

Abstract
The emergence of the highly pathogenic SARS coronavirus (SARS-CoV) has reignited interest in coronavirus biology and pathogenesis. An emerging theme in coronavirus pathogenesis is that the interaction between specific viral genes and the host immune system, specifically the innate immune system, functions as a key determinant in regulating virulence and disease outcomes. Using SARS-CoV as a model, we will review the current knowledge of the interplay between coronavirus infection and the host innate immune system in vivo, and then discuss the mechanisms by which specific gene products antagonize the host innate immune response in cell culture models. Our data suggests that the SARS-CoV uses specific strategies to evade and antagonize the sensing and signaling arms of the interferon pathway. We summarize by identifying future points of consideration that will contribute greatly to our understanding of the molecular mechanisms governing coronavirus pathogenesis and virulence, and the development of severe disease in humans and animals.
Keywords: SARS coronavirus, Innate immunity, Interferon, RNA virus
1. Introduction
Viral interactions with the innate immune system play a central role in determining the outcome of infection. Early control of viral replication by type I interferons (IFN), complement proteins, and other innate immune mediators limit viral spread within the host during the early phases of the disease (Katze et al., 2002). The early innate response also plays an important role in shaping the downstream adaptive immune response, however an overactive innate immune response can also result in immune pathology and subsequent tissue damage (reviewed in Garcia-Sastre and Biron, 2006). Within the last decade, it is clear that many viruses encode specific gene products that antagonize both the innate and acquired arms of the immune response (Andrejeva et al., 2004, Basler et al., 2000, Cruz et al., 2006, Gale et al., 1997, Meylan et al., 2005, Parisien et al., 2001, Park et al., 2003, Symons et al., 1995, Xiang et al., 2002, Ye et al., 2007). Therefore, a detailed knowledge of how specific viruses interact with the host innate immune system is essential for understanding the molecular mechanisms regulating virulence, pathogenesis and disease outcomes.
Coronavirus interactions with the adaptive immune system have been studied in great detail, however, surprisingly little is known about how these viruses interact with the innate immune system (La Bonnardiere and Laude, 1983). Although early studies indicated that mutations in the M glycoprotein of transmissible gastroenteritis virus (TGEV) modulated type I IFN responses, suggesting that coronaviruses may encode a novel set of gene functions that interface with the host innate immune response, little effort focused on unraveling the details of coronavirus innate immune interactions (Charley and Laude, 1988, La Bonnardiere and Laude, 1983). Early experiments showed that variants of mouse hepatitis virus (MHV) are differentially susceptible to IFN. This may contribute to different pathogenic outcomes, however little additional experimentation was performed (Taguchi and Siddell, 1985). The SARS coronavirus (SARS-CoV) epidemic of 2003 rekindled a high level of interest in how coronaviruses interact with the host and whether interactions with the host innate immune system are important in both the control of viral infection or if these interactions lead to virus-induced disease.
In this article, we will discuss two components of the innate immune response that are clearly important for SARS-CoV induced disease: (1) interactions with macrophages (MP) and dendritic cells (DC), which shape the early innate and adaptive immune responses within the lung, while also potentially contributing to virus-induced immune pathology, and (2) the type I IFN system, an essential component of the early innate response to viral infections that appears to be blocked or evaded by SARS-CoV and other coronaviruses.
2. Dendritic cells and macrophages
Dendritic cells and macrophages are first line components of the innate immune network. DCs, which can be grouped into plasmacytoid (pDC) and myeloid types (mDC), play important roles in driving both innate and adaptive immune responses to viral pathogens (Akira and Hemmi, 2003, Ito et al., 2005, Nakano et al., 2001). pDCs rapidly respond to viruses or their derivatives to produce large amounts of type I IFN, which can induce direct antiviral responses and also modulate other components of the innate and adaptive immune response, such as natural killer cells and CD8 T cells (Colonna et al., 2004, Diebold et al., 2003, Cella et al., 1999, Siegal et al., 1999). Though less robust than pDCs, mDCs can also secrete large amounts of type I IFN (Laiosa et al., 2006). However, mDCs also play a major role in stimulating acquired immune responses through their capacity as antigen presenting cells and producers of a wide array of immuno-modulatory cytokines (Laiosa et al., 2006). MPs are potent producers of type I IFNs and other pro-inflammatory cytokines that induce antiviral protection while also potentially contributing to immune pathology associated with viral infections (Diamond et al., 2003). Analysis of the impact of DCs and MPs on SARS-CoV infection will be discussed below.
3. The type I IFN system
Since its discovery 50 years ago by Isaacs and Lindemann, the IFN system has come to be recognized as a crucial frontline defense against viral infection (Isaacs and Lindenmann, 1957). IFNs mediate direct antiviral effects that limit viral replication by activating/up-regulating several well defined antiviral effectors, including PKR and RNaseL, while also modulating other aspects of the innate and adaptive immune responses through the induction of a wide array of IFN inducible genes (ISGs) (Stark et al., 1998, Takaoka and Yanai, 2006). The number of pathogens that neutralize IFN, or other key components of the IFN system, illustrate that this system is essential for the control of a diverse array of viruses and bacteria. Perhaps the clearest indicator of how important this pathway is to antiviral defense is the sheer number of IFN avoidance/antagonism strategies that viruses have evolved. These viral IFN evasion strategies can be roughly segregated into three categories: (1) avoidance, where the virus shields itself or its byproducts from recognition by the host cell sensors that activate the IFN system (Cardenas et al., 2006), (2) suppression of IFN induction, where the virus actively inhibits the host cell sensor machinery or its downstream signaling molecules to prevent the initiation of IFN transcription (Cardenas et al., 2006, Hiscott et al., 2006a, Hiscott et al., 2006b, Lin et al., 2006, Li et al., 2005a, Li et al., 2005b, Meylan et al., 2005), or (3) suppression of IFN signaling, where viral gene products block signaling events at or downstream of the type I IFN receptor complex to prevent activation of an antiviral state within the infected cell or the enhancement of the IFN response by activating late type I IFN genes (Parisien et al., 2001, Rodriguez et al., 2002). This raises the question of how coronaviruses, which include human (SARS-CoV, NL63, OC43 and 229E) and animal (MHV, infectious bronchitis virus (IBV, TGEV, etc.) strains, interact with the type I IFN system. Of these viruses, only 229E has been shown to induce IFN in infected cells in culture, while viruses such as SARS-CoV and MHV fail to induce type I IFN responses in cell culture. However, the mechanism for how coronaviruses evade the innate immune system is largely unexplored.
There are two major pathways through which cells sense invading viruses and activate the IFN pathway (reviewed in Sen, 2001). Toll like receptors (TLRs), which include TLR3, TLR7, TLR8 and TLR9, can detect viruses in endosomal compartments as they enter cells, while cytoplasmic CARD domain containing RNA helicases, RIG-I and Mda5, sense viral RNA in the cytoplasm. Both pathways are based on sensor interactions with pathogen associated molecular patterns (PAMPs), such as double stranded RNA which is a byproduct of viral replication, or structured single stranded RNAs associated with incoming viral genomes, being common targets. The TLR and cytoplasmic IFN induction pathways do utilize different adaptor proteins to mediate signaling, with the TLR dependent pathways utilizing TRIF and/or MyD88, and the cytoplasmic induction pathway utilizing the mitochondrial adaptor protein MAVS/IPS-1/VISA/CARDIF (Fig. 1 ). Downstream of the differing adapters, both pathways share many common signaling molecules and transcription factors, with both pathways ultimately activating IRF-3, NFκB, and AP-1 to initiate type I IFN gene transcription. Each of these pathways will be discussed further with respect to its potential role in coronavirus infection (Table 1 ).
Fig. 1.
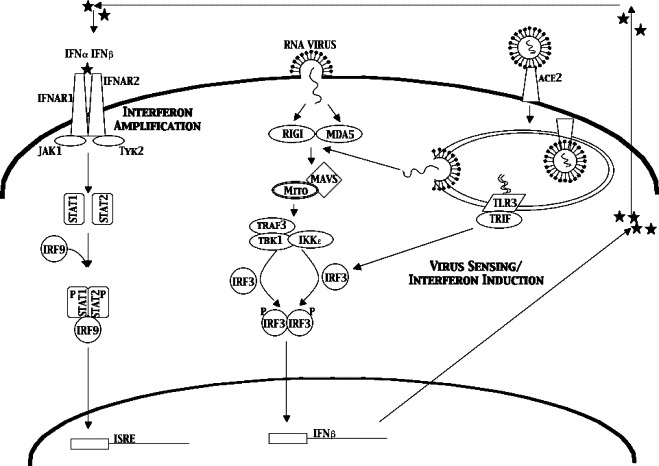
IFN sensing and signaling pathway. RNA viruses are internalized through several mechanisms, either fusion with the plasma membrane or binding to a surface receptor (ACE2 for SARS-CoV, carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) for MHV). That internalization exposes the genomic RNA to the dsRNA sensing machinery in the cell; TLR3, RIGI and MDA5. These proteins signal the IRF-3 cascade leading to induction of IFNb and production of secreted IFNβ protein. That IFNβ protein can then bind IFNα/β receptors (INFAR1) on the surface of the same cell or surrounding cells. This activates the Stat1 signaling pathway to activate the many anti-viral genes found with ISRE promoter elements.
Table 1.
Summary of selected papers cited in this review
| Author | Cells | Results | Type I IFN↑ |
|---|---|---|---|
| Zhang et al. | PBMC from infected patients | IL-6↑, IL-10↑, IL-4↑, IFNγ↑, IL-8↓, TGF-beta↓ | ND |
| Regunathan et al. | PBMC from infected patients | IP-10↑, IL-6↑, IL-8↑, MCP-1↑ | ND |
| Castilletti et al. | Ex vivo infected PBMCs | IFNα↑, IFNβ↑ | Yes |
| Law et al. | Ex vivo infected DCs | IP-10↑, MIP1α↑, Rantes↑, MCP-1↑ | ND |
| Tang et al. | Huh7 cells | No IFNα and IFNβ induction | No |
| Cinatl et al. | Caco2 and CL-14 | CXC chemokines↑, OAS2↑, IL-18↓, MIF↓ | No |
| Speigel et al. | Caco2 and 293 | IP-10↑, IL-8↑ both ↑ in Caco2 but not 293 | No |
| Okabayashi et al. | Caco2 | IFNα↑, IFNβ↑, IRF-7↑, OAS↑… | Yes |
4. Host response to viral infection
The human impact of the SARS-CoV epidemic in 2003 led to an intensive research effort to understand the pathogenesis of SARS-CoV induced disease. Much of this effort focused on interactions between the virus and immune response within the lung. This effort will be broken down into patient studies, which are informative, but by their very nature, complex, studies in animal models, and cell culture based analysis of viral interactions with primary cells or cell lines, which provide greater experimental control at the cost of potentially oversimplifying the system.
Several groups analyzed the serum of SARS-CoV infected patients to determine which classes of cytokines were up-regulated during infection (Jiang et al., 2005, Yu et al., 2005). Zhang et al. found that IL-6 was up-regulated in serum while IL-8 and TGF-beta were decreased in SARS patients. They also found that IFNγ, IL-4 and IL-10 were increased only in convalescent SARS patients (Zhang et al., 2004). Reghunathan et al. found that IP-10 was induced in SARS-CoV infected patients and there was a correlation between high IP-10 levels and poor patient outcome (Reghunathan et al., 2005). The authors also found that IL-6, IL-8 and monocyte chemoattractant protein-1 were highly induced in super-infected patients, with high levels indicative of a high risk of death. Additionally, microarray-based analysis of RNA derived from PBMCs from 10 SARS-CoV infected patients demonstrated that the cells were highly activated and expressing high levels of inflammatory cytokines. However, no IFNα or β induction was detected in these patients. Though these studies provide valuable information on the kinetics and type of host cytokine response that occurs in infected humans during SARS-CoV infection, drawing general conclusions from these studies is difficult due to differences in both the timing of study, the patient population analyzed, and the types of assays used to evaluate cytokine responses.
Several experiments were performed on lung tissue from fatal SARS cases. Most reports used only a single or a few patients with different times of death, whether it was from acute disease or a secondary infection, trying to compare their study to a control patient. Chen et al. identified carbon containing MPs while To et al. found only infection of epithelial cells by in situ and immunofluorescence (Chen and Hsiao, 2004). Another publication by Chow et al. identifies lung pneumocytes, but not MPs as the site of virus replication, while yet another identifies a different lymphocyte population as the main replication target (Chow et al., 2004). Each of these differs in the type and length of time of infection of tissues used, the fixation technique for tissue preservation, the type of probe and the technique used to identify viral proteins and RNA. A recent paper by Nicholls et al. attempts to combine a large set of patient samples using immunohistochemistry, in situ hybridization, and RT-PCR to build a comprehensive picture of the infection cycle (Nicholls et al., 2006). They also comment on how false positive or negatives can arise for several reasons, including: (1) not all lung tissue is homogenous, so using tissue from one part of the lung versus another can critically change your interpretation, (2) different probes and techniques have varying levels of sensitivity depending on the age of the tissue and how it was handled, (3) the age of the individual will alter the immune response and clearance time of infection, (4) the overall immune status of the individual at the time of infection (immuno-compromised versus healthy) would impact on both the overall levels of viral replication and the subsequent host inflammatory response to the virus, and (5) the timeframe from initial symptoms to death may differ significantly between individuals, with individuals that rapidly progress showing higher levels of virus at the time of death, while individuals that succumb to infection after an extended period may have little virus present and die due to secondary causes. Based on their analysis of this large set of patient samples, Nicholls et al., concluded that alveolar epithelium and MPs are the primary targets of SARS-CoV in the lung. They also suggest that pneumocytes may be the initial site of infection, but that MPs take up virions and disseminate the virus within the lungs. Interestingly, in the 25 patients they tested that died within 2 weeks of symptoms, no virus was detected by in situ hybridization or immunohistochemistry, even in tissues with clear damage. This suggests that viral replication may not be directly responsible for death in these individuals, raising the possibility that virus-induced immune pathology may contribute to SARS-CoV induced disease. In fact, since high levels of pro-inflammatory cytokines and chemokines correlate with poor SARS-CoV outcome (Reghunathan et al., 2005, Zhang et al., 2004), it is possible that SARS-CoV infection of MPs or other potent producers of pro-inflammatory cytokines, without high levels of viral replication, may ultimately lead to virus-induced immune pathology within the lungs of patients with poor disease outcomes.
5. SARS-CoV in macrophages and dendritic cells
Given their role in maintaining homeostasis within the lungs, as well as their ability to mount robust innate immune responses, it is important to consider the role of MPs and DCs during SARS-CoV infection. Several conflicting reports of SARS-CoV replication and IFN induction in MPs and DCs have been published over the last few years. Early reports showed that SARS-CoV did not replicate efficiently in purified monocytes and MPs (Yilla et al., 2005). The infection however was donor dependent, with 100% infection in some donors and less than 5% infection efficiency in others. This study also showed that there was a correlation between the donor cells that produced high amounts of IFNα in response to SARS-CoV also being less permissive for productive infection. However, this is not the case for all their donors, with some producing high levels of IFN and being infected while other produce low levels of IFN and were not infected. Castilletti et al. also found IFNα and γ induced from PBMCs infected with SARS-CoV, with the most robust responses occurring when the PBMCs were exposed to fixed SARS-CoV infected Vero cells (Castilletti et al., 2005). These results suggest that PBMCs are poorly permissive for SARS-CoV and the authors propose that PBMCs may be able to detect viral glycoproteins on the surface of cells and that this may be a mechanism for the apparent immuno-pathology seen in SARS patients lungs. Results from Spiegel et al., also demonstrated that both live and UV inactivated SARS-CoV could induce type I IFN responses and cellular maturation in DC cultures (Spiegel et al., 2006), suggesting that productive viral replication is not required to active anti-viral and/or pro-inflammatory responses in these cells.
Similarly, Law et al. find no replication in their isolated DCs but did find a different cytokine induction profile (Law et al., 2005). The authors infected DCs with SARS-CoV and analyzed the effects on cell surface markers and maturation. By electron microscopy virions are seen inside both immature and mature DCs and small amounts of negative strand RNA were detectable in those cells, suggesting that some level of viral gene expression was occurring. However the level of RNA decreased over the course of several days indicating that there was no productive replication. These studies found no evidence of apoptosis or antiviral cytokines such as IFNα, β, γ and IL-12; however, they did find significant up-regulation of IP-10, MIP1α, Rantes and monocyte chemoattractant protein-1 (MCP-1). The differences in cytokine profiles between the Castilleti and Law studies may reflect infection of different cell types, however, both groups suggest that the up-regulation of pro-inflammatory chemokines may recruit monocytic cells to sites of infection and be a major cause of lung pathology in patients.
Several other groups have also evaluated whether MPs and/or DCs are targets of SARS-CoV infection and whether this infection leads to production of pro-inflammatory cytokine responses that might contribute to virus-induced immune pathology within the lung. Tseng et al. characterized the effect of SARS-CoV infection on human MPs and DCs (Tseng et al., 2005). They find that neither cell is permissive to SARS-CoV replication, which is consistent with the results described above, as well as studies by Ziegler et al. (2005). In the studies by Tseng et al., the MPs and DCs were phenotypically altered after SARS-CoV exposure. They found no increase of cell death but exposure increases the production of IL-6 and IL-12 upon exposure to a suboptimal dose of lipopolysaccharide (LPS), indicating that SARS primes the cells to respond to TLR ligands, but did not directly activate the cells. They also demonstrated that SARS-CoV decreased the phagocytic activity of MPs to FITC-Dextran while at the same time increasing the ability of DCs to stimulate naïve T cells.
Overall, these studies suggest that neither MPs nor DCs are highly permissive for SARS-CoV replication. While some groups did not see SARS-CoV associated inflammatory cytokine induction in MPs or DCs, the overall consensus from this work is that SARS-CoV infection can lead to either the direct activation or the priming of pro-inflammatory cytokine responses in these cell types, which may ultimately contribute to the development of virus-induced immune pathology within the lungs. The effect of viral infection on the induction of antiviral type I IFN responses in these cell types is less clear cut, since type I IFN induction has been observed by some groups and not others. This difference may reflect sensitivity differences in their assays, the cell types that are being evaluated, or even genetic differences in their donor cohorts. Therefore, further work is needed to determine whether or not SARS-CoV is an activator of antiviral responses in these cells.
6. Coronavirus interactions with the type I IFN system
Type I IFN induction has been observed in some studies of SARS-CoV infected MPs and DCs (Castilletti et al., 2005). These cell types are capable of mounting type I IFN responses in the absence of active viral replication, it is unclear whether cells that are productively infected with SARS-CoV can mount type I IFN responses, and by extension, whether SARS-CoV actively blocks type I IFN induction or signaling (Yen et al., 2006). The cell line used for most virus growth experiments and protein analysis is VeroE6, which lacks a functional IFNβ gene. Other cell lines have been used to identify the IFN pathways induced during SARS-CoV infection such as MA104 and Caco2 cells which are highly permissive for infection (>90% infected, yields ∼107 PFU/ml) and 293 cells which are semi-permissive at best (∼5–10% infected; yields of ∼104–5 PFU/ml). Several laboratories have been using these cell lines to identify which, if any, cytokines and chemokines are induced during infection.
We have analyzed the type I IFN response in several cell lines. To investigate whether IFNβ was induced by SARS-CoV infection we transfected 293T cells with a plasmid containing the IFNβ promoter followed by luciferase. We found that IFNβ promoter activity was not induced during the course of an infection, from as early as 6 h to 48 h post infection (Fig. 2 ). During this time frame, Sendai Virus (SeV) infected cells induced large amounts of IFNβ promoter activity. We also tested whether MA104 cells, African green monkey kidney cells that have an intact IFN system secrete IFN when infected with SARS-CoV. We find no Type I IFN to be secreted from either Urbani infected or Tor2 infected cells (Fig. 3 ). As a control, PIV infected MA104 cells produced large amounts of Type I IFN over the course of the infection.
Fig. 2.
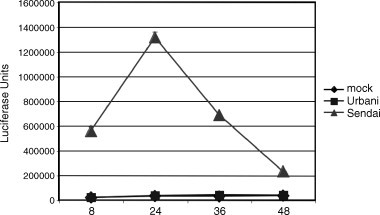
Lack of an IFN beta induction by SARS-CoV. SARS-CoV was tested for its ability to activate an IFN beta promoter by transfection of 293T cells with a plasmid containing the IFNβ promoter driving expression of firefly luciferase. After 24 h post transfection either PBS, SARS-CoV or Sendai Virus was added at an MOI of 5. Samples were taken at 8, 24, 36 and 48 h post infection and analyzed for luciferase production. We find no induction of IFNβ resulting from SARS-CoV infection however a robust expression level is seen in the Sendai Virus infected cells. The depletion of luciferase seen in the Sendai infections after 24 h is due to cell death.
Fig. 3.
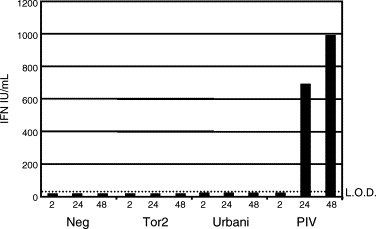
Lack of type I IFN secreted from infected cells. A type I IFN bioassay was performed on media from infected MA104 cells (IFN competent for signaling and production). Cells were infected with an MOI of 5 for either SARS-CoV strains TOR2 and Urbani. Parainfluenza Virus was used as a positive control. Over a timecourse of 2, 24 and 48 h of infection no type I IFN was produced from SARS-CoV infected cells while PIV produced large amounts of IFN. LOD means level of detection for the assay.
Cinatl Jr. et al. compared intestinal cell lines Caco2 and CL-14 for their cytokine profile via microarray analysis (Cinatl et al., 2004). They find the SARS-CoV replicates to very high titer, ∼108 TCID50/ml and infected cells show cytopathic effect (CPE) commensurate with virus titer. Microarray analysis was performed on cells at a single 24 h time point post infection. They find no change in IFNα and β but an up-regulation of CXC chemokines, OAS2 and MX. Interestingly there was no difference in the double stranded RNA activated protein kinase (PKR). IL-18 and MP migration inhibitory factor (MIF) are down-regulated. They extend this correlation to data seen in some patient samples where they find an up-regulation of IP-10 and IL-8. Spiegel et al. also used Caco2 as well as 293 cells to characterize the innate immune response to SARS infection, comparing the effects of virus on permissive (Caco2) and less permissive cells (293T cells) (Spiegel et al., 2006). Although SARS-CoV infection did not induce IFNs, antiviral genes or IL-6; IP-10 and IL-8 were induced in the permissive Caco2 cells, but not 293 cells. They conclude that early virus growth is probably able to expand rapidly while suppressing anti-viral genes but still secreting IP-10 and IL-8 to recruit immune cells.
Tang et al. used a human hepatoma cell line Huh7 for comparative infection of the coronaviruses SARS and 229E (Tang et al., 2005). Using an MOI of 100 TCID50 units, gene expression profiles were compared at 2 and 4 h post infection, very early times in the lifecycle of SARS-CoV. At these early timepoints, IL-8 expression levels were increased while neither IP-10 nor IFNα/β were significantly up-regulated. Importantly, Huh7 cells have been shown to be deficient in some antiviral responses which are in conflict with these findings (Lanford et al., 2003). This likely suggests that Huh7 cells are defective for the double stranded RNA sensing response, consisting of RIGI and Mda5, which would hamper the useful interpretation of these results.
In contrast to the results obtained in the studies by Tang, Speigel, and Cinatl, which suggest that SARS-CoV is a poor inducer of type I IFN responses in productively infected cells, Okabayashi (Okabayashi et al., 2006) found that SARS-CoV infection of Caco2 cells led to high levels of IFNα/β, L, IRF-7, OAS, ISG20 and Mx transcripts at 1–2 days post infection using real-time PCR. They also reported significant inductions of IL-8, IL-6, SOCS3 and TLRs 4, 7 and 9 transcripts. The discrepancy between these different studies might reflect differences in timing, assay sensitivity, or even different passage histories of the cell types in question. However, with the exception of the results from Okabayashi, most reports argue that SARS-CoV is a poor inducer of type I IFN in productively infected cells. We cannot reproduce the results reported by Okabayashi et al., as SARS-CoV infection in Caco2 and 293 cells did not induce expression of IFNα, IFNβ, NFκB or p56 (Fig. 4 ). However in infected cultures, we also reproduce the noted induction of IP-10 in both cell lines supporting reports by Speigel et al. At this time, the preponderance of data argue that SARS-CoV infection does not induce type I IFNs following productive infection in cell culture.
Fig. 4.
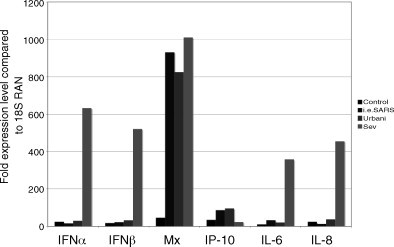
Real-time PCR for innate immune response genes. Caco2 cells were infected with the infectious clone for SARS-CoV (icSARS), the Urbani strain of SARS or Sendai Virus at an MOI of 5 for each. After 18 h cells were harvested for RNA extraction and used for real-time analysis of the denoted genes. Low levels expression was seen in SARS-CoV infected samples for IFNα, IFNβ, IL-6 and IL-8 although large inductions were seen in the Sendai Virus infected samples. Mx was found to be induced by all three viruses to a very large extent while IP-10 was increased up to 100-fold for SARS infected cells and minimal for Sendai infection. All expression levels are shown relative to an 18S standard.
7. Coronavirus replication cycle
SARS-CoV generally does not induce type I IFN in productively infected cells in culture, which suggests that the virus either suppresses or avoids the induction of type I IFN. In light of these possibilities, we will discuss the coronavirus replication cycle and then identify several specific stages where the virus or its derivatives might activate the type I IFN system. Since specific interactions between coronaviruses and the type I IFN system are poorly understood, much of this discussion will be based on known mechanisms employed by other viruses.
The SARS-CoV is a single-stranded, positive polarity enveloped virus. The genome is approximately 29.7 kb long and is associated with the nucleocapsid (N) protein, forming a ribonucleoprotein (RNP) helical N; the RNP is surrounded by a lipid envelope derived from internal cellular membranes of the host cell. The envelope possesses three major envelope proteins: the S glycoprotein, responsible for the receptor recognition and fusion, and the small envelope protein (E) and the M glycoprotein, which are involved in viral budding and release (Marra et al., 2003, Rota et al., 2003) (Fig. 5 ). Minor virion components include ORF7a, ORF7b, ORF6 and ORF3a but these are not essential (Huang et al., 2006, Huang et al., 2007, Schaecher et al., 2007, Shen et al., 2005). The replicase ORF 1a and ORF 1b encode critical functions for virus replication and likely encode important virulence determinants (Eckerle et al., 2006, Sperry et al., 2005).
Fig. 5.
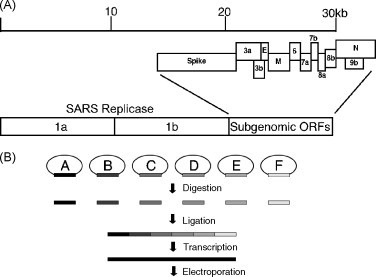
(A) SARS-CoV genome organization. The SARS-CoV genome is shown divided into two main regions, the replicase consisting of ORFs 1a and 1b and the subgenomic ORFs comprising the structural and accessory ORFs. (B) Generation of SARS-CoV infectious clone. The SARS-CoV genome is broken into 6 fragments noted A through F. Each fragment is cloned so as to encode a type II restriction site at either end allowing for directed ligation of all fragments while not changing the amino acids of the encoded proteins. The plasmids are digested and ligated all together to form a single 30 kb fragment. This is used as the template for transcription by T7 polymerase which the 5′ most piece has a start site encoded in it. The resulting RNA is electroporated into Vero cells and virus is collected in the media 24 h after electroporation.
During the coronavirus life cycle (Fig. 6 ), there are several steps where cellular proteins could detect viral components (Weiss and Navas-Martin, 2005). Coronavirus entry is thought to be a three stage process including binding of S glycoprotein to the Angiotensin I converting enzyme (ACE2) protein, cleavage by cathepsin L and activation of a fusion peptide in S2 that mediates entry via fusion through endocytic compartments (Simmons et al., 2005). Fusion, which for SARS happens in an endosomal type structure after cathepsin L cleaves the viral S glycoprotein, viral RNA, which is tightly bound by the N, is released into the cytoplasm of the cell. Following viral genome release into the cytosol, the genome is translated into the viral replicase proteins ORF1a and 1b. These polyproteins are then cleaved by two proteases, a papain like protease (PLP) and the main protease (Mpro), into the individual proteins necessary for replication. The incoming genomic RNA then serves as a template for the synthesis of full length and subgenomic length negative strand RNAs that serves as template for mRNA synthesis. Subgenomic RNA synthesis occurs from discontinuous transcription that joins leader RNA sequences encoded at the 5′ end of the genome to the body sequences of each subgenomic RNA. The model of transcription attenuation argues that the replicase binds to the 3′ end of the genomic RNA, transcribes incomplete negative strands that terminate in highly conserved transcription regulatory sequences (TRS) defined by the sequence ACGAAC, dissociates and re-associates with the full length template near the 5′ end of the genome to prime transcription of subgenomic negative strand RNAs containing anti-leader RNA sequences. The eight different subgenomic negative strands serve as template for the synthesis of like sized subgenomic mRNA (Brian and Baric, 2005). Replicase, structural and accessory proteins are produced during this phase of replication at which time the structural proteins M and E localize to the golgi apparatus and transverse to a ER/golgi intermediate compartment (ERGIC) where budding occurs. Also during this phase, accessory proteins are produced that localize throughout the cell.
Fig. 6.
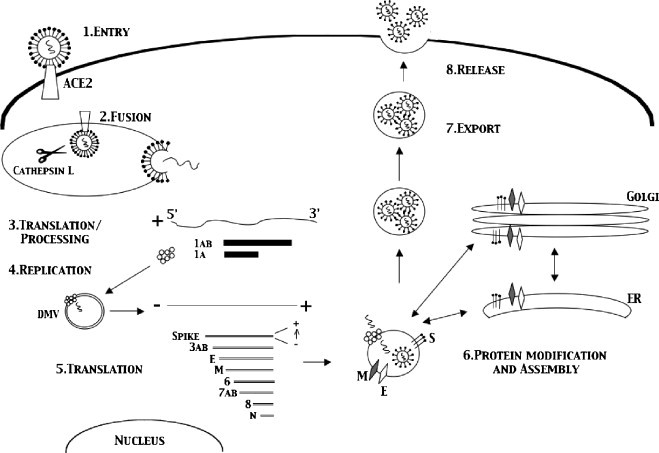
The coronavirus life cycle. Coronavirus entry is mediated by binding of S glycoprotein to the ACE2 receptor, cleavage by cathepsin L and activation of a fusion peptide in S2 that mediates entry via fusion through endocytic compartments [1]. Following fusion with the endosomal compartment the viral genome release into the cytosol where it is translated into the viral replicase proteins ORF1a and 1b [2]. These polyproteins are then cleaved by 2 proteases, Main Protease (Mpro) and Papain like protease, PLP, into the individual proteins necessary for replication [3]. Subgenomic RNA synthesis occurs from discontinuous transcription which joins leader RNA sequences encoded at the 5′ end of the genome to the body sequences of each subgenomic RNA. The eight different subgenomic negative strands serve as template for the synthesis of like sized subgenomic mRNA [4]. Subgenomic RNAs are then translated into viral proteins which localize to their relevant compartments [5]. Assembly of virions occurs in an ERGIC like compartment in the cell. Here S, E, M and N bound to genomic viral RNA are assembled into virions in vesicles [6]. The vesicles are then exported to the cell surface where fusion occurs with release of virions into the exterior environment [7,8].
During virus production, replicase proteins have been shown to localize to double membrane vesicles in the cell that are induced during viral replication in both MHV and SARS-CoV (Brockway et al., 2003, Goldsmith et al., 2004, Gosert et al., 2002, Snijder et al., 2006, van der Meer et al., 1999). The membranes of which are the site of replication and viral assembly. How the structural E, M, and S proteins re-localize to these structures is unknown. It has recently been shown that SARS-CoV E and M proteins do not re-localize to the double membrane structures while for MHV they do (Snijder et al., 2006). Whether this difference is cell specific, virus specific or experimentally specific is unknown. Also whether the replicase proteins are functioning on the inside of the vesicles or in the cytoplasm is unclear.
Once assembly occurs, the virus is secreted using the secretory apparatus of the cell, however, whether coronaviruses pass through the Golgi stacks or use specialized vesicular routes is unknown. Once at the cell membrane, the vesicles carrying the virions fuse with the cell surface and release mature virus into the extracellular space.
8. IFN sensing of coronaviruses
During the replication of coronaviruses, many sentinel sites in the cell's antiviral machinery are potentially impacted, yet with the exception of 229E, the majority of coronaviruses that have been evaluated, fail to induce type I IFN responses (Pitkaranta and Hovi, 1993). Though the specific mechanisms underlying this immune evasion are not well understood, some potential mechanisms will be discussed below.
During entry, the fusion of the virion either at the membrane or in a vacuole, releases genomic RNA into the cytoplasm. Endosome associated TLRs, such as TLR3 or TLR7 may detect SARS-CoV genomic RNA during entry; alternatively the cytoplasmic RNA sensors, RIG-I and Mda5 might detect the RNA as it enters into the cytoplasm. However, a specific role for TLRs or RIG-I/Mda5 in detecting coronaviruses has not been reported, yet several virus families encode products that antagonize signaling at this level (Kash et al., 2006, Zhou et al., 2005). Coronaviruses may also encode proteins which block the viral RNA signaling and sensing pathways. Although specific mechanisms have not been elucidated, it is possible that the viral genome is sequestered, perhaps by the viral N protein, in such a way that the viral RNA is shielded from host sensor proteins. Other viral proteins, such as NS1 of influenza and VP35 of Ebola virus have been shown to block type I IFN induction by interfering with the ability of host sensor proteins (IRF-3, Stat1, RigI, MDA5) to detect incoming virus (Hartman et al., 2004, Kash et al., 2006).
For many coronaviruses, there is no known mechanism of how they evade the host innate immune system. It is hypothesized that it is by either (1) actively producing IFN antagonist proteins, (2) using their own replicase proteins to modify host proteins or by (3) the formation of double membrane vesicles and compartmentalizing replication and perhaps other coronavirus RNAs. The use of double membrane vesicles could hide the RNAs produced by protecting them from the RNA sensing machinery. There may also be a role for N in shielding the viral RNAs from the dsRNA and ssRNA sensing pathways. Many of these possibilities are being actively investigated.
In addition to simply avoiding the activation of type I IFN responses by either masking the viral RNA or sequestering the viral replication complexes into specialized compartments, it is possible that SARS-CoV or other coronaviruses actively inhibit type I IFN induction or signaling. Speigel et al. demonstrated that SARS-CoV infection failed to activate IFNβ promoter activity, but that IRF-3 was translocated to the nucleus in SARS-CoV infected cells (Spiegel et al., 2005). However, SARS-CoV infection interfered with IRF-3 hyper-phosphorylation, dimer formation, and interactions with its essential co-factor, chromatin binding protein (CBP). The role for a specific viral gene or genes in mediating this antagonism has not been described and needs to be evaluated in further detail.
To investigate this concept further, two recent papers have shown that MHV and SARS do not block the IRF-3 signaling pathway in infected cells. Zhou et al. demonstrated that although MHV does not induce nuclear translocation of IRF-3 or IFNβ gene induction it does not block these pathways either (Zhou and Perlman, 2007). Subsequent treatment of cells with poly-I:C or Sendai Virus post infection allows for proper nuclear import of IRF-3 and induction of IFNβ mRNA. They also show that RIGI, MDA5 and TLR3 are also not inhibited by MHV infection. Similar results were shown by Versteed et al. for MHV and SARS-CoV (Versteeg et al., 2007). They also show that MHV does not induce IRF-3 nuclear translocation and IFNα gene induction but that each of these is activated when cells are treated with poly-I:C or Sendai Virus. In addition they show that SARS acts the same way in culture; it does not induce the IFN sensing pathway but does not block the pathways either. Each hypothesizes that there may protection via compartmentalization of the replication complexes and replication RNA in the cell that protects it from being sensed by the anti-viral machinery. Versteeg et al. also proposes that since the 5′ end of SARS RNA is capped it is protected from recognition by RIGI since it was recently shown that RIGI specifically binds to free phosphates at the 5′ end of the RNA (Hornung et al., 2006, Pichlmair et al., 2006). This may be another level of protection SARS-CoV uses to block its sensing from the cellular anti-viral machinery.
We have shown similar data for SARS infections in vitro. We find that SARS infection does not induce IFNβ or NFκB gene induction, however, they do not block those pathways either (Fig. 7A and B). Promoters of IFNβ and NFκB were assayed for their ability to drive luciferase expression after infection by SARS. Wild-type SARS infection of cells does not activate the induction of the either of the promoters however each could be activated post infection by poly-I:C (data not shown), exogenous IFNβ (data not shown) or Sendai Virus (Fig. 7A and B) similar to Zhou et al. and Versteed et al. We created recombinant SARS viruses deleted for each of the SARS accessory ORFs: ORF3a, 3b, 3ab, 3ab/6, 6, 7a, 7b, 8b and 9b. We hypothesized a SARS-CoV accessory ORF may be mediating the IFN sensing block in vivo. However we find that similar to wildtype SARS-CoV, all the deletions fail to induce any of the promoters analyzed above. All of the deletions also allowed for induction post infection by Sendai Virus and poly-I:C of the IFNβ, NFκB and p56 promoters assayed. Our data combined with Zhou et al. and Versteeg et al. lead us to hypothesize that there is a mechanism by which MHV and SARS-CoV evade detection of by innate immune system by (1) sequestering the viral genome on membranous replication complexes, (2) capping viral RNA to evade detection by one arm of the dsRNA sensing machinery and (3) potentially actively inhibiting the innate immune system by the function of virally encoded proteins. One caveat to this work is that SARS-CoV could be inhibiting IFN signaling pathways that are detrimental to its own replication and survival but not inhibiting different pathways inducible by SeV and poly-I:C. In vivo experiments where putative viral antagonists are debilitated or where replication complexes are retargeted to new sites (e.g. autophagy mutants that do not produce double membrane vescicles) may provide insight into which of these hypotheses are most likely.
Fig. 7.
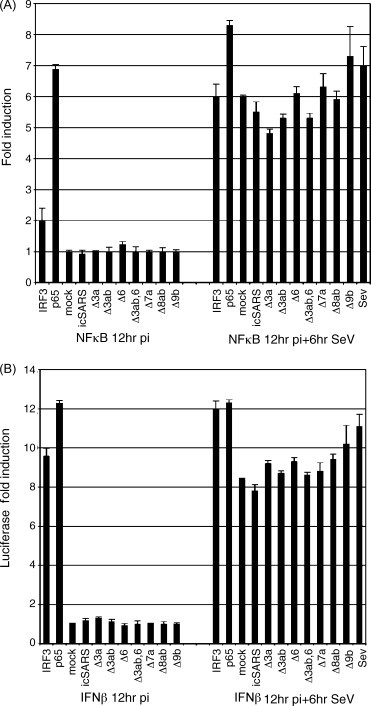
SARS does not block NFκB and IFNβ promoter induction. Vero cells were transfected with either plasmids containing the NFκB (A) or the IFNβ (B) promoter expressing luciferase. 24 h post transfection cells were infected with the designated viruses. The 12 h post infection cells were then infected with Sendai Virus and luciferase was assayed 6 h later using Steady-Glo Luciferase Reagent (Promega). Triplicate wells were averaged and then compared to mock infected wells to graph the fold induction values.
Recent work by Kopecky-Bromberg et al. has identified three SARS proteins, ORF3b, ORF6 and the N protein, that interact with the different elements of the IFN sensing machinery and may add another layer of protection from the innate immune system (Kopecky-Bromberg et al., 2007). When expressed in 293 cells from a plasmid, each viral ORF not only blocked expression from an IFNβ and IRF-3 promoter reporter plasmid but also blocked IRF-3 phosphorylation. N expression blocked NFκB promoter induction while ORF3b and 6 did not, demonstrating an interesting difference in their IFN blocking mechanism. One other distinction between the three genes is that all three blocked expression from an ISRE promoter reporter construct when the cells were stimulated with Sendai Virus, but only 3b and 6 blocked induction of the ISRE reporter when the cells were treated with IFNβ. Shown in Fig. 8a, ORF6 expressed in 293 cells blocked induction of an ISRE promoter expressing luciferase in response to IFNβ treatment. These data suggested a direct block of the IFN amplification side of the innate immune pathway; from the IFNα/β receptor to STAT1 nuclear translocation to induction of STAT1 transcribed genes (assayed by the ISRE promoter construct). ORF6 expression does not effect STAT1 phosphorylation but ORF6 blocked nuclear localization of Stat1 after the cells were treated with IFNβ (Fig. 8b and c). This data shows that each of the 3 genes blocks IFN sensing and signaling in a different way, enabling a reduction in the anti-viral induction pathways from several different pathways.
Fig. 8.
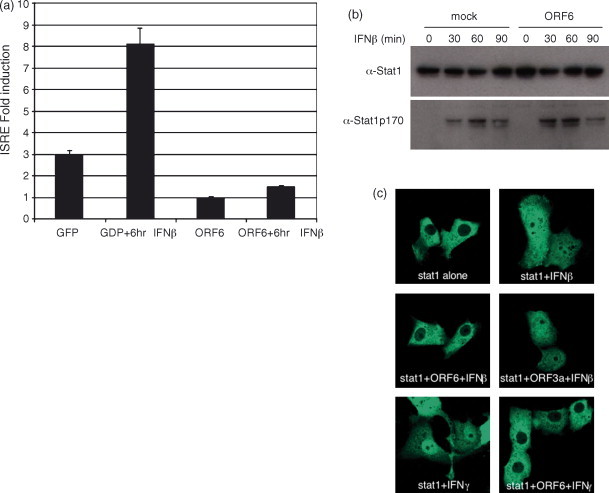
SARS-CoV ORF6 blocks nuclear import of Stat1. (a) Two hundred and ninety three cells were transfected with a plasmid containing an ISRE promoter driving luciferase and either CAGGS/GFP or CAGGS/ORF6. The 24 h post transfection half of the wells were treated with IFNβ for 4 h and then assayed by Steady-Glo Luciferase Reagent (Promega) for ISRE promoter induction. (b) Two hundred and ninety three cells were transfected with HA tagged ORF6. The 24 h post transfection cells were treated with 100 IU/ml of IFNβ for 1 h. Cells were then lysed and assayed by western blot for the presence of total STAT1 (top panel) or phosphorylated STAT1 (bottom panel). (c) Vero cells were transfected with either STAT1/GFP alone or co-transfected with the designated plasmids for 24 h. After 24 h, cells were either untreated or treated with IFNβ or IFNγ as designated. Notice STAT1 is cytoplasmic when untreated but is transported to the nucleus when treated with either IFNβ or IFNγ. Co-expression of SARS ORF6 retains STAT1 in the cytoplasm while SARS 3a expression does not. Also notice that SARS ORF6 blocks IFNγ induced STAT1 nuclear translocation as well.
A report from Cervantes-Barragan et al. may also connect the differences seen in cell lines versus in vivo experiments (Cervantes-Barragan et al., 2007). They have found that there is a rapid induction of type I IFNs upon MHV infection of pDCs compared to cDCs that coincided with a block in virus replication. They also show that SARS-CoV reacts similarly as MHV to different types of DCs (cDC versus pDC). They identify a potential mediator of the IFN induction as well. They find that TLR7 seems to be key to the rapid induction seen in pDCs since TLR7−/− pDCs could produce IFN in response to CpG DNA and not MHV. This result shows that TLR7 is not needed for CpG DNA induced IFN induction but the lack of TLR7 inhibited the induction of IFN by MHV. TLR7 may be sensing the MHV virion or replication products to activate the IFN response in cells.
9. Remaining questions about coronavirus evasion of the innate immune system
Many issues remain unanswered concerning how coronaviruses evade the immune response during infection. With the technology of reverse genetic systems for MHV, SARS-CoV, HCV 229E and TGEV, specific mutations can be made in the viruses to assess individual proteins’ role in immune evasion and pathogenesis. The continued identification of proteins in the signaling pathways described above only adds to the abundance of potential host proteins that may be targeted by viral proteins. Additionally, increased knowledge of pathway inter-regulation and protein:protein interaction networks expand the possible ways that viral pathogens modulate the host response to infection. With these new tools we will be able to gain insight into the critical questions:
-
1.
Which host sensor proteins recognize SARS-CoV genomic RNA or mRNA or viral dsRNA?
-
2.
How do coronaviruses protect their RNA from RNA sensing enzymes in the cell? Does compartmentalization of the replication process keeps the viral RNA in double membrane vesicles and away from the sensing machinery of the host or does an active process of antagonism block sensing of incoming viral genomes? Do these key viral mediators function as virulence alleles and influence disease outcomes?
-
3.
Although coronavirus replicase proteins are highly conserved, the accessory proteins are heterogeneous. Has each virus evolved a different set of IFN antagonizing proteins or are there common mechanisms and protein(s) that functions as an antagonist during coronavirus infection? How many viral antagonist of type I IFN are encoded in the coronavirus genome? Are they species specific? What is their role in pathogenesis?
-
4.
Why is 229E unique in its induction of IFN in culture while the other coronaviruses do not? Does the availability of specific innate immune antagonists modulate the pathogenesis of other human coronaviruses like NL63 and HKU1?
-
5.
Does SARS-CoV or other CoVs induce IFN in monocyte derived cells in a host? In vitro the results are quite different depending on the cell type, origin and preparation. What happens in vivo?
-
6.
Are coronaviruses killed in MPs when they are engulfed during infection? Many SARS-CoV infected lungs show MPs containing SARS-CoV particles. Is this a mechanism for clearance of the virus or is SARS using the MP to evade the immune system and carry the virus deeper into the lung tissue? Do coronaviruses have ways of evading death induced by the MP? Are innate immune responses blocked in during replication in the lung?
-
7.
Are viral proteins acting as intracellular or extracellular IFN antagonists during infection and what stages in the pathways are targeted for inactivation?
-
8.
How does aging impact the innate immunity response and contribute to the increased pathogenesis noted in elderly patients following SARS-CoV infection?
It is anticipated that the interactions of coronavirus genomes and gene products with the host innate immune system will provide a robust research agenda over the next several years, yielding critical information that should elucidate many molecular mechanisms that contribute to coronavirus virulence and pathogenesis in human and animal hosts.
References
- Akira S., Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 2003;85(2):85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- Andrejeva J., Childs K.S., Young D.F., Carlos T.S., Stock N., Goodbourn S., Randall R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA. 2004;101(49):17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler C.F., Wang X., Muhlberger E., Volchkov V., Paragas J., Klenk H.D., Garcia-Sastre A., Palese P. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. USA. 2000;97(22):12289–12294. doi: 10.1073/pnas.220398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian D.A., Baric R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005;287:1–30. doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockway S.M., Clay C.T., Lu X.T., Denison M.R. Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA-dependent RNA polymerase. J. Virol. 2003;77(19):10515–10527. doi: 10.1128/JVI.77.19.10515-10527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas W.B., Loo Y.M., Gale M., Jr., Hartman A.L., Kimberlin C.R., Martinez-Sobrido L., Saphire E.O., Basler C.F. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 2006;80(11):5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilletti C., Bordi L., Lalle E., Rozera G., Poccia F., Agrati C., Abbate I., Capobianchi M.R. Coordinate induction of IFN-alpha and -gamma by SARS-CoV also in the absence of virus replication. Virology. 2005;341(1):163–169. doi: 10.1016/j.virol.2005.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M., Jarrossay D., Facchetti F., Alebardi O., Nakajima H., Lanzavecchia A., Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999;5(8):919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- Cervantes-Barragan L., Zust R., Weber F., Spiegel M., Lang K.S., Akira S., Thiel V., Ludewig B. Control of coronavirus infection through plasmacytoid dendritic cell-derived type I interferon. Blood. 2007;109(3):1131–1137. doi: 10.1182/blood-2006-05-023770. Epub 2006 Sep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charley B., Laude H. Induction of alpha interferon by transmissible gastroenteritis coronavirus: role of transmembrane glycoprotein E1. J. Virol. 1988;62(1):8–11. doi: 10.1128/jvi.62.1.8-11.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P.C., Hsiao, C.H. Re: To, K.F., Tong, J.H., Chan, P.K., et al., 2004. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in-situ hybridization study of fatal cases. J. Pathol. 203 (2), 729–730. [DOI] [PMC free article] [PubMed]
- Chow K.C., Hsiao C.H., Lin T.Y., Chen C.L., Chiou S.H. Detection of severe acute respiratory syndrome-associated coronavirus in pneumocytes of the lung. Am. J. Clin. Pathol. 2004;121(4):574–580. doi: 10.1309/C0EDU0RAQBTXBHCE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinatl J., Jr., Hoever G., Morgenstern B., Preiser W., Vogel J.U., Hofmann W.K., Bauer G., Michaelis M., Rabenau H.F., Doerr H.W. Infection of cultured intestinal epithelial cells with severe acute respiratory syndrome coronavirus. Cell. Mol. Life Sci. 2004;61(16):2100–2112. doi: 10.1007/s00018-004-4222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M., Trinchieri G., Liu Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004;5(12):1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- Cruz C.D., Palosaari H., Parisien J.P., Devaux P., Cattaneo R., Ouchi T., Horvath C.M. Measles virus V protein inhibits p53 family member p73. J. Virol. 2006;80(11):5644–5650. doi: 10.1128/JVI.02400-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond M.S., Shrestha B., Mehlhop E., Sitati E., Engle M. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral. Immunol. 2003;16(3):259–278. doi: 10.1089/088282403322396082. [DOI] [PubMed] [Google Scholar]
- Diebold, S.S., Montoya, M., Unger, H., Alexopoulou, L., Roy, P., Haswell, L.E., Al-Shamkhani, A., Flavell, R., Borrow, P., Reis e Sousa, C., 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424 (6946), 324–328. [DOI] [PubMed]
- Eckerle L.D., Brockway S.M., Sperry S.M., Lu X., Denison M.R. Effects of mutagenesis of murine hepatitis virus nsp1 and nsp14 on replication in culture. Adv. Exp. Med. Biol. 2006;581:55–60. doi: 10.1007/978-0-387-33012-9_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M.J., Jr., Korth M.J., Tang N.M., Tan S.L., Hopkins D.A., Dever T.E., Polyak S.J., Gretch D.R., Katze M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230(2):217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A., Biron C.A. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312(5775):879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Goldsmith C.S., Tatti K.M., Ksiazek T.G., Rollin P.E., Comer J.A., Lee W.W., Rota P.A., Bankamp B., Bellini W.J., Zaki S.R. Ultrastructural characterization of SARS coronavirus. Emerg. Infect. Dis. 2004;10(2):320–326. doi: 10.3201/eid1002.030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002;76(8):3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A.L., Towner J.S., Nichol S.T. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology. 2004;328(2):177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Hiscott J., Lacoste J., Lin R. Recruitment of an interferon molecular signaling complex to the mitochondrial membrane: disruption by hepatitis C virus NS3-4A protease. Biochem. Pharmacol. 2006 doi: 10.1016/j.bcp.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Hiscott J., Lin R., Nakhaei P., Paz S. MasterCARD: a priceless link to innate immunity. Trends Mol. Med. 2006;12(2):53–56. doi: 10.1016/j.molmed.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M., Endres S., Hartmann G. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Huang C., Ito N., Tseng C.T., Makino S. Severe acute respiratory syndrome coronavirus 7a accessory protein is a viral structural protein. J. Virol. 2006;80(15):7287–7294. doi: 10.1128/JVI.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Peters C.J., Makino S. Severe acute respiratory syndrome coronavirus accessory protein 6 is a virion-associated protein and is released from 6 protein-expressing cells. J. Virol. 2007 doi: 10.1128/JVI.02307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs A., Lindenmann J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B: Biol. Sci. 1957;147(927):258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- Ito T., Wang Y.H., Liu Y.J. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR9. Springer Semin. Immunopathol. 2005;26(3):221–229. doi: 10.1007/s00281-004-0180-4. [DOI] [PubMed] [Google Scholar]
- Jiang Y., Xu J., Zhou C., Wu Z., Zhong S., Liu J., Luo W., Chen T., Qin Q., Deng P. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 2005;171(8):850–857. doi: 10.1164/rccm.200407-857OC. [DOI] [PubMed] [Google Scholar]
- Kash J.C., Muhlberger E., Carter V., Grosch M., Perwitasari O., Proll S.C., Thomas M.J., Weber F., Klenk H.D., Katze M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006;80(6):3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze M.G., He Y., Gale M., Jr. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2002;2(9):675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Kopecky-Bromberg S.A., Martinez-Sobrido L., Frieman M., Baric R.A., Palese P. Sars coronavirus proteins Orf 3b, Orf 6, and nucleocapsid function as interferon antagonists. J. Virol. 2007;81(2):548–557. doi: 10.1128/JVI.01782-06. Epub 2006 Nov 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Bonnardiere C., Laude H. Interferon induction in rotavirus and coronavirus infections: a review of recent results. Ann. Rech. Vet. 1983;14(4):507–511. [PubMed] [Google Scholar]
- Laiosa C.V., Stadtfeld M., Graf T. Determinants of lymphoid-myeloid lineage diversification. Annu. Rev. Immunol. 2006;24:705–738. doi: 10.1146/annurev.immunol.24.021605.090742. [DOI] [PubMed] [Google Scholar]
- Lanford R.E., Guerra B., Lee H., Averett D.R., Pfeiffer B., Chavez D., Notvall L., Bigger C. Antiviral effect and virus–host interactions in response to alpha interferon, gamma interferon, poly(i)-poly(c), tumor necrosis factor alpha, and ribavirin in hepatitis C virus subgenomic replicons. J. Virol. 2003;77(2):1092–1104. doi: 10.1128/JVI.77.2.1092-1104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law H.K., Cheung C.Y., Ng H.Y., Sia S.F., Chan Y.O., Luk W., Nicholls J.M., Peiris J.S., Lau Y.L. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106(7):2366–2374. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Foy E., Ferreon J.C., Nakamura M., Ferreon A.C., Ikeda M., Ray S.C., Gale M., Jr., Lemon S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA. 2005;102(8):2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.D., Sun L., Seth R.B., Pineda G., Chen Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA. 2005;102(49):17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R., Lacoste J., Nakhaei P., Sun Q., Yang L., Paz S., Wilkinson P., Julkunen I., Vitour D., Meurs E., Hiscott J. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 2006;80(12):6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300(5624):1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Nakano H., Yanagita M., Gunn M.D. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 2001;194(8):1171–1178. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls J.M., Butany J., Poon L.L., Chan K.H., Beh S.L., Poutanen S., Peiris J.S., Wong M. Time course and cellular localization of SARS-CoV nucleoprotein and RNA in lungs from fatal cases of SARS. PLoS Med. 2006;3(2):e27. doi: 10.1371/journal.pmed.0030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabayashi T., Kariwa H., Yokota S., Iki S., Indoh T., Yokosawa N., Takashima I., Tsutsumi H., Fujii N. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J. Med. Virol. 2006;78(4):417–424. doi: 10.1002/jmv.20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien J.P., Lau J.F., Rodriguez J.J., Sullivan B.M., Moscona A., Parks G.D., Lamb R.A., Horvath C.M. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology. 2001;283(2):230–239. doi: 10.1006/viro.2001.0856. [DOI] [PubMed] [Google Scholar]
- Park M.S., Shaw M.L., Munoz-Jordan J., Cros J.F., Nakaya T., Bouvier N., Palese P., Garcia-Sastre A., Basler C.F. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 2003;77(2):1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Pitkaranta A., Hovi T. Induction of interferon in human leukocyte cultures by natural pathogenic respiratory viruses. J. Interf. Res. 1993;13(6):423–426. doi: 10.1089/jir.1993.13.423. [DOI] [PubMed] [Google Scholar]
- Reghunathan R., Jayapal M., Hsu L.Y., Chng H.H., Tai D., Leung B.P., Melendez A.J. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J.J., Parisien J.P., Horvath C.M. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 2002;76(22):11476–11483. doi: 10.1128/JVI.76.22.11476-11483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Schaecher S.R., Mackenzie J.M., Pekosz A. The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS-CoV) is expressed in virus-infected cells and incorporated into SARS-CoV particles. J. Virol. 2007;81(2):718–731. doi: 10.1128/JVI.01691-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen G.C. Viruses and interferons. Annu. Rev. Microbiol. 2001;55:255–281. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- Shen S., Lin P.S., Chao Y.C., Zhang A., Yang X., Lim S.G., Hong W., Tan Y.J. The severe acute respiratory syndrome coronavirus 3a is a novel structural protein. Biochem. Biophys. Res. Commun. 2005;330(1):286–292. doi: 10.1016/j.bbrc.2005.02.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal F.P., Kadowaki N., Shodell M., Fitzgerald-Bocarsly P.A., Shah K., Ho S., Antonenko S., Liu Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., van der Meer Y., Zevenhoven-Dobbe J., Onderwater J.J., van der Meulen J., Koerten H.K., Mommaas A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006;80(12):5927–5940. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry S.M., Kazi L., Graham R.L., Baric R.S., Weiss S.R., Denison M.R. Single-amino-acid substitutions in open reading frame (ORF) 1b-nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J. Virol. 2005;79(6):3391–3400. doi: 10.1128/JVI.79.6.3391-3400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M., Pichlmair A., Martinez-Sobrido L., Cros J., Garcia-Sastre A., Haller O., Weber F. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 2005;79(4):2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M., Schneider K., Weber F., Weidmann M., Hufert F.T. Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells. J. Gen. Virol. 2006;87(Pt 7):1953–1960. doi: 10.1099/vir.0.81624-0. [DOI] [PubMed] [Google Scholar]
- Stark G.R., Kerr I.M., Williams B.R., Silverman R.H., Schreiber R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Symons J.A., Alcami A., Smith G.L. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81(4):551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Taguchi F., Siddell S.G. Difference in sensitivity to interferon among mouse hepatitis viruses with high and low virulence for mice. Virology. 1985;147(1):41–48. doi: 10.1016/0042-6822(85)90225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A., Yanai H. Interferon signalling network in innate defence. Cell. Microbiol. 2006;8(6):907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- Tang B.S., Chan K.H., Cheng V.C., Woo P.C., Lau S.K., Lam C.C., Chan T.L., Wu A.K., Hung I.F., Leung S.Y., Yuen K.Y. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. J. Virol. 2005;79(10):6180–6193. doi: 10.1128/JVI.79.10.6180-6193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C.T., Perrone L.A., Zhu H., Makino S., Peters C.J. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J. Immunol. 2005;174(12):7977–7985. doi: 10.4049/jimmunol.174.12.7977. [DOI] [PubMed] [Google Scholar]
- van der Meer Y., Snijder E.J., Dobbe J.C., Schleich S., Denison M.R., Spaan W.J., Locker J.K. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 1999;73(9):7641–7657. doi: 10.1128/jvi.73.9.7641-7657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg G.A., Bredenbeek P.J., van den Worm S.H., Spaan W.J. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology. 2007;361(1):18–26. doi: 10.1016/j.virol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005;69(4):635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Condit R.C., Vijaysri S., Jacobs B., Williams B.R., Silverman R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002;76(10):5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Hauns K., Langland J.O., Jacobs B.L., Hogue B.G. Mouse hepatitis coronavirus a59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007;81(6):2554–2563. doi: 10.1128/JVI.01634-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen Y.T., Liao F., Hsiao C.H., Kao C.L., Chen Y.C., Wu-Hsieh B.A. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J. Virol. 2006;80(6):2684–2693. doi: 10.1128/JVI.80.6.2684-2693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilla M., Harcourt B.H., Hickman C.J., McGrew M., Tamin A., Goldsmith C.S., Bellini W.J., Anderson L.J. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005;107(1):93–101. doi: 10.1016/j.virusres.2004.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.Y., Hu Y.W., Liu X.Y., Xiong W., Zhou Z.T., Yuan Z.H. Gene expression profiles in peripheral blood mononuclear cells of SARS patients. World J. Gastroenterol. 2005;11(32):5037–5043. doi: 10.3748/wjg.v11.i32.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Li J., Zhan Y., Wu L., Yu X., Zhang W., Ye L., Xu S., Sun R., Wang Y., Lou J. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect. Immun. 2004;72(8):4410–4415. doi: 10.1128/IAI.72.8.4410-4415.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Perlman S. Mouse hepatitis virus does not induce Beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 2007;81(2):568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Hoebe K., Du X., Jiang Z., Shamel L., Beutler B. Antagonism between MyD88- and TRIF-dependent signals in B7RP-1 up-regulation. Eur. J. Immunol. 2005;35(6):1918–1927. doi: 10.1002/eji.200525971. [DOI] [PubMed] [Google Scholar]
- Ziegler T., Matikainen S., Ronkko E., Osterlund P., Sillanpaa M., Siren J., Fagerlund R., Immonen M., Melen K., Julkunen I. Severe acute respiratory syndrome coronavirus fails to activate cytokine-mediated innate immune responses in cultured human monocyte-derived dendritic cells. J. Virol. 2005;79(21):13800–13805. doi: 10.1128/JVI.79.21.13800-13805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]