

Although it might be ideal to have a single system for defining and reporting serious adverse events that would be applicable to both academic trials investigating established drugs and industry-sponsored trials of novel pharmaceutical agents, such a system would be unlikely to satisfy all stakeholders involved in the conduct, regulation and oversight of clinical research. We advocate that academic trials involving medications commonly used in practice, whether or not for an approved indication, should be considered differently than industry trials.
Accurate and transparent reporting of serious adverse events in randomized trials is crucial to evaluating the benefits and harms of various health care interventions. The Guideline for Good Clinical Practice1 of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use has defined a serious adverse event related to a drug as “any untoward medical occurrence that at any dose, results in death, or is life-threatening, or requires hospitalization or prolongation of existing hospitalization, or is a congenital anomaly or birth defect.” The ideal system for reporting serious adverse events should encourage documentation of important consequences of trial interventions, protect the safety of current and future trial participants, and avoid being unnecessarily burdensome.
When trials are in progress, clinicians, investigators, funders and research ethics boards often find that labelling and interpreting serious adverse events is challenging, particularly for events occurring in critically ill patients.2 Although other acutely ill patients and patients requiring complex care may have comparable difficulties, clinical research involving critically ill patients illustrates several concerns with the existing system for monitoring adverse events. Morbidity and mortality rates are high among patients in the intensive care unit (ICU). For example, the mortality rates for conditions such as septic shock and acute respiratory failure exceed 30%. Critical illness itself often reflects a series of established or acquired complications that evolve, resolve or persist. Therefore, whether enrolled in a trial or not, ICU patients are particularly likely to experience clinical events that fall within the definition of a serious adverse event. These events include death, nosocomial infection and laboratory test results indicating potentially dangerous physiologic abnormalities (Figure 1). Thus, if the foregoing definition is strictly applied, a high proportion of ICU patients may experience a serious adverse event, as occurred in an industry-sponsored sepsis trial, in which 54% of participants in both arms had a serious adverse event.3
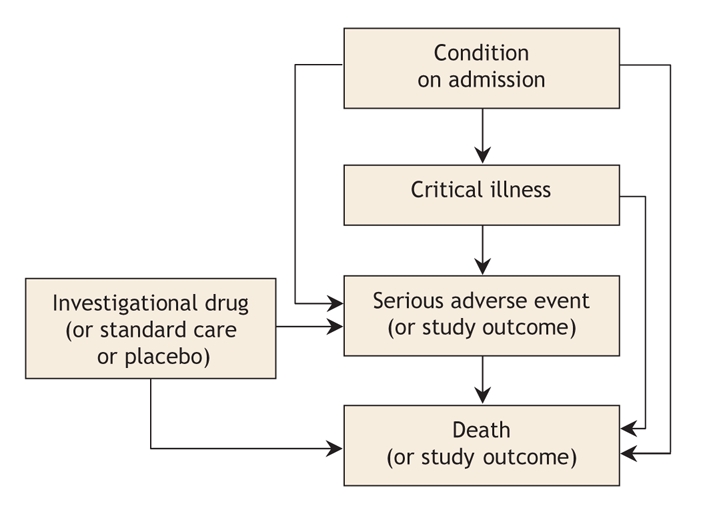
Figure 1: Possible relationships between the condition on admission, a patient's critical illness, the study drug, serious adverse events and death in academic trials of drugs in common use in critical care.
In this commentary, we outline 5 challenges and propose 5 solutions (summarized in Box 1) for more rational reporting of serious adverse events in critical care, focusing on investigator-initiated, peer-review-funded trials of drugs in common use, rather than industry-initiated trials of investigational drugs or established drugs for which a new licensing indication is being sought. Many of these proposed solutions may well be more widely applicable.
Box 1.

Challenge: Variable definition and reporting of serious adverse events
Ambiguous terminology leads to difficulty in defining serious adverse events in critical care research; this in turn creates variable thresholds for reporting such events across trials, as well as across centres within a trial.4 In some jurisdictions, such as Australia, the accepted definition requires investigators to consider including “important medical events that might not be immediately life-threatening or result in death or hospitalisation but might jeopardise the patient or might require intervention to prevent one of the other [adverse] outcomes.”5
Regarding the definition of serious adverse events, death, life-threatening complications and prolonged hospital stay are common, and these are frequently used as prespecified efficacy or safety outcomes reported to an independent data-monitoring committee. For example, in a trial testing corticosteroids for head injury, death was an expected complication and was reported as the primary outcome rather than as a serious adverse event.6 Regarding the reporting of a serious adverse event, the Guideline for Good Clinical Practice1 requires reporting of adverse events that are “serious and unexpected.” It is unclear whether the term “unexpected” means “not anticipated in this patient” or “occurring at a very low baseline prevalence rate.”
Proposed solution
We propose that, before commencing academic critical care trials of medications or devices in common use, investigators should clearly describe the serious adverse events that they plan to identify and report in their protocol, for review by local research ethics boards and data-monitoring committees. Investigators should consider labelling the most concerning and expected of these events as primary, secondary or tertiary outcomes. Adverse events already defined and reported as study outcomes should not be labelled and reported a second time as serious adverse events.
Challenge: Interpretation of serious adverse events in light of the natural history of critical illness
Reporting events considered to be unusual in the natural history of a critical illness (e.g., grand mal seizures occurring in a patient with septic shock) may be important. However, patients may have adverse events as a consequence of the condition requiring admission to the ICU or as a consequence of treatments that are unrelated to the trial intervention. For example, a trauma patient enrolled in a trial of intensive insulin therapy7 might experience tension pneumothorax secondary to chest-wall injury, transfusion-related acute lung injury secondary to initial resuscitation8 and nosocomial pneumonia developing over the course of prolonged mechanical ventilation.9 All 3 events might appear to satisfy the criteria for reportable serious adverse events, but all are most likely due to the primary disease process or its treatment. As such, none of the events is definitively related to the study drug.
Proposed solution
In academic critical care trials of drugs in common use, it is important to consider the natural history of the critical illness affecting each patient enrolled, the expected complications of this illness and the relevance of the complications to the drug being tested. The labelling of a serious adverse event should largely be limited to serious events that are not already labelled as primary, secondary or tertiary outcomes yet which might reasonably occur as a consequence of the study drug. Events that are part of the natural history of the primary disease process or expected complications of critical illness should not be reported as serious adverse events.
Challenge: Attribution of serious adverse events to the drug being tested
Some critically ill trial patients experience adverse events that could be attributed to the study drug. However, it is often difficult to know in real time whether such an event is related to the study drug. For example, bleeding is a common complication among critically ill patients, with 10% of medical– surgical patients suffering major bleeding10 regardless of prophylactic heparin use.11 Therefore, in a trial testing 2 types of heparin thromboprophylaxis, bleeding is expected and may be difficult to attribute to the drugs being compared when the trial is in progress.12 In contrast, hypoglycemia developing during a trial of intensive insulin therapy would likely be attributed to the intensive insulin therapy, except perhaps in a patient in whom severe hepatic failure develops.
Proposed solution
We propose that, in academic critical care trials of drugs in common use, caution be exercised to avoid definitively attributing adverse events to the study drug as the trial is unfolding; this attribution is more sensible when serious adverse events are reported in the 2 arms of the trial and are examined during interim analyses or when the trial is complete.
Challenge: Attribution of death to serious adverse events
Some critically ill trial patients experience serious adverse events that precede death. Because critically ill patients are at high risk of death regardless of their trial enrolment, it is often difficult to attribute death to a serious adverse event. For example, if a bleeding episode precedes death in a trial testing 2 types of heparin thromboprophylaxis, the label “fatal bleeding” is nonspecific. There are 2 possibilities for a patient who dies while experiencing bleeding. Most such patients die with bleeding (e.g., a patient with ongoing melena and concurrent renal failure who experiences hyperkalemic cardiac arrest), although some die because of bleeding (e.g., a patient who dies from hypovolemic shock due to massive uncontrolled upper gastrointestinal hemorrhage).
Proposed solution
We propose that all-cause mortality be reported in all ICU studies. We also propose that, in academic critical care trials of drugs in common use, caution be exercised to avoid attributing deaths to serious adverse events related to the study drug. Cause-specific mortality is usually assessed by central adjudication, using calibrated duplicate independent data review by experts blinded to treatment allocation. Despite the apparent rigour of such adjudication, there are no validated methods to ascertain cause-specific mortality in critically ill patients requiring complex care, most of whom die with multiorgan dysfunction following withdrawal of life-support.13
Challenge: Interpretation of serious adverse events by research ethics boards
Meaningful interpretation of serious adverse events as trials unfold is difficult for local research ethics boards.14 These boards often review such events without specific clinical expertise or knowledge of recent studies in the field, without data on local or global enrolment or distribution of serious adverse events across centres or arms of the trial (in blinded trials), and without knowledge of other outcomes (which are crucial to the consideration of potential harm in light of potential benefit).
Proposed solution
We propose that, in academic critical care trials of drugs in common use, a key role for research ethics boards receiving real-time single-centre reports of serious adverse events is to work with investigators to identify problems requiring remediation of the protocol or its implementation. For example, if a patient fulfilling an exclusion criterion was inadvertently enrolled in a trial and experienced a serious adverse event, the event report should outline the circumstances, consequences and strategies put in place to avoid such an error in the future.
We also propose that independent data-monitoring committees be blinded to allocation, at least initially, when they are monitoring and interpreting individual serious adverse events and that they undertake these activities in the context of emerging literature, in the context of the number of events and patients in each arm (enrolled locally and across all centres) and in the context of other outcomes. We propose that these periodic reports of the data-monitoring committee be sent to each local research ethics board according to the trial protocol.
Discussion
From the clinical perspective, adverse events are the rule rather than the exception in critically ill patients. From the research perspective, faithfully recording serious adverse events is crucial to ensuring that all relevant harms are documented. Accordingly, the call for more transparent reporting of harms in randomized trials distinguishes between studies enrolling participants with non-life-threatening conditions (for whom any harm may be important) and other populations (for whom serious and life-threatening adverse events are the most important).15
Trials involving vulnerable populations need research oversight to ensure that patients' safety, rights and well-being are protected, and to ensure public trust.16 However, overinclusive attempts to identify and definitively interpret serious adverse events in real time during trials involving critically ill patients can create an inappropriate sense of alarm or, worse, a facade of safety. The need for clarity and context-specific reporting of serious adverse events in academic trials of drugs commonly used in critical care reflects a pressing need to avoid spending scarce peer-review resources on paperwork that increases neither the validity nor the safety of the research.
The challenges and solutions we have outlined may be relevant for studies in other settings. Clearly, context is important; thus, the interpretation of serious adverse events will differ for trials of primary prevention involving healthy citizens, for trials in the outpatient clinic setting and for trials involving hospital inpatients. Our proposed solutions for academic critical care trials of drugs in common use focus on interpreting serious adverse events in light of the natural history of critical illness, its complications and management; the designated primary, secondary, and tertiary trial outcomes; the expected frequencies of adverse events; the number of patients enrolled in both arms of the trial in each centre and overall; and whether the existing research oversight evaluates benefits in light of harms and is meaningful and actionable.
Acknowledgments
Deborah Cook is a Research Chair of the Canadian Institutes for Health Research. François Lauzier is supported by a postdoctoral fellowship award from Université Laval.
Footnotes
This article has been peer reviewed.
Contributors: Deborah Cook conceived and drafted the paper. Marcelo Rocha, Mary Jane Sayles and François Lauzier contributed substantially to the conception and design. Marcelo Rocha, Mary Jane Sayles, François Lauzier and Simon Finfer revised the draft for important intellectual content. All of the authors gave final approval of the version to be published. Deborah Cook is the guarantor.
Competing interests: None declared. The authors are dedicated to the care of seriously ill patients and conducting clinical trials to safely improve their outcomes.
Correspondence to: Dr. Deborah Cook, Department of Clinical Epidemiology and Biostatistics, McMaster University Health Sciences Centre, Rm. 2C11, 1200 Main St. W, Hamilton ON L8N 3Z5; fax 905 524-3841; debcook@mcmaster.ca
REFERENCES
- 1.ICH harmonised tripartite guideline. Guideline for good clinical practice E6 (R1). Geneva: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use;1996 Jun 10. Available: www.ich.org/LOB/media/MEDIA482.pdf (accessed 2008 Jan 15).
- 2.Cook D, Moore-Cox A, Xavier D, et al. Randomized trials in vulnerable populations. Clin Trials 2008;5:61-9. [DOI] [PubMed]
- 3.Angus DC, Birmingham MC, Balk R, et al. E4 murine monoclonal antiendotoxin antibody in gram-negative sepsis: a randomized controlled trial. E5 Study Investigators JAMA 2000;283:1723-30. [DOI] [PubMed]
- 4.Santiago LM, Debanne SM, Neuhauser D. Tracking adverse events in RCTs: lack of agreement among regulatory institutions. Qual Saf Health Care 2003;12:234-5. [DOI] [PMC free article] [PubMed]
- 5.Guidelines on the reporting of adverse drug reactions by drug sponsors. Woden (Australia): Australian Government, Department of Health and Ageing, Therapeutic Goods Administration. Available: www.tga.gov.au/docs/html/adrguide.htm (accessed 2008 Jan 15).
- 6.Roberts I, Yates D, Sandercock P, et al; CRASH Trial Collaborators. Effect of intravenous corticosteroids on death within 14 days in 10 008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo controlled trial. Lancet 2004;364:1321-8. [DOI] [PubMed]
- 7.Normoglycaemia in intensive care evaluation and survival using glucose algorithm regulation (NICE - SUGAR STUDY) [study registration record]. Bethesda (MD): National Institutes of Health, ClinicalTrials.gov; 2005. Available: www.clinicaltrials.gov/ct/show/NCT00220987 (accessed 2007 Aug 18).
- 8.Hudson LD, Milberg JA, Anardi D, et al. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med 1995;151:293-301. [DOI] [PubMed]
- 9.Cook DJ, Walters SD, Cook RJ, et al. Incidence of and risk factors for ventilator-associated pneumonia in critically ill patients. Ann Intern Med 1998;129:433-40. [DOI] [PubMed]
- 10.Arnold DM, Donahoe L, Clarke FJ, et al. Bleeding during critical illness: a prospective cohort study using a new measurement tool. Clin Invest Med 2007;30:E93-102. [DOI] [PubMed]
- 11.Douketis J, Cook D, Meade M, et al. Prophylaxis against deep vein thrombosis in critically ill patients with severe renal insufficiency with the low-molecular-weight heparin dalteparin: an assessment of safety and pharmacodynamics. The DIRECT (Dalteparin's Influence on REnally Compromised anti-Ten-A) Study. Arch Intern Med. In press. [DOI] [PubMed]
- 12.Cook DJ, Rocker G, Meade M, et al; PROTECT Investigators; Canadian Critical Care Trials Group. Prophylaxis of ThromboEmbolism in Critical Care Trials (PROTECT): a pilot study. J Crit Care 2005;20:364-72. [DOI] [PubMed]
- 13.Cook D, Rocker G, Marshall J, et al; Level of Care Study Investigators; Canadian Critical Care Trials Group. Withdrawal of mechanical ventilation in anticipation of death in the intensive care unit. N Engl J Med 2003;349:1123-32. [DOI] [PubMed]
- 14.DeMets DL, Fost N, Powers M. An institutional review board dilemma: responsible for safety monitoring but not in control. Clin Trials 2006;3:142-8. [DOI] [PubMed]
- 15.Ioannidis JPA, Evans SJW, Gotzsche PC, et al; CONSORT Group. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med 2004;141:781-8. [DOI] [PubMed]
- 16.Luce JM, Cook DJ, Martin TR, et al; American Thoracic Society. The ethical conduct of clinical research involving critically ill patients in the United States and Canada: principles and recommendations [published erratum in Am J Respir Crit Care Med 2005;172:787]. Am J Respir Crit Care Med 2004;170:1375-84. [DOI] [PubMed]