

Abstract
The specificity of thrombin for procoagulant and anticoagulant substrates is regulated allosterically by Na+. Ordered cleavage of prothrombin (ProT) at Arg320 by the prothrombinase complex generates proteolytically active, meizothrombin (MzT), followed by cleavage at Arg271 to produce thrombin and fragment 1.2. The alternative pathway of initial cleavage at Arg271 produces the inactive zymogen form, the prethrombin 2 (Pre 2)·fragment 1.2 complex, which is cleaved subsequently at Arg320. Cleavage at Arg320 of ProT or prethrombin 1 (Pre 1) activates the catalytic site and the precursor form of exosite I (proexosite I). To determine the pathway of expression of Na+-(pro)exosite I linkage during ProT activation, the effects of Na+ on the affinity of fluorescein-labeled hirudin-(54–65) ([5F]Hir-(54–65)( ) for the zymogens, ProT, Pre 1, and Pre 2, and forthe proteinases, MzT and MzT-desfragment 1 (MzT(−F1)) were quantitated. The zymogens showed no significant linkage between proexosite I and Na+, whereas cleavage at Arg320 caused the affinities of MzT and MzT(−F1) for [5F]Hir-(54–65)( ) to be enhanced by Na+ 8- to 10-fold and 5- to 6-fold, respectively. MzT and MzT(−F1) showed kinetically different mechanisms of Na+ enhancement of chromogenic substrate hydrolysis. The results demonstrate for the first time that MzT is regulated allosterically by Na+. The results suggest that the distinctive procoagulant substrate specificity of MzT, in activating factor V and factor VIII on membranes, and the anticoagulant, membrane-modulated activation of protein C by MzT bound to thrombomodulin are regulated by Na+-induced allosteric transition. Further, the Na+ enhancement in MzT activity and exosite I affinity may function in directing the sequential ProT activation pathway by accelerating thrombin formation from the MzT fast form.
The blood coagulation proteinase, thrombin, is generated after cleavage of two peptide bonds in its zymogen precursor, prothrombin (ProT),3 by the membrane-bound factor Xa·factor Va (prothrombinase) complex (1). The rate of thrombin generation from ProT catalyzed by factor Xa alone is accelerated ~300,000-fold by calcium ion-dependent assembly of factor Xa with its protein cofactor, factor Va. Factor Va in the prothrombinase complex directs the order of peptide bond cleavage in the activation pathway, resulting in sequential cleavage of ProT first at Arg320 in the catalytic domain to form the catalytically active intermediate, meizothrombin (MzT), followed by cleavage at Arg271 between the catalytic domain and ProT fragment 1.2 to produce the reaction products, thrombin and fragment 1.2 (2–7). In the absence of factor Va, the alternative, slower pathway of bond cleavage predominates, in which initial cleavage at Arg271 forms the inactive intermediate prethrombin 2 (Pre 2), bound noncovalently to fragment 1.2, followed by cleavage at Arg320 to form thrombin. Recent studies of the sequential pathway of ProT activation indicate that substrates are bound to prothrombinase in two alternate conformations that present the two peptide bonds sequentially to factor Xa for cleavage (7–14). Conformational changes accompanying catalytic site expression in the conversion of ProT to MzT direct the activation pathway by switching substrate bound to prothrombinase between alternate zymogen and proteinase conformations (8).
On conversion of ProT to thrombin, two electropositive surface sites, exosites I and II, become expressed on thrombin, which provide recognition sites for specific physiological substrates, inhibitors, and regulatory molecules (7, 15–18). Exosite I mediates fibrinogen and protease-activated receptor-1 and -4 substrate interactions, fibrin binding, and binding of the COOH-terminal hirudin-(54 – 65)-peptide (Hir-(54 – 65) ( )). Exosite II binds heparin and the fragment 2 domain of ProT. Both exosites are involved in factor V and VIII activation, glycoprotein Ib binding, thrombomodulin binding, and inactivation of thrombin by heparin cofactor II (15–18). The substrate specificity of MzT is different from that of thrombin, in that it exhibits ≤10% activity toward fibrinogen and ≤2% activity in activating platelets (19, 20). MzT, however, is a potent procoagulant activator of factor V and factor VIII in reactions that are dependent on phospholipid membranes (21, 22). MzT bound to thrombomodulin activates the anticoagulant, protein C, at a rate equal to or greater than thrombin. Protein C activation by thrombomodulin-bound MzT is also stimulated by membranes (19, 20).
Exosite I interactions are allosterically linked to binding of a single sodium ion to thrombin (23, 24). Binding of Na+ converts thrombin from a “slow” to a “fast” form (25). The fast form has higher catalytic efficiency (kcat/Km) for the procoagulant substrates, fibrinogen and protease-activated receptor-1, greater reactivity toward antithrombin, and increased amidol-ytic chromogenic substrate activity, notably toward D-Phe-Pro-Arg-pNA (15, 24). Alanine substitution mutagenesis and analysis of high resolution crystal structures of the fast and slow forms in their free states have identified four structural features critical to the allosteric transition to the fast form (23, 26–28): (a) alignment of residues stabilizing the Na+ binding site; (b) a change in orientation of Asp189 in the primary substrate specificity site; (c) a shift in the Glu192 side chain that links a network of water molecules to Ser195; and (d) a change in Ser195 orientation, all of which optimize catalysis. Comparison of these structures with those of thrombin, either inactivated with the transition state analog, D-Phe-Pro-Arg-CH2Cl (FPR-CH2Cl), or in which exosite I is occupied by the COOH-terminal hirudin peptide, hirugen (hirudin-(53–64)), shows that thrombin adopts the fast conformation in these complexes (27, 29). Recent structural studies (30, 31), consistent with kinetic studies of Na+ binding (31–33), suggest that the catalytically active Na+-free slow form is in equilibrium with a low level of an inactive thrombin in which the catalytic site and Na+ site are occluded.
Previous studies of exosite I expression on ProT activation intermediates characterized a precursor form of exosite I on ProT (proexosite I), with a ~100-fold lower affinity for the model exosite I-specific ligand Hir-(54–65)( ) (34–36). Removal of fragment 1 from ProT to form prethrombin 1 (Pre 1) is accompanied by a ~7-fold increase in proexosite I affinity (35). The further 11- to 20-fold increase in affinity, representing full exosite I expression, occurs in concert with expression of catalytic activity during formation of MzT and thrombin (35, 36). Quantitative analysis of binding of the ProT activation fragments 2 and 1.2 to exosite II on Pre 2 and thrombin indicates that this exosite becomes unmasked upon cleavage at Arg271 and subsequent dissociation of the fragments (35, 36). How linkage between the Na+ site and (pro)exosite I is expressed during ProT activation has not been investigated.
In the present studies, fluorescein-labeled Hir-(54–65)( ) ([5F]Hir-(54–65)( )) was used as a (pro)exosite I-specific probe to examine the linkage between Na+ binding and (pro)exosite I affinity (34–37). The goal was to determine if Na+ regulates the affinity of proexosite I for ProT, Pre 1, and Pre 2 zymogen forms, and the catalytically active intermediates of ProT and Pre 1 activation, MzT and meizothrombin desfrag-ment 1 (MzT(−F1)), respectively. The results demonstrate no significant effect of Na+ on ProT, Pre 1, or Pre 2 proexosite I affinity for [5F]Hir-(54–65)( ). Allosteric linkage between Na+ binding and exosite I affinity occurred concomitantly with activation to MzT and MzT(−F1). The Na+ enhancement of the rate of chromogenic substrate hydrolysis for MzT compared with MzT(−F1) and thrombin, however, followed a kinetically different mechanism. The results demonstrate for the first time that MzT and MzT(−F1) are allosterically regulated by Na+-exosite I interactions, suggesting that the unique substrate specificity of MzT may be Na+-regulated.
EXPERIMENTAL PROCEDURES
Protein and Peptide Purification and Characterization
Human ProT, factor Xa, and thrombin were purified and characterized as described previously (38, 39). Pre 1 was purified from products of ProT cleavage by thrombin (38). FPR-MzT(−F1) was prepared by activation of 20 μM Pre 1 with 2 units/ml (based on the manufacturer’s specifications) of purified ecarin from Echis carinatus venom (Sigma) in the presence of a 10-fold excess of FPR-CH2Cl (40). FPR-MzT was prepared from ProT at the same reactant concentrations followed by chromatography on Resource Q (Amersham Biosciences) in 20 mM sodium citrate buffer, pH 6.0. The column was washed with 10 ml of equilibration buffer and eluted with a 50-ml NaCl gradient in the same buffer up to 0.5 M to remove ecarin. Pre 2 was purified from factor Xa-ProT activation reactions as described (36). The R155A substitution mutant of ProT (ProTR155A) was prepared as described (21). Protein concentrations were determined by absorbance at 280 nm using the following absorption coefficients ((mg/ml)−1cm−1) and molecular weights (Mr) (4, 41, 42): ProT, ProTR155A, and FPR-MzT, 1.47, 71,600; Pre 1 and FPR-MzT(−F1), 1.78, 49,900; Pre 2, 1.73, 37,000; and thrombin, 1.74, 36,600. [5F]Hir-(54–65)( ) was prepared as described previously (37).
Fluorescence Titrations
Binding of [5F]Hir-(54–65)( ) was measured by titrating the labeled peptide with the protein of interest and monitoring the change in fluorescence at 520 nm with excitation at 491 nm (4 or 8 nm band passes), using acrylic cuvettes coated with polyethylene glycol (PEG) 20,000. Fluorescence changes expressed as (Fobs − Fo)/Fo = ΔF/Fo, as a function of total protein concentration were fit by the quadratic binding equation to obtain the maximum fluorescence change ((Fmax −Fo)/Fo = ΔFmax/Fo), and the dissociation constant (KD), with the stoichiometric factor fixed at 1 (37). Fluorescence measurements were corrected for background signal by subtraction of a buffer blank, and for dilution, which was not more than 10%.
Exosite Linkage Analysis
The effect of Na+ on binding of [5F]Hir-(54–65)( ) to (pro)exosite I was characterized in fluorescence titrations with ProT species in 50 mM Tris-Cl, 110 mM NaCl or 110 mM choline chloride (ChCl), 5 mM CaCl2, 1 mg/ml PEG 8000, pH 7.4, at 25 °C. Linkage between Na+ and [5F]Hir-(54–65)( ) (H) binding to thrombin, MzT(−F1), and MzT (designated P) was analyzed according to the thermodynamic cycle in Scheme 1, where Na+ binding affects the affinity of peptide binding and vice versa.
SCHEME 1.
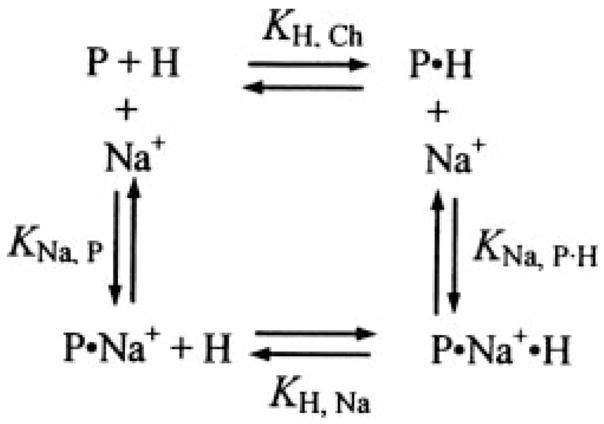
Because of the relatively small differences in fluorescence between titrations in NaCl and ChCl, it was not possible to determine accurately the dissociation constant for Na+ binding from the Na+ concentration dependence at a fixed thrombin concentration. Titrations in 110 mM NaCl and 110 mM ChCl were fit independently by the quadratic binding equation to obtain the maximum fluorescence change ΔFmax, P·H/Fo and KH, Ch from the titration in ChCl, and ΔFmax, P·Na·H/Fo and KH, Na from the titration in the presence of NaCl.
Kinetic and End Point Fluorescence Titrations
To quantitate the effects of Na+ on binding of [5F]Hir-(54–65)( ) to exosite I on active MzT(−F1) and MzTR155A, it was necessary to employ a kinetic approach to separate the ecarin-catalyzed formation of MzT(−F1) and MzTR155A from slower autocatalytic cleavages that process the products further (43). Experiments were performed for Pre 1 by measuring Fo for 20 nM [5F]Hir-(54–65)( ), the instantaneous decrease in fluorescence on addition of Pre 1 due to peptide binding to (pro)exosite I, and the time dependence of the subsequent fluorescence decrease initiated by addition of 2 units/ml of ecarin. The ecarin-initiated fluorescence time courses were fit by the sum of a single exponential decay and a straight line to determine the fluorescence end point corrected for any slow linear drift, and the results were expressed as ΔF/Fo. For analysis of the progress curves, the integrated Michaelis-Menten equation was used to represent the ecarin (E)-catalyzed conversion of Pre 1 or ProTR155A (C) into MzT(−F1) or MzTR155A (P), respectively (Scheme 2). Comparison of this analysis with the integrated rate equation including competitive product inhibition showed no evidence for product inhibition with MzT(−F1), whereas including product inhibition for MzTR155A improved the fit modestly.
SCHEME 2.
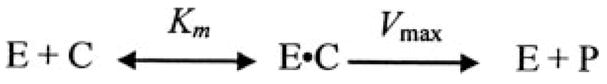
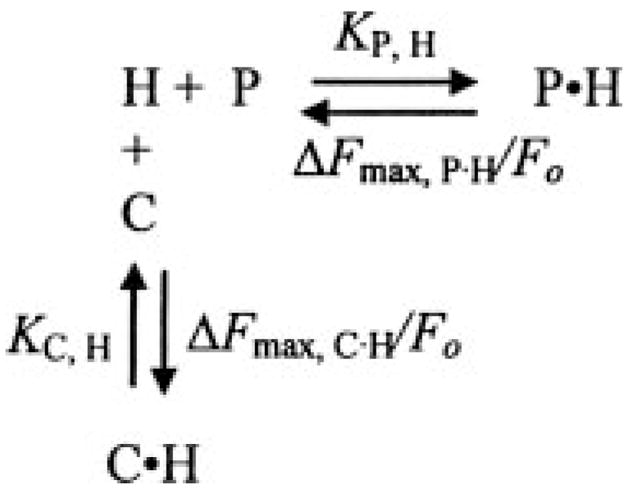
Under the experimental conditions used, the fluorescence change accompanying peptide (H) binding is not a linear function of the concentration of P or C. Moreover, these species compete for the peptide, and binding is accompanied by unequal fluorescence changes for the P·H and C·H complexes (Scheme 3). To analyze the time courses, the cubic competitive binding equation derived previously for Scheme 3 (44) in combination with the integrated Michaelis-Menten equation was used for nonlinear least-squares fitting. Fluorescence versus time data, collected as a function of the concentration of C (Pre 1) in NaCl- or ChCl-containing buffers, were analyzed with the parameters determined independently for H ([5F]Hir-(54–65)( )) binding to C (Pre 1) fixed. The fitted parameters were the maximum fluorescence change for the MzT(−F1) · [5F]Hir-(54–65)( ) complex (ΔFmax, P·H/Fo), KP·H, Km, and Vmax. The fluorescence and binding parameters determined from this analysis were compared with those obtained by fitting of the quadratic binding equation to the reaction end points as a function of C (Pre 1) concentration determined in a model-independent manner by the exponential fit described above. The same approaches were used in kinetic and end point titrations of the activation of ProTR155A to MzTR155A by ecarin and FPR-MzT(−F1) from activation of Pre 1 in the presence of 24 μM FPR-CH2Cl. The presence of 20 nM [5F]Hir-(54–65)( ) in these reactions did not influence the results, because ≤6% of the total Pre 1 or ProTR155A was bound at any time, over the protein concentration range studied.
SCHEME 3.
Pre 1 Activation Assay
Assays specific for MzT(−F1) were performed by modifications of the published methods (2, 5). This approach enables differential measurement of thrombin and MzT(−F1) based on the much faster rate of thrombin inactivation by antithrombin·heparin complex compared with MzT(−F1). Aliquots of activation reactions of Pre 1 in the buffers containing NaCl or ChCl initiated at 25 °C by addition of ecarin were quenched by dilution to 10 nM Pre 1 in 250 mM Hepes, 150 mM NaCl, 75 mM EDTA, 1 mg/ml PEG 8000, pH 7.70 (for NaCl reactions) or 250 mM Hepes, 590 mM NaCl, 75 mM EDTA, 1 mg/ml PEG 8000, pH 7.65 (for ChCl reactions) containing 50 nM antithrombin and 18 μg/ml heparin (Sigma) for exactly 30 s. An aliquot of the quenched reaction was diluted 10-fold in 50 mM Hepes, 125 mM NaCl, 1 mM EDTA, 1 mg/ml PEG 8000, pH 7.4, containing 100 μM D-Phe-Pip-Arg-pNA, and the initial rate of chromogenic substrate hydrolysis was measured from the linear increase in absorbance at 405 nm. Thrombin was confirmed to be >99.9% inactivated during the 30-s quenching time. To correct for the differences in kinetic parameters of thrombin and MzT(−F1) and the small loss of MzT(−F1) during quenching, the rates obtained for Pre 1 fully activated by incubation with 2 units/ml ecarin for 60 min were measured as a function of time after quenching, relative to an equivalent concentration of thrombin. Extrapolation of the slow, linear decrease in MzT(−F1) activity with time due to antithrombin·heparin inactivation and possible autocatalytic thrombin formation to zero time was taken to represent the total MzT(−F1) concentration. The extrapolated chromogenic substrate activity of MzT(−F1) relative to thrombin was determined to be 1.21 and 1.23 in the NaCl and ChCl experiments, respectively, and the correction for loss of MzT(−F1) activity was 10 and 6% in the NaCl and ChCl experiments, respectively.
Na+ Dependence of Chromogenic Substrate Hydrolysis
Initial rates of hydrolysis of 200 μM (saturating) D-Phe-Pip-Arg-pNA were measured at 25 °C as a function of Na+ concentration using mixtures of buffers containing 110 mM NaCl or ChCl. These experiments measured the Na+ dependence of kcat, reflecting Na+ binding to the enzyme·substrate complex. The initial rates at zero and saturating Na+ concentration, and the apparent KD for Na+ binding were obtained by least-squares fitting of the hyperbolic titrations. To estimate the affinities of Na+ for the free enzymes, kcat, Km, and Ki for product were determined by full progress curve analysis of D-Phe-Pip-Arg-pNA hydrolysis as a function of Na+ concentration (45). Five progress curves at 5–15 μM substrate were fit simultaneously to obtain the kinetic parameters. The KD for Na+ binding to thrombin and MzT(−F1) was obtained by fitting the hyperbolic dependence of kcat/Km on Na+ concentration (45). In the case of MzTR155A for which this was not possible, an estimate of the KD for Na+ binding to the free enzyme was obtained from the dependence of the observed Km (Km,obs) on Na+ concentration, where Km was assumed to be equivalent to the dissociation constant for substrate binding. Equation 1, described by Segel (46) for the rapid equilibrium, nonessential activator mechanism, was fit to the data to obtain Km, KNa, and α, which represents the linkage factor by which KNa is changed by substrate binding.
| (Eq. 1) |
RESULTS
Na+ -exosite I Linkage for ProT Zymogen Forms
To assess the linkage between Na+ binding and proexosite I, the effect of Na+ on the affinity of proexosite I for the 5-fluorescein-labeled Tyr63-sulfated hirudin-(54 – 65)-peptide ([5F]Hir-(54 – 65) ( )) was determined in fluorescence titrations in buffers containing 110 mM NaCl or 110 mM ChCl at constant 0.17 M ionic strength. As shown in Fig. 1, ProT, Pre 1, and Pre 2 bound the peptide with virtually the same affinity in NaCl and ChCl. Although the titrations consistently showed a small increase in affinity by Na+ (1.3- to 1.6-fold), this was within the experimental error of the binding parameters (Table 1).
FIGURE 1. Effect of Na+ on [5F]Hir-(54 – 65)( ) binding to ProT zymo-gen forms.
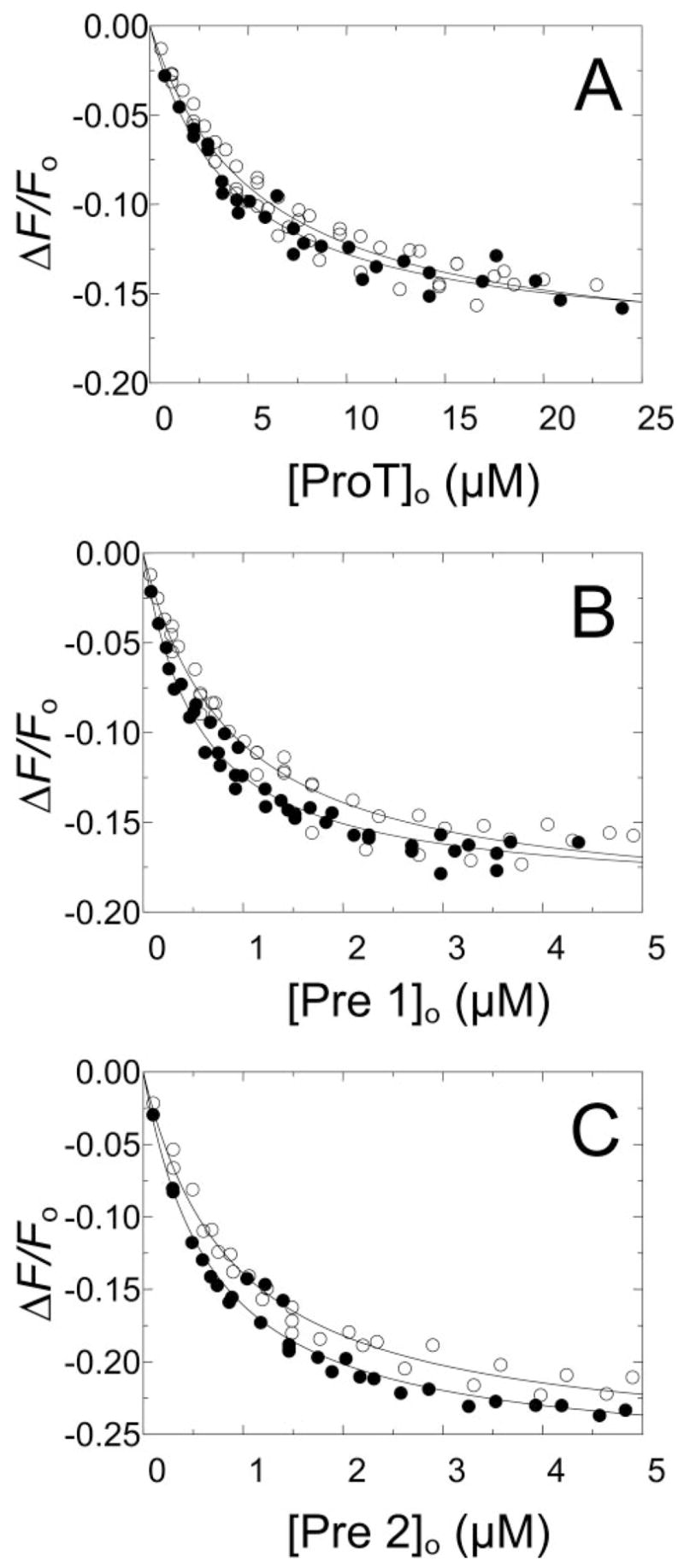
Changes in fluorescence (Δ F/Fo) of 50 nM [5F]Hir-(54 – 65)( ) as a function of the total concentrations of ProT ([ProT]o) (A), Pre 1 ([Pre 1]o) (B), and Pre 2 ([Pre 2]o) (C) are shown for experiments in buffers containing NaCl (●) or ChCl (○). The solid lines represent the nonlinear least-squares fits by the quadratic binding equation with the parameters listed in Table 1. Titrations were performed and analyzed as described under “Experimental Procedures.”
TABLE 1.
Summary of binding parameters Dissociation constants are listed for binding of [5F]Hir-(54–65)( ) to ProT and Pre 1 species in buffers containing 110 mM NaCl (KH, Na), 110 mM ChCl (KH, Ch), their ratio (KH, Ch/KH, Na), and the corresponding maximum fluorescence changes ((ΔFmax/Fo)Na and (ΔFmax/Fo)Ch). Results were obtained by direct, kinetic, and end point titrations as described under “Experimental Procedures.”
| Protein | Method | Dissociation constant
|
KH, Ch/KH, Na | Fluorescence change
|
||
|---|---|---|---|---|---|---|
| KH, Na | KH, Ch | (ΔFmax/Fo)Na | (ΔFmax/Fo)Ch | |||
| nM | % | |||||
| ProT | Direct titration | 4000 ± 700 | 5400 ± 900 | 1.4 | −18 ± 1 | −19 ± 1 |
| ProTR155A | Direct titration | 1100 ± 300 | 1900 ± 300 | 1.7 | −20 ± 3 | −20 ± 2 |
| Pre 1 | Direct titration | 510 ± 70 | 830 ± 140 | 1.6 | −19 ± 1 | −20 ± 1 |
| Pre 2 | Direct titration | 640 ± 90 | 850 ± 120 | 1.3 | −27 ± 1 | −26 ± 1 |
| Thrombin | Direct titration | 48 ± 4 | 270 ± 20 | 5.6 | −30 ± 1 | −32 ± 1 |
| FPR-thrombin | Direct titration | 35 ± 6 | 51 ± 2 | 1.5 | −31 ± 1 | −30 ± 1 |
| MzT(−F1) | Kinetic titration | 41 ± 1 | 210 ± 5 | 5.1 | −29 ± 1 | −29 ± 1 |
| MzT(−F1) | Endpoint titration | 35 ± 6 | 200 ± 4 | 5.7 | −28 ± 1 | −29 ± 2 |
| FPR-MzT(−F1) | Direct titration | 26 ± 5 | 34 ± 3 | 1.3 | −35 ± 1 | −34 ± 1 |
| FPR-MzT(−F1) | Kinetic titration | 40 ± 2 | −32 ± 1 | |||
| FPR-MzT(−F1) | Endpoint titration | 40 ± 20 | −32 ± 3 | |||
| MzTR155A | Kinetic titration | 80 ± 3 | 830 ± 40 | 10.4 | −33 ± 1 | −35 ± 2 |
| MzTR155A | Endpoint titration | 100 ± 30 | 770 ± 120a | 7.7 | −34 ± 5 | −35a |
| FPR-MzT | Direct titration | 35 ± 4 | 41 ± 5 | 1.2 | −34 ± 1 | −34 ± 1 |
The KD for peptide binding to MzTR155A from the reaction end points in the absence of Na+ was obtained by fixing ΔFmax/Fo at −35%.
Na+ -exosite I Linkage for Thrombin and FPR-Thrombin
As expected, thrombin affinity for [5F]Hir-(54 – 65)( ) increased 5.6-fold from dissociation constants of 270 × 20 nM in the absence of Na+ to 48 × 4 nM in NaCl (Fig. 2 and Table 1). Inactivation of thrombin with FPR-CH2Cl virtually obliterated the effect of Na+ on affinity and normalized the dissociation constants in the presence and absence of Na+ to values indistinguishable from those of the fast form of native thrombin (Fig. 2 and Table 1).
FIGURE 2. Effect of Na+ on [5F]Hir-(54 – 65)( ) binding to thrombin and FPR-thrombin.
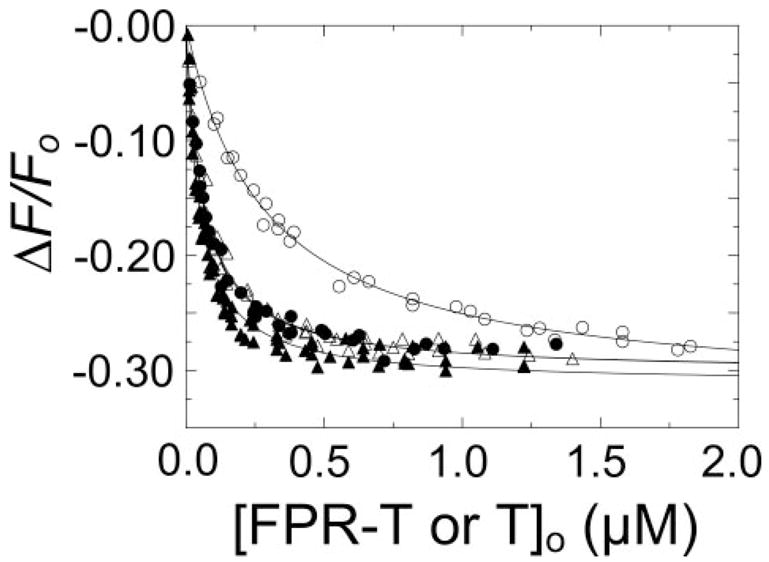
Titrations of the change in fluorescence (ΔF/Fo) of 20 nM [5F]Hir-(54 – 65)( ) as a function of total concentrations of FPR-thrombin or thrombin ([FPR-T or T]o). Titrations with native thrombin in buffers containing NaCl (●) or ChCl (○), and FPR-thrombin in NaCl (▲) or ChCl (△). The solid lines represent the non-linear least-squares fit by the binding equation with the parameters listed in Table 1. Titrations were performed and analyzed as described under “Experimental Procedures.”
Analysis of Na+ -exosite I Linkage for MzT(− F1)
MzT and MzT(− F1) are unstable in their active forms as a result of auto-catalytic cleavage (43). To characterize the effect of Na+ on active MzT(− F1), a kinetic approach was used to quantitate [5F]Hir-(54–65)( ) binding to MzT(− F1) during its generation by ecarin activation of Pre 1 (see “Experimental Procedures”). The integrated Michaelis-Menten equation was used to calculate the full time course of ecarin-catalyzed MzT(− F1) formation. The fluorescence change accompanying peptide binding is not a linear function of MzT(− F1) or Pre 1 concentration, and it was also necessary to account for the fluorescence changes due to competitive [5F]Hir-(54–65)( ) binding to Pre 1, because its concentration decreased during the reaction. A binding equation for a model in which two ligands (Pre 1 and MzT(− F1)) bind competitively to a common fluorescent acceptor ([5F]Hir-(54–65)( )) with unequal fluorescence changes (44) was used to convert the fluorescence data into the concentrations of MzT(− F1) and Pre 1 with time. Fig. 3 shows fluorescence traces in reaction mixtures containing NaCl or ChCl during activation of 1 μM Pre 1 by ecarin in the presence of 20 nM [5F]Hir-(54–65)( ). The initial decrease in fluorescence corresponds to binding of the peptide to Pre 1 and the subsequent time-dependent decrease to generation of a product with a higher affinity for the peptide. To validate the experiments, a chromogenic substrate assay, which enables quantitation of MzT(− F1) formation in the presence of thrombin, was used to follow MzT(− F1) formation. Progress curves of MzT(− F1) generation in the absence and presence of Na+ were in good agreement with the calculated progress curves (Fig. 3).
FIGURE 3. Time courses of [5F]Hir-(54–65)( ) fluorescence changes and MzT(−F1) activity accompanying activation of Pre 1 by ecarin.
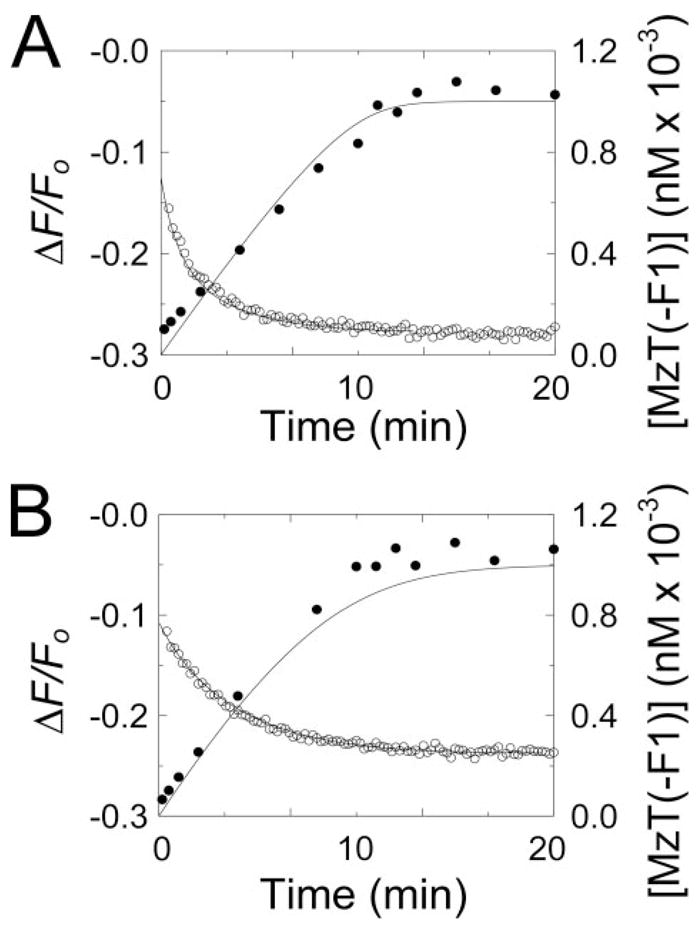
A, the initial change in fluorescence (ΔF/Fo) of 20 nM [5F]Hir-(54–65)( ) on addition of 1μM Pre 1 and the subsequent time course of the fluorescence change initiated by 2 units/ml ecarin (○) are shown for a reaction in the presence of Na+. The solid line represents the nonlinear least-squares fit of the data by the model described under “Experimental Procedures” with the parameters obtained from the simultaneous analysis of the progress curves as a function of Pre 1 concentration listed in Table 1. The time course of MzT(−F1) formation ([MzT(−F1)]o) measured by activity assays (●) is shown with the progress curve (solid line) calculated with the same parameters listed in Table 1. B, progress curves of the fluorescence change (○) and increase in MzT(−F1) concentration measured by activity (●) are shown for reactions in ChCl, with the other conditions as described in A. The lines repre-sentthefitsofthefluorescencedatawiththeparametersdeterminedfromsimul-taneous analysis of progress curves in ChCl listed in Table 1, and the calculated progress curve of MzT(−F1) formation using the same parameters. Fluorescence and activity measurements and data analysis were performed as described under “Experimental Procedures.”
To confirm that the product of Pre 1 activation was MzT(−F1), SDS-gel time courses were done under the same experimental conditions in the absence and presence of Na+ (Fig. 4). The reduced samples showed the disappearance of Pre 1 and formation of the MzT(−F1) B-chain and fragment 2-A-chain. The nonreduced samples showed a single band representing Pre 1 and co-migrating MzT(−F1) for at least 20 min, corresponding to the longest activation reactions performed (Fig. 4). Traces of thrombin only appeared after 40-min incubation in the reaction mixture containing Na+ as can be seen by the faint band at Mr ~ 36,000 in Fig. 4A. The results demonstrated that the observed fluorescence changes were well described by [5F]Hir-(54–65)( ) binding with increased affinity for MzT(−F1) and that the results could be analyzed quantitatively.
FIGURE 4. Time courses of Pre 1 activation monitored by SDS gel-electrophoresis.
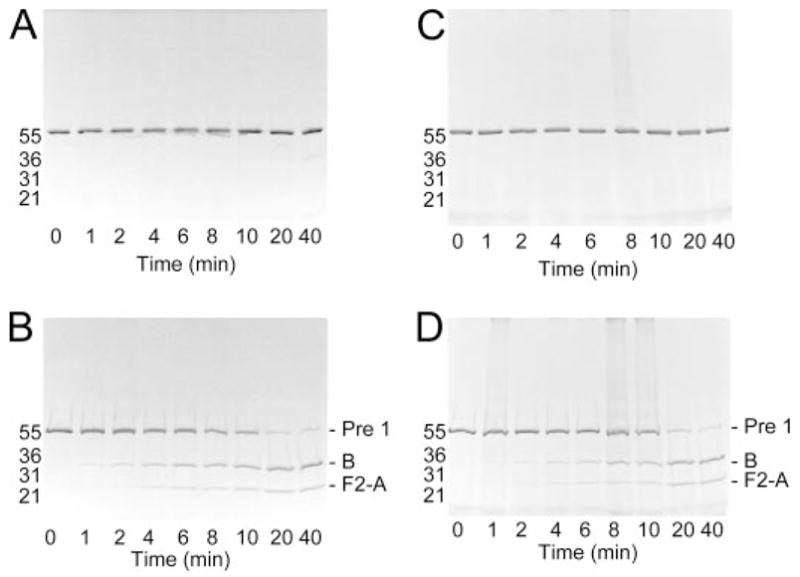
A and B, activation of 1 μM Pre 1 initiated with 2 units/ml ecarin in buffer containing NaCl. Aliquots (−1 μg) were removed at the indicated reaction times, denatured under non-reducing (A) or reducing (B) conditions, and analyzed using 4 –15% SDS gradient gels. Protein-stained bands corresponding to Pre 1, the MzT(−F1) B-chain (B), and fragment 2-A-chain (F2-A) are indicated for the reduced samples. Migration of molecular weight markers is indicated by the molecular weights in thousands. C and D, SDS gel-electrophoresis of non-reduced (C) and reduced (D) samples from Pre 1 activation in buffer containing ChCl as described in A and B. Reactions were performed as described under “Experimental Procedures.”
Regulation of MzT(−F1) Exosite I Affinity by Na+
Fluorescence-monitored time courses at varied Pre 1 concentration in NaCl or ChCl (Fig. 5, A and C) were fit simultaneously as described under “Experimental Procedures.” The independently determined dissociation constant and maximum fluorescence change for [5F]Hir-(54–65)( ) binding to Pre 1 (see Fig. 1) were fixed parameters, whereas Km and Vmax for the ecarin-catalyzed reaction, the maximum fluorescence change, and the dissociation constant for labeled peptide binding to MzT(−F1) were fitted parameters. The Km and Vmax for MzT(−F1) formation were 128 ± 8 nM and 2.1 ± 0.03 nM/s in NaCl and 470 ± 60 nM and 2.9 ± 0.2 nM/s in ChCl. [5F]Hir-(54–65)( ) bound to MzT(−F1) with a dissociation constant of 210 ± 5 nM in ChCl, which was decreased 5.1-fold to 41 ± 1 nM in NaCl, with indistinguishable changes in fluorescence (Table 1).
FIGURE 5. Kinetic and end point titrations of [5F]Hir-(54 – 65)( ) binding to MzT(−F1) in the absence and presence of Na+.
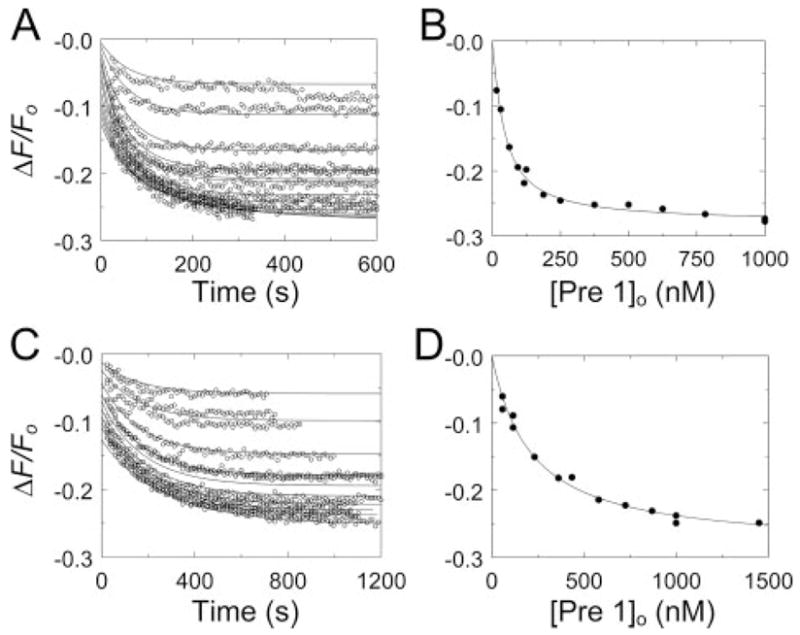
A, kinetic titration of fluorescence changes (ΔF/Fo) of 20 nM [5F]Hir-(54 – 65)( ) during activation of Pre 1 at concentrations ranging from 58 nM to 1000 nM, initiated by 2 units/ml ecarin in buffer containing NaCl (○). The lines represent the fit of the fluorescence data with the parameters listed in Table 1. B, analysis of the reaction end points as a function of the total Pre 1 concentration ([Pre 1]o). The line represents the fit by the quadratic binding equation with the parameters listed in Table 1. C, kinetic titrations for activation of 116 –1450 nM Pre 1 in buffer containing ChCl (○) otherwise as described in A, along with the simultaneous fit with the parameters listed in Table 1. D, end point titration for the reactions in buffer containing ChCl as described in B. Reactions were performed and analyzed as described under “Experimental Procedures.”
Model-independent values for the affinity and fluorescence change maxima for MzT(−F1) were obtained by fitting of the quadratic binding equation to the reaction end points, representing a titration of [5F]Hir-(54 – 65)( ) with MzT(−F1) (Fig. 5, B and D). The parameters from this analysis were in excellent agreement with those obtained for the kinetic analysis and indicated that the affinity of exosite I for [5F]Hir-(54 – 65)( ) increased 5.7-fold in the presence of Na+ (Table 1).
Na+-exosite I Linkage for FPR-MzT and FPR-MzT(−F1)
As was the case for FPR-thrombin, FPR-MzT, and FPR-MzT(−F1) had essentially the same affinities for [5F]Hir-(54–65)( ) determined in direct titrations in the absence and presence of Na+ (Fig. 6). The dissociation constants corresponded closely to the affinity of the peptide for the Na+-bound active MzT(−F1) and the thrombin fast form (Table 1).
FIGURE 6. Effect of Na+ on [5F]Hir-(54 – 65)( ) binding to FPR-MzT and FPR-MzT(−F1).
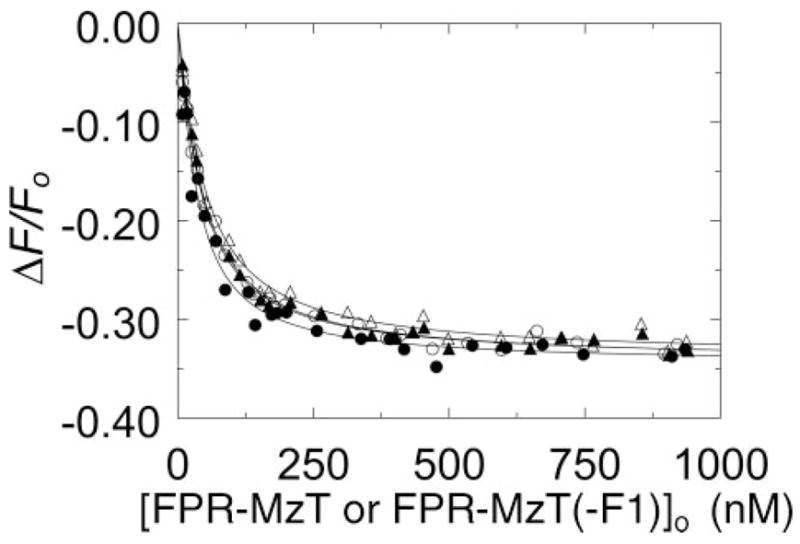
Titrations of the change in fluorescence (ΔF/Fo) of 20 nM [5F]Hir-(54 – 65)( ) in buffers containing NaCl (closed symbols) or ChCl (open symbols) as a function of the total concentrations of FPR-MzT (● and ○) and FPR-MzT(−F1) (▲ and △) ([FPR-MzT or FPR-MzT(−F1)]o). The solid lines represent the nonlinear least-squares fits of the titrations by the quadratic binding equation with the parameters listed in Table 1. Titrations were performed and analyzed as described under “Experimental Procedures.”
Effect of Na+ on Exosite I of Active MzT
Intact, native MzT generated from ProT could not be studied because of rapid autocatalytic conversion to MzT(−F1) during ecarin activation (results not shown). A ProT mutant in which the fragment 1 cleavage site at Arg155 was substituted with Ala was activated to stable MzTR155A, which allowed Na+-exosite I linkage to be characterized. In direct titrations (not shown), ProTR155A bound [5F]Hir-(54–65)( ) with KD 1.9 ± 0.3 μM in ChCl and 1.1 ± 0.4 μM in NaCl. Results of kinetic titrations of MzTR155A formation in Na+ and Ch+ are shown in Fig. 7, A and C, respectively. SDS-gel electrophoresis confirmed that MzTR155A was the product of the reactions and was stable at concentrations and incubation times in excess of the experimental conditions (not shown). Full-length MzTR155A behaved similarly to MzT(−F1) but with a few distinguishing features. Unlike the results for MzT(−F1), the fit of the kinetics of MzTR155A formation was improved modestly by including competitive product inhibition of ecarin. The fitted KD for [5F]Hir-(54–65)( ) binding to MzTR155A was 81 ± 3 nM in NaCl and was 10-fold higher (830 ± 40 nM) in ChCl. The kinetic parameters for ProTR155A activation by ecarin in NaCl were Vmax 2.1 ± 0.2 nM/s, Km 110 ± 30 nM, and Ki 100 ± 50 nM, and were indistinguishable for Vmax, Km, and Ki, determined in ChCl at 2.0 ± 0.3 nM/s, 80 ± 40 nM, and 210 ± 160 nM, respectively. Analysis of the reaction end points for [5F]Hir-(54–65)( ) binding to MzTR155A in NaCl gave a KD of 100 ± 30 nM (Fig. 7B). The end point results in ChCl could not be fit independently due to continued slow decreases in fluorescence at high ProT concentrations and long reaction times. Assuming an end point equivalent to that determined kinetically gave a KD of 770 ± 120 nM (Fig. 7D). Together, the results demonstrated slightly larger effects of Na+ on the affinity of [5F]Hir-(54–65)( ) for MzTR155A (8- to 10-fold) compared with MzT(−F1) (5- to 6-fold).
FIGURE 7. Kinetic and end point titrations of [5F]Hir-(54 – 65)( ) binding to MzTR155A in the absence and presence of Na+.
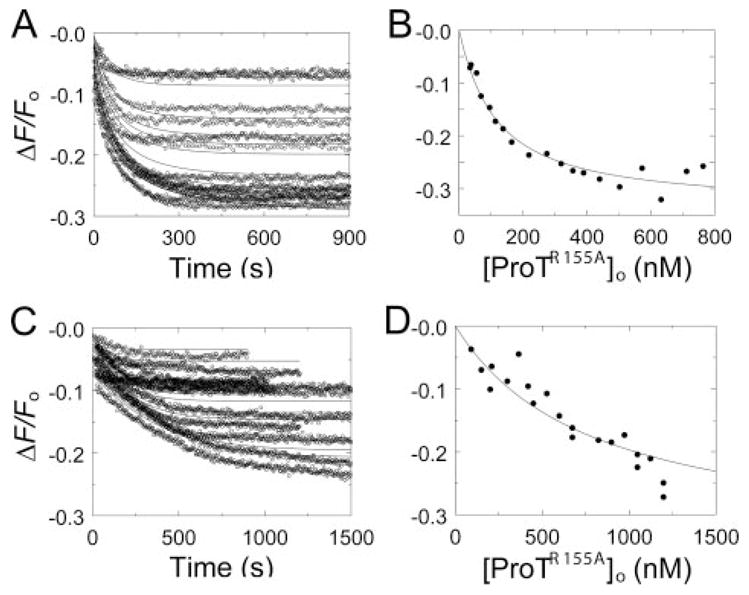
A, kinetic titration of fluorescence changes (ΔF/Fo) of 20 nM [5F]Hir-(54 – 65)( ) during activation of ProTR155A at concentrations ranging from 28 to 760 nM, initiated by 2 units/ml ecarin in buffer containing NaCl (○). 13 reactions out of a total of 19 analyzed are shown. The lines represent the simultaneous fit of the fluorescence data with the parameters listed in Table 1. B, analysis of the reaction end points as a function of the total ProTR155A concentration ([ProTR155A]o). The line represents the fit by the quadratic binding equation with the parameters listed in Table 1. C, kinetic titrations for activation of 91–1198 nM ProTR155A in buffer containing ChCl (○) otherwise as described in A, along with the fit with the parameters listed in Table 1. 12 reactions out of a total of 21 analyzed are shown. D, end point titration for the reactions in buffer containing ChCl as described in B except that the line represents the fit of the binding equation with ΔFmax/Fo fixed at −35%. Reactions were performed and analyzed as described under “Experimental Procedures.”
Na+ Binding Affinity Estimated from the Kinetics of Chromo-genic Substrate Hydrolysis
To compare the apparent affinity of Na+ for thrombin, MzT(−F1), and MzTR155A, the initial rate of 200 μM (saturating) D-Phe-Pip-Arg-pNA hydrolysis was determined as a function of Na+ concentration. Na+ produced hyperbolic increases in rate of 4.9-fold, 3.1-fold, and 3.3-fold, from which apparent KD values of 12 ± 4, 9 ± 2, and 14 ± 4 mM were obtained for the substrate-bound forms of thrombin, MzT(−F1), and MzTR155A, respectively. The apparent affinities were the same and in reasonable agreement with the published KD of 10 mM for Na+ binding to the chromogenic substrate·thrombin complex at 25 °C but at higher pH and ionic strength (25).
The KD values for Na+ binding to the free forms of thrombin and MzT(−F1) were estimated from the dependence of kcat/Km on Na+ concentration (45). Despite the analysis of five progress curves at each Na+ concentration, the error in the dissociation constants was large (Fig. 8A). This was due to the necessity of fitting three parameters (kcat, Km, and Ki) to the progress curves and three parameters to kcat/Km as a function of Na+ concentration (kcat/Km in the absence and at saturating Na+ and KD). The KD values for thrombin and MzT(−F1) were both 26 ± 20 mM, roughly consistent with the value of 22 mM, determined at higher ionic strength and pH (25, 45). By contrast, kcat/Km for MzTR155A increased linearly up to 110 mM NaCl. This was accompanied by a ~12-fold decrease in Km, whereas this parameter decreased only ~2- to 3-fold for thrombin and MzT(−F1) (Fig. 8B). Analysis of the dependence of Km on Na+ concentration according to the non-essential activation mechanism as described under “Experimental Procedures,” gave an estimate of ~185 mM for the dissociation constant for Na+ binding to free MzTR155A and a linkage factor (α) of ~0.085, corresponding to a ~12-fold increase in Na+ affinity at saturating substrate concentration. The value of αKNa of ~16 mM from this analysis was in agreement with 14 ± 4 mM determined directly at saturating substrate concentration. The dependence of kcat on Na+ concentration for all of the enzymes was consistent with the directly measured dissociation constants for the substrate-saturated forms described above. The results demonstrated distinctly different kinetic behavior and a much higher KD for Na+ binding to MzTR155A, which contains the fragment 1 domain, compared with thrombin and MzT(−F1), which do not.
FIGURE 8. Effects of Na+ on chromogenic substrate hydrolysis by thrombin, MzT(−F1), and MzTR155A.
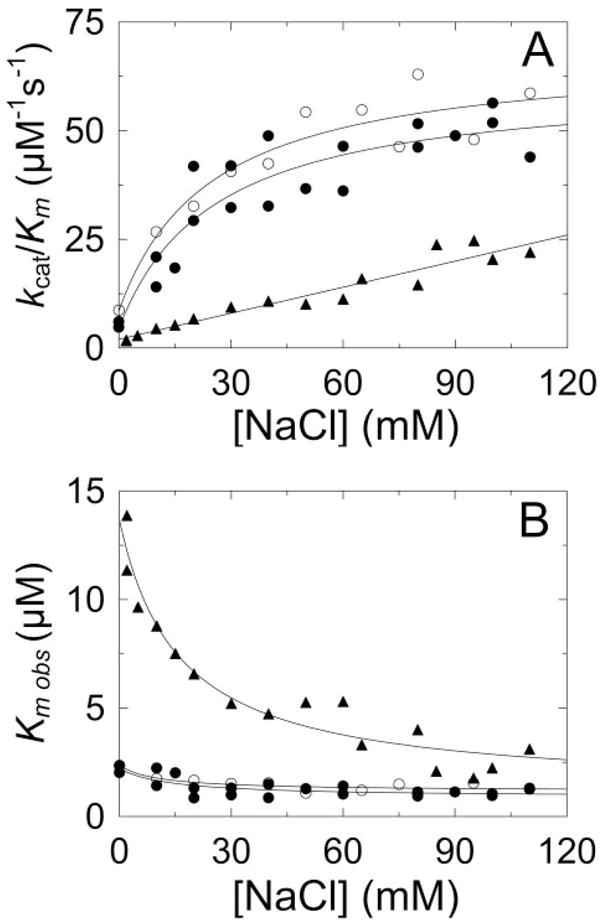
A, dependence of kcat/Km for D-Phe-Pip-Arg-pNA hydrolysis on Na+ concentration for thrombin (●), MzT(−F1) (○), and MzTR155A (▲). The solid lines for thrombin and MzT(−F1) represent the least squares fits of the data as described under “Experimental Procedures,” with indistinguishable KD values for Na+ binding of 26 ± 20 mM. The fit of a straight line is shown for MzTR155A. B, Na+ concentration dependence of the observed Km (Km,obs) for hydrolysis of D-Phe-Pip-Arg-pNA by thrombin (●), MzT(−F1) (○), and MzTR155A (▲). The solid lines represent the fits of Equation 1 with the parameters Km, α, and KNa of 2.2 μM, 0.39, and 34 mM for thrombin; 2.4 μM, 0.5, and 21 mM for MzT(−F1); and 14 μM, 0.085, and 185 mM for MzTR155A. Experiments were performed and analyzed as described under “Experimental Procedures.”
DISCUSSION
Examination of the appearance of Na+-exosite I linkage on ProT and its activation products supports the conclusion that proexosite I on ProT, Pre 1, and Pre 2 is not linked to Na+ binding. Expression of linkage accompanies activation of the thrombin, MzT(−F1), and MzT catalytic sites by cleavage at Arg320. Exosite I affinity and catalytic activity of MzT and MzT(−F1) are shown for the first time to be allosterically regulated by Na+. The well established coupling of protein substrate specificity to Na+ binding for thrombin suggests that the distinctly different specificity of MzT will be regulated by the MzT slow form to fast form transition. Increasing MzT exosite I affinity and catalytic activity in the allosteric transition by Na+ binding may also affect recognition of MzT as a substrate of the prothrombinase complex.
Linkage between Na+ binding and exosite I affinity for [5F]Hir-(54–65)( ) is expressed after cleavage at Arg320 of ProT and the ensuing conformational changes that activate the catalytic site and (pro)exosite I. The magnitudes of the effects of Na+ on the enhancement of [5F]Hir-(54–65)( ) affinity for exosite I on thrombin (5.6-fold) and MzT(−F1) (5.1- to 5.7-fold) were not significantly different, whereas MzTR155A showed a slightly larger effect (7.7- to 10.4-fold). The 5.6-fold increase in affinity for thrombin is in reasonable agreement with previously reported values for acetyl-Hir-(55–65) (2-fold (47)), Hir-(55–65) (2.3- and 6.4 fold (24, 48)), and acetyl-Hir-(53–64) (5.4-fold (32)), determined at different ionic strength, pH, and temperature.
The affinity of [5F]Hir-(54–65)( ) for (pro)exosite I on the zymogen forms, ProT, ProTR155A, Pre 1, and Pre 2 was consistently 1.3- to 1.7-fold higher in the presence of Na+. This difference is well within the experimental error and may not be significant. As shown in the crystal structure of the Pre 2·hirugen complex, the Na+ site is not properly formed on Pre 2 (49). On this basis, the results demonstrate that the absence of linkage between Na+ and proexosite I is due to the inability of all zymogen forms to bind Na+. The conformational changes that complete the folding of the catalytic domain and formation of the catalytic site are necessary for expression of allosteric linkage.
In a recent structural analysis of human thrombin, crystal packing interactions were identified that have influenced interpretation of previous structures of the fast, slow, and inactive forms (31). The structure of the D102N thrombin mutant in the absence of Na+ lacks crystal-packing interactions and is thought to represent more closely the inactive slow form in equilibrium with the active slow form and fast form. In the D102N thrombin structure, the catalytic site is blocked by large movements of Trp215 and Arg221a to occlude the catalytic and primary specificity sites, whereas the guanidinium group of Arg187 occupies the Na+ site (31). Rapid-reaction kinetic studies of Na+ binding indicated that 23% of wild-type thrombin is in the inactive slow form at 15 °C and that this form is not significantly populated at 37 °C (31, 33). Presumably this form was also present at some level in the studies done here at 25 °C, suggesting that binding of [5F]Hir-(54–65)( ) to exosite I contains some contribution from inactive slow forms. Whether such inactive slow forms exist for MzT(−F1) is not known.
Inactivation of thrombin, MzT(−F1), and MzT with FPR-CH2Cl virtually obliterated Na+-exosite I linkage as probed by [5F]Hir-(54–65)( ) binding affinity and normalized all of the affinities to values indistinguishable from that of the fast form of thrombin. This is fully consistent with an analysis of the structures of the thrombin slow and fast forms, free and FPR-inactivated, which demonstrated that active site labeling locks thrombin in a fast form (26), and the essentially identical structures of FPR-thrombin and the Hir-(53–64)·thrombin complex (29). The Na+ independence of the exosite I affinities is explained by binding of the transition state analog to the S1–S44 specificity sites, formation of a hemiketal with Ser195, and subsequent alkylation of His57. Although FPR-thrombin does bind Na+ (26), the relaxation of Asp189 in the S1 site, Ser195, and His57 to their conformations in the slow form in the absence of Na+ cannot take place. It should be noted that this finding does not compromise the conclusions of our previous studies of exosite I expression using FPR-inactivated thrombin and MzT(−F1), because, as shown here, these inactivated enzymes have affinities for the hirudin peptide essentially the same as the active fast forms. The affinities of MzTR155A for the peptide, however, are 2- to 3-fold lower at 110 mM NaCl.
Kinetic analysis of Na+ binding to MzTR155A revealed a lower affinity compared with thrombin and MzT(−F1), which was associated with a larger decrease in Km for the chromogenic substrate, and not significantly larger increases in kcat. This unexpected behavior suggests that the presence of the fragment 1 domain attenuates affinity for Na+ binding. The large effect of the presence of fragment 1 on the Na+-dependent apparent substrate binding affinity may be due to steric or conformational differences affecting the active site or the Na+ binding site, and indirectly exosite I. Further studies are needed to define the kinetic and molecular mechanism of MzT regulation by Na+.
Although the mechanism of Na+ regulation of MzTR155A is not resolved, the results demonstrate for the first time that MzT(−F1) and MzTR155A are Na+-regulated proteinases exhibiting linkage to exosite I. Na+ regulates the specificity of thrombin for procoagulant and anticoagulant substrates and effectors (15, 17, 18, 23, 24). The fast form has higher specificity (kcat/Km) than the slow form for the procoagulant substrates, fibrinogen and protease-activated receptors, whereas relative to the fast form, the slow form is more specific for protein C activation (48). Compared with thrombin, MzT(±F1) has ≤10% activity toward fibrinogen and ≤2% activity in platelet activation despite having an active catalytic site and exosite I (19, 20). Thrombin activates factor V in solution and factor V bound to phospholipid membranes in exosite I- and II-dependent reactions (22, 44, 50–52), whereas MzT activates factor V at a significant rate only when the substrate and enzyme are membrane-bound (21, 22). It has been suggested that MzT may play an important procoagulant role as a specific physiological activator of membrane-bound factor V during initiation of blood coagulation (21, 22). Based on the present results, and the observation that factor V activation is catalyzed more efficiently by the thrombin fast form (52), MzT-catalyzed factor V activation is predicted to be regulated by Na+. The dissociation constant for Na+ binding to thrombin under physiological conditions is 110 mM, indicating an equilibrium of 60% fast and 40% slow forms (25, 53, 54). The KD for Na+ binding to MzTR155A at 37 °C was not determined for the reasons cited above, but, depending on the temperature dependence of Na+ binding, MzT may be similarly poised for Na+ regulation physiologically.
In the thrombin·thrombomodulin complex, the slow form of thrombin becomes a potent anticoagulant proteinase due in part to the loss of specificity for fibrinogen and other procoagu-lant substrates, whereas the capacity for protein C activation is greatly increased (15–18, 48, 55). Activation of protein C by MzT and MzT(−F1) is also stimulated by thrombomodulin to rates comparable to thrombin and even faster rates when MzT is bound to phospholipid membranes (19, 20). This has been suggested to represent an important physiological role for MzT as an anticoagulant (19, 20). The present studies suggest that, like thrombin, Na+-bound and -free forms of MzT may exhibit higher procoagulant and anticoagulant activities, respectively.
ProT activation is dependent on expression of exosites on factor Xa in the factor Xa·Va·membrane complex that mediate a substrate recognition mechanism consisting of initial exosite binding and a subsequent conformational change engaging the catalytic site in peptide bond cleavage (7–14). Other studies support the hypothesis that (pro)exosite I on ProT also contributes to substrate recognition by mediating productive ProT binding and/or affecting the subsequent conformational change through interactions with the ProT binding site on factor Va in the prothrombinase complex (56–59). ProT activation catalyzed by prothrombinase proceeds sequentially, with initial cleavage at Arg320 to form MzT, followed by cleavage at Arg271 to generate thrombin and fragment 1.2 (2–7). Recent studies of the activation pathway support a mechanism in which substrates in the zymogen and proteinase states bind in alternate conformations that direct sequential cleavage at the two activation sites (8). The ProT zymogen is positioned for optimal cleavage at Arg320, whereas the product, MzT in the proteinase conformation, ratchets to a different bound conformation for cleavage at Arg271. On the basis of the evidence for the role of (pro)exosite I in substrate recognition through factor Va (56–59), and the differential expression of the exosite on ProT zymogen and proteinase forms (35, 36), expression of exosite I on MzT may contribute to ratcheting of the bound substrate for presentation of Arg271 for cleavage. The present studies show that the chromogenic substrate activity and affinity of MzT exosite I are enhanced by Na+, suggesting that coupling of Na+ binding and exosite I expression upon cleavage at Arg320 may contribute to recognition of this substrate. Previous studies of the role of Na+ on the rate of Pre 1 activation in the absence of membranes by the Na+-insensitive factor Xa mutant, Y225P, also lacking the γ-carboxyglutamic acid domain, showed a 10-fold enhancement by Na+, which was correlated with a modest increase in affinity for factor Va (60). On the other hand, with wild-type factor Xa assembled into prothrombinase the Na+ enhancement was only <1.5-fold (60). Whether Na+ affects the rate of MzT cleavage by pro-thrombinase remains an open question.
Acknowledgments
We thank Sarah Stuart for excellent technical assistance and Dr. Ingrid Verhamme for comments on the manuscript.
Footnotes
This work was supported in part by NHLBI, National Institutes of Health Grant HL038779 (to P. E. B.), and the Edmond Hustinx Foundation, Cardiovascular Research Institute Maastricht, Dept. of Biochemistry, University Maastricht. This work was also supported by the Dutch Organization for Scientific Research (VIDI Grant 916-046-330 to G. A. F. N.).
The abbreviations used are: ProT, prothrombin; Pre 1, prethrombin 1; Pre 2, prethrombin 2; F1, fragment 1; MzT, meizothrombin; MzT(−F1), meizo-thrombin-desfragment 1; T, thrombin; FPR-CH2Cl, D-Phe-Pro-Arg-CH2Cl; Hir-(54 – 65)( ), Gly-Asp-Phe-Glu-Glu-Ile-Pro-Glu-Glu-Tyr( )-Leu-Gln; [5F]Hir-(54 – 65)( ), Hir-(54 – 65)( ) labeled at the amino terminus with 5-carboxy(fluorescein); hirugen, hirudin-(53– 64); PEG, polyethylene glycol; pNA, p-nitroaniline.
Schechter-Berger (61) notation referring to the residues of a substrate (from the NH2-terminal end) as …P4-P3-P2-P1-P1′-P2′ … with the scissile bond at P1-P1′. The corresponding specificity subsites on the proteinase are designated ...S4-S3-S2-S1-S1′-S2′...
References
- 1.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 2.Rosing J, Zwaal RF, Tans G. J Biol Chem. 1986;261:4224–4228. [PubMed] [Google Scholar]
- 3.Krishnaswamy S, Mann KG, Nesheim ME. J Biol Chem. 1986;261:8977–8984. [PubMed] [Google Scholar]
- 4.Krishnaswamy S, Church WR, Nesheim ME, Mann KG. J Biol Chem. 1987;262:3291–3299. [PubMed] [Google Scholar]
- 5.Tans G, Janssen-Claessen T, Hemker HC, Zwaal RF, Rosing J. J Biol Chem. 1991;266:21864–21873. [PubMed] [Google Scholar]
- 6.Rosing J, Tans G. Thromb Haemost. 1988;60:355–360. [PubMed] [Google Scholar]
- 7.Krishnaswamy S. J Thromb Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 8.Bianchini EP, Orcutt SJ, Panizzi P, Bock PE, Krishnaswamy S. Proc Natl Acad Sci U S A. 2005;102:10099–10104. doi: 10.1073/pnas.0504704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krishnaswamy S, Betz A. Biochemistry. 1997;36:12080–12086. doi: 10.1021/bi970979+. [DOI] [PubMed] [Google Scholar]
- 10.Boskovic DS, Krishnaswamy S. J Biol Chem. 2000;275:38561–38570. doi: 10.1074/jbc.M006637200. [DOI] [PubMed] [Google Scholar]
- 11.Betz A, Krishnaswamy S. J Biol Chem. 1998;273:10709–10718. doi: 10.1074/jbc.273.17.10709. [DOI] [PubMed] [Google Scholar]
- 12.Wilkens M, Krishnaswamy S. J Biol Chem. 2002;277:9366–9374. doi: 10.1074/jbc.M110848200. [DOI] [PubMed] [Google Scholar]
- 13.Orcutt SJ, Pietropaolo C, Krishnaswamy S. J Biol Chem. 2002;277:46191–46196. doi: 10.1074/jbc.M208677200. [DOI] [PubMed] [Google Scholar]
- 14.Orcutt SJ, Krishnaswamy S. J Biol Chem. 2004;279:54927–54936. doi: 10.1074/jbc.M410866200. [DOI] [PubMed] [Google Scholar]
- 15.Page MJ, Macgillivray RT, Di Cera E. J Thromb Haemost. 2005;3:2401–2408. doi: 10.1111/j.1538-7836.2005.01456.x. [DOI] [PubMed] [Google Scholar]
- 16.Huntington JA. J Thromb Haemost. 2005;3:1861–1872. doi: 10.1111/j.1538-7836.2005.01363.x. [DOI] [PubMed] [Google Scholar]
- 17.Bode W. J Thromb Haemost. 2005;3:2379–2388. doi: 10.1111/j.1538-7836.2005.01356.x. [DOI] [PubMed] [Google Scholar]
- 18.Bode W. Blood Cells Mol Dis. 2006;36:122–130. doi: 10.1016/j.bcmd.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 19.Doyle MF, Mann KG. J Biol Chem. 1990;265:10693–10701. [PubMed] [Google Scholar]
- 20.Cote HC, Stevens WK, Bajzar L, Banfield DK, Nesheim ME, MacGillivray RT. J Biol Chem. 1994;269:11374–11380. [PubMed] [Google Scholar]
- 21.Tans G, Nicolaes GA, Thomassen MC, Hemker HC, van Zonneveld AJ, Pannekoek H, Rosing J. J Biol Chem. 1994;269:15969–15972. [PubMed] [Google Scholar]
- 22.Bukys MA, Orban T, Kim PY, Beck DO, Nesheim ME, Kalafatis M. J Biol Chem. 2006;281:18569–18580. doi: 10.1074/jbc.M600752200. [DOI] [PubMed] [Google Scholar]
- 23.Di Cera E. C R Biol. 2004;327:1065–1076. doi: 10.1016/j.crvi.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Di Cera E, Dang QD, Ayala YM. Cell Mol Life Sci. 1997;53:701–730. doi: 10.1007/s000180050091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wells CM, Di Cera E. Biochemistry. 1992;31:11721–11730. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- 26.Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. J Biol Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- 27.Pineda AO, Savvides SN, Waksman G, Di Cera E. J Biol Chem. 2002;277:40177–40180. doi: 10.1074/jbc.C200465200. [DOI] [PubMed] [Google Scholar]
- 28.Prasad S, Cantwell AM, Bush LA, Shih P, Xu H, Di Cera E. J Biol Chem. 2004;279:10103–10108. doi: 10.1074/jbc.M312614200. [DOI] [PubMed] [Google Scholar]
- 29.Vijayalakshmi J, Padmanabhan KP, Mann KG, Tulinsky A. Protein Sci. 1994;3:2254–2271. doi: 10.1002/pro.5560031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrell CJ, Bush LA, Mathews FS, Di Cera E. Biophys Chem. 2006;121:177–184. doi: 10.1016/j.bpc.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Pineda AO, Chen ZW, Bah A, Garvey LC, Mathews FS, Di Cera E. J Biol Chem. 2006;281:32922–32928. doi: 10.1074/jbc.M605530200. [DOI] [PubMed] [Google Scholar]
- 32.Lai MT, Di Cera E, Shafer JA. J Biol Chem. 1997;272:30275–30282. doi: 10.1074/jbc.272.48.30275. [DOI] [PubMed] [Google Scholar]
- 33.Bah A, Garvey LC, Ge J, Di Cera E. J Biol Chem. 2006;281:40049–40056. doi: 10.1074/jbc.M608600200. [DOI] [PubMed] [Google Scholar]
- 34.Anderson PJ, Nesset A, Dharmawardana KR, Bock PE. J Biol Chem. 2000;275:16428–16434. doi: 10.1074/jbc.M001254200. [DOI] [PubMed] [Google Scholar]
- 35.Anderson PJ, Bock PE. J Biol Chem. 2003;278:44489–44495. doi: 10.1074/jbc.M306916200. [DOI] [PubMed] [Google Scholar]
- 36.Anderson PJ, Nesset A, Bock PE. J Biol Chem. 2003;278:44482–44488. doi: 10.1074/jbc.M306917200. [DOI] [PubMed] [Google Scholar]
- 37.Bock PE, Olson ST, Björk I. J Biol Chem. 1997;272:19837–19845. doi: 10.1074/jbc.272.32.19837. [DOI] [PubMed] [Google Scholar]
- 38.Bock PE. J Biol Chem. 1992;267:14963–14973. [PubMed] [Google Scholar]
- 39.Bock PE, Craig PA, Olson ST, Singh P. Arch Biochem Biophys. 1989;273:375–388. doi: 10.1016/0003-9861(89)90496-7. [DOI] [PubMed] [Google Scholar]
- 40.Bock PE. J Biol Chem. 1992;267:14974–14981. [PubMed] [Google Scholar]
- 41.Fenton JW, 2nd, Fasco MJ, Stackrow AB. J Biol Chem. 1977;252:3587–3598. [PubMed] [Google Scholar]
- 42.Mann KG, Elion J, Butkowski RJ, Downing M, Nesheim ME. Methods Enzymol. 1981;80:286–302. doi: 10.1016/s0076-6879(81)80025-0. [DOI] [PubMed] [Google Scholar]
- 43.Petrovan RJ, Govers-Riemslag JW, Nowak G, Hemker HC, Tans G, Rosing J. Biochemistry. 1998;37:1185–1191. doi: 10.1021/bi971948h. [DOI] [PubMed] [Google Scholar]
- 44.Dharmawardana KR, Olson ST, Bock PE. J Biol Chem. 1999;274:18635–18643. doi: 10.1074/jbc.274.26.18635. [DOI] [PubMed] [Google Scholar]
- 45.Di Cera E, Hopfner KP, Dang QD. Biophys J. 1996;70:174–181. doi: 10.1016/S0006-3495(96)79558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley & Sons, Inc; New York: 1993. pp. 227–228. [Google Scholar]
- 47.Ayala YM, Vindigni A, Nayal M, Spolar RS, Record MT, Jr, Di Cera E. J Mol Biol. 1995;253:787–798. doi: 10.1006/jmbi.1995.0591. [DOI] [PubMed] [Google Scholar]
- 48.Dang OD, Vindigni A, Di Cera E. Proc Natl Acad Sci U S A. 1995;92:5977–5981. doi: 10.1073/pnas.92.13.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang E, Tulinsky A. Biophys Chem. 1997;63:185–200. doi: 10.1016/s0301-4622(96)02227-2. [DOI] [PubMed] [Google Scholar]
- 50.Dharmawardana KR, Bock PE. Biochemistry. 1998;37:13143–13152. doi: 10.1021/bi9812165. [DOI] [PubMed] [Google Scholar]
- 51.Esmon CT, Lollar P. J Biol Chem. 1996;271:13882–13887. doi: 10.1074/jbc.271.23.13882. [DOI] [PubMed] [Google Scholar]
- 52.Myles T, Yun TH, Hall SW, Leung LL. J Biol Chem. 2001;276:25143–25149. doi: 10.1074/jbc.M011324200. [DOI] [PubMed] [Google Scholar]
- 53.Guinto ER, Di Cera E. Biochemistry. 1996;35:8800–8804. doi: 10.1021/bi9608828. [DOI] [PubMed] [Google Scholar]
- 54.Griffon N, Di Stasio E. Biophys Chem. 2001;90:89–96. doi: 10.1016/s0301-4622(01)00129-6. [DOI] [PubMed] [Google Scholar]
- 55.Esmon CT, Esmon NL, Harris KW. J Biol Chem. 1982;257:7944–7947. [PubMed] [Google Scholar]
- 56.Anderson PJ, Nesset A, Dharmawardana KR, Bock PE. J Biol Chem. 2000;275:16435–16442. doi: 10.1074/jbc.M001255200. [DOI] [PubMed] [Google Scholar]
- 57.Beck DO, Bukys MA, Singh LS, Szabo KA, Kalafatis M. J Biol Chem. 2004;279:3084–3095. doi: 10.1074/jbc.M306850200. [DOI] [PubMed] [Google Scholar]
- 58.Arocas V, Lemaire C, Bouton MC, Bezeaud A, Bon C, Guillin MC, Jandrot-Perrus M. Thromb Haemost. 1998;79:1157–1161. [PubMed] [Google Scholar]
- 59.Chen L, Yang L, Rezaie AR. J Biol Chem. 2003;278:27564–27569. doi: 10.1074/jbc.M302707200. [DOI] [PubMed] [Google Scholar]
- 60.Rezaie AR, He X. Biochemistry. 2000;39:1817–1825. doi: 10.1021/bi992006a. [DOI] [PubMed] [Google Scholar]
- 61.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]