resulted in losses of 2–7-fold in affinity for [Glu]Pg in the absence of 6-AHA, with no major differences between dissociation constants for the mutants (Table I). The residual affinities of the mutants were near totally lost in the presence of 6-AHA (Fig. 1B) because of ≥3–10-fold further reduced affinities. Conservative estimates of the upper limit of the affinity in the presence of 6-AHA using an assumed titration end point equal to that for wtSK indicated that the dissociation constants for SK-(55–414), SK-(70–414), and SK-(152–414) were ≥25,000 nM (Fig. 1B), representing a loss of ≥35-fold in affinity compared with wtSK. LBS-independent interactions contributed essentially all of the binding free energy of SKαβγ for [Glu]Pg (Table II). Deletion of the α-domain resulted in loss of ≥25% of the free energy of LBS-independent binding and 10–15% of the total binding energy. This compensating effect indicated a relative gain in LBS-dependent interactions or in coupling free energy of ≥1.2 kcal/mol for SKβγ compared with SKαβγ (Table II). Deletion of the α-domain converted binding of SK to [Glu]Pg from a LBS-independent interaction into one near totally LBS-dependent.
Fig. 1. Binding of SK α-domain mutants to fluorescein-labeled [Glu]Pg.
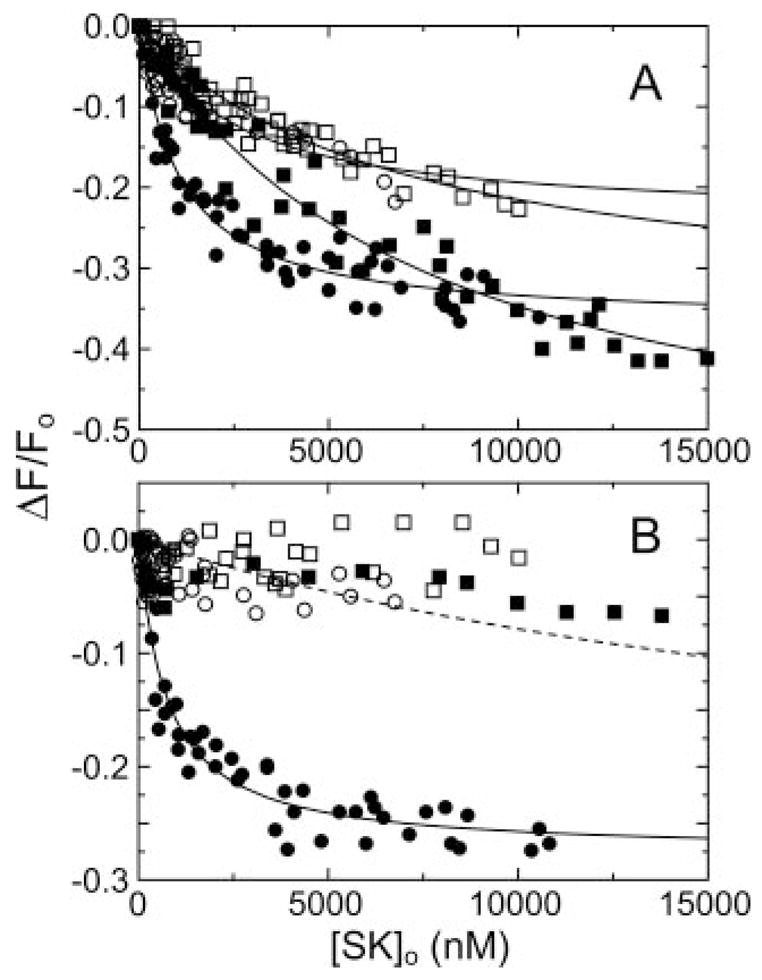
A, changes in the fluorescence (ΔF/Fo) of 15 nM [5F]FFR-[Glu]Pg as a function of the total SK concentration ([SK]o) for wtSK (●), SK-(55–414) (○), SK-(70–414) (■), and SK-(152–414) (□). The lines represent the least squares fits to the data with the parameters listed in Table I. B, titrations of [5F]FFR-[Glu]Pg with wtSK and SK mutants in the presence of 10 mM 6-AHA, as described in A. The dashed line is a simulated curve for a 25,000 nM dissociation constant assuming a titration end point equivalent to that for wtSK. Titrations were performed and analyzed as described under “Experimental Procedures.”
Table I. Parameters for binding of α-domain SK mutants to fluorescein-labeled Pg and Pm analogs.
Binding parameters were determined by analysis of titrations of the fractional change in fluorescence as a function of total SK concentration as described in “Experimental Procedures” to obtain the listed dissociation constants (KD) and maximum fluorescence changes (ΔFmax/Fo) in the absence and presence of 10 mM 6-AHA as indicated.
| Pg | SK |
KD |
ΔFmax/Fo |
||
|---|---|---|---|---|---|
| No 6-AHA | 10 mM 6-AHA | No 6-AHA | 10 mM 6-AHA | ||
| nM | % | ||||
| [Glu]Pg (125 mM Cl−) | wtSK | 1000 ± 200 | 720 ± 170 | −37 ± 2 | −28 ± 2 |
| SK-(55–414) | 2400 ± 1300 | ≥25,000 | −24 ± 1 | ||
| SK-(70–414) | 7400 ± 2000 | ≥25,000 | −60 ± 7 | ||
| SK-(152–414) | 6700 ± 2200 | ≥25,000 | −36 ± 7 | ||
| [Glu]Pg (1 mM Cl−) | wtSK | 47 ± 8 | 183 ± 44 | −35 ± 1 | −25 ± 2 |
| SK-(55–414) | 106 ± 24 | ≥100,000 | −27 ± 1 | ||
| SK-(70–414) | 141 ± 28 | ≥100,000 | −33 ± 1 | ||
| SK-(152–414) | 87 ± 8 | ≥100,000 | −34 ± 1 | ||
| [Lys]Pg | wtSK | 72 ± 18 | 900 ± 300 | −24 ± 1 | −27 ± 3 |
| SK-(55–414) | 104 ± 25 | ≥25,000 | −35 ± 2 | ||
| SK-(70–414) | 124 ± 28 | ≥25,000 | −38 ± 2 | ||
| SK-(152–414) | 73 ± 15 | ≥25,000 | −26 ± 1 | ||
| [Lys]Pm | wtSK | 0.03 ± 0.01 | 0.30 ± 0.06 | −53 ± 2 | −52 ± 3 |
| SK-(55–414) | 5 ± 2 | 1000 ± 200 | −22 ± 1 | −38 ± 2 | |
| SK-(70–414) | 11 ± 4 | 310 ± 60 | −25 ± 1 | −28 ± 2 | |
| SK-(152–414) | 10 ± 2 | 500 ± 100 | −31 ± 1 | −31 ± 2 | |
Table II. Free energy changes for binding of α-domain SK mutants to fluorescein-labeled Pg and Pm analogs.
Free energy changes for total LBS-independent and -dependent binding ( ), LBS-independent binding ( ), the coupling and LBS-dependent binding free energy ( ), and the difference in coupling and LBS-dependent free energies for the SK truncation mutants (SKβγ) and SKαβγ ( ) are listed for the indicated Pg species and SK mutants. Mean values for the mutants are shown in parentheses. Calculations were performed as described in “Results.”
| Pg | SK | ||||
|---|---|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | kcal/mol | ||
| [Glu]Pg (125 mM Cl−) | WtSK | −8.2 | −8.4 | −0.2 | |
| SK-(55–414) | −7.7 | ≥−6.3 | ≥1.4 | ≥1.6 | |
| SK-(70–414) | −7.0 | ≥−6.3 | ≥0.7 | ≥0.9 | |
| SK-(152–414) | −7.1 | ≥−6.3 | ≥0.8 | ≥1.0 | |
| Mutants (mean) | (−7.3) | (≥−6.3) | (≥1.0) | (≥1.2) | |
| [Glu]Pg (1 mM Cl−) | wtSK | −10.0 | −9.2 | 0.8 | |
| SK-(55–414) | −9.5 | ≥−5.5 | ≥4.0 | ≥3.2 | |
| SK-(70–414) | −9.3 | ≥−5.5 | ≥3.8 | ≥3.0 | |
| SK-(152–414) | −9.6 | ≥−5.5 | ≥4.1 | ≥3.3 | |
| Mutants (mean) | (−9.5) | (≥−5.5) | (≥4.0) | (≥3.2) | |
| [Lys]Pg | wtSK | −9.7 | −8.2 | 1.5 | |
| SK-(55–414) | −9.5 | ≥−6.3 | ≥3.2 | ≥1.7 | |
| SK-(70–414) | −9.4 | ≥−6.3 | ≥3.1 | ≥1.6 | |
| SK-(152–414) | −9.7 | ≥36.3 | ≥3.4 | ≥1.9 | |
| Mutants (mean) | (−9.5) | (≥−6.3) | (≥3.2) | (≥1.7) | |
| [Lys]Pm | wtSK | −14.3 | −13.0 | 1.3 | |
| SK-(55–414) | −11.3 | −8.2 | 3.1 | 1.8 | |
| SK-(70–414) | −10.9 | −8.9 | 2.0 | 0.7 | |
| SK-(152–414) | −10.9 | −8.6 | 2.3 | 1.0 | |
| Mutants (mean) | (−11.0) | (−8.6) | (2.5) | (1.2) |
Binding of SK α-domain Truncation Mutants to [Lys]Pg
The affinity of wtSK for [Lys]Pg was 13-fold greater than for [Glu]Pg, and blocking LBS interactions with 6-AHA increased the dissociation constant to 900 ± 300 nM (Fig. 2, A and B). This value was indistinguishable from that for [Glu]Pg in the presence of 6-AHA, indicating an equivalent binding free energy for LBS-independent interactions (9, 11) (Table II). Truncation of the SK α-domain had only a small effect (≤2-fold) on the affinity of SK for [Lys]Pg in the absence of 6-AHA, which was characterized by dissociation constants of 72–124 nM (Fig. 2, A and B and Table I). The affinities of the SK mutants were decreased ≥200–350-fold in the presence of 6-AHA compared with its absence, to weak affinities similar to those obtained for [Glu]Pg in the presence of 6-AHA (Fig. 2B and Table I). The effect of α-domain truncation on [Lys]Pg and [Glu]Pg binding in the presence of 6-AHA was thus very similar. The total free energy of SK binding to [Lys]Pg was −9.7 kcal/mol, with 85% of the free energy due to LBS-independent interactions equivalent to those of [Glu]Pg (Table II). Truncation of the α-domain resulted in loss of ≥1.9 kcal/mol (≥23%) of the LBS-independent free energy, as conservatively estimated from the ≥25,000 nM dissociation constants for binding of the mutants in the presence of 6-AHA. This contribution to binding was not reflected in the total binding energy of SK for [Lys]Pg (in the absence of 6-AHA) because of a compensating increase in binding energy of the βγ-fragment of ≥1.7 kcal/mol from the sum of increased LBS interactions or coupling of these interactions with LBS-independent binding (Table II).
Fig. 2. Binding of SK α-domain mutants to fluorescein-labeled [Lys]Pg.
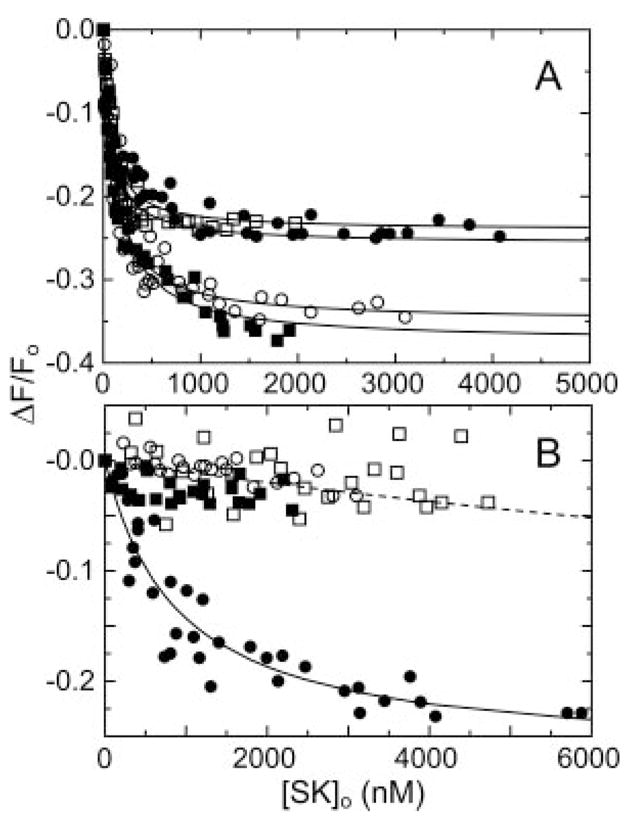
A, fluorescence changes (ΔF/Fo) of 15 nM [5F]FFR-[Lys]Pg as a function of the total SK concentration ([SK]o) for wtSK (●), SK-(55–414) (○), SK-(70–414) (■), and SK-(152–414) (□). The lines represent the least squares fits to the data with the parameters listed in Table I. B, titrations of [5F]FFR-[Lys]Pg with wtSK and SK mutants in the presence of 10 mM 6-AHA, as described in A. The dashed line is a simulated curve for a 25,000 nM dissociation constant assuming a titration end point the same as that for wtSK. Titrations were performed and analyzed as described under “Experimental Procedures.”
Binding of SK α-domain Mutants to the Extended Conformation of [Glu]Pg
Binding of wtSK and mutant SK species to [Glu]Pg in ionic strength 0.15 M buffer containing 1 mM NaCl and 124 mM sodium acetate were compared with binding in the preceding experiments at 125 mM NaCl to assess the interactions with the extended conformation of [Glu]Pg formed at low chloride concentration (11, 47). In the absence of 6-AHA, wtSK showed a 21-fold enhanced affinity for [Glu]Pg, which was reduced 4-fold by 6-AHA to a dissociation constant 4-fold lower than that of [Glu]Pg in the compact conformation (Fig. 3). Truncation of the α-domain resulted in loss of 2–3-fold affinity in the absence of 6-AHA, representing only 4–7% of the total binding energy. The mutant SK species lost essentially all affinity for the extended form of [Glu]Pg in the presence of 6-AHA, with a minimum estimate of the dissociation constants of ≥100,000 nM (Fig. 3B), representing loss of ≥40% of the LBS-independent contribution to the binding energy. As in the cases of [Glu]Pg in 125 mM NaCl and [Lys]Pg, the loss of ≥4.0 kcal/mol accompanying truncation of the α-domain was countered by a gain of ≥3.2 kcal/mol for the βγ-fragment from LBS-dependent interactions or their coupling with LBS-independent binding (Table II). The results demonstrated that interactions of the α-domain contribute primarily to LBS-independent interactions with both the compact and extended conformations of [Glu]Pg and that in both cases the βγ-fragment showed increased LBS interactions with Pg or coupling of LBS-dependent and -independent interactions.
Fig. 3. Binding of SK α-domain mutants to fluorescein-labeled [Glu]Pg at low chloride ion concentration.
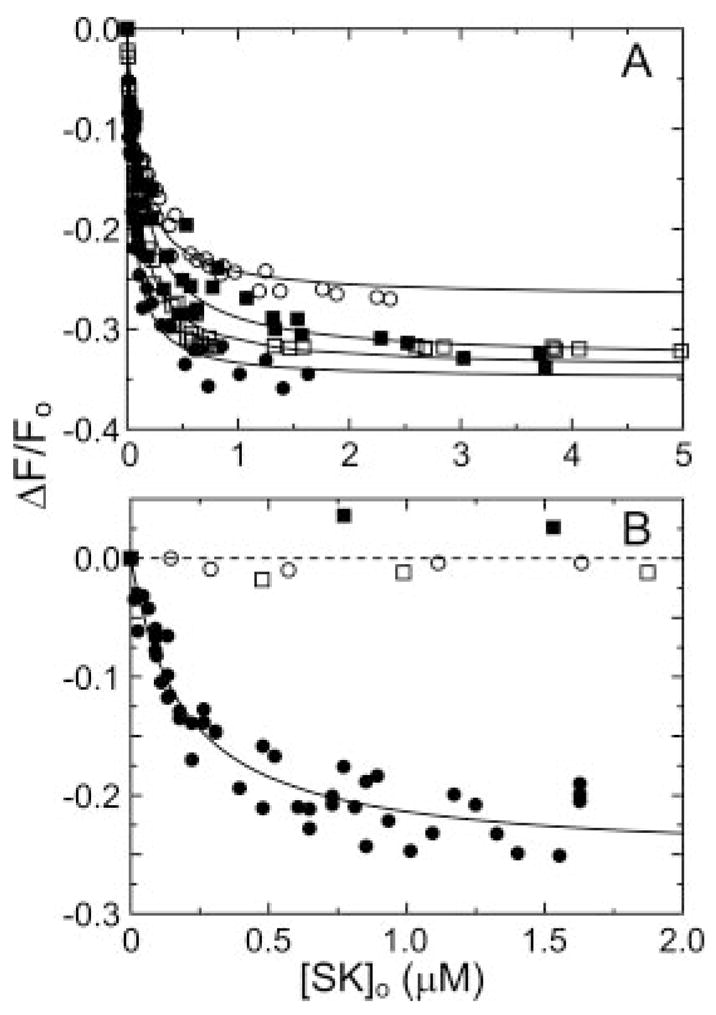
A, fluorescence changes (ΔF/Fo) of 15 nM [5F]FFR-[Glu]Pg as a function of total SK concentration ([SK]o) for wtSK (●), SK-(55–414) (○), SK-(70–414) (■), and SK-(152–414) (□) in buffer containing 1 mM NaCl and 124 mM sodium acetate. Lines represent the fits to the data with the parameters listed in Table I. B, fluorescence titrations of [5F]FFR-[Glu]Pg with SK species at 1 mM chloride ion in the presence of 10 mM 6-AHA, as described in A. The dashed line represents binding with a KD of ≥100,000 nM. Experiments were performed as described under “Experimental Procedures.”
Effect of SK-(152–414) on Binding of wtSK to [Glu]Pg and [Lys]Pg in the Presence of 6-AHA
The possibility that the decreased fluorescence changes seen in titrations of [Glu]Pg and [Lys]Pg with the SK mutants in the presence of 6-AHA were caused by loss of the fluorescence signal rather than loss of affinity was addressed in competitive binding experiments. Titrations of [Glu]Pg (Fig. 4A) and [Lys]Pg (Fig. 4B) with wtSK in the absence and presence of 5 μM SK-(152–414) were indistinguishable under conditions where a similar affinity of the SK species would have produced a large competitive effect (Fig. 4). The titrations of [Glu]Pg and [Lys]Pg in the presence of 6-AHA gave equivalent dissociation constants of 840 ± 160 nM and 820 ± 140 nM, respectively. Analysis of the results in the presence and absence of SK-(152–414) with a competitive binding model (48) yielded lower limit estimates of ≥100,000 and ≥15,000 nM for binding of SK-(152–414) to [Glu]Pg and [Lys]Pg, respectively (Fig. 4). These results demonstrated that the decreased fluorescence changes for the SK mutants reflected loss of affinity.
Fig. 4. Effect of SK-(152–414) on binding of wtSK to fluorescein-labeled [Glu]Pg and [Lys]Pg.
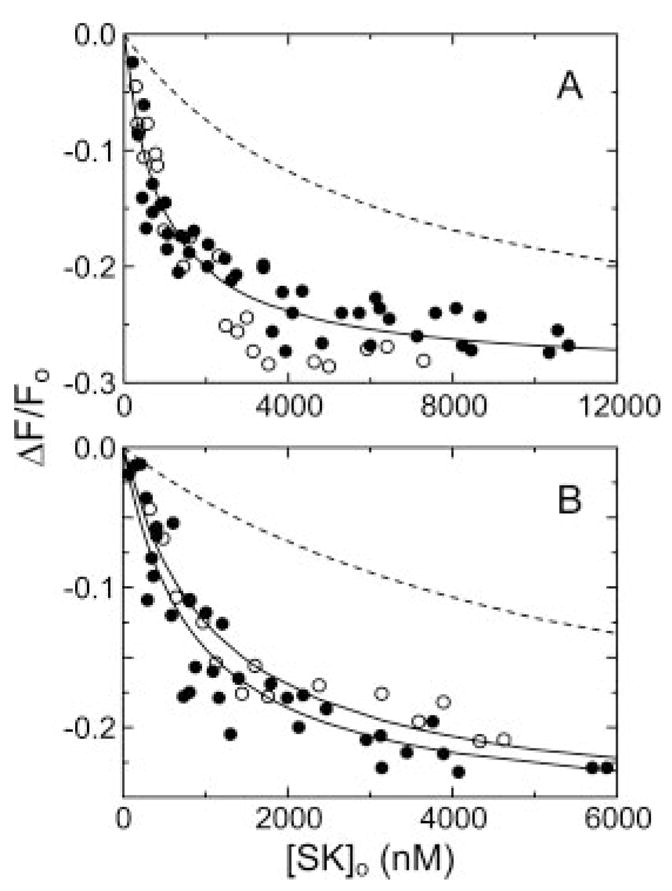
A, changes in fluorescence (ΔF/Fo) in titrations of 15 nM [5F]FFR-[Glu]Pg in the absence (●) and presence (○) of 5 3M SK-(152–414) are shown. The solid lines represent the fits by the competitive binding equation with the parameters given in the text. The dashed line shows a simulated competitive binding titration assuming an equal affinity of wtSK and SK-(152–414) for fluorescein-labeled [Glu]Pg. B, fluorescence titrations of 15 nM [5F]FFR-[Lys]Pg in the absence (●) and presence (○) of 5 μM SK-(152–414), as described in A. Titrations were performed and analyzed as described under “Experimental Procedures.”
Binding of SK α-domain Mutants to [Lys]Pm
wtSK bound fluorescein-labeled Pm with 2400-fold higher affinity than [Lys]Pg (Table I), consistent with the ~3000-fold higher affinity reported previously for native SK (11, 16). 6-AHA decreased the affinity of wtSK for labeled Pm 10-fold, similar to previous results with native SK (11). This indicated that LBS interactions contributed indistinguishable −1.5 and −1.3 kcal/mol to the binding energy of wtSK for [Lys]Pg and [Lys]Pm, respectively. The LBS-independent contribution to the affinity of wtSK for Pm represented 91% of the total binding energy. Truncation of the SK α-domain weakened the overall affinity in the absence of 6-AHA 170–370-fold, and the LBS-independent affinity measured in 6-AHA by 1000–3000-fold (Fig. 5). This demonstrated a major role of LBS-independent interactions of the α-domain in the large difference in affinity for [Lys]Pm compared with [Lys]Pg. The loss of free energy of SK binding with deletion of the α-domain represented 34% of the total LBS-independent free energy of Pm binding. These results demonstrated that the βγ-domains also contributed substantially to the enhanced affinity accompanying activation of the catalytic domain of [Lys]Pm compared with [Lys]Pg through LBS-independent interactions. LBS-dependent interactions and the coupling free energy of SK binding to Pm accounted for 2.5 and 1.3 kcal/mol contributions to the affinity for Pm of SK α-domain mutants and wtSK, respectively, indicating a favorable free energy of coupling or increased contribution of LBS-dependent interactions of 1.2 kcal/mol for SKβγ (Table II).
Fig. 5. Binding of SK α-domain mutants to [Lys]Pm.
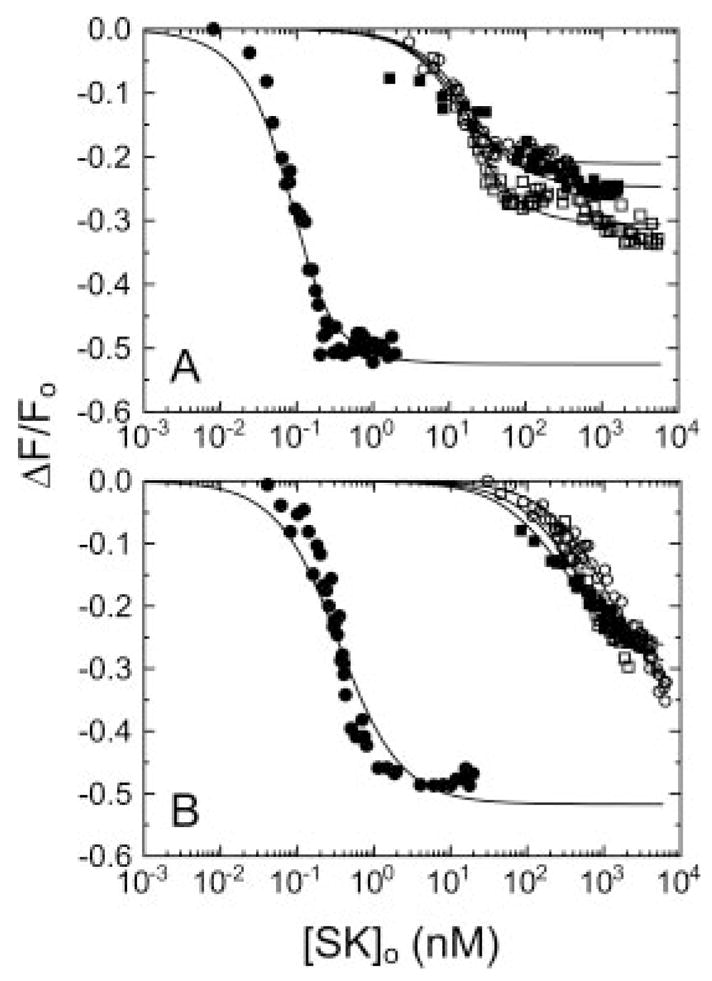
A, titrations of the fluorescence changes (ΔF/Fo) of 75 pM [5F]FFR-[Lys]Pm for wtSK and 15 nM for the mutants as a function of the total SK concentration ([SK]o) are shown for wtSK (●), SK-(55–414) (○), SK-(70–414) (■), and SK-(152–414) (□). Lines represent the least-squares fits to the data with the parameters listed in Table I. B, fluorescence titrations of 15 nM [5F]FFR-[Lys]Pm in the presence of 10 mM 6-AHA as described in A. Titrations were performed and analyzed as described under “Experimental Procedures.”
DISCUSSION
Analysis of the binding of SK α-domain truncation mutants to [Glu]Pg, [Lys]Pg, and [Lys]Pm shows that the α-domain makes a major contribution to LBS-independent binding free energy for all Pg and Pm species. In the cases of [Glu]Pg and [Lys]Pg, conservative estimates of the free energy contribution were ≥23–25%, and for the extended conformation of [Glu]Pg ≥40%, whereas for [Lys]Pm a 34% contribution was better defined. It should be recognized that the values for [Glu]Pg and [Lys]Pg represent minimum estimates, because of the absence of detectable binding in the presence of 6-AHA. LBS-independent interactions of SK principally reflect binding to the catalytic domain of Pg and Pm, based on the structure of the SK·micro-Pm complex (17). Because the structure only shows the Pg catalytic domain; however, LBS-independent interactions of segments of SK that are disordered in the structure could contribute to binding to intact Pg and Pm. Such interactions are thought to represent a minor contribution because most of the structure is defined. Assessment of the LBS-independent and -dependent interactions with [Lys]Pg compared with [Lys]Pm indicates that the 2400-fold higher affinity of SK for the activated conformation of the Pg catalytic domain is a result of increased LBS-independent interactions involving not only the α-domain but all three SK domains. LBS-dependent interactions of SK occur through Pg and Pm kringle domains (8, 11, 12, 14, 32). Our results do not support a previous hypothesis (17) and observations (25) implicating the SK-(1–59) residue sequence of the α-domain in LBS interactions with Pg. The results indicate that residues of SK that enhance the affinity of Pg and Pm binding SK through kringle interactions to form the catalytic SK·Pg* and SK·Pm complexes are located in the βγ-domains.
A limitation of the analysis was that the free energy difference between LBS-independent binding and the total binding energy represented the sum of the free energies of LBS-dependent binding and coupling of LBS-dependent and -independent interactions. This could not be resolved further because of the absence of reliable dissociation constants for LBS-dependent binding of SK mediated by Pg and Pm kringles. Literature values for binding of SK proteolytic fragments and isolated domains to kringles and multiple kringle fragments vary from 13 nM to 7 μM, and include conflicting results for kringle 5 (14, 25, 32). It is also unclear precisely which kringles are involved. Consequently, further analysis of the observed free energy differences was not possible.
The most striking feature of the results was the expression of a substantial increase in the contribution of LBS-dependent free energy or coupling free energy for Pg and Pm interactions accompanying truncation of the SK α-domain. No significant differences were observed between the affinities of SK-(55–414), SK-(70–414), and SK-(152–414) for all Pg and Pm species, consistent with the previous finding that deletion of SK-(1–59) results in unfolding of the α-domain (10, 26, 31). The results for both the compact and extended forms of [Glu]Pg, and [Lys]Pg showed that deletion of the α-domain transformed these interactions from totally (compact [Glu]Pg) or substantially (extended [Glu]Pg and [Lys]Pg) LBS-independent to greatly LBS-dependent. This was due not only to loss of LBS-independent interactions of the α-domain but also to a compensating increased contribution from LBS-dependent or coupling of LBS-dependent and -independent interactions. The compensating free energy change was estimated to be ≥1.2 and ≥1.7 kcal/mol for the compact conformation of [Glu]Pg and [Lys]Pg, respectively, and a larger ≥3.2 kcal/mol for [Glu]Pg in the extended conformation. The larger value was due primarily to the complete absence of detectable binding in the presence of 6-AHA. For Pm, the compensating free energy change was more clearly defined as 1.2 kcal/mol. The results support the conclusion that the SKβγ·Pg and SKβγ·Pm complexes are stabilized more by LBS-dependent interactions than are the SKαβγ complexes.
The redistribution of contributions to the binding energy implies that the structures of the SKαβγ·Pg*/Pm and SKβγ·Pg/Pm complexes are distinctly different. This can be explained by the three domains of SKαβγ being constrained when bound, whereas SKβγ assumes a different structure that bridges differently the LBS-dependent interactions with Pg kringles and LBS-independent interactions with the Pg catalytic domain. This difference in bound conformation can be interpreted in terms of the flexibility of the linked SK domains (18). SK behaves in solution as three independent domains connected by flexible segments, allowing SK to assume a variety of different conformations. As seen in the crystal structure of the SK·micro-Pm complex (17), when bound, SK assumes a well defined conformation in which the three domains surround the micro-Pm catalytic site. These observations indicate that the domains arrange on forming complexes with the Pg and Pm catalytic domains. The finding that the α-domain by itself under crystallization conditions binds to a different location on micro-Pg compared with micro-Pm may be an artifact of crystallization, or it may illustrate the need for covalent linkage between the domains to restrict alternative modes of binding (24). The observed redistribution of binding free energy between SKαβγ and SKβγ and the resulting change in bound conformations are thought to be allowable because of the flexible linkers and the loss of SK α-domain LBS-independent interactions that constrain the bound SKαβγ conformation.
Previous studies of the properties of SK α-domain truncation and point mutants have concluded several roles for this domain in Pg activation. The overall losses in binding affinity accompanying α-domain deletion may explain some of the reported differences in Pg activation associated with these mutations. In studies of single and two-domain SK constructs, dissociation constants for Pm were not taken into account in analysis of [Glu]Pg activation by mixtures of the mutant SKs and Pm (15). The large differences in apparent kcat seen in these studies may be due in part to loss of affinity for Pm. Because of the critical role of SK Ile1 in conformational Pg activation, truncation of the α-domain renders SK unable to activate Pg non-proteolytically (15, 20, 21, 25, 33). The observation that SK-(60–414) has lower activity in activating [Glu]Pg, but shows an enhanced fibrin dependence of its activation (25, 33) may be explained in part by the results of the present studies. Because of the loss of Ile1, Pg activation by SK α-domain truncation mutants depends on adventitious Pm in Pg preparations or on addition of Pm (15, 20, 21, 25, 33). The loss of binding affinity for Pm likely contributes to the very low activity of SK-(60–414) in activating [Glu]Pg in the presence of Pm. It is possible that differences in the structure of the SKβγ·Pm complex and enhanced LBS-mediated binding contribute to enhanced fibrin binding by the complex, although the mechanism for this is unknown. Mixtures of SK-(60–414) and Pm have also been shown to be more susceptible to inhibition by antiplasmin compared with SKαβγ (25, 33). This probably reflects the lower affinity of SK α-domain truncation mutants for Pm, which allows antiplasmin to react with the higher concentration of free Pm in equilibrium, resulting in dissociation of the complex, rather than an intrinsically greater reactivity of the SK-(60–414)·Pm complex. In addition to effects caused by loss of affinity, the apparent difference in bound conformation of native SK and α-domain truncation and deletion mutants suggests that complex effects on the kinetics of Pg activation can be expected. Further studies of Pg activation initiated by the SK mutants will be needed to determine the kinetic consequences of the differences in the bound SK structures.
Based on the results of the present studies, alteration of the functional properties of other SK domain deletion and point mutants in which individual domain interactions are affected may also result in rearrangement of the mutant-SK·Pg/Pm interactions and contribution from LBS-independent and -dependent binding to complex stability. This suggests that loss of function accompanying such mutations may not be easily interpreted as loss of a specific function of a particular domain without consideration of the interdependency of the domain interactions with Pg and Pm.
Acknowledgments
We thank Karen Godfrey for help with two of the titrations.
Footnotes
This work was supported by National Institutes of Health Grant HL56181 (to P. E. B.).
The abbreviations used are: SK, native streptokinase; [Glu]Pg, intact native plasminogen; [Lys]Pg, native Pg lacking the N-terminal 77 residues; wt, wild type; Pm, [Lys]plasmin; wtSK, recombinant wild-type streptokinase; SKαβγ, native or wtSK; SKβγ, α-domain-truncated SK; 6-AHA, 6-aminohexanoic acid; FFR-CH2Cl, (D-Phe)-Phe-Arg-CH2Cl; ATA-FFR-CH2Cl, Nα-[(acetylthio)acetyl]-(D-Phe)-Phe-Arg-CH2Cl; PEG, polyethylene glycol 8000; MES, 4-morpholineethanesulfonic acid; LBS, lysine binding site.
References
- 1.Collen D, Lijnen HR. Thromb Haemost. 1995;74:167–171. [PubMed] [Google Scholar]
- 2.McClintock DK, Englert ME, Dziobkowski C, Snedeker EH, Bell PH. Biochemistry. 1974;13:5334–5344. doi: 10.1021/bi00723a013. [DOI] [PubMed] [Google Scholar]
- 3.Boxrud PD, Verhamme IM, Bock PE. J Biol Chem. 2004;279:36633–36641. doi: 10.1074/jbc.M405264200. [DOI] [PubMed] [Google Scholar]
- 4.Boxrud PD, Bock PE. J Biol Chem. 2004;279:36642–36649. doi: 10.1074/jbc.M405265200. [DOI] [PubMed] [Google Scholar]
- 5.Reddy KN, Markus G. J Biol Chem. 1972;247:1683–1691. [PubMed] [Google Scholar]
- 6.Schick LA, Castellino FJ. Biochem Biophys Res Commun. 1974;57:47–54. doi: 10.1016/s0006-291x(74)80355-4. [DOI] [PubMed] [Google Scholar]
- 7.Lin LF, Oeun S, Houng A, Reed GL. Biochemistry. 1996;35:16879–16885. doi: 10.1021/bi961531w. [DOI] [PubMed] [Google Scholar]
- 8.Lin LF, Houng A, Reed GL. Biochemistry. 2000;39:4740–4745. doi: 10.1021/bi992028x. [DOI] [PubMed] [Google Scholar]
- 9.Bock PE, Day DE, Verhamme IM, Bernardo MM, Olson ST, Shore JD. J Biol Chem. 1996;271:1072–1080. doi: 10.1074/jbc.271.2.1072. [DOI] [PubMed] [Google Scholar]
- 10.Conejero-Lara F, Parrado J, Azuaga AI, Dobson CM, Ponting CP. Protein Sci. 1998;7:2190–2199. doi: 10.1002/pro.5560071017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boxrud PD, Bock PE. Biochemistry. 2000;39:13974–13981. doi: 10.1021/bi000594i. [DOI] [PubMed] [Google Scholar]
- 12.Dhar J, Pande AH, Sundram V, Nanda JS, Mande SC, Sahni G. J Biol Chem. 2002;277:13257–13267. doi: 10.1074/jbc.M108422200. [DOI] [PubMed] [Google Scholar]
- 13.Chaudhary A, Vasudha S, Rajagopal K, Komath SS, Garg N, Yadav M, Mande SC, Sahni G. Protein Sci. 1999;8:2791–2805. doi: 10.1110/ps.8.12.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young KC, Shi GY, Wu DH, Chang LC, Chang BI, Ou CP, Wu HL. J Biol Chem. 1998;273:3110–3116. doi: 10.1074/jbc.273.5.3110. [DOI] [PubMed] [Google Scholar]
- 15.Sundram V, Nanda JS, Rajagopal K, Dhar J, Chaudhary A, Sahni G. J Biol Chem. 2003;278:30569–30577. doi: 10.1074/jbc.M303799200. [DOI] [PubMed] [Google Scholar]
- 16.Boxrud PD, Fay WP, Bock PE. J Biol Chem. 2000;275:14579–14589. doi: 10.1074/jbc.275.19.14579. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Lin X, Loy JA, Tang J, Zhang XC. Science. 1998;281:1662–1665. doi: 10.1126/science.281.5383.1662. [DOI] [PubMed] [Google Scholar]
- 18.Damaschun G, Damaschun H, Gast K, Gerlach D, Misselwitz R, Welfle H, Zirwer D. Eur Biophys J. 1992;20:355–361. doi: 10.1007/BF00196594. [DOI] [PubMed] [Google Scholar]
- 19.Boxrud PD, Verhamme IM, Fay WP, Bock PE. J Biol Chem. 2001;276:26084–26089. doi: 10.1074/jbc.M101966200. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Reed GL, Hedstrom L. Biochemistry. 1999;38:5232–5240. doi: 10.1021/bi981915h. [DOI] [PubMed] [Google Scholar]
- 21.Wang S, Reed GL, Hedstrom L. Eur J Biochem. 2000;267:3994–4001. doi: 10.1046/j.1432-1327.2000.01434.x. [DOI] [PubMed] [Google Scholar]
- 22.Terzyan S, Wakeham N, Zhai P, Rodgers K, Zhang XC. Proteins. 2004;56:277–284. doi: 10.1002/prot.20070. [DOI] [PubMed] [Google Scholar]
- 23.Friedrich R, Panizzi P, Fuentes-Prior P, Richter K, Verhamme I, Anderson PJ, Kawabata S, Huber R, Bode W, Bock PE. Nature. 2003;425:535–539. doi: 10.1038/nature01962. [DOI] [PubMed] [Google Scholar]
- 24.Wakeham N, Terzyan S, Zhai P, Loy JA, Tang J, Zhang XC. Protein Eng. 2002;15:753–761. doi: 10.1093/protein/15.9.753. [DOI] [PubMed] [Google Scholar]
- 25.Sazonova IY, Robinson BR, Gladysheva IP, Castellino FJ, Reed GL. J Biol Chem. 2004;279:24994–25001. doi: 10.1074/jbc.M400253200. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Sazonova IY, Turner RB, Chowdhry SA, Tsai J, Houng AK, Reed GL. J Biol Chem. 2000;275:37686–37691. doi: 10.1074/jbc.M003963200. [DOI] [PubMed] [Google Scholar]
- 27.Kim DM, Lee SJ, Kim IC, Kim ST, Byun SM. Thromb Res. 2000;99:93–98. doi: 10.1016/s0049-3848(00)00225-5. [DOI] [PubMed] [Google Scholar]
- 28.Brockway WJ, Castellino FJ. Biochemistry. 1974;13:2063–2070. doi: 10.1021/bi00707a010. [DOI] [PubMed] [Google Scholar]
- 29.Parrado J, Conejero-Lara F, Smith RA, Marshall JM, Ponting CP, Dobson CM. Protein Sci. 1996;5:693–704. doi: 10.1002/pro.5560050414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siefring GE, Jr, Castellino FJ. J Biol Chem. 1976;251:3913–3920. [PubMed] [Google Scholar]
- 31.Conejero-Lara F, Parrado J, Azuaga AI, Smith RA, Ponting CP, Dobson CM. Protein Sci. 1996;5:2583–2591. doi: 10.1002/pro.5560051221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loy JA, Lin X, Schenone M, Castellino FJ, Zhang XC, Tang J. Biochemistry. 2001;40:14686–14695. doi: 10.1021/bi011309d. [DOI] [PubMed] [Google Scholar]
- 33.Mundada LV, Prorok M, DeFord ME, Figuera M, Castellino FJ, Fay WP. J Biol Chem. 2003;278:24421–24427. doi: 10.1074/jbc.M301825200. [DOI] [PubMed] [Google Scholar]
- 34.Reed GL, Houng AK, Liu L, Parhami-Seren B, Matsueda LH, Wang S, Hedstrom L. Proc Natl Acad Sci U S A. 1999;96:8879–8883. doi: 10.1073/pnas.96.16.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castellino FJ, Powell JR. Methods Enzymol. 1981;80:365–378. doi: 10.1016/s0076-6879(81)80031-6. [DOI] [PubMed] [Google Scholar]
- 36.Nieuwenhuizen W, Traas DW. Thromb Haemost. 1989;61:208–210. [PubMed] [Google Scholar]
- 37.Dharmawardana KR, Bock PE. Biochemistry. 1998;37:13143–13152. doi: 10.1021/bi9812165. [DOI] [PubMed] [Google Scholar]
- 38.Fay WP, Bokka LV. Thromb Haemost. 1998;79:985–991. [PubMed] [Google Scholar]
- 39.Taylor FB, Jr, Botts J. Biochemistry. 1968;7:232–242. doi: 10.1021/bi00841a028. [DOI] [PubMed] [Google Scholar]
- 40.Jackson KW, Tang J. Biochemistry. 1982;21:6620–6625. doi: 10.1021/bi00269a001. [DOI] [PubMed] [Google Scholar]
- 41.Violand BN, Castellino FJ. J Biol Chem. 1976;251:3906–3912. [PubMed] [Google Scholar]
- 42.Sjoholm I. Eur J Biochem. 1973;39:471–479. doi: 10.1111/j.1432-1033.1973.tb03146.x. [DOI] [PubMed] [Google Scholar]
- 43.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 44.Bock PE. J Biol Chem. 1992;267:14974–14981. [PubMed] [Google Scholar]
- 45.Bock PE. Methods Enzymol. 1993;222:478–503. doi: 10.1016/0076-6879(93)22030-j. [DOI] [PubMed] [Google Scholar]
- 46.Bock PE. J Biol Chem. 1992;267:14963–14973. [PubMed] [Google Scholar]
- 47.Chibber BA, Castellino FJ. J Biol Chem. 1986;261:5289–5295. [PubMed] [Google Scholar]
- 48.Dharmawardana KR, Olson ST, Bock PE. J Biol Chem. 1999;274:18635–18643. doi: 10.1074/jbc.274.26.18635. [DOI] [PubMed] [Google Scholar]
- 49.Jencks WP. Proc Natl Acad Sci U S A. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]