

Abstract
In the early stages of lung development, the endoderm undergoes extensive and stereotypic branching morphogenesis. During this process, a simple epithelial bud develops into a complex tree-like system of tubes specialized for the transport and exchange of gas with blood. The endodermal cells in the distal tips of the developing lung express a special set of genes, have a higher proliferation rate than proximal part, undergo shape change and initiate branching morphogenesis. In this study, we found that of the four p38 genes, only p38α mRNA is localized specifically to the distal endoderm suggesting a role in the regulation of budding morphogenesis. Chemical inhibitors specific for the p38α and p38β isoforms suppress budding of embryonic mouse lung explants and isolated endoderm in vitro. Specific knockdown of p38α in cultured lung endoderm using shRNA also inhibited budding morphogenesis, consistent with the chemical inhibition of the p38 signaling pathway. Disruption of p38α did not affect proliferation or expression of the distal cell markers, Sox9 and Erm. However, the amount of E-cadherin protein increased significantly and ectopic expression of E-cadherin also impaired budding of endoderm in vitro. These results suggest that p38α modulates epithelial cell-cell interactions and possibly cell rearrangement during branching morphogenesis. This study provides the first evidence that p38α is involved in the morphogenesis of an epithelial organ.
Keywords: lung, p38, branching morphogenesis, E-cadherin
INTRODUCTION
Mouse lung branching morphogenesis starts from embryonic day 9.5 (E9.5) when two primordial buds appear at the ventral lateral side of the foregut. The inner layer of the primordial bud is composed of endodermal epithelial cells and is surrounded by mesenchymal cells derived from mesoderm. Between E9.5 and E16.5 (the pseudoglandular stage), the primordial bud undergoes repetitive rounds of budding and branching to give rise to a tree-like structure. This process is regulated by reciprocal interactions between the epithelial cells and the surrounding mesenchyme and is mediated by extracellular signaling molecules, cell membrane receptors and intracellular signaling pathways. In response to signals from the mesenchyme, the epithelial cells at the distal tips of the developing lung undergo several changes, including increased proliferation rate, coordinated cell shape changes and the generation of the precursors of different cell types along the proximal-distal axis. Thus, the epithelial cells in the distal tip and proximal stalk have distinct phenotypes, including different proliferation rates and cell shape (Cardoso, 2001; Hogan and Yingling, 1998; Warburton et al., 2000). In addition, these two populations of cells express different sets of genes. For example, Bmp4, Erm, Zdhhc6 and Sox9 are expressed at the distal tip while Netrin-1 and -4 are expressed in more proximal regions (Liu and Hogan, 2002; Liu et al., 2003; Liu et al., 2004).
Members of several families of conserved signaling molecules, including Fibroblast Growth Factor (FGF), Sonic hedgehog, Bone Morphogenetic Protein/Transforming Growth Factor-β, Wnt, Epidermal Growth Factor and Platelet-Derived Growth Factor families, are expressed in lung during the pseudoglandular stage (Hogan and Yingling, 1998). Previous studies using genetically modified mice have highlighted the role of FGF signaling during lung development. For example, FGF receptor isoform 2 IIIb (FGFR2IIIb) in the endoderm is essential for normal branching morphogenesis during lung development. Pulmonary agenesis is seen in FGFR2IIIb-null mice in addition to other developmental defects (De Moerlooze et al., 2000) and transgenic expression of a dominant negative form of FGFR2IIIb in lung epithelium inhibits branching morphogenesis (Peters et al., 1994). Activating ligands of FGFR2IIIb include FGF7 and FGF10, which are released from the mesenchyme (Bellusci et al., 1997; Ornitz et al., 1996), bind receptors and initiate downstream signals. Although activation of FGFR2IIIb is required for normal branching morphogenesis and lung development, the specific downstream signaling events involved are largely uncharacterized. It is well established that mitogen-activated protein kinases (MAPKs) are key downstream components of FGF signaling pathways in numerous cell types. The MAPK family is divided into three subfamilies based on their activation motifs: extracellular signal-regulated protein kinase (ERK) has a Thr-Glu-Tyr motif; Jun NH2-terminal kinase (JNK) has a Thr-Pro-Tyr motif; and p38 kinase has a Thr-Ala-Tyr motif. MAPKs have been shown to regulate several critical cellular functions including proliferation, differentiation, and migration (Pearson et al., 2001). ERK1 and ERK2 are MAPKs typically activated by FGFs in several cell types and previous studies suggest that they are important for normal lung development (Liu et al., 2004). In this study we provide evidence supporting a functional role for p38 MAPK in lung branching morphogenesis.
Initially, p38 kinase was characterized as an inflammatory or stress activated kinase in a wide variety of cell types. Subsequently, p38 activation has been seen in response to growth factors including FGFs, as well as cytokines, UV light, heat and osmotic stress. Like all MAPKs, p38 is activated through a cascade of kinases broadly defined as MAPK kinase kinases (MKKKs) and MAPK kinases (MKKs). Included in these groups of upstream activators are Mixed-Lineage Kinase 3 (MLK3), Dual Leucine Zipper-Bearing Kinase (DLK) and Mitogen-Activated Protein Kinase Kinase 3/6 (MKK3/6). The downstream targets of p38 include MAPK-Activated Protein Kinases 2/3 (MAPKAP2), Heat Shock Protein 25/27 (HSP25/27) and several transcription factors such as Activating Transcription Factor-2 (ATF-2) and Myocyte Enhancer Factor 2 (MEF2) (Herlaar and Brown, 1999). Recently, p38 signaling has been implicated in normal developmental processes, such as wing development in Drosophila, and muscle formation in Xenopus (Adachi-Yamada et al., 1999; Keren et al., 2005; Nebreda and Porras, 2000). Four p38 genes have been identified in mice: p38α/MAPK14, p38β/MAPK11, p38δ/MAPK13 and p38γ/MAPK12 (Kuida et al., 2004). Although all four isoforms are activated by stress and mitogenic stimuli, distinct functional roles for each is suggested, especially during mammalian development. Disruption of p38β or p38γ does not cause a detectable developmental defect (Kuida and Boucher, 2004) and a p38δ mutant mouse has not been reported. On the other hand, p38α null mutant mice die at E12.5 apparently due to a defect in placental angiogenesis (Tamura et al., 2000). Recently, an embryo-specific deletion of p38α was generated by crossing mice with floxed p38α allele to mice with a MORE-cre allele. These p38α-null mice die shortly after birth with distorted alveolar structures and massive infiltration of hematopoietic cells in the lung. Whether a defect in branching morphogenesis coexists is not clear since morphometric analysis of the lungs prior to the massive hematopoietic cell infiltration at E17.5 was not performed (Hui et al., 2007). Postnatal deletion of p38α revealed an important regulatory role for p38α in lung stem and progenitor cell proliferation and differentiation (Ventura et al., 2007).
Additional evidence suggests that the p38α isoform has several important roles during development. A chemical inhibitor SB203580 that specifically inhibits p38α and β impairs cavity formation in mouse blastocysts before implantation (Maekawa et al., 2005). Recently, a mouse mutation disrupting p38-interacting protein (p38IP or droopy eye) was shown to result in neural tube, eye and gastrulation defects. p38IP appears to specifically interact with p38α, but not other p38 isoforms, resulting in its activation. In the absence of normal p38IP activation of p38 signaling, E-cadherin down regulation and mesoderm migration away from the primitive streak are impaired during gastrulation (Zohn et al., 2006).
In this study, we found that the expression pattern of p38α suggests a role in mouse lung branching morphogenesis. Chemical inhibition of p38α/β reduced distal bud formation in embryonic whole lung explants. Using a three-dimensional culture system, we disrupted p38α in the embryonic lung endoderm with either chemical inhibitors or short hairpin RNA (shRNA). Both methods of targeting the p38α signaling pathway blocked budding morphogenesis. In addition, disruption of p38α and inhibition of budding was associated with increased levels of E-cadherin protein. Ectopic expression of E-cadherin was sufficient to inhibit secondary budding of isolated endoderm. We propose that p38α plays a role in regulating E-cadherin levels related to epithelial branching morphogenesis.
MATERIALS AND METHODS
In situ hybridization
Whole-mount in situ hybridization was performed by using digoxygenin-labeled antisense RNA probes as described (Liu et al., 2003). The probes for p38α, δ, γ were generated from IMAGE clones 5100700 (GenBank Number BI220793), 6397838 (GenBank Number BQ926696), 6333323 (GenBank Number BQ899725), respectively. The probes for p38β were derived from RT-PCR products using primer 5′-CCATGAAATTGAGCAGTG-3′ and 5-AGTTACTTGGTCAGCTCC-3′. For all probes, sense RNA was used as control and none of them show detectable signal.
Embryonic whole lung explant culture
Lungs were dissected from ICR mice at E11.5 (noon on the day of plug is E0.5) and cultured in Costar Transwell plates (3 μm pores) containing BGJb medium supplemented with 0.2 mg/ml ascorbic acid and penicillin/streptomycin (P/S) in the lower chamber as described (Kling et al., 2002). Media was changed daily. Lung explants were photographed using a QImaging MicroPublisher 5.0 camera attached to a Leica MZ9.5 stereoscope. Images were captured using QCapture Pro software and imported into Adobe Photoshop to count the terminal buds. Heparin-acrylic beads (Sigma) were soaked in FGF10 or BSA solutions (100 ng/μl) for 1 hour at RT prior to use.
Lung endodermal culture
Lungs were dissected from ICR mice at E11.5. Distal endoderm was separated from mesenchyme and cultured in growth factor reduced Matrigel as described (Nogawa and Ito, 1995). The endoderm was cultured in a serum-free medium consisting of Ham’s F12:DMEM 1:1, 1% BSA and 2 mM glutamine. The media was supplemented with recombinant FGF7 (30 ng/ml) or FGF10 (250 ng/ml) as indicated (R&D Systems). In some experiments, chemical inhibitors SB203580, SB202190 or SP600125 (Calbiochem) were added at 10 μM, U0126 was added at 5 μM, at 24 hours after culture initiation. In other experiments, the endoderm was infected with lentivirus at the time of isolation and initial culturing. The endoderm was cultured for a designated time, released from Matrigel and processed for immunohistochemistry, Western blot analysis or real-time RT-PCR analysis. Each culture condition was repeated at least three times with about ten endoderm samples per experiment.
Cell Culture
LA-4 cells (a generous gift from Marc B. Hershenson, Univ. of Michigan) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and P/S at 37°C in 5% CO2 (Stoner et al., 1975). The cells were serum-starved for 24 hours in DMEM containing 0.2% BSA and P/S before treatment with FGF7 or FGF10.
Lentiviral Constructs, Production and Transduction
The shRNA constructs were generated using the lentiviral transfer vector pLVTHM (a generous gift from Dr. Didier Trono at Swiss Institute of Technology Lausanne, Switzerland). This vector contains a self-inactivating long terminal repeat (SIN LTR), a human H1 promoter for expression of the shRNA and a GFP marker expressed from an EF1α promoter (Wiznerowicz and Trono, 2003). The targeting sequences for shRNA constructs were as follow: 5′-GGTCACTGGAGGAATTCAA-3′ and 5′-GGCATCGTGTGGCAGTTAA-3′ for mouse p38α; and 5′-GAATGTTATAGGCATCCGA-3′ for mouse ERK1.
PCR was used to add a carboxy-terminal FLAG epitope tag to full-length mouse E-cadherin cDNA (a generous gift from Masatoshi Takeichi, RIKEN Center for Developmental Biology, Japan). The FLAG-tagged E-cadherin was cloned into the lentiviral transfer vector pWPI (a generous gift from Dr. Didier Trono at Swiss Institute of Technology Lausanne, Switzerland) which contains a SIN LTR, a human EF1α promoter for expression of the gene of interest and GFP expressed from an internal ribosomal entry site (Pham et al., 2004).
Lentivirus was produced by co-transfecting HEK293T cells with a transfer vector together with a second generation packaging vector, psPAX2 (a generous gift from Didier Trono) and the VSV-G envelope protein vector, pHCMV-G (a generous gift from Dr. Garry Nolan, Stanford) (Wolkowicz et al., 2004). The medium containing virus was harvested 48-hours post-transfection and the virus was concentrated by centrifugation at 6000×g for 24 hours at 4°C (Nanmoku et al., 2003). The titer of the viral supernatant was determined using serial dilutions to infect NIH 3T3 cells. Successfully transduced cells were identified by the expression of GFP detected by fluorescence microscopy. The percentage of GFP positive cells was determined by manual counting and normalized to the total number of cell present in one field. A titer of 107 to 108 transducing units/ml was typically obtained. Experiments were repeated using at least three independently prepared batches of control and shRNA expressing virus with matched viral titers.
Immunohistochemistry and immunoblotting
Whole-mount antibody staining of cultured lung endoderm was performed as previously described (Liu et al., 2004). Anti-phospho-histone H3 (Ser10) (Upstate Cell Signaling) was used at 1 μg/ml and anti-E-cadherin (BD Transduction Laboratories) was used at 0.25μg/ml. Cy3 conjugated secondary antibodies (Jackson Immunoresearch) were used at concentrations recommended by manufacturer. In certain experiment, the tissues were also labeled with 0.13 μM Alexa Fluor 488 phalloidin or propidium iodide as previously described (Liu et al., 2004). The fluorescence images were captured using a Zeiss LSM 510 confocal microscopy (University of Chicago Cancer Research Center Integrated Microscopy Facility). Immunoblotting of the cultured endoderm was performed as previously described (Liu et al., 2004). Specifically, protein extracts of 30 endoderm samples were loaded on each lane and the following primary antibodies were used: anti-p38α (Chemicon) at 0.5μg/ml; anti-β-actin (Abcam) at 0.1μg/ml; anti-phospho-histone H3 (Ser10) (Upstate Cell Signaling) at 1 μg/ml; anti-E-cadherin (BD Transduction Laboratories) at 0.25μg/ml; and anti-Sox9 (Abcam) at 0.5μg/ml. Total and phospho-specific p42/44 kinase antibodies (Cell Signaling) were used at a 1:1000 dilution.
Protein quantification by Western analysis was determined using densitometric scanning (SigmaScan Pro, Jandel Corp., San Rafael, CA) as previously described (Abe et al., 2001) with normalization to β-actin protein levels.
RNA isolation, reverse transcription and real-time RT-PCR
Total RNA isolation and reverse transcription (RT) were performed using the RNAqueous-4PCR kit (Ambion) according to the manufacturer’s protocol. DNAse I treated total RNA from about 30 cultured lung endoderm samples was used per RT reaction with an oligo-dT primer and either MMLV-RT or water. Real-time RT-PCR (qRT-PCR) reactions were performed on an Applied Biosystems 7300 Real Time PCR System running Sequence Detection Software v1.2.3. Real time primer sets were designed using the Primer Express software (Applied Biosystems): ERM, 5′-GCCGAGGCATGGAATTTAAG-3′ and 5′-AGAGCGGCTCAGCTTGTCATA-3′; E-cadherin, 5′-AAAATCCATCTCAAGCTCGCG-3′ and 5′-ATTCCCGCCTTCATGCAGTT-3′; Rab7, 5′-AAGGAGGCCATCAATGTGGAG-3′ and 5′-CCCGGTCATTCTTGTCCAGTT-3′; Snail, 5′-TTCCTGCTTGGCTCTCTTGGT-3′ and 5′-TATGGCTCGAAGCAGCTGTGT-3′; Slug, 5′-TGATGCCCAGTCTAGGAAATCG-3′ and 5′-GCCACAGATCTTGCAGACACAA-3′; and β-actin, 5′-CACTATTGGCAACGAGCGGT-3′ and 5′-TTCTGCATCCTGTCAGCAATG-3′. Primers were used at a final concentration of 200 nM with SYBR Green Master Mix (Applied Biosystems). Following a single denaturation step for 10 min at 95°C, 40 cycles of two-step PCR was performed consisting of 15 seconds at 95°C and 1 min at 60°C. Melting curve analysis and detection by gel electrophoresis with ethidium bromide staining were performed to determine specificity of the PCR products. Data were analyzed using the relative quantification RQ Study Software (Applied Biosystems) using the comparative CT method. The amount of target mRNA was nomalized to β-actin. Relative quantification to the control sample calibrator was calculated using the formula 2−ΔΔCT. For each sample set, the baseline and threshold (CT) were selected automatically with visual verification.
RESULTS
Expression patterns of p38 isoforms in embryonic lung
To understand the role of p38 during branching morphogenesis, we compared the expression patterns of all four p38 isoforms in embryonic mouse lung at the pseudoglandular stage by in situ hybridization. p38α is expressed as early as E10.5 (Fig. 1A). From E11.5 to E13.5, when the lung undergoes active branching morphogenesis, p38α transcript is localized to the endodermal epithelial cells and is enriched at the distal tips. Epithelial cells in this region are most actively engaged in budding and branching morphogenesis and have a higher proliferation rate compared to the proximal region. They also contain precursors of alveolar type I and II cells (Liu et al., 2003; Perl et al., 2002; Weaver et al., 1999). Thus this pattern of expression suggests that p38α is involved one or more of the distal specific developmental processes such as proliferation, differentiation, budding/branching. In contrast, at E11.5, p38β and p38δ transcripts are more uniformly distributed throughout the proximal and distal endoderm (Fig. 1D and E). Expression of p38γ appears to be relatively low in both the endoderm and mesoderm without a difference between proximal and distal regions (Fig. 1F). Unlike p38α, the expression patterns of p38β, δ and γ suggest that they are not involved in distal specific processes.
Fig. 1. Expression pattern of genes encoding four p38 isoforms during lung branching morphogenesis.

Whole mount in situ hybridization was performed in embryonic lungs during branching morphogenesis using anti-sense RNA probes for p38α (A–C), p38β (D), p38δ (E) and p38γ (F). Three time points during the pseudoglandular stage were analyzed: E10.5 (A); E11.5 (B, D, E, F); and E13.5 (C). (A) p38α is expressed at E10.5 shortly after the initiation of lung development. At E11.5 (B) and E13.5 (C), when lung endoderm is actively engaged in branching morphogenesis, p38α transcripts remain localized at the distal tip of epithelium, the sites of active budding and branching. (D) p38β is expressed in both the mesenchyme and epithelium with similar levels throughout proximal and distal regions. (E) p38δ transcript is located in the endoderm with similar levels of expression throughout proximal and distal regions. (F) The expression level of p38γ is very low and does not appear to have a specific proximal-distal pattern. Scale bar = 50 μm
Inhibition of p38α/β signaling impairs branching morphogenesis of embryonic lung explants
To test whether p38α is involved in early embryonic lung branching morphogenesis, whole lungs from E11.5 mice were cultured in the presence of a pyridinyl imidazole inhibitor, SB203580, which specifically inhibits the α and β isoforms of p38 by binding to their activation pockets and competing with ATP (Fitzgerald et al., 2003). Embryonic lungs cultured in the presence of 10 μM of SB203580 displayed evidence of impaired branching morphogenesis with a reduced distal bud count after 48 hours compared to untreated and vehicle (DMSO) treated explants (Fig. 2). These results suggest that p38α/β signaling plays a role in embryonic lung branching morphogenesis.
Fig. 2. Inhibition of p38α/β reduces branching morphogenesis in embryonic lung explants.
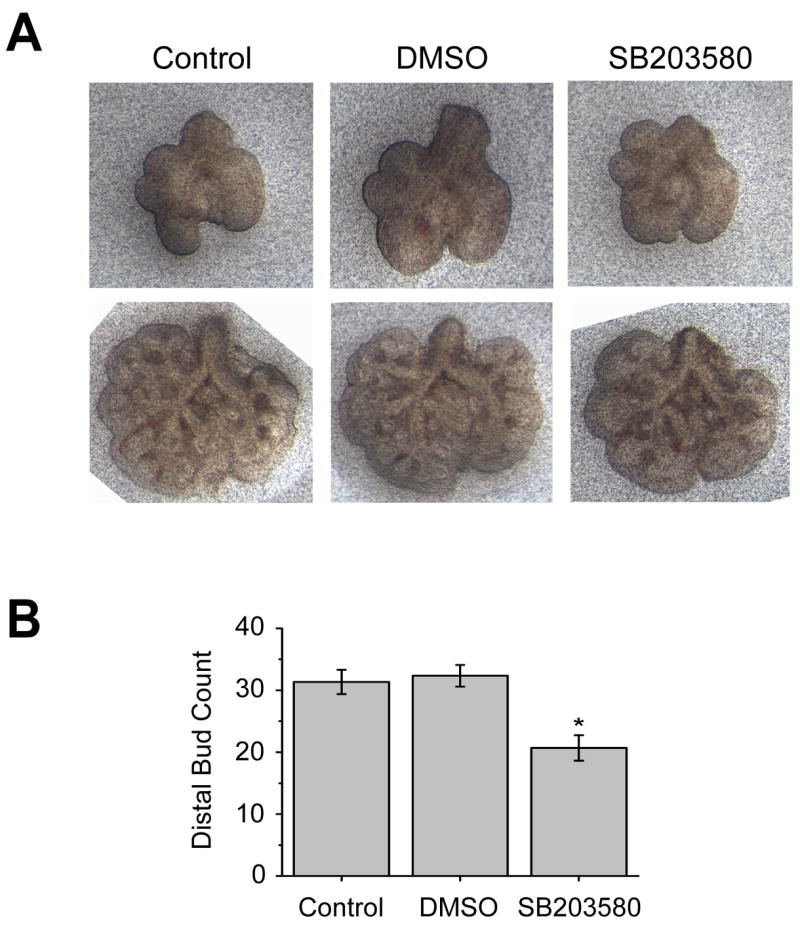
(A) Whole lung explants isolated from E11.5 mice were treated with either vehicle (DMSO) or the p38α/β inhibitor SB203580 (10 μM). Untreated explants were used as a Control. Lungs are shown at the time of isolation (top row) and after 48 hours in culture (bottom row). (B) Graphical representation of the distal bud count after 48 hours in culture. Data are the mean ± SEM from six explants per group. *P<0.01 compared with either Control or DMSO groups (One way ANOVA followed by Bonferroni t-test).
p38α is required for budding morphogenesis in vitro
To study the function of p38 in early lung branching morphogenesis and specifically in the developing endoderm, a three-dimensional in vitro culture system was utilized in which distal lung endoderm is isolated from surrounding mesenchyme and cultured in growth factor reduced Matrigel in serum-free medium supplemented with 30 ng/ml FGF7 (Fig. 3A) (Liu et al., 2004).
Fig. 3. Morphological responses of cultured endoderm to the treatments of chemical inhibitors of MAPKs.

(A) A diagram illustrating the method used to culture mesenchyme-free lung endoderm. E11.5 embryonic mouse lung endoderm was dissected from mesenchyme, embedded in growth factor reduced Matrigel and cultured in serum-free media containing 30 ng/ml FGF7. (B) DIC image of endoderm treated with vehicle (DMSO). (C) Whole mount in situ hybridization of an isolated endoderm sample grown in the presence of FGF7 (30 ng/ml) using a probe for p38α. Note the presence of numerous secondary buds in response to FGF7 (B) and the enrichment of p38α transcripts in the secondary buds (arrows) (C). (D) DIC image of endoderm treated with SB203580 (10 μM). In the presence of SB203580, a chemical inhibitor of p38α/β, the endodermal epithelium forms a cyst that expands but fails to generate secondary buds. (E) Endoderm cultured in the presence of FGF10 (250 ng/ml) instead of FGF7 and treated with vehicle (DMSO) or (F) treated with SB203580 (10 μM). (G–H) DIC images of the endoderm cultured in the presence of FGF7 (30 ng/ml) and treated with U0126 (5 μM) (G) or SP600125 (10 μM) (H). In the presence of U0126, chemical inhibitor of ERKs, the endodermal epithelium forms a cyst with folds inside and no secondary buds are formed (G). In the presence of SP600125, an inhibitor of JNK, the endodermal epithelium forms a cyst, but the lumen fills with necrotic appearing cells (H). Scale bar = 40μm.
Within hours of isolation in culture, the endoderm seals and forms a vesicle with the apical cell surface facing the lumen. Later, the endoderm grows and expands into a cyst. At 24–36 hours after culture initiation, numerous secondary buds start to form at the surface of the cyst (Fig. 3B). Previously, the transcripts of multiple distal specific genes, such as Bmp4, Erm and Sox9, have been shown to be preferentially expressed in the secondary buds (Liu et al., 2003; Liu et al., 2004) indicating that these secondary buds mimic the distal budding endoderm in developing lung. Since expression of p38α transcript is also localized to the distal endoderm (Fig. 1), in situ hybridization was performed to determine its expression pattern in cultured lung endoderm. Consistently, p38α transcript is enriched in the secondary buds (Fig. 3C). This finding further supports the idea that this three-dimensional embryonic lung endoderm culture system faithfully imitates budding and branching morphogenesis and proximal-distal patterning seen in vivo making it an excellent model system to study of the function of p38α in the embryonic lung.
To determine the functional role of p38α in developing lung endoderm, the p38α/β inhibitor, SB203580 was utilized. In the presence of this p38α/β inhibitor, the isolated lung endoderm cultured with FGF7 (30 ng/ml) formed hollow cysts, but consistently failed to develop secondary buds (Fig. 3D). Inhibition of FGF10 (250 ng/ml) induced secondary budding was also seen with SB203580 (Figs. 3E–F). While both FGF7 and FGF10 initiate signals downstream of FGFR2IIIb and induce expression of distal endoderm markers, their budding phenotypes differ (Bellusci et al., 1997; Izvolsky et al., 2003; Liu et al., 2003; Weaver et al., 2000). An identical phenotype was seen with SB202190, another pyridinyl imidazole inhibitor specific for p38α/β (data not shown). The phenotype seen with inhibition of p38α/β was distinct from that seen following treatment with U0126 and SP600125, chemical inhibitors of the ERK and JNK signaling pathways, respectively (Figs. 3G–H). In the presence of U0126, an inhibitor to ERK1, ERK2 and ERK5, FGF7 stimulated endodermal epithelium formed a cyst with folds inside and no secondary buds (Fig. 3G). In the presence of SP600125, the FGF7 stimulated endodermal epithelium formed a cyst, but the lumen filled with necrotic appearing cells (Fig. 3H). These inhibitor studies suggest that p38α and/or p38β play a role during lung epithelial budding morphogenesis.
While SB203580 and SB202190 are generally thought to be relatively specific for p38α/β compared to other chemical inhibitors of protein kinases (Bain et al., 2003; Davies et al., 2000), RNA interference was used to confirm the findings seen with these inhibitors as well as determine whether p38α or p38β is the major isoform involved in the regulation of bud morphogenesis. In order to achieve stable knockdown at high efficiency in the cultured lung endoderm, a lentiviral delivery system for shRNA was used. As shown in Fig. 4, infection rates of 100% within the entire epithelial cyst could be achieved with this viral delivery system. Furthermore, ectopic genes introduced using lentivirus express as early as 36 hours post infection and continue to increase for as long as 60 hours post infection (Figs. 4A–D). The ectopic gene starts to express at approximately the same time as secondary buds initiation. Localized expression of p38α to regions of active branching in the developing lung (Fig. 1) and the lack of a developmental phenotype in p38β null mice, suggest that p38α is the isoform most likely involved in the regulation of secondary budding. Therefore, p38α was knocked down using lentiviral delivered shRNA in lung endoderm cultured in the presence of FGF7 (Figs. 4E–F). A total of 102 p38α shRNA treated lung endoderm cysts were generated using several independent lentiviral preparations and 80.4% showed a phenotype similar to those treated with the chemical inhibitor of p38α/β, with the formation of cysts that lack secondary bud formation. In contrast, only 6 of 90 control lung buds (6.7%) failed to develop secondary buds (Table 1). The failure to see an identical phenotype in 100% of the p38α shRNA treated lung buds could be the result of residual p38α expression due to the infection efficiency and/or shRNA effectiveness. A second shRNA construct that targets a different region of p38α also resulted in endodermal cysts that failed to develop secondary budding (data not shown). In contrast, specific shRNA targeting of ERK1 that left ERK2 expression intact did not cause a morphological change in the cultured endoderm (Figs. 4H–J). While combined inhibition of ERK1 and ERK2 with U0126 affected budding morphogenesis (Fig. 3G), this result is consistent with the lack of a developmental phenotype in the ERK1-null mouse (Kuida and Boucher, 2004). Importantly, it indicates that the observed budding morphogenesis defect seen with p38α shRNA is the result of p38α knockdown rather than a nonspecific off-target effect of saturating the endogenous RNAi machinery. Taken together, the impaired formation of secondary buds resulting from treatment with either SB203580 or p38α shRNA strongly suggest that p38α is involved in FGF induced budding/branching morphogenesis.
Fig. 4. Disrupting p38α in cultured endoderm using lentiviral delivered shRNA.

(A–D) GFP was transduced into embryonic lung endodermal epithelium using lentivirus. (A) GFP expression is barely detectable 24 hours post infection. (B) GFP expression is clearly visible 36 hours post infection. (C) The level of GFP expression continues to increase at 60 hours post infection. (D) Using this method, transduction of the entire epithelium can be achieved. GFP transduced cells (green) are shown with nuclei identified by propidium iodide (red). (E–G) Knockdown of p38α using lentiviral delivered shRNA inhibits budding morphogenesis in cultured lung endoderm. Lentivirus without (E) or with p38α shRNA (F) was used to infect isolated lung endoderm. Note the presence of secondary buds in the lentivirus control (E). Endoderm transduced with p38α shRNA appear smaller and fail to generate secondary buds (F). (G) Western blot analysis of lung endoderm treated with p38α shRNA indicates that the levels of p38α are significantly reduced. Protein quantification using densitometric scanning and β–actin as a loading control revealed a 70% reduction in p38α expression. (H–J) Knockdown of ERK1 using the same lentiviral delivery system does not impair secondary budding. Lentivirus without (H) or with Erk1 shRNA (I) was used to infect isolated lung endoderm. (J) Western blot analysis of lung endoderm treated with Erk1 shRNA indicates that the levels of ERK1 are significantly reduced while ERK2 is unchanged. Scale bar = 40μm.
Table 1.
Phenotypic score of p38α shRNA treated endoderm samples
| High (>10) | Moderate (5–10) | Low (<5) | No 2nd buds | Total | % (no 2nd/total) | |
|---|---|---|---|---|---|---|
| con | 43 | 27 | 14 | 6 | 90 | 6.7 |
| p38sh | 0 | 2 | 18 | 82 | 102 | 80.4 |
Extracellular FGF can regulate p38
While activation of p38 signaling can occur in response to a wide variety of extracellular stimuli including growth factors, it is classically thought of as a pathway mediating stress-related signals. In addition, only limited data exist suggesting that FGFs can activate p38 signaling. To determine whether activation of p38 can occur downstream of FGFs known to signal through the FGFR2IIIb receptor, a mouse lung epithelial cell line (LA-4) was serum-starved and treated with either FGF7 (Fig. 5A) or FGF10 (Fig. 5B). The level of p38 activation was determined by Western analysis using an antibody that specifically recognizes the activated form of the kinase that is dually phosphorylated on the threonine and tyrosine residues within its activation loop (Fig. 5A–B) (Kang et al., 2006). Both FGF7 and FGF10 stimulation activated p38 in cultured mouse lung epithelial cells within 5 minutes. These results suggest that p38 signaling can be activated downstream of FGF-FGFR2IIIb in lung epithelial cells.
Fig. 5. Extracellular FGF can regulate p38 phosphorylation and p38α expression.
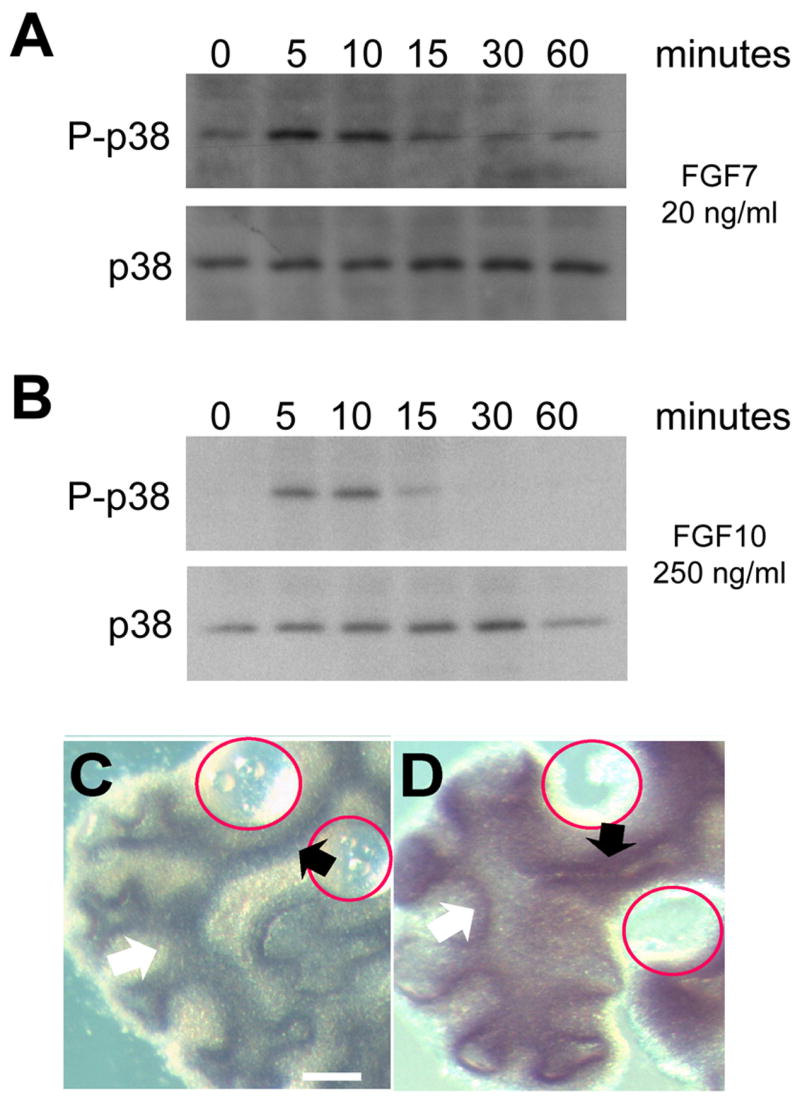
(A–B) FGF7 and FGF10 activate p38 in the LA-4 lung epithelial cell line. Serum-starved LA-4 cells were treated with either FGF7 (20 ng/ml) or FGF10 (250ng/ml) for 0, 5, 10, 15, 30 and 60 minutes. Lysate were analyzed by Western blotting for TGY phosphorylated p38 (P-p38) and total p38 (p38). (C–D) FGF10 induce ectopic p38α expression in the intact lung. BSA (C) or FGF 10 (D) saturated beads were implanted in the mesoderm of the bronchial region, where normally only low level of p38α is expressed. After 48 hours, p38α expression was assayed by whole-mount in situ hybridization. Red circles indicate the position of the implanted beads. Compare the p38α levels in the regions close to the beads (black arrows) with regions away from the beads (white arrows).
Enhanced p38α expression in the distal endoderm of developing lung (Fig. 1) and in secondary buds of isolated distal endoderm (Fig. 3), also suggests that p38α expression might be regulated by FGF-FGF2IIIb signaling. To evaluate this possibility, beads saturated with either BSA (Fig. 5C) or FGF10 (Fig. 5D) were engrafted in the mesoderm adjacent to the proximal bronchial region of isolated whole embryonic lung where only low levels of p38α transcript are normally expressed. After 48 hours, an increase in p38α expression was seen by whole-mount in situ hybridization in the regions adjacent to the FGF10, but not BSA soaked beads (Fig. 5D and 5C, respectively). Detection of p38α expression in the proximal endoderm required longer development of the in situ hybridization. Nonetheless, these results suggest p38α expression can be induced by FGF-FGFR2IIIb initiated signaling.
p38α is not required for epithelial proliferation and the expression of distal specific markers
To understand how p38α directs budding morphogenesis, the effect of impaired p38α signaling on cellular proliferation of the cultured embryonic lung endoderm was determined since increased proliferation is seen in the distal regions of active branching morphogenesis in vivo (Weaver et al., 2000). Immunohistochemical detection of phospho-histone H3, as a marker of mitosis and cell proliferation, did not detect an apparent difference when p38α signaling was disrupted by either SB203580 or p38α shRNA in the presence of FGF7 (Figs. 6A–F). The mitotic index of vector control versus p38α shRNA treated endoderms was calculated as the fraction of phospho-histone H3 positive nuclei among total number of nuclei, and no significant difference was seen between the control (0.156 = 191/1223) and p38α shRNA (0.174 = 224/1284) treatment. In addition, the proliferating cells were distributed randomly in the endoderm, instead of concentrated within the secondary buds. This distribution pattern is consistent with previous data indicating that proliferation is not an initiating factor in secondary budding (Nogawa et al., 1998). Thus, it is likely that p38α directs budding morphogenesis via a mechanism independent of proliferation.
Fig. 6. Filamentous actin (F-actin) distribution, cell proliferation and distal marker expression in control and p38α-disrupted endoderm.
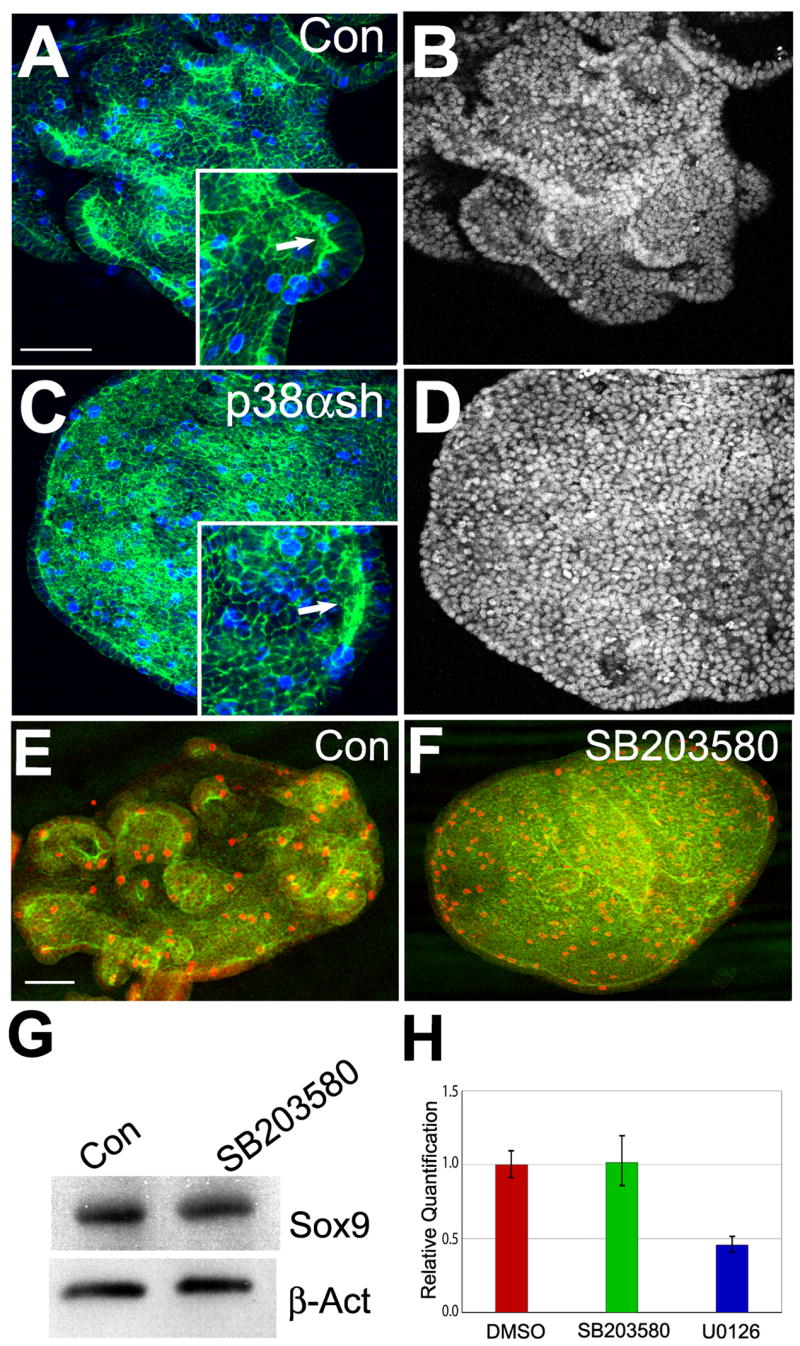
(A–D) F-actin distribution and cell proliferation in control (A and B) and p38α shRNA treated (C and D) endoderm was evaluated by triple labeling with propidium iodide (B and D), Alexa Fluor 488 phalloidin (A and C, green) and antibody to phospho-histone H3 (A and C, blue). Insets show the localized F-actin enrichment. (E and F) Cell proliferation of endoderm grown in the presence of FGF7 and treated with either vehicle (DMSO) or SB203580 (10 μM). Proliferating cells were detected using an anti-phospho-histone H3 antibody (red) and counterstained with Alexa Fluor 488 phalloidin (green). (G) Western analysis of lysates from cultured lung endoderm for Sox9 expression (top panel) following treatment with DMSO (Con) or 10μM SB203580. The protein level of Sox9 in SB203580 treated sample was 90% of control as determined by protein quantification using densitometric scanning. Expression of β-actin (bottom panel) was measured as a loading control. (H) Transcripts levels of Erm in control (red) and SB203580 (green) or U0126 (blue) treated sample. Cultured embryonic lung endoderm was treated with DMSO (Con), p38α/β inhibitor SB203580 (10 μM) or the ERK1/2 inhibitor U0126 (5 μM). Relative Quantification of Erm was determined by two-step qRT-PCR. Data are the mean ± SEM. Scale bar = 40μm.
To determine whether p38α plays a role in regulating the expression of distal endoderm specific genes, two distal markers Sox9 and Erm were analyzed in control versus p38α disrupted endoderm. Western blot analysis suggests that p38α does not regulate the expression of Sox9 as neither SB203580 nor p38α shRNA treatment affects its expression level in response to FGF7 (Fig. 6G and data not shown). Similarly, Erm expression was evaluated by qRT-PCR. Although the development of FGF7 induced secondary buds was impaired in SB203580 treated endoderm, Erm transcript levels remained relatively unaffected (Fig. 6H). Taken together, these results suggest that p38α regulation of budding morphogenesis occurs independently of changes in cell proliferation or expression of certain distal lung endoderm markers.
p38α is required for regulating the E-cadherin protein level in endodermal cells
Other possible mechanisms involved in the initiation and regulation of budding morphogenesis include the modulation of cytoskeletal components and/or cell-cell adhesion molecules. During branching morphogenesis, epithelial cells undergo coordinated and dynamic shape changes, which may involve actin cytoskeleton rearrangement. Alexa 488 labeled phalloidin was used to visualize the distribution of filamentous actin (F-actin) in the FGF7-stimulated isolated embryonic lung endoderm following disruption of p38α signaling. Using this method, F-actin was detected within the cell cortex region in control as well as in p38α shRNA and SB203580 treated lung endoderm. A dramatic difference in the overall intensity of the staining was not seen with the disruption of p38α (Figs. 6A and C). F-actin appeared to be enriched in the budding regions of the control lung endoderm and in certain regions of the p38α shRNA or SB203580 treated endoderm (Figs. 6A and C and data not shown). Whether these regions represent sites of failed secondary buds in the p38α disrupted endoderm is not clear. Nonetheless, these results suggest that p38α can regulate secondary budding through a mechanism other than F-actin rearrangement.
Regulation of cell-cell interaction is an important mechanism in embryonic development. The amount and distribution of cell adhesion molecules, such as E-cadherin, in distal tip cells need to be tightly regulated during branching morphogenesis (Hirai et al, 1989). Recently, a p38α binding protein, p38IP, has been shown to be necessary to control E-cadherin protein levels during mouse gastrulation (Zohn et al., 2006). To determine whether p38α regulates E-cadherin levels during budding morphogenesis of the lung, isolated embryonic lung endoderms cultured in the presence of FGF7 or FGF10 were treated with vehicle (DMSO) or SB203580 to inhibit p38α/β signaling. The levels of E-cadherin protein were compared by Western blot analysis using β-actin as a loading control. SB203580 treatment resulted in a two-fold increase in the amount of E-cadherin protein (Fig. 7A–B). In contrast, treatment with PD98059 or U0126, chemical inhibitors of ERKs, did not affect the level of E-cadherin protein expression in isolated lung endoderm (data not shown).
Fig. 7. Disruption of p38α in cultured endoderm increases E-cadherin protein level.
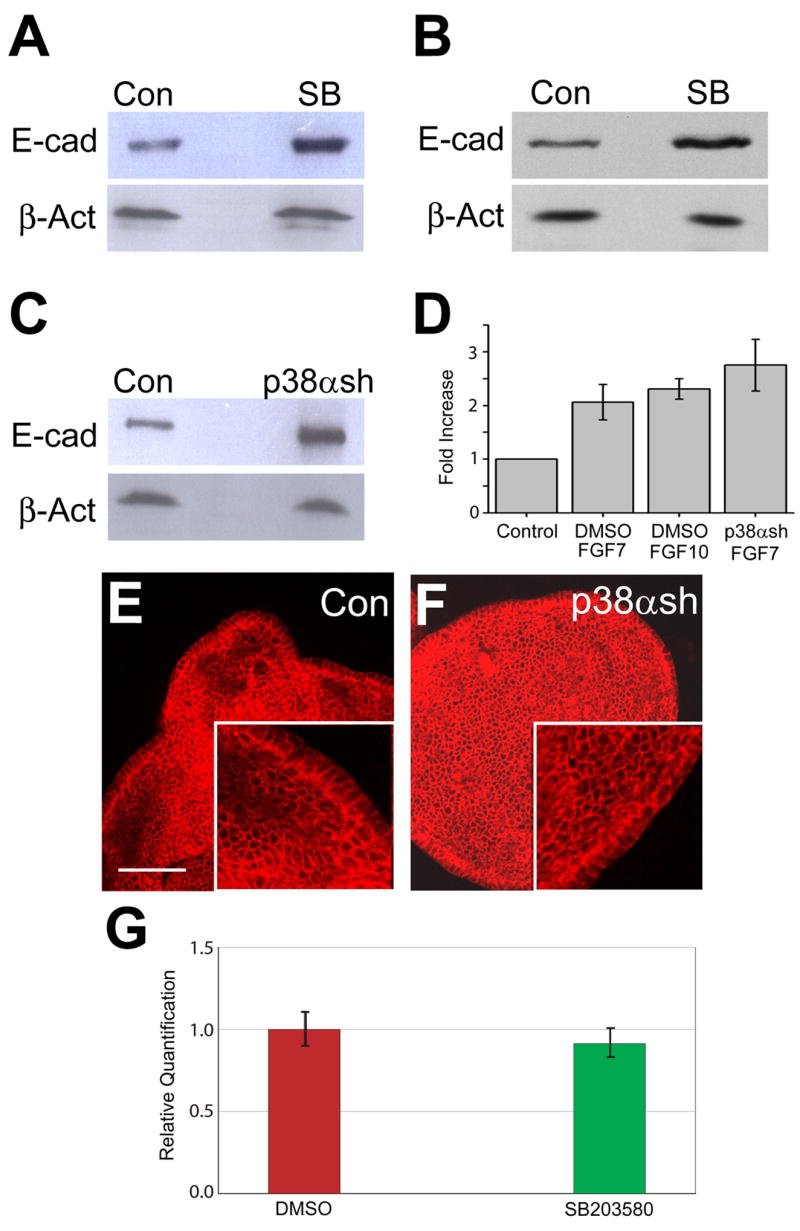
(A) Lysates from embryonic lung endoderm cultured in the presence of FGF7 (30 ng/ml) and treated with DMSO (Con) or 10 μM SB203580 (SB) were immunoblotted for expression of E-cadherin (top panel) and β-actin (bottom panel). (B) Lysates from embryonic lung endoderm cultured in the presence of FGF10 (250 ng/ml) and treated with DMSO (Con) or 10 μM SB203580 (SB) were immunoblotted for expression of E-cadherin (top panel) and β-actin (bottom panel). (C) Lysates from lung endoderm cultured in the presence of FGF7 (30 ng/ml) and transduced using control lentivirus (Con) or p38α shRNA lentivirus (p38αsh) were immunoblotted for expression of E-cadherin (top panel) and β-actin (bottom panel). (D) Graphical representation of Western analyses of E-cadherin protein levels resulting from treatment with SB203580 or p38α shRNA. Quantification was performed by densitometric scanning and normalized to β-actin expression as a loading control. Relative expression is depicted with the expression level of the control group in each case arbitrarily defined as 1.0. Data are the mean ± SEM. Each Western blot image is representative of three independent experiments. (E and F) E-cadherin localization in control (E) and p38α shRNA (F) transduced endoderm was detected by fluorescence confocal microscopy. (G) Transcripts levels of E-cadherin in control and SB203580 treated endoderm cultured in the presence of FGF7 (30 ng/ml). Cultured embryonic lung endoderm from E11.5 mice was treated with DMSO (red) or p38α/β inhibitor SB203580 (green). Relative quantification of E-cadherin was determined by two-step qRT-PCR. Data are the mean ± SEM. Scale bar = 40μm.
To verify the role of p38α in the regulation of E-cadherin expression, embryonic lung endoderm cultured in the presence of FGF7 was transduced with control virus or virus expressing p38α shRNA. Consistent with the chemical inhibitor data, the expression of E-cadherin protein was more than two-fold higher in endodermal cells in which p38α was knockdown by shRNA (Fig. 7C). The overall distribution of E-cadherin, however, did not appear to be affected as immunohistochemical staining suggests that it is localized at epithelial adherens junctions (Figs. 7E–F). In addition, qRT-PCR using RNA from control and SB203580 treated endoderm did not detect a significant difference in E-cadherin transcripts levels (Fig. 7G). Taken together, these results suggest that p38α regulates E-cadherin at a post-transcriptional level, which is consistent with the regulation of E-cadherin by p38α and p38IP during gastrulation (Zohn et al., 2006).
E-cadherin based adherens junctions undergo dynamic assembly and turnover that is tightly regulated in order to allow coordinated cell movement without complete cell dispersion during development (Gumbiner, 2005). Inhibiting the ability of E-cadherin to form intercellular junctions has been shown to disrupt morphogenetic movement and impair embryonic lung branching morphogenesis (Halbleib and Nelson, 2006; Hirai et al., 1989). Our results suggest that altered E-cadherin homeostasis resulting in elevated levels of E-cadherin can also impair budding morphogenesis. To determine whether increased E-cadherin expression is sufficient to inhibit secondary budding, isolated embryonic lung endoderm was transduced with lentivirus expressing E-cadherin containing a C-terminal FLAG-tag. The presence of an epitope tag on the C-terminal end of E-cadherin does not impair its ability to form intercellular junctions (Adams et al., 1998). Endoderm transduced with the vector control lentivirus underwent budding morphogenesis in response to FGF10 (Fig. 8A). On the other hand, budding morphogenesis was severely impaired in isolated endoderm transduced with FLAG-tagged E-cadherin (Fig 8A). Ectopic expression of E-cadherin was verified by Western analysis (Fig. 8B). This finding suggests that alterations in normal E-cadherin homeostasis may be sufficient to impair embryonic lung endoderm budding morphogenesis.
Fig. 8. E-cadherin over-expression impairs budding morphogenesis.
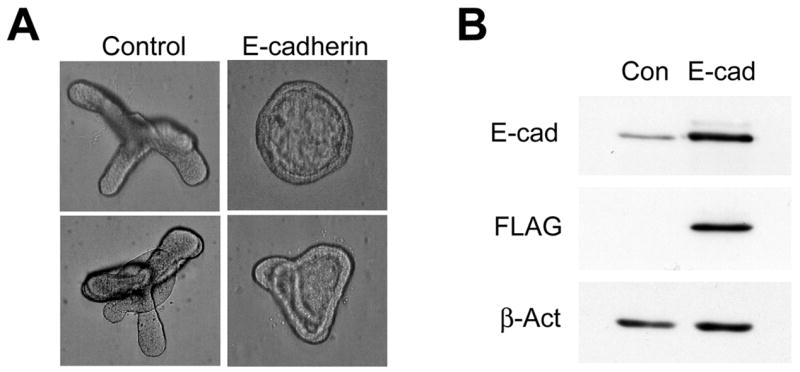
(A) Isolated embryonic lung endoderm was transduced with lentivirus containing either the empty vector (Control) or E-cadherin-FLAG (E-cadherin) in the presence of FGF10 (250 ng/ml) for 48 hours. (B) Lysates were immunoblotted for E-cadherin, the FLAG epitope tag, or β-actin (top, middle and bottom panels, respectively).
DISCUSSION
Branching morphogenesis is a major process during embryonic lung development that lays the foundation for the three-dimensional organization of epithelial-lined airways and alveoli. While several critical extracellular factors, their cell surface receptors and the downstream nuclear transcription factors involved in the regulation of lung branching morphogenesis have been identified, little is actually known about the specific intracellular signaling pathways. This deficit is largely due to the difficulty in applying techniques used to define signaling pathways in cultured cells to model systems of complex organ development. Here, we used a three-dimensional primary endoderm culture coupled with shRNA and chemical inhibition to identify a role for p38α MAPK in budding and branching morphogenesis. This study provides the first evidence that p38 kinase has a specific functional role in epithelial branching morphogenesis during organogenesis.
In our three-dimensional primary mesenchyme-free endodermal lung epithelium culture system, both chemical inhibition of p38α/β signaling as well as targeted knockdown of p38α through RNA interference inhibit the development of secondary budding. Analysis of developing whole embryonic lung using in situ hybridization revealed that p38α is the only p38 isoform specifically expressed in distal endoderm. The relatively restricted expression of p38α to the regions of active budding and branching suggests a role for branching morphogenesis. In contrast, the other p38 isoforms, β, δ, and γ, all have a more ubiquitous expression pattern in developing lung. In addition, while a p38δ mutant mouse has not been reported, the lack of a detectable developmental defect when p38β or p38γ is disrupted in mice supports the idea that these p38 isoforms may not have a specific role in branching morphogenesis (Kuida and Boucher, 2004).
The mechanistic regulation of bud morphogenesis in developing lung endodermal epithelium is not fully understood, but appears to involve the coordination of a wide variety of cellular processes including proliferation, cytoskeletal rearrangement and cell-cell adhesion. In isolated lung endoderm, regulation of bud morphogenesis by p38α dependent signaling does not appear to involve proliferation. Both chemical inhibition and RNAi knockdown of p38α did not alter cellular proliferation as measured by histone H3 phosphorylation. Even though increased rates of proliferation have been detected in the branching regions of whole lung endoderm (Weaver et al., 2000), our finding is consistent with previous data indicating that proliferation, as measured by BrdU staining, is not involved in the initiation of bud formation (Nogawa et al., 1998). The lack of an effect on proliferation may seem surprising since p38 signaling has been implicated in the negative regulation of the cell cycle in several model systems. In the heart, p38 activation appears to inversely correlate with normal embryonic cardiac growth and cardiomyocyte proliferation is increased in cardiac specific p38α null mice (Engel et al., 2005). Postnatal deletion of p38α leads to increased proliferation of lung stem and progenitor cells (Ventura et al., 2007). On the other hand, the effect of p38α on cellular proliferation is not universal and may depend on environmental or developmental context or may be cell type specific. While embryonic p38α inactivation leads to an increased number of SP-C expressing immature type II cells in newborn mouse lung, a difference in lung cell proliferation was not seen with Ki67 and phospho-histone H3 staining (Hui et al., 2007). Similarly, proliferation was increased in only a subset of hematopoietic lineages in the embryonic p38α null mice (Hui et al., 2007).
While impaired p38α signaling inhibited secondary bud formation, it did not appear to prevent the expression of previously identified distal endoderm markers, Sox9 and Erm, in response to FGF7. This finding suggests that cells that normally form secondary buds undergo certain aspects of differentiation independent of physical bud formation. Inhibition of p38α signaling also did not appear to alter actin rearrangement dramatically in FGF7 induced budding. This finding is somewhat surprising since p38 signaling is known to play a significant role in the regulation of actin polymerization through its downstream targets MAPKAP2 and Hsp25/27 (Herlaar and Brown, 1999). It is possible, however, that some subtle changes in actin rearrangement were not detected.
While p38α does not appear to regulate cellular proliferation, differentiation or actin cytoskeletal rearrangement in early bud morphogenesis, it does appear to regulate E-cadherin protein level in developing lung endoderm. Western analysis detected a 2–3 fold increase in E-cadherin protein in isolated endoderm following both chemical inhibition of p38α/β and specific p38α knockdown. Although indirect immunofluoresence staining suggested that inhibition of p38α did not alter the overall distribution of E-cadherin, a clear increase in signal intensity was not seen by this method. The reason for this discrepancy is not clear. It is possible that indirect immunofluoresence did not detect total cellular E-cadherin seen with whole cell lysate Western blotting or other technical issues prevented detection of an overall 2–3 fold difference in expression. On the other hand, inhibition of secondary budding with over-expression of E-cadherin is consistent with the notion that p38α can control budding morphogenesis through regulation of E-cadherin.
The ability of p38α to regulate E-cadherin levels in developing lung endoderm appears to occur at a post-translational level since E-cadherin transcript levels were not affected significantly with chemical inhibition of p38α. This is further supported by our finding that expression of two transcriptional repressors of E-cadherin, Snail and Slug, were not affected by chemical inhibition of p38α as measured by qRT-PCR (Fig. S1A). These findings are consistent with the recent report that p38 signaling is critical for down-regulation of E-cadherin during mouse gastrulation (Zohn et al., 2006). In that study, a p38 interacting protein, p38IP, that appears to interact specifically with the p38α isoform is required for the activation of p38 leading to the post-translational down regulation of E-cadherin expression during the epithelial to mesenchymal transition (EMT) as cells migrate away from the primitive streak during gastrulation (Zohn et al., 2006).
While there is no evidence of complete EMT during branching morphogenesis where cells lose all intercellular adhesion and polarization, a partial EMT process may occur where cell-cell adhesion could be regulated to allow coordinated movement of epithelial cells without complete cell scattering (Gumbiner, 2005). Steady-state regulation of E-cadherin at adherens junctions typically involves transient removal of E-cadherin from the cell membrane through endocytosis and recycling. Signaling involving p38 has been implicated in the regulation of endosome mediated turnover leading to lysosomal degradation. Both activation of the small GTPase Rab5, which facilitates the fusion of early endosomes, and expression of the small GTPase Rab7, which is responsible for the transportation from early endosome to late endosomes and lysosomes, is facilitated by p38 signaling (Bhattacharya et al., 2006). Pharmacological inhibition of p38α/β signaling reduced Rab7 mRNA by 50% compared to vehicle (DMSO) treatment in cultured embryonic lung endoderm (Fig. S1B). This finding suggests that p38 signaling is required for maximal Rab7 expression and lysosomal degradation of E-cadherin. The overall E-cadherin level may regulate the strength of cell-cell adhesion. The ability of ectopic E-cadherin to impair secondary budding supports this concept. Accumulation of E-cadherin from impaired turnover could alter the ability of the developing lung epithelial cells to undergo collective movement to form buds. Conversely, reduced expression of E-cadherin mediated by p38α signaling resulting in fewer adherens junctions could facilitate coordinated epithelial cell movement and bud formation.
Regulation of E-cadherin mediated cell-cell interactions is important for normal development and has been shown to be important for bud morphogenesis. In developing salivary glands, down-regulation of E-cadherin occurs in epithelial cells immediately adjacent to the fibronectin fibrils forming clefts suggesting that E-cadherin mediated cell-cell interactions are replaced with cell-matrix interactions (Sakai et al., 2003). In developing whole embryonic lung explants, antibody interference of E-cadherin mediated cell-cell interactions disrupted normal budding (Hirai et al., 1989). Our finding that p38α signaling affects E-cadherin levels and bud morphogenesis together with these previous findings suggest that tightly controlled E-cadherin homeostasis is important for bud morphogenesis.
While it is an in vitro system, our three-dimensional culture closely mimics certain aspects of in vivo lung development in that the secondary buds in our endodermal culture show the characteristic cell shape change and express all distal differentiation markers examined (Liu et al., 2004). Even though the endoderm produces certain signaling molecules such as Bmp4, our mesoderm-free culture is a simplified system in which the signaling pathways downstream of isolated mesenchyme-derived signaling molecules, such as FGF, can be studied in culture. Reduced terminal bud count in whole lung explants treated with SB203580 suggests that p38α signaling has a role in branching morphogenesis even in the presence of additional mesenchyme derived signals. Recently, an embryo-specific deletion of p38α was reported to result in perinatal lethality with distorted alveolar structures and massive infiltration of hematopoietic cells in the lung (Hui et al., 2007). Whether a defect in branching morphogenesis also exists is not clear since detailed morphometric analysis of the lungs prior to the massive hematopoietic cell infiltration was not performed (personal communication). Targeted deletion in vivo in the developing distal lung endoderm should allow additional characterization of the role of p38α in lung branching morphogenesis.
Branching morphogenesis is a process critical for normal embryonic lung development. In this study, we specifically identify p38α as an important signaling intermediate in the regulation of bud morphogenesis in developing lung. The apparent mechanism involves the regulation of E-cadherin expression and presumably epithelial cell-cell interactions. Given the recent report showing that p38IP regulates gastrulation through p38α and through a post-transcriptional down regulation of E-cadherin (Zohn et al., 2006), our findings suggest a broader role for p38α in the development of epithelial–derived tissues.
Supplementary Material
Cultured embryonic lung endoderm from E11.5 mice was treated with either DMSO or the p38α/β inhibitor SB203580. Relative Quantification (RQ) of Snail and Slug (A) and Rab7 (B) was determined by two-step qRT-PCR. Data are the mean ± SEM.
Acknowledgments
This work was supported by grants from NIH HL07605 (Y. L.) and the Childrens’ Research Foundation (Y. L.). We thank Drs. Brigid Hogan Marsha Rosner for discussing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe MK, Kahle KT, Saelzler MP, Orth K, Dixon JE, Rosner MR. ERK7 is an autoactivated member of the MAPK family. J Biol Chem. 2001;276:21272–9. doi: 10.1074/jbc.M100026200. [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T, Nakamura M, Irie K, Tomoyasu Y, Sano Y, Mori E, Goto S, Ueno N, Nishida Y, Matsumoto K. p38 mitogen-activated protein kinase can be involved in transforming growth factor beta superfamily signal transduction in Drosophila wing morphogenesis. Mol Cell Biol. 1999;19:2322–9. doi: 10.1128/mcb.19.3.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CL, Chen YT, Smith SJ, Nelson WJ. Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J Cell Biol. 1998;142:1105–1119. doi: 10.1083/jcb.142.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124:4867–78. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- Bhattacharya M, Ojha N, Solanki S, Mukhopadhyay CK, Madan R, Patel N, Krishnamurthy G, Kumar S, Basu SK, Mukhopadhyay A. IL-6 and IL-12 specifically regulate the expression of Rab5 and Rab7 via distinct signaling pathways. EMBO J. 2006;25:2878–88. doi: 10.1038/sj.emboj.7601170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso W. Molecular regulation of lung development. Annu Rev Physiol. 2001;63:471–94. doi: 10.1146/annurev.physiol.63.1.471. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest J, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptro 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–92. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald CE, Patel SB, Becker JW, Cameron PM, Zaller D, Pikounis VB, O’Keefe SJ, Scapin G. Structural basis for p38alpha MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat Struct Biol. 2003;10:764–9. doi: 10.1038/nsb949. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- Halbleib JM, Nelson WJ. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006;20:3199–3214. doi: 10.1101/gad.1486806. [DOI] [PubMed] [Google Scholar]
- Herlaar E, Brown Z. p38 MAPK signalling cascades in inflammatory disease. Mol Med Today. 1999;5:439–47. doi: 10.1016/s1357-4310(99)01544-0. [DOI] [PubMed] [Google Scholar]
- Hirai Y, Nose A, Kobayashi S, Takeichi M. Expression and role of E- and P-cadherin adhesion molecules in embryonic histogenesis. I. Lung epithelial morphogenesis. Development. 1989;105:263–270. doi: 10.1242/dev.105.2.263. [DOI] [PubMed] [Google Scholar]
- Hogan BL, Yingling JM. Epithelial/mesenchymal interactions and branching morphogenesis of the lung. Curr Opin Genet Dev. 1998;8:481–6. doi: 10.1016/s0959-437x(98)80121-4. [DOI] [PubMed] [Google Scholar]
- Hui L, Bakiri B, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Komnenovic V, Scheuch H, Beug H, Wagner EF. p38 alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nature Genetics. 2007;39:741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- Izvolsky K, Shoykhet D, Yang Y, Yu Q, Nugent M, Cardoso W. Heparan sulfate-FGF10 interactions during lung morphogenesis. Dev Biol. 2003;258:185–200. doi: 10.1016/s0012-1606(03)00114-3. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Seit-Nebi A, Davis RJ, Han J. Multiple Activation Mechanisms of p38{alpha} Mitogen-activated Protein Kinase. J Biol Chem. 2006;281:26225–26234. doi: 10.1074/jbc.M606800200. [DOI] [PubMed] [Google Scholar]
- Keren A, Bengal E, Frank D. p38 MAP kinase regulates the expression of XMyf5 and affects distinct myogenic programs during Xenopus development. Dev Biol. 2005;288:73–86. doi: 10.1016/j.ydbio.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Kling DE, Lorenzo HK, Trbovich AM, Kinane TB, Donahoe PK, Schnitzer JJ. MEK-1/2 inhibition reduces branching morphogenesis and causes mesenchymal cell apoptosis in fetal rat lungs. Am J Physiol Lung Cell Mol Physiol. 2002;282:L370–378. doi: 10.1152/ajplung.00200.2001. [DOI] [PubMed] [Google Scholar]
- Kuida K, Boucher DM. Functions of MAP kinases: insights from gene-targeting studies. J Biochem (Tokyo) 2004;135:653–6. doi: 10.1093/jb/mvh078. [DOI] [PubMed] [Google Scholar]
- Liu Y, Hogan BL. Differential gene expression in the distal tip endoderm of the embryonic mouse lung. Mech Dev, Gene Expression Paterns. 2002;2:229–233. doi: 10.1016/s1567-133x(02)00057-1. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jiang H, Crawford HC, Hogan B. Role for ETS domain transcription factors Pea3/Erm in mouse lung development. Dev Biol. 2003;261:10–24. doi: 10.1016/s0012-1606(03)00359-2. [DOI] [PubMed] [Google Scholar]
- Liu Y, Stein E, Oliver T, Li Y, Brunken WJ, Koch M, Tessier-Lavigne M, Hogan BL. Novel role for Netrins in regulating epithelial behavior during lung branching morphogenesis. Curr Biol. 2004;14:897–905. doi: 10.1016/j.cub.2004.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Yamamoto T, Tanoue T, Yuasa Y, Chisaka O, Nishida E. Requirement of the MAP kinase signaling pathways for mouse preimplantation development. Development. 2005;132:1773–83. doi: 10.1242/dev.01729. [DOI] [PubMed] [Google Scholar]
- Nanmoku K, Kawano M, Iwasaki Y, Ikenaka K. Highly efficient gene transduction into the brain using high-titer retroviral vectors. Dev Neurosci. 2003;25:152–61. doi: 10.1159/000072264. [DOI] [PubMed] [Google Scholar]
- Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–60. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- Nogawa H, Ito T. Branching morphogenesis of embryonic mouse lung epithelium in mesenchyme-free culture. Development. 1995;121:1015–1022. doi: 10.1242/dev.121.4.1015. [DOI] [PubMed] [Google Scholar]
- Nogawa H, Morita KWVC. Bud formation precedes the appearance of differential cell proliferation during branching morphogenesis of mouse lung epithelium in vitro. Dev Dyn. 1998;213:228–35. doi: 10.1002/(SICI)1097-0177(199810)213:2<228::AID-AJA8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Ornitz D, Xu J, Colvin J, McEwen D, MacArthur C, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–7. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Perl AT, Wert S, Nagy A, Lobe CG, Whitsett J. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A. 2002;99:10482–87. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L. Targeted expression of a dominant negative FGF receptor blocks branching morphogenesis and epithelial differentiation of the mouse lung. Embo J. 1994;13:3296–301. doi: 10.1002/j.1460-2075.1994.tb06631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham HM, Arganaraz ER, Groschel B, Trono D, Lama J. Lentiviral vectors interfering with virus-induced CD4 down-modulation potently block human immunodeficiency virus type 1 replication in primary lymphocytes. J Virol. 2004;78:13072–13081. doi: 10.1128/JVI.78.23.13072-13081.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Larsen M, Yamada KM. Fibronectin requirement in branching morphogenesis. Nature. 2003;423:876–81. doi: 10.1038/nature01712. [DOI] [PubMed] [Google Scholar]
- Stoner GD, Kikkawa Y, Kniazeff AJ, Miyai K, Wagner RM. Clonal isolation of epithelial cells from mouse lung adenoma. Cancer Res. 1975;35:2177–2185. [PubMed] [Google Scholar]
- Tamura K, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Requirement for p38alpha in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102:221–31. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M, Pasparakis M, Nebreda AR. p38 alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nature Genetics. 2007;39:750–758. doi: 10.1038/ng2037. [DOI] [PubMed] [Google Scholar]
- Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV. The molecular basis of lung morphogenesis. Mech Dev. 2000;92:55–81. doi: 10.1016/s0925-4773(99)00325-1. [DOI] [PubMed] [Google Scholar]
- Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development. 2000;127:2695–704. doi: 10.1242/dev.127.12.2695. [DOI] [PubMed] [Google Scholar]
- Weaver M, Yingling JM, Dunn NR, Bellusci S, Hogan BL. Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development. 1999;126:4005–15. doi: 10.1242/dev.126.18.4005. [DOI] [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–61. doi: 10.1128/JVI.77.16.8957-8961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolkowicz R, Nolan GP, Curran MA. Lentiviral vectors for the delivery of DNA into mammalian cells. Methods Mol Biol. 2004;246:391–411. doi: 10.1385/1-59259-650-9:391. [DOI] [PubMed] [Google Scholar]
- Zohn IE, Li Y, Skolnik EY, Anderson KV, Han J, Niswander L. p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell. 2006;125:957–69. doi: 10.1016/j.cell.2006.03.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cultured embryonic lung endoderm from E11.5 mice was treated with either DMSO or the p38α/β inhibitor SB203580. Relative Quantification (RQ) of Snail and Slug (A) and Rab7 (B) was determined by two-step qRT-PCR. Data are the mean ± SEM.