

Abstract
Mitochondrial membrane potential (ΔΨm) depolarization contributes to cell death and electrical and contractile dysfunction in the post-ischemic heart. An imbalance between mitochondrial reactive oxygen species production and scavenging was previously implicated in the activation of an inner membrane anion channel (IMAC), distinct from the permeability transition pore (PTP), as the first response to metabolic stress in cardiomyocytes. The glutathione redox couple, GSH/GSSG, oscillated in parallel with ΔΨm and the NADH/NAD+ redox state. Here we show that depletion of reduced glutathione is an alternative trigger of synchronized mitochondrial oscillation in cardiomyocytes and that intermediate GSH/GSSG ratios cause reversible ΔΨm depolarization, although irreversible PTP activation is induced by extensive thiol oxidation. Mitochondrial dysfunction in response to diamide occurred in stages, progressing from oscillations in ΔΨm to sustained depolarization, in association with depletion of GSH. Mitochondrial oscillations were abrogated by 4′-chlorodiazepam, an IMAC inhibitor, whereas cyclosporin A was ineffective. In saponin-permeabilized cardiomyocytes, the thiol redox status was systematically clamped at GSH/GSSG ratios ranging from 300:1 to 20:1. At ratios of 150:1-100:1, ΔΨm depolarized reversibly, and a matrix-localized fluorescent marker was retained; however, decreasing the GSH/GSSG to 50:1 irreversibly depolarized ΔΨm and induced maximal rates of reactive oxygen species production, NAD(P)H oxidation, and loss of matrix constituents. Mitochondrial GSH sensitivity was altered by inhibiting either GSH uptake, the NADPH-dependent glutathione reductase, or the NADH/NADPH transhydrogenase, indicating that matrix GSH regeneration or replenishment was crucial. The results indicate that GSH/GSSG redox status governs the sequential opening of mitochondrial ion channels (IMAC before PTP) triggered by thiol oxidation in cardiomyocytes.
The dual nature of oxygen as a vital electron acceptor in oxidative phosphorylation and as a dangerously reactive molecule has created pressure for the cell to evolve powerful antioxidant defenses that convert reactive oxygen species (ROS)3 into harmless products to maintain a predominantly reduced redox environment. This depends on the following two factors: the reduction potential of the electron carriers, and the reducing capacity (i.e. the total concentration of the reduced species) of linked redox couples present in the cytoplasm or in the intraorganellar compartments (e.g. the mitochondrial matrix) of the cell. In addition to the redox couples involved in mitochondrial electron transport (the nicotinamide adenine dinucleotides NADH/NAD+, and the flavins FADH2/FAD), the three main cellular redox pairs participating in intracellular reactions include reduced/oxidized glutathione (GSH/GSSG), thioredoxin (Trx(SH)2/TrxSS), and NADPH/NADP+, with the latter providing the thermodynamic driving force behind the glutathione and thioredoxin systems (1). The GSH/GSSG pool is the largest of the cell (2, 3) and is considered to be a major indicator of the cellular redox status. GSH is a tripeptide (l-γ-glutamyl-l-cysteinyl-glycine) that is converted from its disulfide form, GSSG, by an NADPH-dependent reductase (glutathione reductase (GR)) whose activity increases in response to an increase in GSSG concentration. GSH provides reducing power for a variety of thiol modifications of disulfide bridges, thioethers, and thioesters and is a substrate for protein glutathionylation (4, 5). Overall, the balance of GSH and GSSG provides a dynamic indicator of oxidative stress (6).
Mitochondria, as the central site of oxygen consumption in the cell during the process of oxidative metabolism, are also a main source of ROS production. At the level of the respiratory chain, the highly positive reduction potential of the oxygen/superoxide anion redox pair combined with the proximity of other redox pairs such as the ubiquinone (CoQ)/ubisemiquinone radical can result in . generation (1). Under normal conditions, there is a balance between ROS formation and antioxidant activity; however, under pathological conditions oxidative stress can occur as a consequence of either increased ROS production or by depletion of the antioxidant pool (6-8).
Higher organisms have adapted to the presence of potentially toxic ROS within cells not only by building up antioxidant defenses but also by harnessing ROS as signaling molecules for regulating the activity of the cell. Changes in the cellular redox environment, both oxidative and reductive, can trigger redox cascades altering signal transduction, DNA, RNA and protein synthesis, enzyme regulation, gene expression, and cell cycle progression (9-13). For some proteins (e.g. phosphofructokinase, 3-hydroxy-3-methylglutaryl-CoA reductase, ribonuclease A, and lysozyme), oxidized thiols (disulfide state) are essential for biological function (14, 15), whereas the activity of other proteins depends on maintaining critical thiols in the reduced state. Such is the case for the ryanodine receptor of the sarcoplasmic reticulum of muscle cells, which possesses sensitive sulfhydryl groups that influence the rate of Ca2+ release depending upon the thiol redox status (16). Considerable evidence also shows that agents altering GSH concentration affect transcription of detoxification enzymes, cell proliferation, and apoptosis (17, 18) or necrosis, depending on the severity of the oxidative challenge.
GSH is synthesized in the cytoplasm and is exchanged with other intracellular compartments, with overall cellular GSH/GSSG ratios ranging between 30:1 and 300:1, but with the exception that the endoplasmic reticulum maintains a relatively oxidative environment with ratios of 1:1 to 3:1 (14, 18). Mitochondria do not synthesize GSH but are able to transport and accumulate up to ∼15% of the total cellular GSH (1, 19). As both a source and target of ROS production, the importance of the antioxidant capacity of mitochondria has been emphasized in recent years. An established target of oxidative stress in mitochondria is the permeability transition pore (PTP), a major player in initiating both apoptotic and necrotic cell death. Oxidative stress through oxidation of intracellular GSH and other critical sulfhydryl groups favors the PTP opening (20-22). Previously, we demonstrated that metabolic stress in the form of substrate deprivation (23, 24) or localized ROS generation (25) can trigger cell-wide oscillations or collapse of ΔΨm in isolated cardiomyocytes and that cyclical GSH oxidation occurred in parallel. The magnitude and extent of oxidative stress on the mitochondrial network was a key predictor of whether synchronized cell-wide mitochondrial oscillations occurred (25, 26). A reproducible threshold of oxidation of an ROS-sensitive reporter preceded the first global ΔΨm transition of the mitochondrial network, and we referred to this sensitive state as “mitochondrial criticality” (27). This complex self-organizing phenomenon was scaled to alter the electrical and Ca2+ handling properties of the whole cell and contributed to the arrhythmias induced by ischemia-reperfusion in the whole heart (28, 29). Further investigation into the mechanism of this phenomenon revealed that several inhibitors of the mitochondrial inner membrane anion channel (IMAC) could reversibly suppress or prevent the mitochondrial ROS-induced ROS release response, thereby stabilizing ΔΨm (28). A computational model also supported a mechanistic scheme involving the activation of IMAC by ROS (26).
In this study, we investigate the role played by the glutathione redox potential in the approach to mitochondrial criticality and the collapse of ΔΨm in the mitochondrial network. We show that the glutathione redox status determines the rate of mitochondrial ROS production and that the changes in the absolute concentrations of GSH and GSSG as well as the GSH/GSSG ratio, trigger, at moderate ratios (150:1 to 100:1), the reversible opening of IMAC and, at a lower ratio (50:1), irreversible PTP activation.
EXPERIMENTAL PROCEDURES
Cardiomyocyte Isolation and Handling
Cardiomyocytes were prepared from adult guinea pig hearts using a Langendorff perfusion system as described before (23). The freshly isolated cells were handled as described before (25, 26). Briefly, after isolation, cells were either immediately used for imaging or stored in Dulbecco’s modified Eagle’s medium (10-013 DMEM, Mediatech, Inc., Herndon, VA) containing 5% fetal bovine serum and 1% penicillin/streptomycin in a 5% CO2 incubator at 37 °C for at least 2 h before assaying them. Experimental recordings of the freshly isolated cells (with or without storage in DMEM) started after suspending them in Tyrode solution containing (in mm) the following: 140 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, 1 CaCl2, pH 7.5 (adjusted with NaOH), supplemented with 10 mm glucose or the addition of diamide when indicated. The perfusion chamber containing the cardiomyocytes was thermostatically controlled at 37 °C with unrestricted access to atmospheric oxygen on the stage of a Nikon E600FN upright microscope.
Fluorescent Probes and Loading Conditions
Tetramethyl-rhodamine ethyl (or methyl) ester (TMRM) accumulates across polarized membranes with a Nernstian distribution. ROS production was monitored with the ROS-sensitive fluorescent probe 5-(-6)-chloromethyl-2′,7′-dichlorohydrofluorescein diacetate (CM-H2DCFDA, Invitrogen) (25).
Reduced glutathione (GSH) was measured in myocytes loaded with the membrane permeant indicator monochlorobimane (MCB) (26, 30). This probe reports the level of GSH as the fluorescent product glutathione S-bimane (GSB) according to the reversible reaction, MCB + GSH ⇔ GSB. We have determined that concentrations up to 50 μm MCB do not deplete the intracellular pool of GSH to a level that compromises cell viability.
The concentrations of the probes were 100 nm TMRM, 2 μm CM-H2DCFDA, and 50 μm MCB, and they were loaded into the cells for at least 20 min at 37 °C. The response of the MCB fluorescence to changes in GSH was quantified by the addition of exogenous GSH (see also below and Supplemental Material).
Two-photon Microscopy
The equipment and methods utilized for imaging isolated cardiomyocytes were as described previously (25, 26). Images were recorded using a two-photon laser scanning microscope (Bio-Rad MRC-1024MP) with excitation at 740 nm (or 780 nm for GSB imaging). The red emission of TMRM was collected at 605 ± 25 nm; the green emission of CM-DCF was recorded at 525 ± 25 nm, and the blue GSB emission was recorded at 480 ± 20 nm. The NADH emission was collected as the total fluorescence <490 nm. Because of the overlap of NADH and GSB emissions (at steady state, NADH emission was approximately one-third that of GSB (26, 31)), separate experiments were conducted in which we monitored either ΔΨm, ROS, and NADH or ΔΨm and GSB, with cells loaded with TMRM and CM-DCF or TMRM and MCB, respectively. Images were collected simultaneously at the indicated time intervals (3.5, 5, or 30 s) and stored as described before (25).
Permeabilization Procedure of Cardiomyocytes
Myocytes, which had been stored in DMEM for at least 2 h (which facilitates repletion of the cellular glutathione pool), were resuspended in the experimental solution, and the fluorescent probes were loaded directly in the perfusion chamber on the stage of the microscope for at least 20 min at 37 °C before starting the experiment. The permeabilizing solution (PS) contained 25 μg/ml saponin (to selectively permeabilize the sarcolemma but not the mitochondrial membranes) and (in mm) 130 potassium methanesulfonate, 20 KCl, 0.5 MgCl2, 10 HEPESNa, 0.1 EGTA, 3 ATP, 5 pyruvate; the pH was adjusted to 7.2 with KOH and was perfused into the chamber for 2.5 min. Controls were performed to determine the efficiency of permeabilization by comparing the effects of solutions with or without saponin but containing the same GSH/GSSG ratios as shown in the results (with GSH constant at 3 mm). In the absence of saponin, there was no response of the mitochondria to lowering the GSH/GSSG ratio to <50:1. In the first set of experiments GSH/GSSG was varied in the presence of a fixed GSH level (at either 1, 3, or 4 mm) by increasing the GSSG concentration. In other experiments, as indicated under “Results,” the GSSG concentration was fixed at 10 μm, and the GSH was varied. The initial permeabilization of the cells was always carried out in the presence of a 300:1 GSH/GSSG ratio. TMRM, CM-H2DCFDA, or MCB was included in the perfusion solution in most experiments to avoid any loss of the mitochondria-localized dyes in the course of the experiment in permeabilized cells subjected to changes in the GSH/GSSG ratio. A main aim of the permeabilization was to preserve mitochondrial integrity along with facilitating the access to cells of the perfusion solution. Several controls were run in order to verify mitochondrial function and the extent of permeabilization. These are described in the Supplemental Material.
Kinetic Experiments with Intact and Permeabilized Cardiomyocytes
Kinetic experiments with intact or permeabilized cardiomyocytes were carried out at 37 °C in a thermostatically controlled flow chamber mounted on the stage of the upright microscope (Nikon E600FN) attached to the multiphoton laser scanning system. A constant flow was controlled with a peristaltic pump. With the fluorescent dye carboxyfluorescein (not taken up by the cells), we determined that the delay time for a compound to increase from 10 to 90% of its final concentration was 60 s for the chamber utilized. TMRM- and CM-DCF-loaded or TMRM- and MCB-loaded cardiomyocytes were perfused with normal modified Tyrode’s solution (as described above for cardiomyocyte isolation) containing 1 mm Ca2+ in the absence or presence of different reagents (e.g. diamide).
Quantification of Intracellular GSH
GSH levels in intact cells were monitored after incubating the cells with MCB. To determine the intracellular concentration of GSH (in mm), we performed in vitro calibrations by serially diluting a 10 mm GSB stock solution that was loaded into glass microcapillaries and placed in the same field as MCB-loaded myocytes. Purified glutathione S-transferase (GST, from rabbit liver; Sigma) was used to catalyze (37 °C) the reaction of GSH and MCB in an assay mixture consisting of the following (in mm): 10 PO4KH2/10 HEPES, pH 7.1, 0.2 MCB, 10 GSH, and 3 units of GST. The time course of the reaction was monitored with fluorometry at maximal emission of the fluorescent adduct GSB (λem = 390 nm; λex = 480 nm). We used a fluorometer (Photon Technology International) equipped with an adjustable photomultiplier and a thermostated cuvette with stirring.
To calibrate the two-photon microscope for the measurement of intracellular GSH, we filled the microcapillaries (50 μm diameter) with known amounts of GSB obtained as described above. The capillaries were imaged by two-photon microscopy in the range 1-10 mm with a linear response obtained in the range 1-5 mm (supplemental Fig. S3A). This calibration standard was used as a reference for measuring intracellular GSH in myocytes loaded with MCB. The two-photon imaging of the myocytes loaded with 50 μm MCB was performed in the presence of a 3 mm GSB-filled microcapillary as an internal standard (supplemental Fig. S3B). The steady state cell GSB fluorescence signal was measured and referred to the internal standard of 3 mm GSB. Using this method we found an ∼2.7 mm concentration of GSH in isolated cardiomyocytes from guinea pig in high K+ or DMEM (see supplemental Fig. S3C).
RESULTS
Effects of Glutathione Oxidation on ΔΨm in Intact Cells
We have previously shown that cell-wide oscillations in mitochondrial energetics in adult ventricular myocytes are preceded by a threshold level of oxidation of the ROS probe CM-H2DCF (25-27). As an alternative method to induce oxidative stress in the myocyte, we tested whether a pro-oxidative shift in the thiol status of glutathione, the largest cellular redox pool, influenced mitochondrial ROS accumulation and triggered ΔΨm oscillations.
Several intracellular variables related to the mitochondrial functional status (ΔΨm, NADH, ROS, and GSH) were monitored in intact cardiomyocytes in the absence or presence of the thiol oxidant diamide (0.1 mm), which effectively depletes the reduced glutathione pool (5, 32). Diamide induced an increased rate of mitochondrial ROS production that, upon reaching a threshold level of oxidative stress, triggered oscillations in ΔΨm and NADH (Fig. 1A, 1st arrow). Further depletion of GSH in the presence of diamide resulted in the eventual irreversible depolarization of ΔΨm, followed ∼800 s later by cell contracture (Fig. 1A, 2nd arrow). A higher concentration of diamide (1 mm) provoked rapid ΔΨm loss and cell contracture without traversing the oscillatory domain (data not shown). Similar results were also obtained with another thiol-oxidizing agent, diallyldisulfide (31), a component of garlic extract (data not shown).
FIGURE 1. Diamide-triggered mitochondrial oscillations in intact cardiomyocytes.
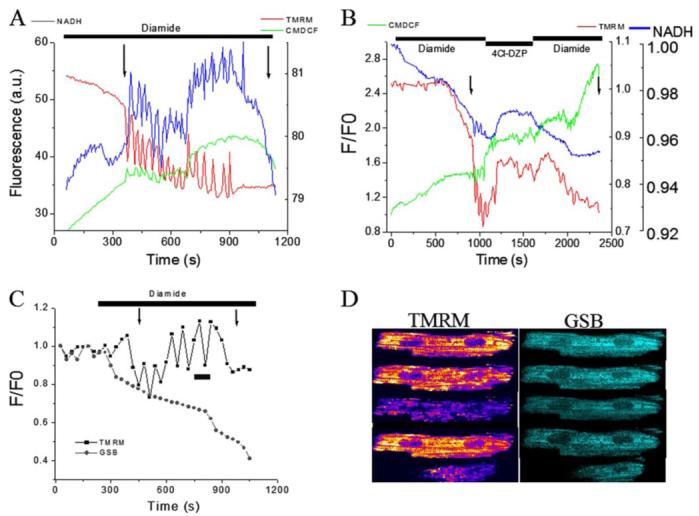
Freshly isolated guinea pig cardiomyocytes were loaded with TMRM (ΔΨm probe) and CM-H2DCFDA (ROS probe) at 37 °C and imaged by two-photon laser scanning fluorescence microscopy in a perfusion chamber. A, after exposure to the thiol-oxidizing agent diamide (0.1 mm), the oxidation of the ROS probe steadily increased until mitochondrial oscillations were spontaneously triggered. Cycles of ΔΨm depolarization-repolarization and NADH oxidation-reduction continued in-phase, usually for more than 10 min before the irreversible collapse of ΔΨm and maximal NADH oxidation occurred followed by rapid cell contracture. The 1st and 2nd arrows mark the onsets of ΔΨm oscillation and sustained depolarization associated with cell contracture, respectively. Images were collected every 3.5 s. B, mitochondrial benzodiazepine receptor antagonist 4Cl-DZP (64 μm) stopped the diamide-elicited oscillations and stabilized ΔΨm. ΔΨm oscillation and cell death resumed after washout in the continued presence of diamide. C, cardiomyocyte loaded with TMRM (100 nm) and MCB (50 μm) was treated with 0.1 mm diamide under similar conditions as in A and B. Mitochondrial oscillations were triggered after an ∼20% decrease in the GSB fluorescence. Images were acquired every 30 s. D, montage of images taken from the time course depicted in C during an oscillatory cycle and during the irreversible collapse of ΔΨm.
Exposure of the cells to 4′-chlorodiazepam (4Cl-DZP) during the diamide-induced ΔΨm oscillations stabilized ΔΨm, reversed the oxidation of the mitochondrial NADH pool, and slowed the rate of CM-H2DCF oxidation (Fig. 1B). This effect was reversible; after washout of 4Cl-DZP in the presence of diamide, the rate ROS production increased, the NADH pool oxidized, and ΔΨm depolarized, subsequently ending in cell contracture. ΔΨm oscillation and sustained loss of ΔΨm were correlated with the gradual depletion of the intracellular GSH pool, as monitored by a continuous decrease in the fluorescence levels of the GSB adduct (Fig. 1C). The effect of 4Cl-DZP to stabilize ΔΨm in the presence of diamide (Fig. 2A) was not reproduced with the PTP inhibitor cyclosporin A (CsA; Fig. 2B), indicating that 4Cl-DZP was inhibiting a distinct target on the inner membrane (25, 33). In fact, although 4Cl-DZP had a repolarizing influence of ΔΨm that could be washed out to re-initiate oscillations, CsA exacerbated mitochondrial depolarization (probably a nonspecific toxic effect that is opposite to what would be expected for inhibition of PTP), with partial restoration of oscillation upon washout. Thus, moderate depletion of GSH with diamide triggers reversible 4Cl-DZP-sensitive oscillations in ΔΨm. Continued exposure to diamide for a long time severely depletes the GSH antioxidant pool and activates the PTP irreversibly (at the arrows in Figs. 1 and 2).
FIGURE 2. Inhibition of diamide-induced ΔΨm oscillations by 4Cl-DZP but not CsA.
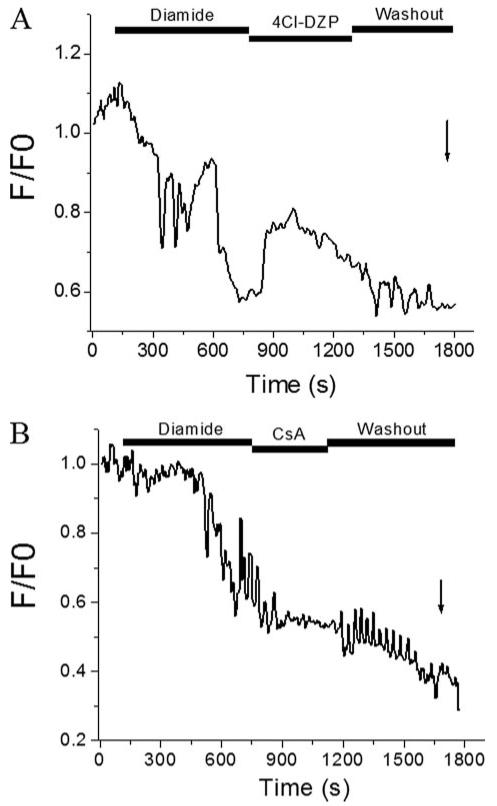
A, freshly isolated cardiomyocytes were loaded with 100 nm TMRM and exposed to 0.1 mm diamide until oscillations were induced. Treatment with 4-Cl-DZP (64 μm) stopped the oscillations and stabilized ΔΨm in the polarized state. After washout of 4Cl-DZP, ΔΨm oscillations returned before the mitochondria irreversibly depolarized and the cell went into contracture. B, at moderate levels of GSH depletion by diamide, ΔΨm oscillations were not reversed by the PTP inhibitor CsA (1 μm). In fact, CsA had a nonspecific depolarizing effect that was the opposite what would be expected for PTP inhibition. More severe GSH depletion activated PTP later in the experiment, as indicated by the arrow. Images were collected every 3.5 s.
Taken together, the results suggest that the mitochondrial oscillations elicited by diamide are consistent with the mechanism we have described previously for other triggers of metabolic stress, i.e. that mitochondrial oxidative stress triggers the activation of IMAC as part of a self-sustaining limit cycle oscillator (25, 26). Importantly, these results strongly suggest that GSH, and likely the glutathione redox potential, are main cellular variables that determine the approach of the mitochondrial network to criticality through an increase in oxidative stress. In the next section, we test this possibility in a permeabilized cell system.
Influence of the GSH/GSSG Ratio and Glutathione Redox Potential on ΔΨm in Partially Permeabilized Myocytes
Varying GSH/GSSG
In the first series of experiments in saponin-permeabilized cardiomyocytes, cells were exposed progressively to GSH/GSSG ratios ranging from 300:1 to 50:1 effected by increasing the concentration of GSSG while keeping the GSH constant at 3 mm. This concentration of GSH was used because it was close to the intracellular [GSH] determined in freshly isolated cardiomyocytes using MCB as a probe (see under “Experimental Procedures” and Supplemental Material). In association with a pro-oxidative shift in the glutathione reduction potential, an increase in both the oxidation rate of the CM-H2DCF (Fig. 3, A and B) and of the NADH pool was observed, together with a graded depolarization of ΔΨm up to the 50:1 GSH/GSSG ratio, at which point ΔΨm collapsed (Fig. 3B; n = 7 cells from six independent experiments). Sharp positive inflections in the ROS signal were evident at the transitions to GSH/GSSG ratios of 100:1 and 50:1 (Fig. 3, A and B). TMRM images revealed that at GSH/GSSG ratios between 150:1 and 100:1, transient depolarizations of ΔΨm with a heterogeneous TMRM distribution could be observed (Fig. 3C, bottom left), similar to that noted above in intact diamide-treated cells. The CM-DCF probe (Mr < 500) was retained in the mitochondrial matrix during ΔΨm oscillation, in keeping with our earlier findings (25) (Fig. 3C, bottom right). At the critical ratio of 50:1, an irreversible collapse of ΔΨm together with complete release of the ROS sensor and oxidation of the NADH pool ensued, involving the entire mitochondrial network (Fig. 3, B and D). This sustained depolarization occurred in association with maximal oxidation of the ROS probe and was followed shortly after by loss of the probe from the mitochondrial matrix, consistent with a massive mitochondrial permeability transition. This permeability transition often took the form of a slow wave (∼30 μm/min) that traveled longitudinally across the cell (Fig. 3D).
FIGURE 3. ROS production, ΔΨm, and NADH in saponin-permeabilized cardiomyocytes.
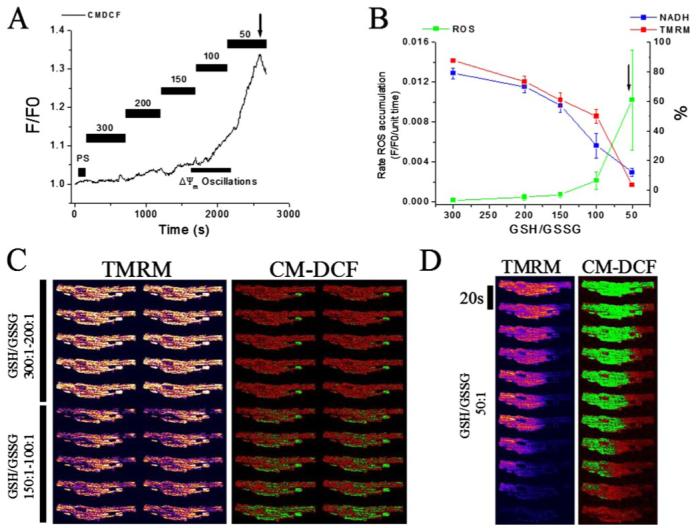
Myocytes stored in DMEM were resuspended in the experimental solution in the perfusion chamber and loaded with TMRM (100 nm) and CM-H2DCFDA (2 μm) for at least 20 min. After loading, the excess dye was washed out, and the cells were briefly superfused with a permeabilizing solution (see “Experimental Procedures”) and then continuously perfused with an intracellular solution containing different GSH/GSSG ratios as indicated. ΔΨm, oxidation of the ROS probe, and NADH redox state were simultaneously monitored using two-photon fluorescence excitation. TMRM was included in the medium to avoid depletion of the probe during depolarization-repolarization cycles. A, representative raw trace of CM-DCF signal at different ratios, from which the rates of ROS production are calculated in the steady portion of the signal. The bar within the range 150 - 100:1 indicates where IMAC-dependent ΔΨm oscillations happen. B, rate of oxidation of the ROS probe (F/F0/unit time), NADH (in % of initial fluorescence before permeabilization), and TMRM (ΔΨm, in % of initial fluorescence before permeabilization) obtained from seven different cells in independent experiments (mean ± S.E.) at different GSH/GSSG ratios (3 mm GSH concentration). The arrows in A and B denote the point at which ΔΨm irreversibly collapsed. C and D, representative montage of images of a permeabilized cardiomyocyte loaded with the ΔΨm and ROS sensors at different GSH/GSSG ratios: 300:1 (C, top left), 200:1 (C, top right), 150:1 (C, bottom left), 100:1 (C, bottom right), and 50:1 (D). Notice the ΔΨm oscillation (in the range 150:1 to 100:1) and the irreversible depolarization with exit of the ROS probe from mitochondria (50:1).
When the same experiment shown in Fig. 3 was performed in the presence of the IMAC inhibitor 4Cl-DZP (60 μm), mitochondrial ROS production increased as compared with the control (Fig. 4A) without affecting significantly the rate of NADH oxidation. Importantly, 4CL-DZP preserved a higher ΔΨm until irreversible depolarization at the 50:1 ratio (Fig. 4B). In contrast, the PTP inhibitor CsA (1 μm) had no effect either on the rate of oxidation of the ROS probe (Fig. 4A) or ΔΨm (Fig. 4B). The GSH/GSSG ratio at which irreversible ΔΨm depolarization occurred (50:1) was not altered by 4CL-DZP or CsA; however, the timing of this event was consistently delayed in the presence of CsA. The findings support the hypothesis that IMAC opens at moderate oxidative stress levels, whereas PTP activation represents a terminal, irreversible event.
FIGURE 4. ROS production, ΔΨm, and NADH in permeabilized cardiomyocytes in the presence of IMAC or PTP inhibitors.
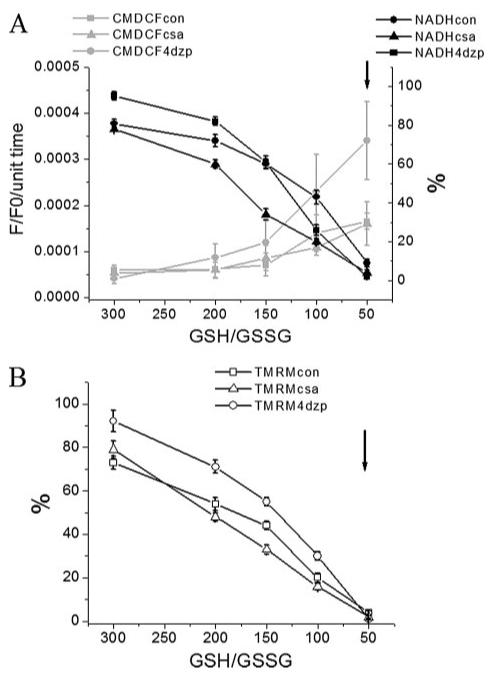
Myocytes were handled, loaded with the ΔΨm and ROS sensors, and permeabilized as described in the legend of Fig. 3. Rate of oxidation of the ROS probe (F/F0/unit time), NADH (in % of initial fluorescence before permeabilization) (A), and ΔΨm (in % of initial TMRM fluorescence before permeabilization) (B) obtained from four cells in the absence or the presence of 60 μm 4CL-DZP or 1 μm CsA (mean ± S.E.; 2 experiments) at different GSH/GSSG ratios (3 mm GSH concentration). The arrows in A and B denote the point at which ΔΨm irreversibly collapsed. Key to symbols: con, control; csa, cyclosporin A; 4dzp, 4Cl-DZP.
GSH and GSSG Concentrations and the Glutathione Redox Potential
NADPH/NADP+, GSH/GSSG, and Trx(SH)2/TrxSS represent the three main redox couples of the cell (1, 14, 34). Because the concentration of GSH is much higher than that of the other two systems, it is often considered the principal redox buffer of the cell, although specific enzymes may exclusively require a particular electron donor for function.
The half-cell reduction potential (Ehc) of the GSSG/GSH redox couple can be determined according to the Nernst Equation 1 (1),
| (Eq. 1) |
Equation 1 was used to calculate the effects of [GSH] on Ehc (Fig. 5), to determine whether the observed effects on mitochondrial function were specifically because of the glutathione reduction potential or if they were influenced by the reductive capacity of the redox pool. The standard reduction potential, ΔE0,of -252 mV was adjusted to the conditions used in our experiments (37 °C and pH 7.2). Under those conditions, RT/nF = 61.51/2, and E7.2 = -240 + ((7.2-7.0) (-61.51)) mV = -252 mV (1) with EpH = 7 = -240 mV.
FIGURE 5. Relationship between glutathione redox potential, GSH/GSSG ratio, and GSH pool size.
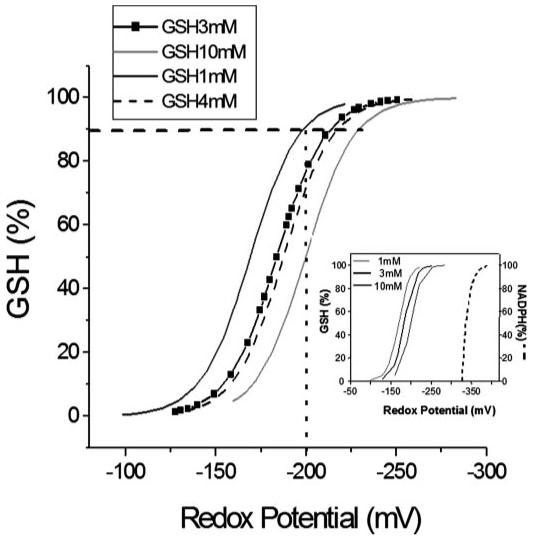
The glutathione redox potential, Ehc, was calculated according to Equation 1 in the text. The main panel shows how the total concentration of GSH influences the Ehc midpoint potential and how the pool size influences the % oxidation of the GSH pool. For example, if 10% of the GSH is oxidized to GSSG (horizontal dashed line), then for an initial GSH concentration of 10 mm, Ehc = -230 mV; for 4 mm, Ehc = -218 mV; for 3 mm, Ehc = -214 mV; and for 1 mm, Ehc = -200 mV. Thus, a cell with 10 mm GSH will have a higher reduction potential and a higher reducing capacity than one containing 1 mm GSH. The inset shows the reduction potential of NADP+/NADPH, whose negative redox potential (-400 mV (6)) makes it a key electron donor for the GSH system, for other redox systems (1), and for biosynthetic reactions. In general, NADPH is a cofactor in reductive (biosynthetic) reactions and serves as a source of electrons, whereas NAD+-dependent reactions are oxidative (catabolic) reactions where NAD+ serves as a sink for electrons. As opposed to the NADH/NAD+ ratio (= 100:1), the NADPH/NADP+ ratio is much lower (1:100) in cells and tissues (for review see Refs. 1, 6). The numbers at the top left of the inset correspond to GSH concentrations whose redox potential is represented in the plot (three continuous lines), and NADPH redox potential is shown in dashed line.
Because the redox potential is affected by the square of the GSH concentration, one can appreciate the important contribution of the total glutathione pool size to Ehc. For example, to maintain a -200-mV intracellular redox potential, a higher fraction of the absolute initial amount of GSH would have to be reduced at low rather than high GSH levels (i.e. compare the %GSH required to maintain -200 mV at 1 (∼80%) versus 10 mm GSH (∼50%) by the intersection of the vertical dashed line in Fig. 5).
By varying the total GSH pool size, additional experiments showed that half-maximal acceleration of the rate of oxidation of the ROS probe and oxidation of mitochondrial NADH were not strictly dependent on the calculated redox potential of the solution (Fig. 6). ΔΨm collapse occurred at redox potentials of -200, -220, or -240 mV for 1, 3, or 4 mm [GSH] added to the bathing solution, respectively (Fig. 6, A-C). Marked changes in the mitochondrial variables occurred even though the percentage oxidation of total GSH pool was small (<5%). Moreover, when GSH was increased to 4 mm, the irreversible ΔΨm collapse occurred at the 100:1 GSH/GSSG ratio rather than at 50:1 as in 3 mm GSH (Fig. 6, B and C). This paradoxical finding that ΔΨm loss occurs at a more reduced GSH/GSSG ratio when the pool size increases could perhaps be explained by the necessary increase in the levels of GSSG; when we increase GSH, we also have to increase the absolute concentration of GSSG to obtain the desired ratios. The GSSG concentrations were 20, 60, and 80 μm for the 50:1 solutions containing 1, 3, or 4 mm GSH, respectively.
FIGURE 6. Effects of glutathione redox status and pool size on mitochondrial ROS production, NADH, and ΔΨm in permeabilized myocytes.
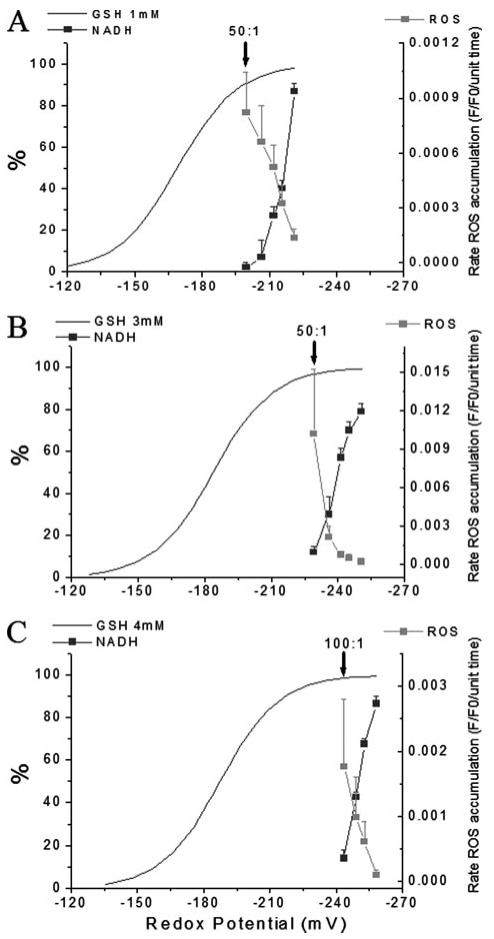
The rates of oxidation of the matrix localized CM-H2DCF (ROS probe) and the NADH autofluorescence at different GSH/GSSG ratios are compared for total GSH concentrations of 1 mm (A), 3 mm (B), and 4 mm (C). Data are plotted as a function of the glutathione reduction potential for the GSH/GSSG solutions, calculated as shown in Fig. 5. Note that the midpoint of the curve describing the acceleration of ROS production lies in a region where there is a relatively small change in Ehc. Moreover, the redox potential at which ΔΨm collapses differs significantly depending on the GSH pool size, indicating that the mechanism of mitochondrial dysfunction is not strictly dependent on the reduction potential of the cytoplasmic solution. The arrows in A-C denote the ratio at which ΔΨm irreversibly collapsed.
To test the interpretation that GSSG was another key factor in altering mitochondrial function, we performed the inverse experiment by keeping GSSG constantly low (10 μm) and changing GSH from 0.2 to 3 mm to achieve GSH/GSSG ratios ranging from 20:1 to 300:1. In this case, oxidation of the ROS probe remained low over the whole range of ratios. Furthermore, mitochondria resisted depolarization up to a ratio of 20:1, distinctly different from the control at 3 mm fixed GSH (Fig. 7).
FIGURE 7. Effects of GSH or GSSG concentration on mitochondrial ROS production in permeabilized myocytes.
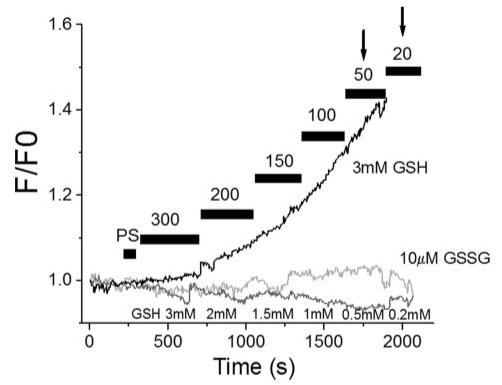
GSH/GSSG ratio was varied between 300:1 and 20:1 with either a GSH fixed at 3 mm or with GSSG fixed at 10 μm. Increasing GSSG at a fixed GSH induced corresponding increases in the rate of oxidation of the ROS probe while clamping GSSG to 10 μm inhibited ROS production and shifted mitochondrial ΔΨm depolarization (arrows) to a lower ratio (20:1).
Role of Mitochondrial Enzymes and GSH Uptake in Controlling the Mitochondrial Glutathione Redox State
The experiments above described how mitochondrial variables react to exogenous modulation of the glutathione redox potential. Next, we examined the intrinsic enzymatic processes in the mitochondrial matrix involved in the control of oxidative stress. The regeneration of GSH from GSSG trapped inside the mitochondria requires GR, which harnesses the more negative reduction potential of NADPH (see inset of Fig. 5) in the process (35) shown in Reaction 1,
| RECATION 1 |
NADPH in turn will be regenerated by the THD, which in animal mitochondria, as well as in bacteria, couples hydride transfer between NADH and NADP to the protonmotive force (36-38) as shown in Reaction 2,
| RECATION 2 |
From these two enzymatic reactions, it can be clearly appreciated that the ability of mitochondria to maintain a highly reduced level of NADPH is essential for detoxification of ROS (38). Progressive inhibition of GR at increasing concentrations of the inhibitor BCNU (carmustine) rendered mitochondria more susceptible to oxidative stress, as can be judged by the increasingly higher GSH/GSSG ratios at which ΔΨm collapsed (Fig. 8A).
FIGURE 8. Mitochondrial redox status and ROS production in response to inhibition of enzymes responsible for mitochondrial GSH regeneration and GSH transport.
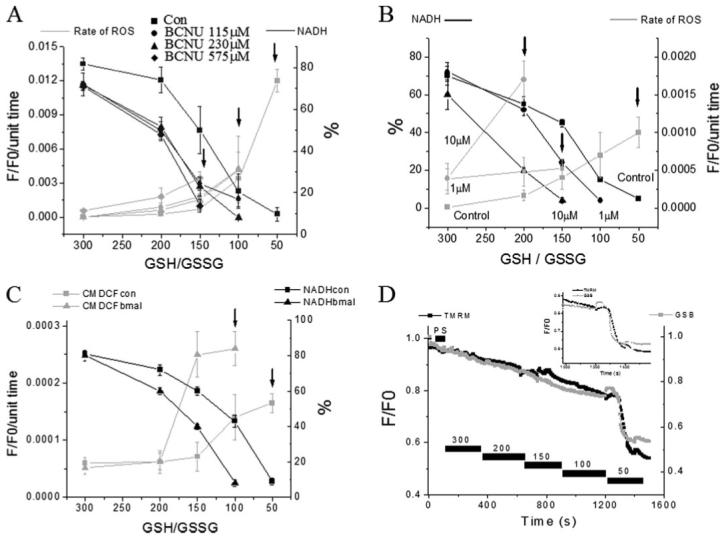
NADH (as % of initial level) and ROS production in saponin-permeabilized cardiomyocytes were exposed to different GSH/GSSG ratios. A, concentration-dependent effects of the glutathione reductase inhibitor BCNU. B, concentration-dependent effects of the NADH/NADPH transhydrogenase inhibitor NBD chloride. C, rate of oxidation of the ROS probe (F/F0/unit time) and NADH (in % of initial fluorescence before permeabilization) in the absence or the presence of 200 μm butyl malonate obtained from four cells (mean ± S.E.; two experiments). The rates of ROS production and NADH oxidation were similar to the control in the presence of 100 μm butyl malonate (not shown). The arrows denote the GSH/GSSG ratio at which ΔΨm collapsed irreversibly. ΔΨm collapses at increasingly higher (more reduced redox potentials) GSH/GSSG ratios in the presence of increasing concentrations of the inhibitors. D, timing of the depletion of reduced glutathione levels and ΔΨm induced by decreasing the GSH/GSSG ratio in saponin-permeabilized cardiomyocytes. Cells were loaded with 100 nm TMRM and 50 μm MCB and then permeabilized to remove the cytoplasmic component of GSB. Inset, note that the rapid oxidation of the mitochondrial GSB signal occurs ∼30 s before the irreversible collapse of ΔΨm. Key to symbols: con, control; bmal, butyl malonate.
Inhibiting THD with 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD chloride) (39) also produced the expected increased rate of oxidation of the ROS probe in the mitochondria and, concomitantly, an increased extent of oxidation of the NAD(P)H pool at any given GSH/GSSG ratio. Diminished tolerance of mitochondria to ROS was evident because ΔΨm collapsed at GSH/GSSG ratios of 150:1 and 200:1 in the presence of 1 or 10 μm NBD chloride, respectively (Fig. 8B), instead of 50:1 (control).
The GSH transport is another way the intra- and extramitochondrial pools of GSH could sense each other. GSH uptake has been shown to be mediated by the dicarboxylate (DIC) and 2-oxoglutarate carriers in mitochondria from rat renal cortex (40). When GSH uptake was inhibited with 200 μm butyl malonate (but not with 100 μm), a selective inhibitor of DIC, we found increased rates of ROS production and NADH oxidation followed by ΔΨm collapse at higher GSH/GSSG ratios; 100:1 instead of 50:1 as in the control (Fig. 8C).
Interestingly, ΔΨm loss correlated with oxidation of the NAD(P)H pool to <20% of the base-line level. This threshold corresponded to rapid depletion of the mitochondrial GSH pool just before the ΔΨm collapse in cells loaded with the GSH fluorescent probe MCB (Fig. 8D).
DISCUSSION
In our previous work, we have presented a model of mitochondrial network oscillation involving ROS-induced ROS release, IMAC, and derived from complex III of the respiratory chain (26, 41). More recently, we have shown that even under physiological conditions, the mitochondrial network behaves as a collection of individual weakly coupled oscillators that, under stress, can undergo a pathological transition to cellwide synchronized, autonomous oscillation (42, 43). A hallmark of this transition is that a critical number of mitochondria in the network experience a threshold level of oxidative stress prior to the collapse of ΔΨm. We referred to the hypersensitive condition at the precipice of oscillation as mitochondrial criticality. A key determinant of the approach to criticality was an imbalance of mitochondrial ROS production and ROS scavenging, which could be triggered by the localized generation of ROS in a small fraction of mitochondria in the network. In the present work, we demonstrate the following: (i) that the critical state can be induced by partial depletion of the reduced glutathione pool; (ii) that the reversible (IMAC-mediated) and irreversible (PTP-mediated) depolarizations of ΔΨm can be distinguished based on the cytoplasmic glutathione redox status, i.e. IMAC-mediated ΔΨm oscillation is triggered at a GSH/GSSG ratio of 150:1-100:1, whereas PTP opening is triggered at a GSH/GSSG of 50:1; (iii) that both the GSH/GSSG ratio and the total pool size of the redox couple influence mitochondrial ion channel opening rather than the absolute glutathione redox potential; and (iv) that the GSH uptake and the mitochondrial matrix enzymes glutathione reductase and NADH/NADPH transhydrogenase modulate the sensitivity of mitochondrial ROS production and ΔΨm depolarization to GSH/GSSG (27-29, 40, 44).
Induction of Mitochondrial Depolarization by Diamide
As expected from studies on other cell types (45), reduced glutathione levels were gradually depleted when intact cardiac cells were exposed to 0.1 mm diamide, reflected by a decrease in the glutathione S-bimane signal. Markers of oxidative stress, in the form of an increase in the oxidation rate of the mitochondrial ROS probe (CM-H2DCF) and a decrease in the NADH signal, were evident immediately upon exposure to diamide; however, ΔΨm was largely maintained over the first 5-7 min. When GSH levels were depleted by 20 -30%, the majority of cells showed sustained oscillations in ΔΨm, NADH, and ROS production, These oscillations were sensitive to the mitochondrial benzodiazepine ligand 4Cl-DZP in a reversible manner, and they did not involve any change in inner membrane permeability to a 500 molecular weight marker. These findings are consistent with the inhibition of a selective IMAC, as characterized previously in mitochondrial swelling assays (46) and mitoplast patch clamp studies (47). At moderate oxidative stress CsA did not repolarize ΔΨm, but severe depletion of the GSH pool ultimately did activate the PTP (Figs. 1 and 2). In permeabilized cells, the latter event usually occurred at GSH/GSSG ratios of 50:1 (Fig. 3D), at which point ΔΨm depolarized irreversibly, and the fluorescent marker in the mitochondrial matrix was lost.
This evidence is the first to demonstrate that mitochondrial criticality can be reached, not only by an increase in mitochondrial ROS generation (25) but by depletion of the cytosolic reduced thiol pool, consistent with the idea that the trigger of mitochondrial oscillation involves the balance between ROS production and scavenging (26, 41). Another novel finding of the present study is that continued depletion of the glutathione pool by diamide eventually induces the activation of the PTP, as evidenced by sustained depolarization and the rapid loss of the low molecular weight mitochondrial matrix marker. This evidence strongly supports the argument that the activation of IMAC and the PTP are distinct processes with different sensitivities to the redox state and to chemical inhibitors. PTP opening required more than 50% depletion of the GSH pool and was correlated with very high rates of ROS production. In this sense, IMAC can be thought of as an upstream “instigator” of the PTP, because it promotes depletion of the mitochondrial glutathione pool and increases ROS loads. Unlike 4CL-DZP, CsA did not affect ROS production or the GSH/GSSG ratio triggering PTP opening, although the latter was delayed or prevented.
In the intact myocyte experiments, it is difficult to determine whether Ca2+ overload, a well known effector of the PTP (48), contributed to the late permeability transition. Although the myocytes were not electrically stimulated, this phase of the response usually involved increased spontaneous contractile activity and the eventual hypercontracture of the cell, suggestive of altered Ca2+ homeostasis. Ca2+ is not a required cofactor for the oscillatory ΔΨm depolarizations (25), but it may be an important cofactor in the transition between the reversible and irreversible events during GSH depletion. In other experiments (results not shown), we also found that the heavy metal divalent cation, Co2+, strongly promotes PTP opening, as well as glutathione depletion, under similar conditions. This effect could be related either to the divalent triggering mechanism of the PTP or to the reported inhibitory effect of Co2+ on superoxide dismutase (49). In the permeabilized cells, divalent cations were less likely to play a role in PTP opening, because the bathing solution contained EGTA.
Effects of Fixed Cytosolic GSH/GSSG Ratios on Mitochondria in Permeabilized Myocytes
Selective permeabilization of the sarcolemmal membrane allowed us to quantitatively examine the influence of glutathione redox status on mitochondrial function while controlling other important factors such as pH, substrates (pyruvate, ATP), and ionic composition (K+, Na+).
In permeabilized cells, clear increases in the rate of oxidation of the mitochondrial ROS probe occurred in a stepwise manner upon lowering the GSH/GSSG ratio over the range from 300:1 to 50:1. The mitochondrial NADH pool was also oxidized in a graded manner. On average, ΔΨm was gradually lost until the limiting GSH/GSSG ratio of 50:1, when a sudden collapse was observed. However, corroborating the findings in intact cells, two distinct phases of ΔΨm depolarization were observed. First, at a fixed GSH/GSSG ratio between 150:1 and 100:1, oscillations in ΔΨm were induced. In the presence of 4CL-DZP, the oscillations were abolished, and the oxidized CM-DCF fluorescence remained localized in the mitochondrial matrix, supporting earlier evidence that the benzodiazepine-sensitive IMAC, and not the PTP, underlies the transient depolarizations (25). A surprising result was that the rate of oxidation of the CM-DCF was enhanced in the presence of 4Cl-DZP. This finding, that matrix oxidation is increased even though ΔΨm is more polarized, can perhaps be explained by inhibition of the efflux rate when IMAC is closed (26), leading to enhanced oxidation of the matrix-localized CM-DCF. We have previously observed an increase in the mitochondrial ROS signal within the laser-excited (flashed) zone of myocytes in the presence of 4Cl-DZP (25). However, it is important to note that in the intact cell, the global rate of mitochondrial ROS production is inhibited (cf. Fig. 1B) when the positive feedback of mitochondrial ROS-induced ROS release is interrupted by IMAC inhibition. The major difference in the permeabilized cell system is that the oxidative stress on the network (imposed by the bath solution redox potential) is exogenous and continuous, i.e. the cytosolic GSH/GSSG is no longer controlled by mitochondrial ROS production rate. Taken together, these data are consistent with the idea that efflux from the matrix to the cytosol is mediated by IMAC and that IMAC opening is also responsible for energy dissipation leading to ΔΨm depolarization.
PTP inhibition did not affect either ROS production or IMAC opening in response to changes in the glutathione redox status. At 50:1 GSH/GSSG, ΔΨm collapse in the entire mitochondrial network was often still observed in the presence of CsA, accompanied by loss of the CM-DCF fluorescence from the matrix; however, this obvious permeability transition was delayed and sometimes prevented by CsA. These data provide an important positive control for PTP opening and illustrate the criteria required to attribute ΔΨm depolarization to PTP opening, i.e. in the absence of marker redistribution or CsA sensitivity, loss of ΔΨm cannot be unequivocally ascribed to the PTP. This raises an important caveat with respect to previous literature suggesting that the PTP may undergo reversible opening or “flicker” (50). Without demonstrating inner membrane permeabilization directly, other mechanisms (including IMAC activation) could explain such results.
A remarkable and somewhat paradoxical finding was that when the total GSH + GSSG pool was larger, the irreversible ΔΨm collapse occurred at a slightly higher (more reduced) ratio (100:1 at 4 mm GSH versus 50:1 at 3 mm GSH), emphasizing that not only the ratio and redox potential of the thiol pool are important, but also the absolute pool size. One possible explanation for this finding could be that the GSSG concentration is, by necessity, higher at 4 mm than at 3 mm, suggesting that perhaps it could be a key regulator contributing to activation of the mitochondrial channels. Although GSSG is poorly permeable through membranes (35, 44, 51), it may affect the oxidation state of thiols on the periplasmic face of the mitochondrial membrane (52), perhaps altering the voltage threshold for channel activation, as reported for the PTP previously (21). Two additional experimental observations support this interpretation as follows: first, an increase in the number of permeabilized cells exhibiting reversible (IMAC-mediated) ΔΨm depolarization at 4 mm GSH versus 3mm GSH at the same ratio of 100:1; and second, the increased stability of ΔΨm, extremely low ROS production, and a shift in the requirement for PTP opening to a more oxidized GSH/GSSG ratio (20:1 instead of 50:1) when GSSG was clamped at 10 μm. These results demonstrate that the triggering of ΔΨm depolarization is not strictly dependent on the actual reduction potential of the solution (as illustrated in Fig. 6), but that total GSH + GSSG pool size and GSH/GSSG ratio are important.
Another point to be considered is that mitochondrial GSH is not synthesized in the matrix but is imported from the cytoplasmic compartment (1), effectively linking changes in the glutathione redox status in the two compartments. Thus, part of the effect of varying GSH/GSSG on mitochondrial ROS production could be secondary to a change in matrix GSH/GSSG. Because of the differential permeability of the two species, this would likely be manifested by the availability of GSH for the glutathione peroxidase. Inhibiting the DIC carrier with butyl malonate, responsible for at least 60% of the mitochondrial GSH uptake in rat renal cortex (40), rendered mitochondria more susceptible to oxidative stress. Another potential mechanism might be given by GSSG stimulation of ROS production by the formation of mixed disulfides between glutathione and complex I of the respiratory chain (44).
Mitochondrial Redox and Criticality
The regeneration of GSH within the mitochondrial matrix is a key process for controlling the rate of ROS production and, in turn, the sensitivity of the mitochondrial network to criticality. As shown in the scheme of Fig. 9, this process depends on GSH uptake by mitochondria and two redox-sensitive steps as follows: (i) NADPH regeneration by THD (36-38), and (ii) GR that connects both the NADPH/NADP+ and GSH/GSSG systems in mitochondria.
FIGURE 9. Coupled glutathione, NADH, and NADPH redox cycles in the mitochondrial matrix.
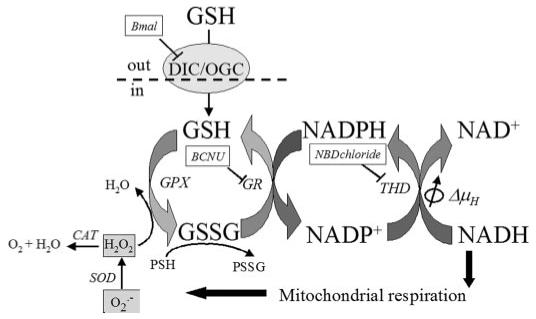
This scheme shows the carrier-mediated mitochondrial GSH uptake and the main enzymatic steps involved in the regeneration of GSH and NADPH in the mitochondrial matrix. DIC, dicarboxylate carrier (its inhibition by butyl malonate, Bmal, is indicated); OGC, 2-oxoglutarate carrier; GPX, glutathione peroxidase; GR, NADPH-dependent glutathione reductase (its inhibition by carmustine, BCNU, is indicated); THD, transhydrogenase (its inhibition by NBD chloride, NBD chloride, is indicated); ΔμH, proton electrochemical gradient; CAT, catalase; SOD, Mn-superoxide dismutase; PSSG, protein-SSG mixed disulfides; PSH, protein-SH. ROS is highlighted in light gray: , superoxide anion; H2O2, hydrogen peroxide.
Although THD is not the only source of NADPH in mitochondria (the NADP-isocitrate dehydrogenase and the malic enzyme are also potential sources (37, 38, 53)), the negative redox potentials of the large NADH pool and the smaller NADPH pool (see plot of NADPH redox potential in inset of Fig. 5) represent strong thermodynamic driving forces for keeping the GSH/GSSG ratio high through the NADPH-dependent GR (Fig. 9). The marked changes in glutathione sensitivity upon inhibition of THD emphasize its crucial role for keeping the exposed -SH groups on proteins reduced in the face of oxidative stress, which favors accumulation of protein-SSG mixed disulfides and correspondingly decreases in the concentration of protein -SH (Fig. 9). GR inhibition also contributed significantly to the sensitivity of ROS production, NADH oxidation, and ΔΨm collapse.
Notably, NADPH regeneration by mitochondrial THD partially uncouples the protonmotive force, because in the presence of the electrochemical proton gradient, the rate of the THD reaction is shifted toward NADPH formation (Fig. 9) (37, 38). Thus, in a scenario of increased oxidative stress, THD activity will end up consuming ΔΨm and aggravating the uncoupling mediated by the ROS-sensitive mitochondrial channels. Moreover, a higher production of the freely permeable H2O2 by the mitochondria will also deplete the extramitochondrial GSH.
Mitochondrial Redox and Criticality in Pathophysiology
Mitochondrial oxidative stress and glutathione depletion are common features of neurodegenerative (54) and cardiac diseases. Several mitochondrial targets are known to be damaged by free radicals, including the activity of electron transport complexes I, III, and IV, ATP synthase, and adenine nucleotide translocase (e.g. Refs. 55-58). A number of tricarboxylic acid cycle enzymes are also susceptible, including α-ketoglutarate dehydrogenase, succinate dehydrogenase, and aconitase (59). In severe neurological disorders such as Parkinson or Huntington disease, it has been shown that complex I in the respiratory chain is selectively damaged. Glutathionylation and irreversible inactivation through the formation of mixed disulfides is one of the molecular mechanisms that has been favored by existing experimental evidence (44). Overall, the evidence available points out that mitochondrial proteins involved in oxidative phosphorylation and in mitochondrial ROS signaling (for review see Ref. 43) are targets of oxidative damage and may be relevant for a range of age-related human pathologies (60). Mitochondrial targeted antioxidants that detoxify mitochondrial ROS or reinforce the ROS scavenging mechanisms are being actively developed (60, 61).
Upon reperfusion after ischemia, when electron flow is restored, NADH levels are high, and the rate of ROS accumulation accelerates (59, 62); the course of cardiomyocyte survival or death is determined in large part by the ability of mitochondria to recover. The activation of mitochondrial ion channels plays a role in this decision. In this study we show that inner mitochondrial membrane ion channel (IMAC and PTP) activation is sequential as a function of the extramitochondrial GSSG/GSH redox potential and the concentrations of both components of the redox couple.
In our previous related studies (25-27), we propose that the conditions leading to mitochondrial criticality and the emergence of self-sustained, cell-wide, high amplitude, low frequency mitochondrial oscillations (42) describe seminal events occurring during reperfusion after ischemic injury. We also showed that the consequences of the critical behavior of the mitochondrial network result in reperfusion-related arrhythmias after ischemia that can be blocked by inhibition of the IMAC, and partially by inhibiting the PTP (23, 28, 63).
In summary, in this work we show intracellular conditions that can lead to the stable depolarized state of ΔΨm and cell death. The results obtained suggest that a critical value of the cellular redox potential represented by the GSH/GSSG ratio determines when oxidative stress will provoke ΔΨm depolarization. In agreement with kinetic experiments performed with living cells, the results obtained also indicate that the absolute concentrations of GSH and GSSG modulate the effects exerted by oxidative stress on ΔΨm. In living cardiomyocytes, mitochondrial oscillations ensue at certain values of GSH titration performed with the thiol prooxidant diamide followed by sudden ΔΨm collapse and death when GSH is further decreased. More specifically, we show that heart cell death involves the progression of mitochondria to a stable depolarized state and oxidized NADH that is attained at high rates of ROS production and compromised regeneration of mitochondrial GSH.
Acknowledgment
We thank Dr. Jan Hoek for helpful suggestions.
Footnotes
This work was supported by National Institutes of Health Grant R37-HL54598 (to B. O’R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S3.
- ROS
- reactive oxygen species
- NBD chloride
- 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole
- CM-H2DCFDA
- 5-(-6)-chloromethyl-2′,7′-dichlorohydrofluorescein diacetate
- GR
- glutathione reductase
- PTP
- permeability transition pore
- IMAC
- inner membrane anion channel
- THD
- transhydrogenase
- 4Cl-DZP
- 4′-chlorodiazepam
- BCNU
- carmustine
- DIC
- dicarboxylate
- Trx
- thioredoxin
- DMEM
- Dulbecco’s modified Eagle’s medium
- TMRM
- tetramethylrhodamine methyl ester
- MCB
- monochlorobimane
- GSB
- glutathione S-bimane
- CsA
- cyclosporin A
- CM-DCF
- chloromethyl dichlorofluorescein.
REFERENCES
- 1.Schafer FQ, Buettner GR. Free Radic. Biol. Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 2.Chance B, Sies H, Boveris A. Physiol. Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 3.Rebrin I, Kamzalov S, Sohal RS. Free Radic. Biol. Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shelton MD, Chock PB, Mieyal JJ. Antioxid. Redox. Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 5.Sies H. Free Radic. Biol. Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 6.Jones DP. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 7.Droge W. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 8.Halliwell B. In: Oxygen Radicals and the Disease Process. Thomas CE, Kalyanaraman B, editors. Harwood Academic, Amsterdam; Netherlands: 1997. pp. 1–14. [Google Scholar]
- 9.Abate C, Patel L, Rauscher FJ, III, Curran T. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 10.Arrigo AP. Free Radic. Biol. Med. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 11.Powis G, Gasdaska JR, Baker A. Adv. Pharmacol. 1997;38:329–359. doi: 10.1016/s1054-3589(08)60990-4. [DOI] [PubMed] [Google Scholar]
- 12.Shackelford RE, Kaufmann WK, Paules RS. Free Radic. Biol. Med. 2000;28:1387–1404. doi: 10.1016/s0891-5849(00)00224-0. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki YJ, Forman HJ, Sevanian A. Free Radic. Biol. Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 14.Gilbert HF. Adv. Enzymol. Relat. Areas Mol. Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 15.Schwaller M, Wilkinson B, Gilbert HF. J. Biol. Chem. 2003;278:7154–7159. doi: 10.1074/jbc.M211036200. [DOI] [PubMed] [Google Scholar]
- 16.Xia R, Stangler T, Abramson JJ. J. Biol. Chem. 2000;275:36556–36561. doi: 10.1074/jbc.M007613200. [DOI] [PubMed] [Google Scholar]
- 17.Hall AG. Eur. J. Clin. Investig. 1999;29:238–245. doi: 10.1046/j.1365-2362.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- 18.Hwang C, Sinskey AJ, Lodish HF. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 19.Meister A, Anderson ME. Annu. Rev. Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 20.Chernyak BV. Biosci. Rep. 1997;17:293–302. doi: 10.1023/a:1027384628678. [DOI] [PubMed] [Google Scholar]
- 21.Costantini P, Chernyak BV, Petronilli V, Bernardi P. J. Biol. Chem. 1996;271:6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 22.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. J. Biol. Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- 23.O’Rourke B, Ramza BM, Marban E. Science. 1994;265:962–966. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- 24.Romashko DN, Marban E, O’Rourke B. Proc. Natl. Acad. Sci. U. S. A. 1998;95:1618–1623. doi: 10.1073/pnas.95.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aon MA, Cortassa S, Marban E, O’Rourke B. J. Biol. Chem. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- 26.Cortassa S, Aon MA, Winslow RL, O’Rourke B. Biophys. J. 2004;87:2060–2073. doi: 10.1529/biophysj.104.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aon MA, Cortassa S, O’Rourke B. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4447–4452. doi: 10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. J. Clin. Investig. 2005;115:3527–3535. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aon MA, Cortassa S, Akar FG, O’Rourke B. Biochim. Biophys. Acta. 2006;1762:232–240. doi: 10.1016/j.bbadis.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kosower NS, Kosower EM. Methods Enzymol. 1987;143:76–84. doi: 10.1016/0076-6879(87)43015-2. [DOI] [PubMed] [Google Scholar]
- 31.Lemar KM, Passa O, Aon MA, Cortassa S, Muller CT, Plummer S, O’Rourke B, Lloyd D. Microbiology. 2005;151:3257–3265. doi: 10.1099/mic.0.28095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosower NS, Kosower EM. Methods Enzymol. 1995;251:123–133. doi: 10.1016/0076-6879(95)51116-4. [DOI] [PubMed] [Google Scholar]
- 33.Beavis AD. J. Bioenerg. Biomembr. 1992;24:77–90. doi: 10.1007/BF00769534. [DOI] [PubMed] [Google Scholar]
- 34.Follmann H, Haberlein I. Biofactors. 1995;5:147–156. [PubMed] [Google Scholar]
- 35.Griffith OW, Meister A. Proc. Natl. Acad. Sci. U. S. A. 1985;82:4668–4672. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoek JB, Rydstrom J. Biochem. J. 1988;254:1–10. doi: 10.1042/bj2540001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson JB. FEBS Lett. 2003;545:18–24. doi: 10.1016/s0014-5793(03)00388-0. [DOI] [PubMed] [Google Scholar]
- 38.Rydstrom J. Trends Biochem. Sci. 2006;31:355–358. doi: 10.1016/j.tibs.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Bragg PD, Hou C. Biochim. Biophys. Acta. 1999;1413:159–171. doi: 10.1016/s0005-2728(99)00090-0. [DOI] [PubMed] [Google Scholar]
- 40.Lash LH. Chem. Biol. Interact. 2006;163:54–67. doi: 10.1016/j.cbi.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aon MA, Cortassa S, O’Rourke B. In: Cellular Oscillatory Mechanisms. Maroto M, Monk N, editors. Landes Bioscience; Geor-getown, TX: 2007. [Google Scholar]
- 42.Aon MA, Cortassa S, O’Rourke B. Biophys. J. 2006;91:4317–4327. doi: 10.1529/biophysj.106.087817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aon MA, Cortassa S, O’Rourke B. In: Molecular System Bioenergetics. Saks V, editor. Wiley Interscience; New York: 2007. [Google Scholar]
- 44.Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP. J. Biol. Chem. 2003;278:19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 45.Brennan P, O’Neill LA. Biochem. J. 1996;320:975–981. doi: 10.1042/bj3200975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beavis AD, Garlid KD. J. Biol. Chem. 1987;262:15085–15093. [PubMed] [Google Scholar]
- 47.Borecky J, Jezek P, Siemen D. J. Biol. Chem. 1997;272:19282–19289. [PubMed] [Google Scholar]
- 48.Di Lisa F, Bernardi P. Cardiovasc. Res. 2005;66:222–232. doi: 10.1016/j.cardiores.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 49.O’Neill P, Fielden EM, Cocco D, Rotilio G, Calabrese L. Biochem. J. 1982;205:181–187. doi: 10.1042/bj2050181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huser J, Blatter LA. Biochem. J. 1999;343:311–317. [PMC free article] [PubMed] [Google Scholar]
- 51.Martensson J, Lai JC, Meister A. Proc. Natl. Acad. Sci. U. S. A. 1990;87:7185–7189. doi: 10.1073/pnas.87.18.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filomeni G, Rotilio G, Ciriolo MR. Biochem. Pharmacol. 2002;64:1057–1064. doi: 10.1016/s0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- 53.Vogel R, Wiesinger H, Hamprecht B, Dringen R. Neurosci. Lett. 1999;275:97–100. doi: 10.1016/s0304-3940(99)00748-x. [DOI] [PubMed] [Google Scholar]
- 54.Jenner P. Acta Neurol. Scand. Suppl. 1993;146:6–13. [PubMed] [Google Scholar]
- 55.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Biochem. J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hardy L, Clark JB, Darley-Usmar VM, Smith DR, Stone D. Biochem. J. 1991;274:133–137. doi: 10.1042/bj2740133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. Am. J. Physiol. 1997;273:H1544–H1554. doi: 10.1152/ajpheart.1997.273.3.H1544. [DOI] [PubMed] [Google Scholar]
- 58.Vuorinen K, Ylitalo K, Peuhkurinen K, Raatikainen P, Ala-Rami A, Hassinen IE. Circulation. 1995;91:2810–2818. doi: 10.1161/01.cir.91.11.2810. [DOI] [PubMed] [Google Scholar]
- 59.Sadek HA, Nulton-Persson AC, Szweda PA, Szweda LI. Arch. Biochem. Biophys. 2003;420:201–208. doi: 10.1016/j.abb.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 60.James AM, Cocheme HM, Murphy MP. Mech. Ageing Dev. 2005;126:982–986. doi: 10.1016/j.mad.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 61.Sheu SS, Nauduri D, Anders MW. Biochim. Biophys. Acta. 2006;1762:256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 62.Marczin N, El-Habashi N, Hoare GS, Bundy RE, Yacoub M. Arch. Biochem. Biophys. 2003;420:222–236. doi: 10.1016/j.abb.2003.08.037. [DOI] [PubMed] [Google Scholar]
- 63.O’Rourke B. J. Physiol. (Lond.) 2000;529:23–36. doi: 10.1111/j.1469-7793.2000.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]