

Abstract
Studies of the mechanisms of blood coagulation zymogen activation demonstrate that exosites (sites on the activating complex distinct from the protease active site) play key roles in macromolecular substrate recognition. We investigated the importance of exosite interactions in recognition of factor IX by the protease factor XIa. Factor XIa cleavage of the tripeptide substrate S2366 was inhibited by the active site inhibitors p-aminobenzamidine (Ki 28 ± 2 μM) and aprotinin (Ki 1.13 ± 0.07 μM) in a classical competitive manner, indicating that substrate and inhibitor binding to the active site was mutually exclusive. In contrast, inhibition of factor XIa cleavage of S2366 by factor IX (Ki 224 ± 32 nM) was characterized by hyperbolic mixed-type inhibition, indicating that factor IX binds to free and S2366-bound factor XIa at exosites. Consistent with this premise, inhibition of factor XIa activation of factor IX by aprotinin (Ki 0.89 ± 0.52 μM) was non-competitive, whereas inhibition by active site-inhibited factor IXaβ was competitive (Ki 0.33 ± 0.05 μM). S2366 cleavage by isolated factor XIa catalytic domain was competitively inhibited by p-aminobenzamidine (Ki 38 ± 14 μM) but was not inhibited by factor IX, consistent with loss of factor IX-binding exosites on the non-catalytic factor XI heavy chain. The results support a model in which factor IX binds initially to exosites on the factor XIa heavy chain, followed by interaction at the active site with subsequent bond cleavage, and support a growing body of evidence that exosite interactions are critical determinants of substrate affinity and specificity in blood coagulation reactions.
Factor IX (fIX)1 is the zymogen precursor of a trypsin-like plasma protease, factor IXaβ (fIXaβ), that contributes to fibrin clot formation through proteolytic activation of factor X (1, 2). fIX conversion to fIXaβ is achieved through two proteolytic cleavages after Arg145 and Arg180, releasing an 11-kDa activation peptide (3–5). During hemostasis, fIX activation occurs through two distinct pathways mediated by the protease factors VIIa and XIa (fXIa) (3, 4, 6–8). Plasma coagulation is initiated when factor VIIa binds to the integral membrane protein tissue factor (TF) at a wound site (8–10). fIXaβ generated by factor VIIa/TF converts factor X to Xa and is probably involved in initial fibrin formation and sustained thrombin production. Activation of fIX by fXIa likely occurs after initial fibrin formation (11, 12) and is required for maintenance of clot stability, particularly in tissues rich in fibrinolytic activity that would otherwise quickly degrade the clot (13, 14).
fXIa differs from other coagulation proteases in several important aspects. fXI is a homodimer (15, 16), whereas other coagulation proteases are monomers (2). Although the C-terminal portion of the fXI polypeptide is a typical trypsin-like protease domain, the N-terminal non-catalytic region contains four repeats called apple domains (A1–A4 from the N terminus) not found on other coagulation proteases (16, 17). Furthermore, fXIa lacks the phospholipid-binding Gla domain characteristic of vitamin K-dependent coagulation proteases. Indeed, although phospholipid lowers Km for most coagulation protease reactions several orders of magnitude, it has little effect on fIX activation by fXIa (4, 7, 18). The molecular mechanism by which fXIa activates fIX is not completely understood, and the unusual structural features of fXIa cited above make it difficult to extrapolate from data obtained for other coagulation proteases.
Enzymes involved in fibrin formation are members of the chymotrypsin family of serine proteases (2, 19, 20). Despite having relatively similar catalytic domains, these enzymes exhibit specific substrate recognition (21). Substrate specificity and affinity for many serine proteases involved in digestive or degradative processes are governed primarily by interactions between the protease catalytic domain and sites on the substrate near the protease cleavage site (22–24). These interactions, which involve active site groups (primarily the S1–S3 substrate-binding subsites) and surface loops on the catalytic domain, are critical for proper alignment of substrate with the active site. More specialized proteases, including those involved in coagulation, frequently have domains outside of the protease domain that are required for proper function (1, 2, 25). A substantial body of evidence obtained by structural biology approaches indicates that binding interactions outside the protease domain are important determinants of substrate affinity and specificity in coagulation reactions (26–30). Studies of prothrombin (31–35) and factor X activation (28) identified a primary role for these exosite interactions in determining binding affinity and specificity and established experimental approaches for determining the functional importance of exosite interactions in formation of productive enzyme-substrate complexes. A two-step model of coagulation protease zymogen activation has been proposed in which substrate binds initially to an exosite followed by docking at the active site and subsequent catalysis (28, 31).
Several lines of evidence suggest that fIX activation involves exosite interactions with the fXIa heavy chain. Km for fIX activation by the isolated fXIa catalytic domain is ~ 25-fold higher than for intact fXIa (36). Recombinant fXIa in which the A3 domain is replaced with the homologous domain from plasma kallikrein also activates fIX with a substantially greater Km when compared with wild type fXIa (37, 38). Although these studies clearly show that the fXIa heavy chain interacts with fIX, the importance of these interactions, relative to those at the protease active site, in productive substrate recognition is not clear. Here, we investigated the importance of exosite binding to substrate recognition of fIX by fXIa.
EXPERIMENTAL PROCEDURES
Materials
S299 (methyl-sulfonyl-D-cyclo-hexyl-glycyl-glycyl-arginine-p-nitroanilide) was from American Diagnostics (Greenwich, CT), and S2366 (L-pyroglutamyl-L-prolyl-L-arginine-p-nitroanilide) was from DiaPharma (West Chester, OH). Aprotinin and p-aminobenzamidine (pAB) were from Sigma.
Plasma Proteins
fIX was prepared from human plasma collected into acid-citrate-dextrose. Plasma (2 liters) at 4 °C was supplemented with benzamidine to 20 mM, and 160 ml of 1 M BaCl2 was slowly added with stirring. After 1 h, the precipitate was pelleted at 10,000 × g for 20 min, washed twice with 1 liter of 10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 20 mM benzamidine, 10 mM BaCl2, and resuspended in 100 ml of 10 mM Tris-HCl, pH 7.5, 20 mM benzamidine, 20% saturated ammonium sulfate, 20 μg/ml soybean trypsin inhibitor. The suspension was dialyzed against 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 20 mM benzamidine, 5 mM EDTA and then against 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 20 mM benzamidine, 2.5 mM CaCl2. After centrifugation, fIX was purified from supernatant by antibody-affinity chromatography by using the calcium-dependent anti-human fIX monoclonal IgG SB 249417 (Dr. John Toomey, GlaxoSmithKline) linked to Affi-Gel-10 (Bio-Rad) (30). After loading, the column was washed with 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM CaCl2, 5 mM benzamidine and then eluted with 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 25 mM EDTA, 5 mM benzamidine. Protein-containing fractions were concentrated by ultrafiltration and dialyzed against 50 mM Tris-HCl, pH 7.5, 100 mM NaCl (TBS). Purity was determined by SDS-PAGE and concentration by colorimetric assay (Bio-Rad). Human fXIa, fIXaβ, and fIXai (fIXaβ with the active site inhibited by Glu-Gly-Arg-chloromethyl ketone) were from Hematologic Technologies (Essex Junction, VT).
Recombinant fXIa Catalytic Domain
The human fXI cDNA (17) was altered by using a QuikChange mutagenesis kit (Stratagene, La Jolla, CA), converting the TGT triplet for Cys362 to TCT (Ser) and converting the TGC triplet for Cys482 to AGC (Ser). The construct (fXI-Ser362,482) codes for a protein lacking the disulfide bond that connects the fXIa catalytic domain to the heavy chain. fXI-Ser362,482 cDNA was ligated into vector pJVCMV (38), and 50 × 106 293 fibroblasts (ATCC CRL 1573) were cotransfected by electroporation (Electrocell Manipulator 600 BTX, San Diego, CA) with 40 μg of construct and 2 μg of pRSVneo (38). Cells were grown in Dulbecco’s modified Eagle’s medium, 5% fetal bovine serum, 500 μg/ml G418. Supernatants from G418-resistant clones were tested by enzyme-linked immunosorbent assay by using goat anti-human fXI antibody (Affinity Biologicals, Hamilton, Ontario, Canada). Expressing clones were expanded in 175-cm2 flasks, and conditioned medium was collected every 48 h. fXI-Ser362,482 was purified from medium on an anti-fXI IgG 1G5 affinity column (38). Purified fXI-Ser362,482 (~ 300 μg/ml) was activated with 5 μg/ml factor XIIa (Enzyme Research Laboratories, South Bend, IN) at 37 °C, and complete activation was confirmed by SDS-PAGE. Activated protein was reapplied to the 1G5 antibody column to separate the fXIa-Ser362,482 catalytic domain (fXIaCD), which binds to the column, from factor XIIa and the fXIa-Ser362,482 heavy chain, which flow through the column.
fXIa Hydrolysis of S2366
fXIa (6 nM active sites, 3 nM protein) or fXIaCD (6 nM active sites, 6 nM protein) was diluted in TBS containing 0.1 mg/ml bovine serum albumin (TBSA), 5 mM CaCl2, and S2366 (50–2000 μM) in the presence or absence of pAB, aprotinin, fIX, or fIXai. Rates of generation of free p-nitroaniline were measured by continuous monitoring of absorbance at 405 nm by using 100-μl reaction volumes (3-mm path length) in a SpectraMax 340 microtiter plate reader (Molecular Devices Corp., Sunnyvale, CA). Assays were performed in triplicate. Peptide p-nitroaniline substrate concentrations were determined by absorbance at 342 nm by using an absorption coefficient of 8,266 M−1 cm−1, and product concentrations were calculated by using an absorption coefficient of 9,933 M−1 cm−1 at 405 nm (39). Active site concentrations for preparations of fXIa were determined by active site titration with human antithrombin in an S2366 cleavage assay.
| (Eq. 1) |
fIX Activation by fXIa
fIX (25–2000 nM) in TBSA containing 5 mM CaCl2 was activated by the addition of fXIa (0.4 nM active sites, 0.2 nM protein). At various time points (0–240 min), 50-μl aliquots were removed and supplemented with aprotinin (final concentration 15 μM) to inhibit fXIa. The steady-state kinetics of hydrolysis of S299 (1 mM) by quenched samples was studied in TBSA containing 5 mM CaCl2 and 33% ethylene glycol. Changes in absorbance at 405 nm were measured on a SpectraMax 340 plate reader. Duplicate assays were run for each fIX concentration. Generation of fIXaβ as a function of time was determined by interpolation from the linear dependence of the initial rate of S299 hydrolysis on known concentrations of fIXaβ. Initial steady-state rates of fIXaβ formation were determined from slopes of plots documenting the linear appearance of fIXaβ with time. Control experiments established that aprotinin completely inhibited fXIa activity without measurable effect on the detection of fIXaβ activity. fIX (50–3000 nM) activation by fXIa was also studied in the presence of aprotinin or fIXai. Aliquots (50 μl) withdrawn at various time points (20–180 s) after the initiation of activation were quenched by the addition of aprotinin (final concentration 15 μM), and fIXaβ generation was determined as described above. All assays were performed in triplicate.
Data Analysis
The apparent steady-state kinetic parameters Km and kcat for fIX activation were obtained both by non-linear least-squares fitting of full progress curves for fIX activation at substrate concentrations ranging from 25 to 1000 nM fIX (0.3–11 × Km), taking into account competitive product inhibition by fIXaβ formed during the time courses (40), and by the initial rate dependence of fIXaβ formation as a function of fIX concentration. Km and kcat for hydrolysis of S2366 by fXIa were obtained by initial rate analysis of p-nitroaniline generation as a function of S2366 concentration. Ki values for binding of pAB and aprotinin to fXIa were obtained by fitting the substrate and inhibitor dependences of the initial rates of S2366 hydrolysis both by the general hyperbolic mixed-type inhibition model (Scheme 1) and by the classical competitive inhibition model in which no ESI complex is formed (41).
Scheme. 1.
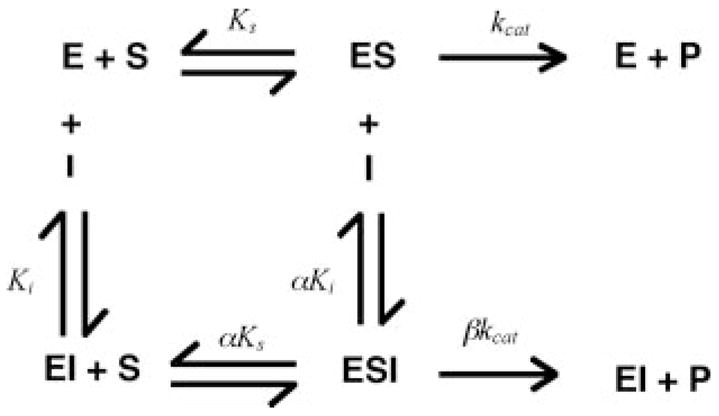
The hyperbolic mixed-type inhibition model describes the conversion of S2366 (S) to product (P) by fXIa (E) in the presence of fIX as an inhibitor (I). fIX binds to both free fXIa (E) and fXIa in complex with S2366 (ES) through an exosite interaction. Ks (≈Km) and Ki are dissociation constants for binding of S and I to E, respectively, and kcat is the rate constant for turnover of S in the ES or ESI complex. α and β are the factors by which Ks and kcat change, respectively, when I (fIX) is bound to E. Hyperbolic mixed-type inhibition (Scheme 1) is defined by Equation 1.
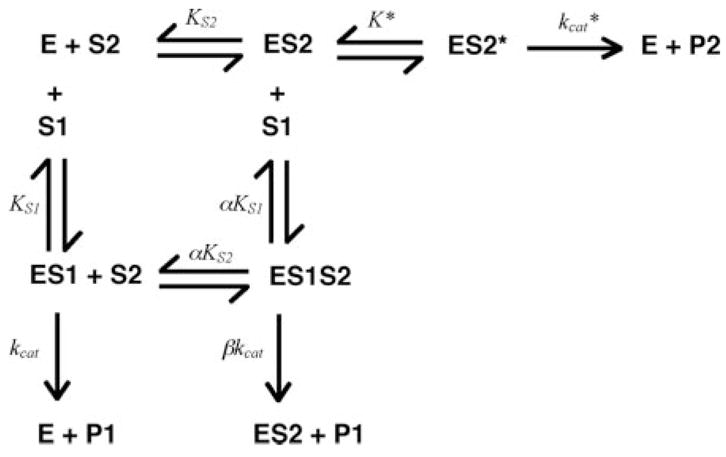
Equation 1 was used to analyze hydrolysis of S2366 (S) by fXIa (E) in the presence of fIX or fIXai (I), and was compared to the non-competitive (β = 0) model to analyze the cleavage of fIX (S) by fXIa (E) in the presence of aprotinin (I). Binding of fIX or fIXai to fXIa and fXIa·S2366 complex was analyzed by the hyperbolic mixed-type inhibition model and by a two-step interaction model proposed by Boskovic and Krishnaswamy (33), shown in Scheme 2.
Scheme. 2.
| (Eq. 2) |
| (Eq. 3) |
In this model, a macromolecular ligand (S2) interacts at the exosite during the first step, and after a conformational rearrangement governed by K*, binds to the active site during the second step. Binding of the macromolecular ligand to the active site is in competition with binding of a small ligand (S1) that reacts with the active site but not with the exosite. Rate equations were derived for this model for turnover of S2366 (S1) proposed to bind only at the active site of fXIa (E) with the dissociation constant KS1 and catalytic constant kcat, and for turnover of fIX (S2) proposed to dock initially at the exosite governed by KS2, and in a second step governed by equilibrium constant K*, with the active site, forming product with the catalytic constant kcat*. The equations were derived without the previous assumption that α = β = 1 (33). The initial rate of hydrolysis of chromogenic substrate in Michaelis-Menten form is given by Equation 2 and the initial rate of cleavage of fIX is given by Equation 3. Least squares fitting was performed with Scientist software (MicroMath Scientific Software, Salt Lake City, UT), and reported estimates of error represent ± 2 S.D.
RESULTS
Inhibition of fXIa-catalyzed Hydrolysis of S2366 by pAB and Aprotinin
fXIa is a homodimer with two active sites (15, 16). For all analyses, it is assumed that the active sites function independently of each other, and all protease concentrations given are active site concentrations. The small molecule inhibitor pAB binds reversibly to the S1 substrate-binding sub-site of arginine-specific serine proteases (42, 43) and is an effective active site inhibitor of factor XIa (44). Given its mechanism of action, pAB is expected to be a classical competitive inhibitor of substrate binding to the fXIa active site. The tripeptide substrate S2366 interacts predominantly with the S1–S3 substrate-binding subsites of the active site and was cleaved by fXIa with Km 233 ± 78 μM and kcat 117 ± 10 s−1 (Table I). As expected, pAB was a purely competitive inhibitor of S2366 cleavage with Ki 28 ± 2 μM (Fig. 1, A and B, and Table I). The Kunitz-type inhibitor aprotinin interacts with the fXIa active site and other sites on the catalytic domain but is not expected to interact appreciably with the fXIa heavy chain (45). Aprotinin behaved as a competitive inhibitor of fXIa cleavage of S2366 with Ki 1.13 ± 0.07 μM (Fig. 1, C and D, and Table I). The data indicate that S2366, pAB, and aprotinin bind to the fXIa active site in a mutually exclusive manner.
Table I. Kinetics of inhibition of fXIa activity by active site inhibitors and by fIX/fIXai.
Kinetics of inhibition were determined from initial velocity measurements using eight concentrations of S2366 or seven concentrations of fIX. Values for Km and kcat for cleavage of S2366 or fIX were fixed for the purpose of determining, α, β and Ki, and K* in inhibition models. α and β were derived from the general hyperbolic mixed-type inhibition model (41) and were fixed when determining K* in the two-step model. All ranges are ± 2 standard deviations (95% confidence intervals).
| Substrate | Inhibitor | Inhibition type | Km | kcat | α | β | Ki | K* |
|---|---|---|---|---|---|---|---|---|
| μM | s−1 | μM | ||||||
| S2366 | None | 233 ± 78 | 117 ± 10 | |||||
| S2366 | pAB | Competitive | 233 ± 78 | 117 ± 10 | 28 ± 2 | |||
| S2366 | aprotinin | Competitive | 233 ± 78 | 117 ± 10 | 1.13 ± 0.07 | |||
| S2366 | fIX | Mixed hyperbolic | 233 ± 78 | 117 ± 10 | 2.7 ± 0.4 | 0.5 ± 0.1 | 0.22 ± 0.05 | |
| S2366 | fIX | Two step conformational | 233 ± 78 | 117 ± 10 | 2.7 ± 0.4 | 0.5 ± 0.1 | ≥5 | |
| S2366 | fIXai | Mixed hyperbolic | 233 ± 78 | 117 ± 10 | 2.5 ± 0.2 | 0.9 ± 0.1 | 0.11 ± 0.02 | |
| S2366 | fIXai | Two step conformational | 233 ± 78 | 117 ± 10 | 2.5 ± 0.2 | 0.9 ± 0.1 | ≥5 | |
| FIX | None | 0.09 ± 0.04 | 0.49 ± 0.05 | |||||
| FIX | Aprotinin | Non-competitive | 0.09 ± 0.04 | 0.49 ± 0.05 | 6.3 ± 4.1 | 0 | 0.89 ± 0.52 | |
| FIX | fIXai | Competitive | 0.09 ± 0.04 | 0.49 ± 0.05 | 0.33 ± 0.05 |
Fig. 1. Inhibition of fXIa cleavage of S2366 by pAB and aprotinin.
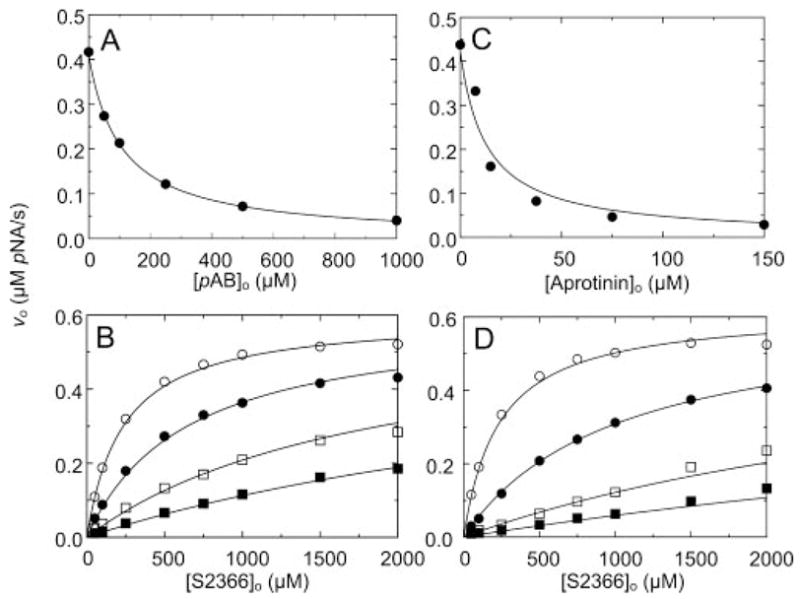
Initial velocities of S2366 hydrolysis by 6 nM fXIa active sites (υo) as a function of pAB concentration ([pAB]o) (A) or aprotinin concentration ([Aprotinin]o) (C) at 0.5 mM S2366; and as a function of S2366 concentration ([S2366]o) at fixed concentrations of pAB of 0 (○), 50 (●), 200 (□), and 500 (■) (B) or aprotinin, 0 (○), 3.8 (●), 15 (□), and 45 μM (■) (D) are shown. Data shown are means for three separate experiments. The lines represent the least-squares fits to the data with the parameters listed in Table I. Initial rates were measured and analyzed as described under “Experimental Procedures.” pNA, p-nitroaniline.
Inhibition of fXIa-catalyzed Hydrolysis of S2366 by fIX and fIXai
In surface plasmon resonance studies, fIX and fIXaβ bind to fXIa with similar affinity (30). The effect of fIX and active site-blocked fIXaβ (fIXai) on fXIa hydrolysis of S2366 was investigated. During reactions with fIX, it is expected that some fIXaβ will be generated. Preliminary experiments showed that 2 μM fIXaβ does not hydrolyze S2366 appreciably. If fIX interacts with fXIa exclusively at the active site, it would be expected to behave as a competitive inhibitor of fXIa hydrolysis of S2366. In this case, fIXai interaction at the active site may be weak because of the absence of the activation peptide. On the other hand, if exosite interactions are involved in fIX and fIXai binding to fXIa, mixed-type inhibition of S2366 cleavage would be expected for both molecules. fIX and fIXai inhibited fXIa cleavage of S2366 in a concentration-dependent manner (Fig. 2), and substantial residual fXIa activity remained at saturating fIX or fIXai concentrations. The data were fit well by the hyperbolic mixed-type inhibition model (Scheme 1). Binding of fIX resulted in a 2.7-fold increase in Km and a 50% reduction in kcat for fXIa cleavage of S2366. Ki for binding of fIX to free and S2366-bound fXIa was 0.22 ± 0.05 and 0.59 ± 0.09 μM, respectively (Table I). Binding of fIXai caused a similar 2.5-fold increase in Km and a less explicit (14%) decrease in kcat for S2366 cleavage. Ki for binding of fIXai to free fXIa and to the fXIa·S2366 complex was 0.11 ± 0.02 and 0.28 ± 0.02 μM, respectively (Table I). The Ki for binding of fIXai to fXIa was in good agreement with the Ki for product inhibition obtained from progress curves of fIX hydrolysis by fXIa (0.075 ± 0.015 μM, Fig. 3).
Fig. 2. Inhibition of fXIa hydrolysis of S2366 by fIX and fIXai.
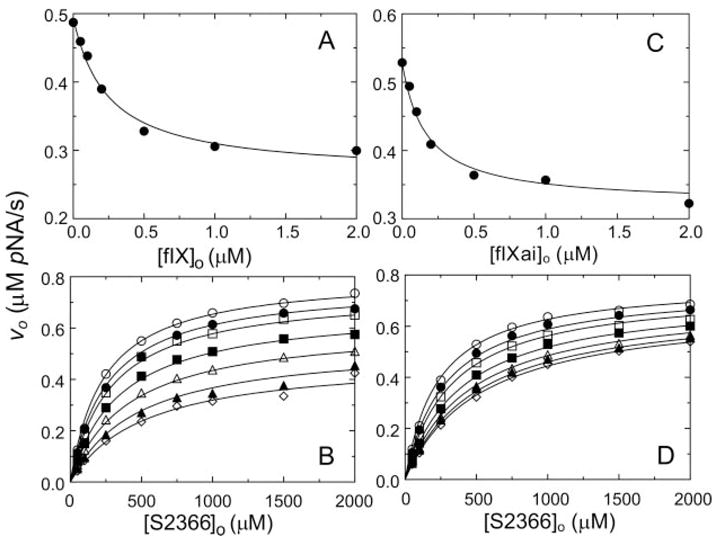
A, initial velocities of S2366 hydrolysis (0.5 mM) by 6 nM fXIa (υo) as a function of fIX concentration ([fIX]o). B, initial velocities of S2366 hydrolysis by 6 nM fXIa active sites (υo) as a function of S2366 concentration ([S2366]o) in the presence of fixed fIX concentrations, 0 (○), 50 (●), 100 (□), 250 (■), 500 (△), 1000 (▲), and 2000 (⋄) nM. C, initial velocities of S2366 hydrolysis (0.5 mM) by 6 nM fXIa (υo) as a function of fIXai concentration ([fIXai]o). D, initial velocities of S2366 hydrolysis by 6 nM fXIa (υo) as a function of S2366 concentration ([S2366]o) in the presence of fixed fIXai concentrations of 0 (○), 50 (●), 100 (□), 250 (■), 500 (△), 1000 (▲), and 2000 (⋄) nM. Data shown are means for three separate experiments. The lines represent the least-squares fits to the data with the parameters listed in Table I. Initial rates were measured and analyzed as described under “Experimental Procedures.” pNA, p-nitroaniline.
Fig. 3. Kinetic parameters for fIX cleavage by fXIa.
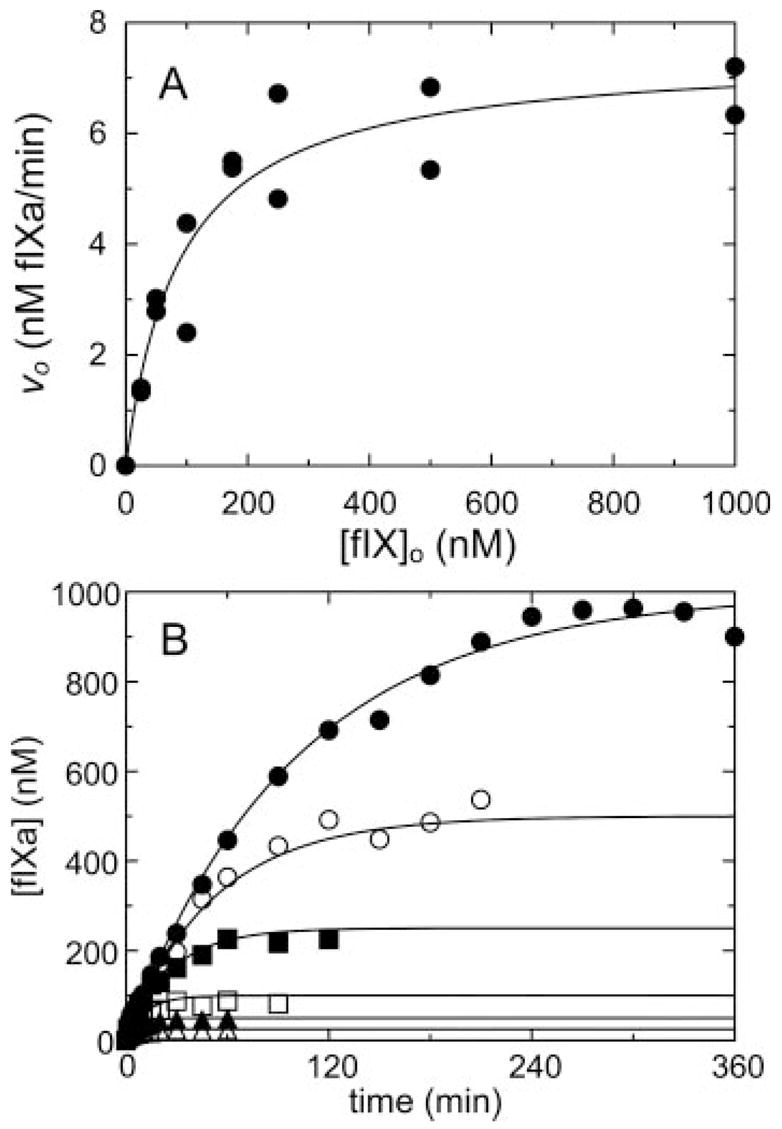
A, initial rates of fIX hydrolysis by 0.4 nM fXIa active sites (υo) as a function of fIX concentration ([fIX]o). B, progress curves of fIXa generation ([fIXa]) from fIX at 25 (△), 50 (▲), 100 (□), 250 (■), 500 (○), and 1000 nM (●). The lines represent the least-squares fits to the data with the parameters listed in Table I. Rates were measured and analyzed as described under “Experimental Procedures.”
The model was expanded to contain a reversible step for exosite binding followed by docking to the active site, governed by an equilibrium constant K* = [ES]/[ES*] in which [ES] is the fXIa·fIX complex engaged only at the exosite, and [ES*] is the complex engaged at the exosite and active site. A similar model has been described by Boskovic and Krishnaswamy (33) (Scheme 2). Using the fixed Ki values from the mixed inhibition model allowed the estimation of a lower limit for K* of ~ 5 for fXIa·fIX and fXIa·fIXai binding. In the latter case, ES2* is a dead-end complex. Ternary complex formation clearly affected Km and kcat, and omitting linkage (assuming α and β = 1) produced poor fits; thus, the linkage was concluded to be essential in the mechanism. The results indicate that fIX binds to fXIa regardless of whether or not the active site is occupied by S2366, consistent with binding to an exosite. fIX exosite binding apparently causes conformational changes in the fXIa active site, affecting kcat for S2366 cleavage and also Km, possibly by interfering with S2366 interactions at the active site.
Inhibition of fXIa-catalyzed Activation of fIX by Aprotinin
Activation of fIX by fXIa was studied by progress curve analysis and from the fIX dependence of the initial rate of fIXaβ formation. Progress curves were fit by the integrated Michaelis-Menten equation, and initial rates were analyzed with the Michaelis-Menten equation (Fig. 3), giving Km 0.09 ± 0.04 μM, kcat 0.49 ± 0.05 s−1, and Ki for product inhibition 0.075 ± 0.015 nM as fitted parameters. The results indicate that fXIa has indistinguishable apparent affinities for substrate (Km) and product (Ki). As the conversion of fIX to fIXaβ involves cleavage at two distinct sites, values for kcat represent an overall constant for cleavage at both sites. If binding affinity for this reaction is mediated primarily by exosites on fXIa, pAB would be expected to behave as a non-competitive inhibitor of fIX activation. Technical considerations with the fIXaβ chromogenic substrate assay precluded the use of pAB in these experiments (pAB interferes with fIXaβ cleavage of chromogenic substrate). Instead, aprotinin, which inhibits fXIa without affecting fIXaβ activity, was used as an active site inhibitor. Initial velocity studies of fIX activation showed that aprotinin inhibits fXIa activation of fIX in a non-competitive manner with Ki 0.89 ± 0.52 μM for the fXIa·aprotinin complex (Fig. 4A and Tables I and II) and αKi of 5.6 ± 2.1 μM for the ternary fXIa·fIX·aprotinin complex. The results imply that the fXIa·fIX·aprotinin complex is a dead-end complex (β = 0) and that aprotinin binding to fXIa weakens the affinity for fIX by ~ 6-fold.
Fig. 4. Inhibition of fXIa activation of fIX by aprotinin and fIXai.
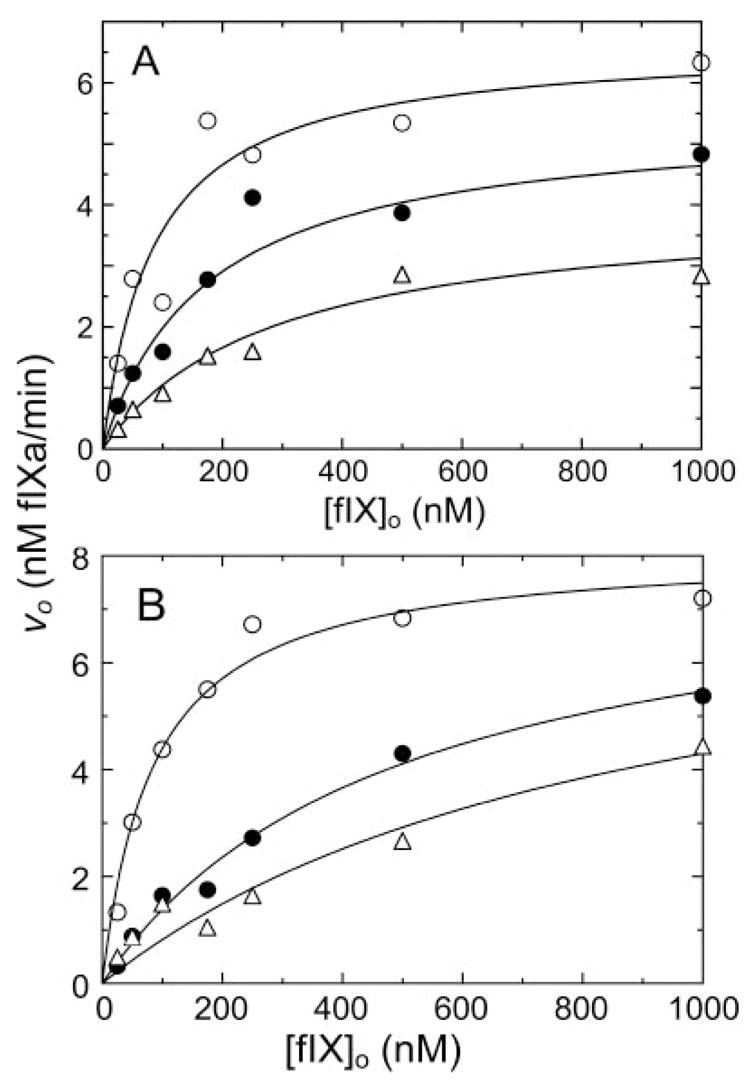
A and B, initial velocities of activation of fIX by 0.4 nM fXIa active sites (υo) as a function of fIX concentration ([fIX]o) in the presence of aprotinin at 0 (○), 1.2 (●), or 3.8 (□) μM (A) or factor IXai at 0 (○), 1.5 (●), and 3.0 (□) μM (B). Lines represent the least-squares fits to the data with the parameters listed in Table I. Data shown are means for two separate experiments. Rates were measured and analyzed as described under “Experimental Procedures.”
Table II. Kinetics of inhibition of fXIaCD activity by pAB and by fIX.
Kinetics of inhibition were determined from initial velocity measurements using eight concentrations of S2366 or seven concentrations of fIX. Values for Km and kcat for cleavage of S2366 were fixed for the purpose of determining Ki in inhibition models. All ranges are ± 2 standard deviations (95% confidence intervals).
| Substrate | Inhibitor | Inhibition type | Km | kcat | Ki |
|---|---|---|---|---|---|
| μM | s−1 | μM | |||
| S2366 | None | 352 ± 42 | 67 ± 2 | ||
| S2366 | pAB | Competitive | 352 ± 42 | 67 ± 2 | 38 ± 14 |
| S2366 | fIX | No inhibition | 352 ± 42 | 67 ± 2 |
Inhibition of fXIa-catalyzed Activation of fIX by fIXai
fIX binding to fXIa exosites presumably involves residues on fIX that are remote from the activation cleavage sites after Arg145 and Arg180. On this basis, the product fIXaβ could behave as a competitive inhibitor of fIX activation by fXIa, if product and substrate recognize the same exosite. Such competitive product inhibition has been demonstrated for factor X activation by factor VIIa/TF (28) and for prothrombin activation by the prothrombinase complex (32, 33). The effects of active site-inhibited fIXaβ (fIXai) on fIX activation by fXIa were studied (Fig. 4B). fIXai inhibits fIX activation in a competitive manner, with Ki 0.33 ± 0.05 μM (Table I) supporting the premise that fIX and fIXaβ bind to fXIa in a mutually exclusive manner.
Effects of pAB and fIX on S2366 Hydrolysis by Isolated fXIa Catalytic Domain (fXIaCD)
fXI-Ser362,482 lacks the disulfide bond that connects the catalytic domain and heavy chain after proteolysis at the activation cleavage site (16, 17). fXI-Ser362,482 and plasma fXI are 160-kDa disulfide-linked dimers under non-reducing conditions (Fig. 5A), and 80-kDa monomers when reduced (Fig. 5B). After activation, plasma fXI is still a dimer on non-reducing SDS-PAGE (Fig. 5A, lane 3), whereas reduction reveals the separate heavy chains and catalytic domains (Fig. 5B). In contrast, unreduced fXIa-Ser362,482 runs as two bands (Fig. 5A), with the 100-kDa band representing heavy chain dimers connected by a disulfide bond, and the 35-kDa band representing the catalytic domains (fXIaCD), which separate from the heavy chains in the absence of the Cys362-Cys482 bond. The absence of the Cys362-Cys482 bond allows fXIaCD to be isolated from the heavy chains by antibody affinity chromatography (Fig. 5C).
Fig. 5. SDS-PAGE of recombinant wild type and Ser362–482 fXI and fXIa.
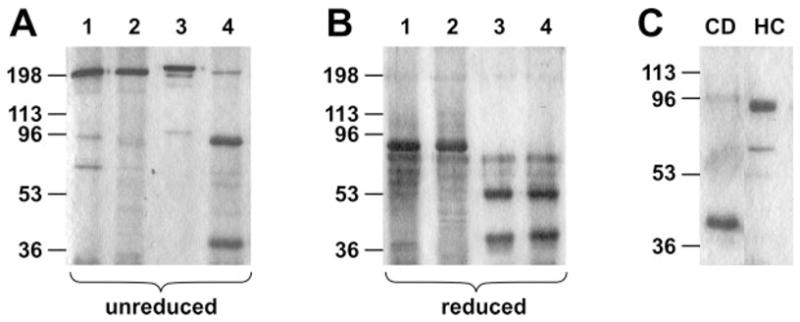
A, non-reducing gel of plasma fXI (lane 1), fXISer362–482 (lane 2), plasma fXIa (lane3), and fXIaSer362–482 (lane 4). B, same proteins as in panel A but under reducing conditions. C, fXIa catalytic domain (CD) purified from activated fXISer362–482 by antibody affinity chromatography. The flow-through of the affinity column contains dimers of fXIa heavy chain (HC). Positions of molecular mass standards in kilodaltons are shown at the left of each panel. Proteins were run on 10% SDS-PAGE and stained with GelCode Blue.
If interactions with exosites on the fXIa heavy chain determine binding affinity for the fIX·fXIa complex, fXIaCD will have reduced affinity for fIX. pAB should be a competitive inhibitor of S2366 cleavage by fXIa because both molecules interact only at the active site. However, the mixed hyperbolic inhibition observed for fIX inhibition of S2366 cleavage would not be observed because of the absence of the exosite. fXIaCD cleaves S2366 with Km 352 ± 42 μM and kcat 67 ± 2 s−1 (Table II), consistent with results for the isolated plasma fXIa catalytic domain (36). pAB inhibited hydrolysis of S2366 by fXIaCD in a competitive manner (Fig. 6, A and B), with Ki 38 ± 14 μM (Table II). In contrast to results obtained with fXIa, fIX at concentrations up to 10 μM did not have an appreciable effect on S2366 cleavage by fXIaCD. The result is consistent with the absence of a fIX-binding exosite due to loss of the fXIa heavy chain. In separate experiments, fXIa-Ser362,482 had no apparent activity in a plasma clotting assay (data not shown), as would be expected with the dissociation of the catalytic domain from the exosite-binding heavy chains after fXI activation.
Fig. 6. Inhibition of fXIaCD hydrolysis of S2366 by pAB.
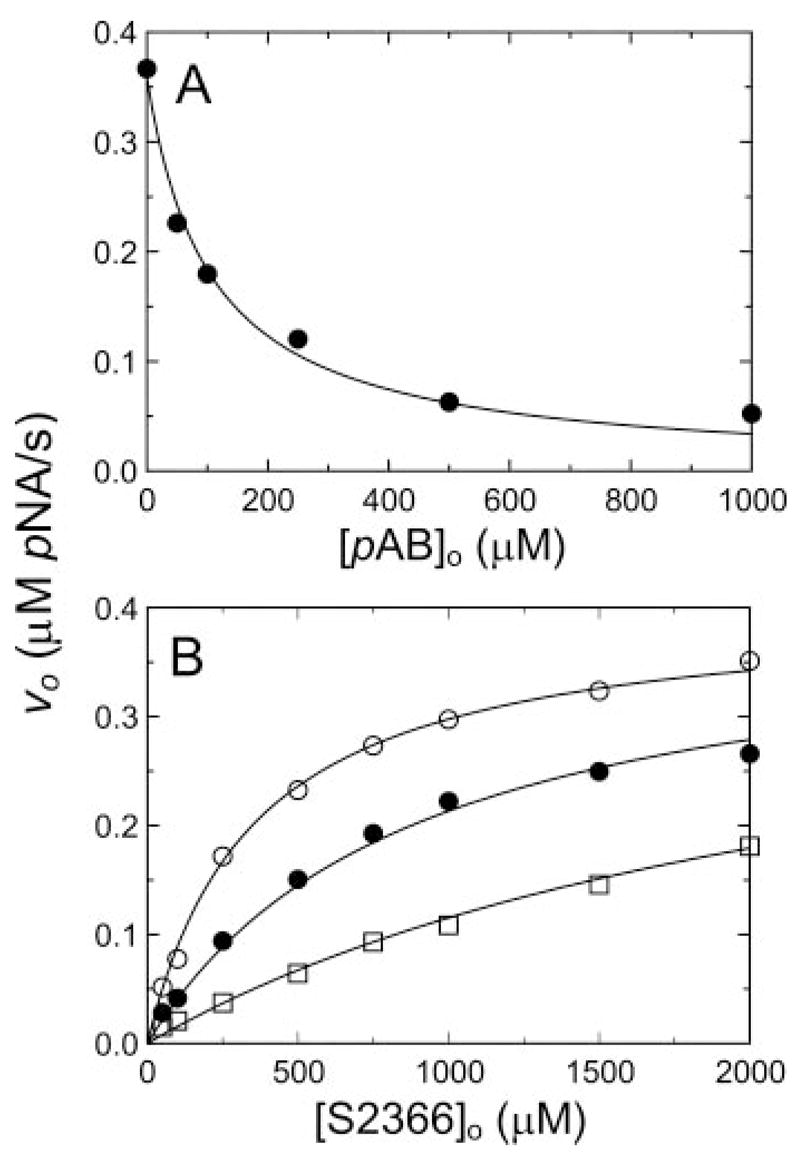
A, initial velocities of S2366 (0.5 mM) hydrolysis by 6 nM fXIaCD (υo) as a function of pAB concentration ([pAB]o). B, initial velocities of S2366 hydrolysis by 6 nM fXIaCD (υo) as a function of S2366 concentration [S2366]o) in the presence of fixed pAB concentrations of 0 (○), 50 (●), and 200 μM (□). Data represent means for three separate experiments. The lines represent the least-squares fits to the data with the parameters listed in Table II. Initial rates were measured and analyzed as described under “Experimental Procedures.” pNA, p-nitroaniline.
DISCUSSION
We used a strategy based on characterizing the effects of active site and macromolecular inhibitors of fXIa to investigate the mechanism by which fXIa recognizes fIX. The results support the conclusion that recognition of fIX as an fXIa substrate is mediated by exosites on fXIa. Our results with active site inhibitors are in agreement with work by Pedicord et al. (46), who used an enzyme-linked immunosorbent assay-based system to measure fIX activation. Their work showed mixed inhibition of fIX activation by the inhibitors aprotinin and leupeptin. These findings and the more extensive results of our studies are consistent with a two-step model first proposed for prothrombin activation (31), and subsequently, for factor X activation (28). Initial binding of macromolecular substrate occurs at an exosite(s) on the enzyme followed by a docking step at the active site and catalysis (Fig. 7). The finding that aprotinin is a non-competitive inhibitor of fIX activation by fXIa demonstrates that exosite binding is primarily responsible for substrate affinity (approximated Km ≈ Ks) and specificity. fIX might have been expected to behave as a competitive inhibitor of fXIa cleavage of S2366, similar to the active site inhibitors pAB and aprotinin. However, data for fIX-mediated inhibition of S2366 cleavage are fit best by a mixed-type inhibition model, supporting the concept that fIX binds to an exosite, resulting in conformational changes in the fXIa active site that may facilitate subsequent docking at the active site.
Fig. 7. Kinetic pathways for fIX activation by fXIa.
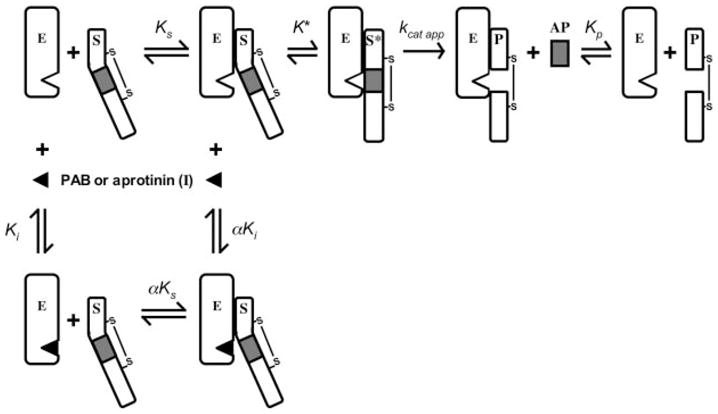
The model illustrates the conversion of fIX (S) to fIXaβ (P) by fXIa (E), with release of the activation peptide (AP) in the presence or absence of the reversible active site inhibitors pAB or aprotinin (I). The equilibrium dissociation constants for binding of S, I, and P to E are designated Ks, Ki, and Kp, respectively. The value for α is 2.7 ± 0.04 for inhibition with pAB and 6.3 ± 4.1 for aprotinin. Formation of the ES complex involves exosites on E remote from the active site. K* is the equilibrium constant for the docking interaction between S and E at the enzyme active site after binding at the exosite, S* is the substrate engaged at the enzyme active site, and kcat app represents catalysis. Note that kcat app is a term representing cleavage at both fIX activation sites and does not give specific information on cleavage at individual sites. The model shows one-half of the fXIa dimer and makes the assumption that each half-dimer activates fIX independently of the other half. An alternative model (not shown) could involve both catalytic domains of the dimer acting on one fIX molecule, with each active site cleaving one fIX activation site (49).
The docking step that follows exosite binding contributes primarily to kcat for the reaction and involves interactions between areas of the substrate near the scissile bonds and the enzyme active site. Changes in K*, the equilibrium binding constant that governs the docking interaction, could affect both the apparent kcat and the apparent Km (28) (Equation 3). In our analysis, K* appeared to be linked to formation of the ES encounter complex, where Kd is fixed by experimental results at 100–300 nM (30).2 These values are in close agreement with those for Km for fIX activation by fXIa, suggesting that K* does not favor the reaction going to product (i.e. K* is > 1). A limitation of our analysis is that we could not establish an upper limit for K*, probably because the conversion of substrate to product is relatively unfavorable. Indeed, the estimated lower limit for K* (~ 5) indicates that conversion to product is unfavorable and would reduce the apparent kcat. This may explanation why the mixed inhibition model that does not take into account the intramolecular K* step (Scheme 1) fits the data so well. If K* favored the reaction going to product (K* ≪ 1), a large discrepancy would have been expected between the models in Scheme I and II.
Activation of fIX involves cleavage after Arg145 and Arg180 (4, 5). During activation by factor VIIa/TF, initial cleavage at Arg145–Ala146 results in accumulation of the intermediate fIXβ followed by cleavage at Arg180-Val181 to form fIXaβ(4, 47–49). In contrast, Wolberg et al. (49) showed that little intermediate is formed during fIX activation by fXIa, a finding consistent with the progress curves for fIX activation in the present study. This could be explained by a mechanism in which the first cleavage is followed by rapid cleavage at the second site (50). However, this possibility is not supported by the observation that fIX and intermediates fIXα and fIXaα (cleaved at Arg180–Val181) are converted by fXIa to fIXaβ at similar rates (49). Wolberg et al. (49) proposed a processive mechanism for fIX activation by fXIa in which both activation sites are cleaved without generation of an intermediate (Fig. 7). This process may be facilitated by the dimeric structure of fXIa, with each catalytic domain acting on one fIX activation site (49). The mechanism involving rapid sequential cleavage of activation sites (intermediates formed) and the processive mechanism (no intermediates formed) are both compatible with exosite-mediated formation of the initial fIX·fXIa encounter complex. Our analysis does not distinguish between these mechanisms but demonstrates that exosite interactions precede docking at the active sites and catalysis. Given the apparent lack of intermediate generation, we treated fIX activation by fXIa as a simple process for the purpose of constructing our models. We appreciate, however, that the actual process may be considerably more complex, and a full understanding of the mechanism of fIX activation by fXIa will require detailed characterization of cleavage at the individual activation sites. The high value for K* (i.e. > 1) for fIX activation in Scheme 2 is consistent with cleavage at one and possibly both sites being unfavorable but does not provide us with information regarding the kinetics of cleavage at the individual sites.
Studies of a variant of fXI with a glutamic acid substitution for glycine 555 provide corroborating evidence for the importance of the docking step in determining apparent kcat for fIX activation (44, 51). Gly555 forms part of the active site oxyanion hole and S2′ substrate-binding site (44, 51, 52). fXIa-Glu555 hydrolyzes S2366, probably because occupancy of the abnormal active site partially restores the conformation required for catalysis (44). However, this protease has a severe defect in fIX activation, manifest as a several hundredfold decrease in apparent kcat, without an effect on Km, likely caused by a steric clash interfering with the S2′-P2′ interaction (44). Kd for binding of fIX to fXIa-Glu555 in surface plasmon resonance experiments is similar to binding to wild type fXIa (51). Thus, a mutation that interferes with substrate binding near the active site has a primary effect on kcat and not on Km, supporting the hypothesis that exosite binding is the major determinant of affinity in the fIX-fXIa interaction.
As previously demonstrated for thrombin (33) and factor Xa (28), fIXaβ is a competitive inhibitor of fIX activation by fXIa. By using surface plasmon resonance techniques, Aktimur et al. (30) demonstrated that fIX and fIXaβ bind to fXIa with similar Kds (100–150 nM). The present study demonstrates that binding of fIX and active site-inhibited fIXaβ to fXIa is mutually exclusive, indicating that substrate and product bind to a common site on the enzyme. As reported for active site-inhibited factor Xa binding to factor VIIa/TF, the affinity of fIXai is modestly reduced (~ 3–4-fold) when compared with Km for fIX activation by fXIa and Ki for product inhibition calculated from analysis of progress curves. The structure of fIXaβ may be altered when the active site is occupied by the chloromethyl ketone, weakening affinity for fXIa. Indeed, we and Pedicord et al. (46) observed that occupation of the fXIa active site by aprotinin causes a modest (~ 6-fold) increase in Kd for fIX binding. The absence of the activation peptide from the fIXai preparation may also have altered the affinity of the protein for the exosite.
The mechanism described above is supported by a large amount of data obtained over the past 20 years on the importance of the fXIa heavy chain in fIX activation. Isolated plasma fXIa catalytic domain (fXIaCD) activates fIX poorly, while retaining the capacity to cleave small chromogenic substrates (36, 48, 53). fXIa and fXIaCD activate fIX similarly in the absence of calcium ions. Ca2+ substantially enhances fIX activation by fXIa but has little effect on activation by fXIaCD (36), indicating that Ca2+-dependent binding of fIX to the fXIa heavy chain is required for fIX activation. This premise is supported by the observation that the Ca2+-binding fIX Gla domain is required for fIX binding to fXIa (30). In the present study, fIX inhibited fXIa cleavage of S2366 by a mixed type of inhibition, indicating that fIX binds to fXIa and the fXIa·S2366 complex at sites remote from the active site. The failure of fIX to inhibit S2366 cleavage by fXIaCD is consistent with loss of an exosite on the heavy chain. Even without exosite binding, fIX might have competed with S2366 for binding to the active site. That this was not observed suggests that binding at the active site is weak in the absence of binding to the heavy chain, possibly because exosite binding changes the conformation of fIX, the fXIa active site, or both to facilitate docking and catalysis.
Areas on the fXIa heavy chain have been identified that may contain fIX-binding exosites. Peptides representing portions of the fXIa A2 domain are competitive inhibitors of fIX activation by fXIa (54). Recombinant fXIa in which apple domains are replaced with corresponding domains from plasma prekallikrein (PK) indicate that the A3 domain is required for fIX activation (37). The chimera fXIa-PKA3 (fXIa with PKA3) activates fIX with a Km ~ 30-fold greater than for wild type fXIa but with similar kcat (37). These results are supported by work with monoclonal antibodies (36, 37, 55). Sinha et al. (36) showed that an antibody against the fXIa heavy chain is a competitive inhibitor of fIX activation by fXIa, whereas an antibody against the catalytic domain is a non-competitive inhibitor. Sun and Gailani (37) demonstrated that anti-A3 antibodies inhibit fXIa activation of fIX in plasma. Cumulatively, published data and the results of the present study support the hypothesis that exosites within the A2 and/or A3 domains of fXIa are critical for initial substrate recognition and formation of a productive enzyme-substrate complex with fIX.
Acknowledgments
We thank Drs. Sriram Krishnaswamy and Danilo Boskovic for assistance and helpful discussions with the two-step enzyme-substrate interaction model.
Footnotes
This work was supported in part by National Heart, Lung, and Blood Institute Grants HL58837 (to D. G.) and HL38779 (to P. E. B.).
The abbreviations used are: fIX, factor IX; fIXaβ, factor IXaβ; fIXai, active site-inhibited factor IXaβ; fXI, factor XI; fXIa, factor XIa; fXIaCD, factor XIa catalytic domain; A, apple domain; TF, tissue factor, pAB, p-aminobenzamidine; PK, prekallikrein.
S. Smith, unpublished observation.
References
- 1.Furie B, Furie B. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 2.Davie E, Fujikawa K, Kisiel W. Biochemistry. 1991;30:10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 3.Fujikawa K, Lagaz M, Kato H, Davie E. Biochemistry. 1974;13:4508–4516. doi: 10.1021/bi00719a006. [DOI] [PubMed] [Google Scholar]
- 4.DiScipio R, Kurachi K, Davie E. J Clin Investig. 1978;61:1528–1538. doi: 10.1172/JCI109073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindquist P, Fujikawa K, Davie E. J Biol Chem. 1978;253:1902–1909. [PubMed] [Google Scholar]
- 6.Osterud B, Rapaport S. Proc Natl Acad Sci (U S A) 1977;74:5260–5264. doi: 10.1073/pnas.74.12.5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osterud B, Bouma B, Griffin G. J Biol Chem. 1978;253:5946–5951. [PubMed] [Google Scholar]
- 8.Broze G, Girard T, Novotny W. Biochemistry. 1990;29:7539–7546. doi: 10.1021/bi00485a001. [DOI] [PubMed] [Google Scholar]
- 9.Rapaport S, Rao L. Arterioscler Thromb. 1992;12:1111–1121. doi: 10.1161/01.atv.12.10.1111. [DOI] [PubMed] [Google Scholar]
- 10.Nemerson Y. Semin Hematol. 1992;29:170–176. [PubMed] [Google Scholar]
- 11.Broze G. Semin Hematol. 1992;29:159–169. [PubMed] [Google Scholar]
- 12.Walsh P. Semin Hematol. 1992;29:189–201. [PubMed] [Google Scholar]
- 13.Asakai R, Chung D, Davie E, Seligsohn U. N Eng J Med. 1991;325:153–158. doi: 10.1056/NEJM199107183250303. [DOI] [PubMed] [Google Scholar]
- 14.Seligsohn U. Thromb Haemostasis. 1993;70:68–70. [PubMed] [Google Scholar]
- 15.Bouma B, Griffin J. J Biol Chem. 1977;252:6432–6437. [PubMed] [Google Scholar]
- 16.McMullen B, Fujikawa K, Davie E. Biochemistry. 1991;30:2056–2060. doi: 10.1021/bi00222a008. [DOI] [PubMed] [Google Scholar]
- 17.Fujikawa K, Chung D, Hendrickson L, Davie E. Biochemistry. 1986;25:2417–2424. doi: 10.1021/bi00357a018. [DOI] [PubMed] [Google Scholar]
- 18.Mannhalter C, Schiffman S, Deutsch E. Br J Haematol. 1984;56:261–271. doi: 10.1111/j.1365-2141.1984.tb03954.x. [DOI] [PubMed] [Google Scholar]
- 19.Neurath H. Science. 1984;224:350–357. doi: 10.1126/science.6369538. [DOI] [PubMed] [Google Scholar]
- 20.Bode W, Brandstetter H, Mather T, Stubbs M. Thromb Haemostasis. 1997;78:501–511. [PubMed] [Google Scholar]
- 21.Mann K, Jenny R, Krishnaswamy S. Annu Rev Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 22.Perona J, Craik C. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perona J, Craik C. J Biol Chem. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- 24.Krem M, Rose T, Di Cera E. J Biol Chem. 1999;274:28063–28066. doi: 10.1074/jbc.274.40.28063. [DOI] [PubMed] [Google Scholar]
- 25.Doolittle R, Feng D. Cold Spring Harbor Symp Quant Biol. 1987;52:869–874. doi: 10.1101/sqb.1987.052.01.095. [DOI] [PubMed] [Google Scholar]
- 26.Duffy E, Parker E, Mutucumarana V, Johnson A, Lollar J. J Biol Chem. 1992;267:17006–17011. [PubMed] [Google Scholar]
- 27.Shobe J, Dickinson C, Edgington T, Ruff W. J Biol Chem. 1999;274:24171–24175. doi: 10.1074/jbc.274.34.24171. [DOI] [PubMed] [Google Scholar]
- 28.Baugh R, Dickinson C, Ruf W, Krishnaswamy S. J Biol Chem. 2000;275:28826–28833. doi: 10.1074/jbc.M005266200. [DOI] [PubMed] [Google Scholar]
- 29.Gale A, Tsavaler A, Griffin J. J Biol Chem. 2002;277:28836–28840. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- 30.Aktimur A, Gabriel M, Gailani D, Toomey J. J Biol Chem. 2003;278:7981–7987. doi: 10.1074/jbc.M212748200. [DOI] [PubMed] [Google Scholar]
- 31.Krishnaswamy S, Betz A. Biochemistry. 1997;36:12080–12086. doi: 10.1021/bi970979+. [DOI] [PubMed] [Google Scholar]
- 32.Betz A, Krishnaswamy S. J Biol Chem. 1998;273:10709–10718. doi: 10.1074/jbc.273.17.10709. [DOI] [PubMed] [Google Scholar]
- 33.Boskovic D, Krishnaswamy S. J Biol Chem. 2000;275:38561–38570. doi: 10.1074/jbc.M006637200. [DOI] [PubMed] [Google Scholar]
- 34.Orcutt S, Pietropaolo C, Krishnaswamy S. J Biol Chem. 2002;277:46191–46196. doi: 10.1074/jbc.M208677200. [DOI] [PubMed] [Google Scholar]
- 35.Boskovic D, Troxler T, Krishnaswamy S. J Biol Chem. 2004;279:20786–20793. doi: 10.1074/jbc.M400469200. [DOI] [PubMed] [Google Scholar]
- 36.Sinha D, Seaman F, Walsh P. Biochemistry. 1987;26:3768–3775. doi: 10.1021/bi00387a005. [DOI] [PubMed] [Google Scholar]
- 37.Sun Y, Gailani D. J Biol Chem. 1996;271:29023–29028. doi: 10.1074/jbc.271.46.29023. [DOI] [PubMed] [Google Scholar]
- 38.Sun M, Zhao M, Gailani D. J Biol Chem. 1999;274:36373–36378. doi: 10.1074/jbc.274.51.36373. [DOI] [PubMed] [Google Scholar]
- 39.Lottenberg R, Hall J, Blinder M, Binder E, Jackson C. Biochim Biophys Acta. 1983;742:539–557. doi: 10.1016/0167-4838(83)90272-8. [DOI] [PubMed] [Google Scholar]
- 40.Duggleby R, Morrison J. Biochim Biophys Acta. 1977;481:297–312. doi: 10.1016/0005-2744(77)90264-9. [DOI] [PubMed] [Google Scholar]
- 41.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems. John Wiley & Sons, Inc; New York: 1993. pp. 178–192. [Google Scholar]
- 42.Bode W, Schwager P. J Mol Biol. 1975;98:693–717. doi: 10.1016/s0022-2836(75)80005-2. [DOI] [PubMed] [Google Scholar]
- 43.Evans S, Olson S, Shore J. J Biol Chem. 1982;257:3014–3017. [PubMed] [Google Scholar]
- 44.Schmidt A, Ogawa T, Gailani D, Bajaj S. J Biol Chem. 2004;279:29485–29492. doi: 10.1074/jbc.M402971200. [DOI] [PubMed] [Google Scholar]
- 45.Navaneetham D, Jin L, Babine R, Abdel-Meguid S, Walsh P. Blood. 2003;102:Abstr. 435. [Google Scholar]
- 46.Pedicord D, Seiffert D, Blat Y. Biochemistry. 2004;43:11883–11888. doi: 10.1021/bi048964g. [DOI] [PubMed] [Google Scholar]
- 47.Bajaj S, Rapaport S, Russell W. Biochemistry. 1983;22:4047–4053. doi: 10.1021/bi00286a009. [DOI] [PubMed] [Google Scholar]
- 48.Lawson J, Mann K. J Biol Chem. 1991;266:11317–11327. [PubMed] [Google Scholar]
- 49.Wolberg A, Morris D, Stafford D. Biochemistry. 1997;36:4074–4079. doi: 10.1021/bi962274y. [DOI] [PubMed] [Google Scholar]
- 50.Morris D, Stevens R, Wright D, Stafford D. J Bio Chem. 1995;270:30491–30498. doi: 10.1074/jbc.270.51.30491. [DOI] [PubMed] [Google Scholar]
- 51.Zivelin A, Ogawa T, Bulvik M, Landau M, Toomey J, Lane J, Seligsohn U, Gailani D. J Thromb Haemost. 2004;2:1782–1789. doi: 10.1111/j.1538-7836.2004.00882.x. [DOI] [PubMed] [Google Scholar]
- 52.Bode W, Mayr I, Baumann U, Huber R, Stone S, Hofsteenge J. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Graaf F, Greengard J, Bouma B, Kerbiriou D, Griffin J. J Biol Chem. 1983;258:9669–9675. [PubMed] [Google Scholar]
- 54.Baglia F, Jameson B, Walsh P. J Biol Chem. 1991;266:24190–24197. [PubMed] [Google Scholar]
- 55.Sinha D, Koshy A, Seaman F, Walsh P. J Biol Chem. 1985;260:10714–10719. [PubMed] [Google Scholar]