

Abstract
SUMMARY
G protein-coupled receptor (GPCR) instability represents one of the most profound obstacles to the structural study of GPCRs that bind diffusible ligands. The introduction of targeted mutations at non-conserved residues that lie proximal to helix interfaces has the potential to enhance the fold stability of the receptor helix bundle while maintaining wild-type receptor function. To test this hypothesis, we studied the effect of amino acid substitutions at Glu-1223.41 in the well studied β2-adrenergic receptor (β2AR), which was predicted from sequence conservation to lie at a position equivalent to the tryptophan interface between transmembrane domains (TMs) 3, 4, and 5. The data indicate that the replacement of Glu-1223.41 with bulky hydrophobic residues, such as tryptophan, tyrosine and phenylalanine increase the yield of functionally folded β2AR by as much as 5-fold. Receptor stability in detergent solution was studied by isothermal denaturation and it was found that the E122W and E122Y mutations enhanced the β2AR thermal half-life by 9.3- and 6.7-fold, respectively at 37 °C. The β1AR was also stabilized by the introduction of tryptophan at Glu-1473.41, and the effect on protein behavior was similar to the rescue of the unstable wild-type receptor by the antagonist propranolol. Molecular modeling of the E122W and E122Y mutants using a previously published β2AR homology model revealed that the tryptophan ring edge and tyrosine hydroxyl are positioned proximal to the helical break in TM5 introduced by the conserved Pro-2115.50, and may stabilize the helix by interacting favorably with the unpaired carbonyl oxygen of Val-2065.45. Conformational flexibility of TM5 is believed to be a general property of rhodopsin-like GPCRs and, therefore, engineering of the TM4-3-5 interface at the 3.41 position may provide a general strategy for the stabilization of other receptors.
Keywords: GPCR, Stability, Membrane, Adrenergic Receptor, Mutagenesis
INTRODUCTION
Detailed structural analysis of the G protein-coupled receptor (GPCR) rhodopsin together with the exhaustive biochemical and biophysical characterization of other receptors suggests that GPCRs share a common seven transmembrane domain (7TM) architecture consisting of a conserved structural core formed by the first four TMs (TM1-TM4) and a dynamic activation domain encompassing TM5-TM7.1,2 The prevailing “global toggle switch” model of GPCR activation holds that the key step in receptor activation is the rotameric isomerization of Trp6.48, which leads to a shift in the conformation of TM6 at the Pro6.50 kink, followed by movements of TM6 and TM7 toward the receptor center.3 Although the prevailing evidence supports the notion that GPCRs share a common activation mechanism, high-resolution structural insight is currently available only for the photoreceptor rhodopsin.
The inactive state structure of rhodopsin revealed that 11-cis-retinal is covalently anchored to TM7 and likely acts as an inverse agonist by restraining movements of the TM helices and through interaction with Trp-2656.48.4 The β-ionone ring of 11-cis-retinal also interacts intimately with Phe-2125.47 on TM5, which has been suggested to serve as a mechanistic “lock” for Trp-2656.48 in the active state conformation.5 Biochemical and spectroscopic studies of rhodopsin have suggested that the initial step in receptor activation involves the translation of the isomerization of 11-cis-retinal to all-trans-retinal and translation of the β-ionone ring toward the interface between TM3 and TM5, which may disrupt the interaction of TM5 with the receptor core.1,6,7 Although TM5 may serve as a key structural and functional bridge between the TM1-4 core with TM6 and TM7, it has the lowest packing value and is stabilized by only one conserved hydrogen-bonding interaction between Glu-1223.37 and the backbone carbonyl of His-2115.46 (Figure 1(a)).6,8 The interaction between Glu-1223.37 and His-2115.46 that mediates TM3-TM5 packing is pair-wise conserved only in the rod cell rhodopsins, and is absent in other GPCRs 5. EPR and cross-linking experiments indicate that while the intracellular half of TM5 remains relatively immobile during activation the hydrogen bonding patterns of Glu-1223.37 and His-2115.46 and a third residue, Trp-1263.41, change significantly, presumably due to a shift in the conformation of TM5 at the helical bulge introduced by the conserved Pro-2155.50.9
Figure 1.
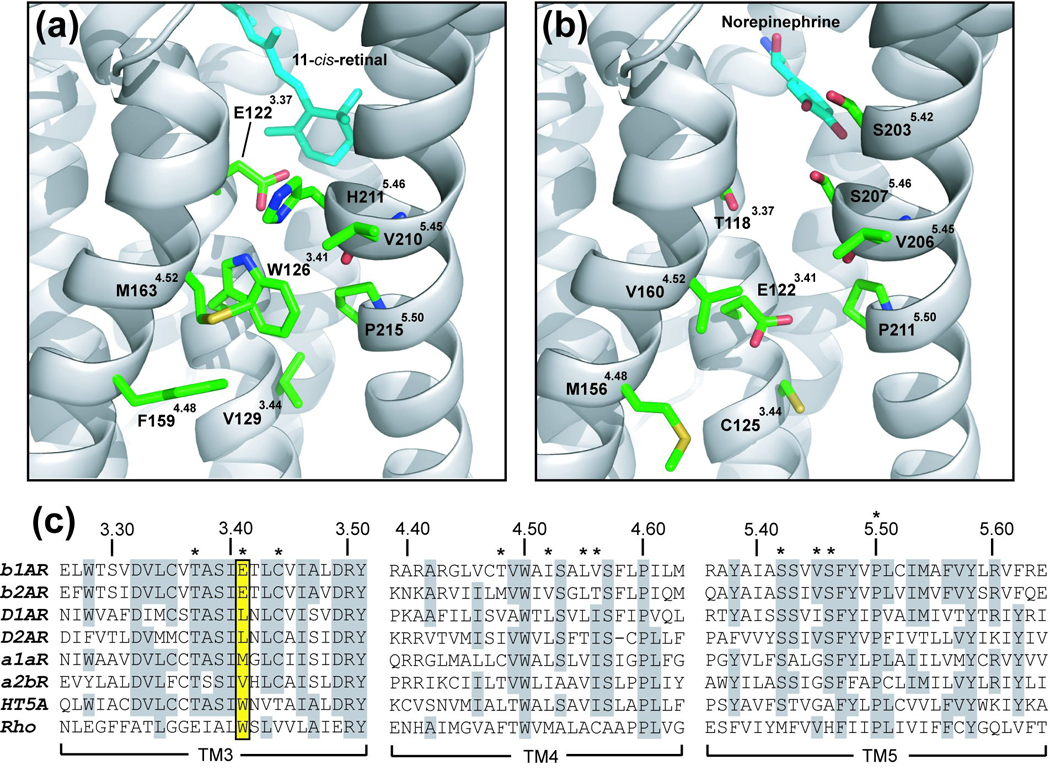
Structural models of the TM4-3-5 interface. (a) Rhodopsin inactive state structure (PDB ID code 1U19) showing residues proximal to Trp-1263.41 5. Residues are numbered according to their gene-specific position in the protein sequence and by a superscript indicating the general indexed position based on the Ballesteros and Weinstein system 38. TM helices are colored grey and side-chains carbon atoms are colored green. (b) Previously published rhodopsin-based homology model of the β2AR displaying residues predicted to be structurally equivalent to the model in (a). (c) Clustal W sequence alignment illustrating the residue conservation in TM3, TM4, and TM5 for the βARs, rhodopsin and several members of the biogenic amine family 35. Identical residues are highlighted in grey. Key residues mentioned in the text are marked with asterisks. Position 3.41 (Ballesteros and Weinstein System) is highlighted in yellow and includes E1223.41 in the β2AR and in W1263.41 in rhodopsin.
Trp-1263.41 is the only residue in TM3 that makes simultaneous contact with both TM4 and TM5, and therefore, may have a profound effect on the conformational dynamics and stability of the receptor. While the Glu-1223.37 and His-2115.46 pair is conserved only in 32% of opsins, Trp-1263.41 is conserved in 86% of opsin receptors, suggesting that like the other buried tryptophan residues in GPCRs, it may serve a critical structural or functional role.10 In the rhodopsin crystal structure, Trp-1263.41 is positioned adjacent to the conserved Pro-2155.50 breakpoint in TM5, and hydrogen bonds with Glu-1223.41.11 Pro-2155.50 breaks the hydrogen bonding pattern within the helix main chain and results in the exposure of the Val-2105.45 main chain carbonyl, which due to the resulting helical bulge points toward the electron deficient edge of Trp-1263.41. In fact the Trp-1263.41 ring edge is only 3.4 Å from the carbonyl oxygen and could therefore participate in a stabilizing CH…O hydrogen bond.12,13 Proximity of the electron rich ring face to the Pro-2155.50 side chain may additionally stabilize the conformation of the helical bulge in TM5 through a CH…π hydrogen-bonding interaction with the aromatic π-cloud.14,15
Although it is suggestive that Trp-1263.41 might serve to regulate the conformational dynamics of TM5, we were surprised to find that only eight (0.5%) of 1739 non-rhodopsin receptor sequences we analyzed share a tryptophan in an equivalent position. Of those, five were found to be representatives of the 5-HT5 subfamily, members of medically important amine family GPCRs. Because the 5-HT5 receptors have evolved to tolerate a tryptophan residue at the TM4-3-5 interface and are closely related to the adrenergic subfamily, we chose to investigate the effect of hydrophobic substitutions on receptor stability using the best characterized non-rhodopsin GPCR, the β-adrenergic receptor (βAR). In marked contrast to the generally hydrophobic consensus at the 3.41 position (92% in class A, excluding rhodopsin), all three βAR subtypes share an invariant Glu3.41.16 This radical divergence from the consensus provided us with a unique opportunity to study the effect of hydrophobic substitutions on receptor stability without a pre-evolved hydrophobic bias. In addition, the wild-type β2AR is known to be conformationally unstable when removed from the membrane, a property that may be related in part to the dynamics of TM5.17,18 Supporting this, the helical stability of the extra-cellular segment of TM5 above Pro5.50 was found to vary markedly between the β2AR and the closely related dopamine D2 receptor, as indicated by the different results obtained with cysteine accessibility studies. In one such study, when the dopamine D2 receptor was labeled by a polar, thiol-reactive probe, only residues facing the expected binding pocket on the inner face of TM5 are labeled.19 By contrast, in the β2AR, nearly all positions around the helix in the corresponding part of TM5 are labeled with a polar cysteine reactive probe suggesting that the structure of the extracellular end of TM5 is more flexible or dynamic.20 The Pro5.50 induced break in TM5 could therefore be envisioned to introduce a high degree of flexibility in the loosely packed TM5, leading to conformational dynamics that lead to constitutive activity and receptor instability.
Using the β2AR system, we set out to study the effect of hydrophobic substitutions at Glu-1223.41 on receptor expression and thermal stability. Using site-directed mutagenesis, we constructed a set of Glu-1223.41 substitution mutants spanning the natural 3.41 residue diversity. Expression, analytical SEC, and binding studies were done to assess the effect of mutations on functional receptor folding, while isothermal denaturation of purified mutants was used to compare their stability. We found that tryptophan and tyrosine substitutions at the β2AR Glu-1223.41 significantly improved protein folding and enhanced the thermal stability of the receptor in detergent solution. Molecular modeling of the mutants revealed that the tryptophan ring and tyrosine hydroxyl are positioned proximal to the proline induced bulge in TM5 and may stabilize the receptor by attenuating conformational dynamics.
RESULTS
Flow Cytometric Analysis of Receptor Expression
To address the effect of mutations at Glu-1223.41 (E122) on expression, Sf-9 cells infected with baculovirus encoding GFP fusions of the wild-type β2AR and E122 mutants were analyzed for total and cell-surface expression using a Guava™ capillary flow-cytometer which can simultaneously detect multiple fluorescence wavelengths.21 In previous studies with the β2AR where GFP was fused C terminally, receptor trafficking, pharmacology, and effector coupling were found to be similar to wild-type.22
Mean GFP fluorescence intensity from the infected cell population was used as a measure of total receptor expression, and this analysis revealed that most of the E122 mutants expressed at levels moderately higher (within 2-fold) than wild-type. β2AR cell surface expression was measured by monitoring the binding of an Alexa Fluor 647 R-PE labeled antibody directed against the N terminus. Upon analysis of the cell surface fluorescence, we found that all E122 mutants were expressed more highly than wild-type β2AR on the cell surface (Figure 2). E122 substitutions with tyrosine, tryptophan, and leucine resulted in approximately a 3.5-fold increase in cell surface expression.
Figure 2.

Flow cytometric analysis of β2AR E122 mutant expression. Sf-9 cells expressing wild-type β2AR-GFP or E122 mutants were stained with a red fluorescent R-PE conjugated FLAG antibody and were analyzed by flow cytometry as described in “Materials and Methods”. Cell surface (grey bars) and total expression (checkered bars) was monitored for at least 2500 individual cells by simultaneous measurement of the fluorescence emission in the red and green channels, respectively. Receptor-GFP expressing cells were gated and analyzed independently from uninfected or non-expressing cells, and the total expression level calculated as the mean GFP fluorescence intensity of the expressing population. The relative cell-surface expression was calculated by dividing the mean red channel of the gated population by the total expression. The data are expressed as multiples of wild-type β2AR (WT = 1) and represent the average ± S.E. of three independent experiments done in triplicate.
Analytical SEC Analysis
Membranes from Sf-9 cells expressing β2AR mutants were solubilized in 1% DDM and characterized by analytical SEC with fluorescence detection. From the chromatograms, it is clear that the majority of receptor elutes as a major single peak termed here “monomer” (Figure 3(a)). Gaussian fitting was used to calculate the area under the monomer fraction (Figure 3(b)) and values were divided by the mean GFP intensity determined previously by flow cytometry (Figure 2). The ratio between extracted monomer peak area and total expression is termed the “relative monomer yield” (Figure 3(c)). Because the amount of monomer species is adjusted for expression, it allows a comparison to be made directly between mutants. When normalized to wild-type receptor, (WT = 1) the amount of monomer extracted by DDM varies over a wide range.
Figure 3.

F-SEC analysis of β2AR E122 mutants extracted by DDM. (a) DDM cellular extracts were resolved by analytical SEC and the soluble β2AR-GFP peaks were detected by inline detection of GFP fluorescence. The GFP fluorescence intensity is plotted as a function of chromatographic retention time (s). Representative chromatograms from a single experiment of the indicated mutants were overlaid for comparison and the monomer was labeled “M” for reference. (b) Chromatograms were deconvoluted into separate peaks by fitting to an exponentially modified Gaussian function (EMG). (c) Areas were computed for the “monomer” peak and were divided by each mutant’s total protein expression (flow cytometry) to arrive at the “relative monomer yield”. Data are the average ± S.E. of three independent experiments done in triplicate and shown as multiples of wild-type (WT=1).
Binding Properties of β2AR E122 Mutants
Functional receptor densities of selected mutants from the set were quantified by saturation binding of [3H] DHA to membrane preparations from baculovirus infected cultures expressing E122 substitution mutants (Figure 4(a)). Bmax and Kd values were computed by non-linear regression analysis (Figure 4(b)). When Bmax values were divided the receptor expression level (Figure 4(c), checkered bars), it was found that the trend in the expression of functional receptors (Figure 4(d)) was closely correlated with the trend in cell surface expression (Figure 4(c), gray bars). When the expression normalized Bmax values were compared, E122Y, E122W, and E122L demonstrated more than a four-fold increase in the yield of functional receptors per unit expression indicating that a substantially greater fraction of receptor molecules were folding properly relative to wild-type β2AR. While the number of functional receptors was affected significantly by the E122 mutations, the affinities for the antagonist [3H] DHA was only slightly affected (Figure 3(b)). Kd values for E122W, E122Y and E122L displayed the greatest loss in affinity (2-fold loss) relative to wild-type perhaps reflecting a slight shift in receptor conformation.
Figure 4.

β2AR E122 mutant saturation binding analysis. (a) Representative curves from the [3H] DHA saturation binding analysis performed on Sf-9 cell membrane homogenates expressing a subset of β2AR-GFP E122 mutants as described under “Materials and Methods”. (b) Bmax and Kd values from saturation binding were determined by non-linear regression analysis and are shown in units of pmol/mg protein and nM, respectively. Data are the average ± S.E. from three independent experiments performed in triplicate. (c) The cell surface expression (grey bars) and total cellular expression (checkered bars) of receptor-GFP fusion constructs was determined by flow-cytometry immediately prior to cell harvest and membrane preparation. The data are shown as multiples of wild-type where WT = 1. (d) Relative Bmax values were calculated by dividing the Bmax values in B by the total receptor expression (mean GFP fluorescence intensity) as determined in (c).
Biochemical Characterization of the β2AR E122W
To ensure that the E122W mutation did not dramatically disrupt normal receptor biochemistry, binding constants for the classical full agonist Isoproterenol and the specific inverse agonist ICI 118,551 were determined. In addition, the signaling response to agonist was investigated using a cAMP accumulation assay in HEK293T cells, which measures activation of adenylyl cyclase through the effector Gs. The relative binding affinities for the agonist Isoproterenol and the inverse agonist ICI 118,551 were determined by competition with the radio-labeled antagonist [3H]DHA. The binding constants (Ki) for Isoproterenol were found to be 323.8 ± 26.3 nM and 622.3 ± 62.30 nM for wild-type and E122W, respectively (Figure 5(a)). The canonical inverse agonist ICI 118,551 also bound to the E122W mutant with a two-fold reduction in affinity relative to wild-type. The Ki values determined for ICI 118,551 were 1.01 ± 0.11 nM and 1.52 ± 0.09 nM for the wild-type and E122W β2AR, respectively (Figure 5(b)). The affinity shifts relative to wild-type were consistent with the values determined by saturation for the antagonist [3H] DHA, and therefore suggests that aberrant ligand selectivity has not been introduced. To ensure that the E122W mutation did not impair receptor signaling, Isoproterenol stimulated cAMP production was measured in HEK293T cells transiently transfected with either the wild-type or mutant β2AR (Figure 5(c)). From the dose-response curves, EC50 values for wild-type β2AR and E122W were found to be 40.6 ± 1.1 nM and 43.1 ± 1.2 nM, respectively.
Figure 5.

Pharmacological characterization of WT and E122W β2AR. Competition binding studies were performed with 1 nM [3H] DHA as described under “Materials and Methods.” (a) Competition of the full agonist Isoproterenol for [3H] DHA binding sites on membranes expressing either the wild-type or E122W β2AR-GFP. (b) Competition of the inverse agonist ICI 118,551 for [3H]DHA binding sites on membranes expressing either the wild-type or E122W β2AR. Data was fitted using a non-linear algorithm in GraphPad Prism, and binding constants (Ki) were determined from the IC50 values using the Chang-Prusoff equation. The Ki values for Isoproterenol were 323.8 ± 26.3 nM and 622.3 ± 62.30 nM for wild-type and E122W, respectively, and 1.01 ± 0.11 nM and 1.52 ± 0.09 nM for the wild-type and E122W β2AR, respectively. (c) Transfected HEK293 cells were assayed for signaling activity by measuring cAMP accumulation in response to the agonist Isoproterenol. From the dose-response curves, EC50 values for β2AR-WT and E122W were found to be 40.6 ± 1.1 nM and 43.1 ± 1.2 nM, respectively. All data are the average ± S.E. of three independent experiments done in triplicate.
Expression of the β2AR E122W in HEK293T Cells and Confocal Microscopy
Using flow cytometry, both the surface and total expression of the GFP-tagged wild-type β2AR and β2AR E122W mutant were assessed in transiently transfected HEK293T cells (Figure 6(a)). Similar to the effect observed in the insect cell system, the surface expression of the E122W was substantially higher (~2-fold) than wild-type while the total expression was virtually unchanged (data not shown). Confocal microscopy of live HEK293T cells transiently transfected with either wild-type β2AR (Figure 6(b)) or E122W (Figure 6(c)) revealed that a large percentage of the receptor population remained in the internal membrane network, while distinct plasma membrane expression was visible only for the E122W mutant.
Figure 6.

Cell surface expression and confocal microscopy of transfected HEK293T cells expressing WT and E122W β2AR -GFP. (a) HEK293T cells expressing wild-type β2AR-GFP or E122W were analyzed by flow cytometry as described in “Materials and Methods”. Cell surface expression (gray bars) was monitored for at least 2500 individual cells. Data are the average ± S.E. of three independent experiments done in triplicate and are shown as multiples of wild-type (WT=1). HEK293T cells were transfected with equal amounts of plasmid DNA containing either wild-type or E122W β2AR-GFP and visualized by laser-scanning confocal microscopy with detection of GFP fluorescence. “N” denotes the cell nucleus and “PM” denotes the plasma membrane. (b) Wild-type β2AR-GFP showing primarily intracellular receptor expression with little receptor visible in the plasma membrane. (c) E122W mutant expressing cells with receptor clearly visible (see arrow) in the plasma membrane.
Effect of E122 Mutations on β2AR Thermal Stability
To study the effects of Glu-1223.41 mutations on β2AR stability, mutants were purified from Sf-9 cells, subjected to isothermal denaturation in 0.1% dodecyl maltoside, bound to the novel Alexa Fluor 532-labeled alprenolol derivative (Falp) and then analyzed by F-SEC to detect the residual to active receptor (see Methods). From initial analytical F-SEC studies, it was determined that more than 90% of the functional DDM extracted receptor migrates as a single peak corresponding to the monomer fraction (data not shown). Therefore, prior to the thermal denaturation studies, the monomer peak areas for all mutants were determined from the F-SEC chromatograms (GFP fluorescence signal) and were used to normalize the concentration of functional receptor for different mutants. A serial dilution series was used to test for linearity of the decrease in binding to ensure the concentration of the active receptor was at least ten-fold below the threshold of ligand depletion (data not shown). Individual receptor aliquots were heated at 37 °C at timed intervals and then cooled to 4 °C and incubated with a saturating concentration (1 µM) of Falp until equilibrium was achieved. Receptor-ligand complexes were then separated from free ligand by SEC under conditions where the separation and detection was complete in 5 min, thus minimizing the time spent in non-equilibrium conditions. Bound Falp was detected by the fluorescence signal at 560 nm and peak areas were calculated by Gaussian fitting. The total integrated fluorescence area at each time point was used to construct a denaturation curve from which the receptor half-life could be estimated from non-linear fitting of the data to a first order exponential decay function (Figure 7). From the decay curves, the wild-type β2AR was found to decay with a half-life of 3.0 ± 0.1 min, while E122W and E122Y decayed more slowly with half-lives of 27.8 ± 1.7 min and 20.0 ± 1.2 min, respectively. E122F, however failed to improve the thermal stability much beyond that of wild-type, increasing the decay half-life to 4.7 ± 0.2 min.
Figure 7.

Thermal Stability of wild-type (WT) and E122W β2AR in DDM. GFP-tagged receptors were purified, subjected to thermal denaturation in 1% dodecyl maltoside at 37 °C, and then analyzed by F-SEC to detect bound Falp. The synthesis of Alexa Fluor 532 labeled alprenolol (Falp) is described in detail in Methods. Chromatographic areas were quantified by Gaussian function fitting (see Methods) and used to determine the amount of functional receptor remaining at each time point. The data were fit to a single exponential decay function by non-linear regression analysis and half-lives (t1/2) were calculated and used for comparison of receptor stability. Wild-type β2AR decays with a t1/2 of 3.0 ± 0.1 min, while E122W and E122Y decay with a substantially increased t1/2 of 27.8 ± 1.7 min and t1/2 of 20.0 ± 1.2 min, respectively. The t1/2 for E122F is marginally greater than WT at 4.7 ± 0.2 min indicating a slightly slower decay rate. All data are the average ± S.E. of three independent experiments done in triplicate.
Propranolol and the Trp3.41 mutation enhance β1AR and β2AR stability
Sequence alignment of all three beta adrenergic subtypes revealed conservation of glutamate at the 3.41 position suggesting a conserved structural or functional role for the residue. We therefore sought to compare the effect of the E147W mutation (equivalent to E122 in the β2AR) on the folding of the β1AR by comparing its impact on the recovery of stable, monomeric protein. In addition, we sought to understand the biological effect of receptor stabilization by comparing the effect of the mutations to the “pharmacochaperone” action of the hydrophobic non-specific βAR antagonist propranolol 23. Propranolol is known to be one of the most highly lipophilic general βAR antagonists and is believed to cross the plasma membrane by passive diffusion where it can bind to receptors in the internal membrane compartments of the cell.24 Wild-type and mutant β1AR or β2AR receptor-GFP fusions were expressed in Sf-9 cells in the absence of antagonist for 24 hr to allow protein expression to commence. Cultures were then split into equal parts and saturating propranolol (10 µM) was added to one of the duplicates. At 48 hr post infection, cells were harvested solubilized in DDM and analyzed by analytical F-SEC. Receptor peak areas were quantified by Gaussian fitting as described in the methods. Propranolol increased the monomer fraction of the wild-type β2AR (Figure 8(a)) and the wild-type β1AR (Figure 8(b)) by 1.44 ± 0.10- and 3.40 ± 0.20-fold, respectively. By contrast, neither mutant was significantly affected. Although propranolol clearly improved the fraction of folded (monomeric) receptor by SEC, this was not evident from the cell surface or total expression data which failed to reveal any substantial (<10%) upregulation of receptor expression or trafficking to the cell surface (data not shown). Monomer peak areas with and without propranolol (Figure 8(a) and (b)) addition were normalized to wild-type, (WT = 1) and the ratio between each mutant receptor and its respective wild-type was calculated and the results shown Figure 8(c). The E147W mutation in the β1AR resulted in a 3.6 ± 0.2-fold increase in soluble monomer relative to wild-type β1AR, which was similar in magnitude to the 4.3 ± 0.3-fold increase in monomer observed for β2AR E122W.
Figure 8.

Rescue of βAR folding by antagonist and Glu3.41 mutation. The effect of propranolol on the yield of solubilized receptor in DDM was determined by co-expression of wild-type or mutant receptors in the presence or absence of propranolol followed by analytical F-SEC. All samples were pre-incubated with 10 µM propranolol prior to extraction to stabilize remaining functional receptor. (a) Representative F-SEC traces for wild-type β2AR and E122W in the presence (+) or absence (−) of propranolol during expression. (b) Parallel experiment with wild-type or E147W β1AR. (c) The histogram displays the yield of DDM solubilized receptor monomer per unit expression (see “Materials and Methods”) relative to the respective wild-type (WT=1) construct expressed in the absence of propranolol. Monomer yields were determined after expression in the presence (dark grey bars) or absence (light grey bars) of 10 µM propranolol. Propranolol increased the monomer fraction of the wild-type β2AR and the wild-type β1AR by 1.44 ± 0.10- and 3.40 ± 0.20-fold, respectively. By contrast, neither mutant was significantly affected. The E147W mutation in the β1AR resulted in a 3.6 ± 0.2-fold increase in soluble monomer relative to wild-type β1AR, which was similar to the 4.3 ± 0.3-fold increase in monomer observed for β2AR E122W.
DISCUSSION
One of the major stumbling blocks in membrane protein structural biology is the need to isolate milligram quantities of pure, well folded receptor that remains stable in the detergent solutions typically used for their purification and crystallization. This issue is particularly vexing for the GPCR superfamily, whose members are notoriously unstable in detergent solution. Intense efforts to overexpress and structurally characterize GPCRs have been met with only a modicum of success. In fact, the only representative member of the family whose structure has been determined is bovine rhodopsin, which is both highly abundant in its natural source and exceedingly stable in detergent solution due to its covalently bound inverse agonist 11-cis-retinal. We believe GPCR instability represents one of the most profound obstacles to the structural study of GPCRs that bind diffusible ligands, and have thus attempted to optimized the stability of the β2AR through the targeted introduction of single point mutations that stabilize the receptor against detergent induced denaturation, while maintaining near wild-type receptor function. A similar strategy was applied effectively for the stabilization of Escherichia coli diacylglycerol kinase, where fairly conservative single mutations in the TM domain resulted in dramatic increases in thermal stability and resistance to detergent induced denaturation.25,26
In this report we have identified a candidate site on TM3 in the β2AR (position 3.41 by the Ballesteros and Weinstein numbering scheme) that was predicted from sequence conservation and homology modeling to lie at the interface between TM3, TM4, and TM5. The βARs contain what would appear to be a highly unfavorable glutamate at this position, which we mutated to a variety of other residues based on class A sequence diversity. A number of hydrophobic substitutions were found to enhance functional receptor expression and stability upon solubilization in DDM. The current study focuses on the characterization of these mutant receptors and provides insight into a potential mechanism for their stabilizing effects. Herein, we present evidence that the thermal stability of a non-rhodopsin GPCR can be significantly enhanced through engineering of the TM domain, while maintaining wild-type function. Additionally, the stability enhancement was found to beneficially affect the properties of the receptor when removed from the membrane by detergent, allowing functionally and structurally stable receptor to be purified in the absence of added ligand. We conclude that engineering of the TM4-3-5 interface may provide a general strategy for the functional stabilization of amine family GPCRs.
Hydrophobic substitutions at Glu1223.41 enhance functional folding of the β2AR
We found that mutations of the β2AR-E1223.41 increased the relative surface expression up to a level of more than 3.5 times wild type (Figure 2). In addition, Bmax values were found to track the increase in surface expression, suggesting that the increase in surface expression is due to a beneficial effect of the mutations on functional folding (Figure 4(d)). When membranes were extracted with DDM and receptors analyzed by SEC, we found that the yield of “monomer” receptor per unit expression reflected the relative increase in functional binding sites, indicating that the active receptor existed in a single oligomeric state. It is important to note that the increases in binding sites and detergent extractable protein reported are adjusted for total expression of the receptor using the GFP signal quantified by flow-cytometry. Therefore, the results indicate that bulky residues such as tryptophan, tyrosine and leucine did not simply increase the expression of protein, but rather imparted a fundamental improvement either in the folding or extraction behavior. This distinction allows us to separate a shift in the properties of the molecule from over-expression that might result from a simple improvement in translation efficiency or mRNA stability.
The improvement in the yield of functionally folded receptor appeared to correlate with both the hydrophobicity and surface area of the residues, with the notable exception of isoleucine, which by our interpretation should have expressed at a level roughly equivalent to leucine. Valine also failed to fit the trend, as the substitution resulted in functional expression roughly equivalent to wild-type. Considering the isoleucine and valine results together, we hypothesized that in β2AR, a β-branched amino acid may not be tolerated due to steric incompatibility. Supporting this idea, our analysis of 1472 aligned GPCR sequences at position 3.41 found that while phenylalanine, leucine and tyrosine were the top three represented residues at 37%, 26%, 10%, of genes, respectively, isoleucine and valine are found much less frequently at 1.3% and 2.9% of the set. While the expression results were generally correlated with the trend in natural diversity, one residue was found to be a clear outlier. Tryptophan, although found at the 3.41 position in only 0.5% of analyzed non-rhodopsin GPCR receptors, was surprisingly well tolerated at the 3.41 position, resulting in both high cell-surface expression and extracted relative monomer yield (Figure 4(c)). We found this to be surprising as buried tryptophan residues are rare in membrane proteins and if present are often involved in critical functional or structural roles. In fact, the biological hydropathy scale devised by Hessa et al. predicts that tryptophan and tyrosine would be the least energetically favored of the hydrophobics at the center of the bilayer, while phenylalanine and isoleucine would be significantly more favored.10,27 Therefore, we hypothesized that tryptophan and tyrosine may be most energetically favored at the TM4-3-5 interface due to their ability to participate in both hydrophobic and polar interactions with proximal residues.
E122W stabilizes the βAR helical bundle
Results of the expression and analytical SEC experiments suggest that tyrosine and tryptophan increase the yield of functionally folded β2AR and stabilize the TM4-3-5 interface more so than other hydrophobic residues. Stability could be envisioned to arise from different phenomena. First, it is possible that aromatics such tyrosine and tryptophan interact favorably with membrane components or with detergent and, thus, could enhance the membrane insertion efficiency, or the DDM extraction efficiency rather than stabilizing the fold itself. Alternatively, tryptophan and tyrosine could interact more favorably with TM3, TM4 and TM5, resulting in a stabilization of the fold. To separate the two possible scenarios, we performed a “rescue” experiment where the antagonist propranolol was added as a pharmacological chaperone. Physically destabilized mutants of the β2AR have been rescued by the addition of antagonists and the degree of recovery was found to correlate with the degree of instability of the mutants both in the membrane and in purified form.28,29 The βAR antagonist binding site is believed to be composed of residues on TM3, TM5 and TM6 that face the interior of the receptor and, therefore, it can be imagined that any observed increase in functional receptor yield would result specifically from stabilization of the TM bundle during folding.30 The β2AR homology model indicates that the 3.41 position faces the membrane exterior and is remote from the antagonist binding site. Therefore, it might be expected that the stabilizing effects of the mutations at 3.41 and the antagonist chaperone would be independent. In fact, we observed an enhancement in folding for both wild-type βARs upon addition of antagonist, while neither mutant receptor was significantly affected. This supports the notion that wild-type GPCRs are inherently unstable and, therefore, prone to misfolding, a property that has been invoked to explain their constitutive activity.9 The enhancement of β1AR folding in response to propranolol or the E147W mutation resulted in similar increases in β1AR protein yields, which suggests that the mutation and the ligand both act to rescue the folding of an inherently unstable receptor. Propranolol however, failed to increase the yield of wild-type β2AR receptor to the same levels observed for E122W, which suggests that the mutation may enhance folding though a parallel but separate mechanism.
E122W and E122Y stabilize the Pro-2115.50 breakpoint in TM5
The results of the thermal stability experiments demonstrate a dramatic stabilization of the folded receptor relative to wild-type for E122W and E122Y, but not for E122F. The E122W provided the greatest structural stabilization of the receptor as suggested by the nine-fold enhancement of thermal half-life over the wild-type β2AR. Comparison of the E122F and E122Y denaturation curves revealed that E122Y also reduced the receptor denaturation rate by more than six-fold relative to wild-type. Surprisingly, the E122F mutant was only marginally more stable (1.6-fold) than wild-type, and substantially less stable than the E122Y mutant, suggesting that the tyrosine hydroxyl was contributing the bulk stabilizing affect.
To understand the origin of the stabilizing effect observed for E122W and E122Y, we modeled the lowest energy conformation of each mutant on a previously published structural homology model of β2AR and compared the models to the E122F mutant that only marginally enhanced receptor stability relative to wild-type (Figure 9(a–c)).31 The Glu-122 side-chain was systematically mutated to tryptophan, tyrosine, or phenylalanine in Pymol and the resultant models were energy minimized in CNS.32 During minimization, side chain conformations were allowed to vary while all backbone atoms were fixed to prevent relaxation of the secondary structure. The lowest energy models were chosen for analysis and the TM4-3-5 interface of each model is shown in Figure 9(a–c). Energy minimization placed the tryptophan ring (Figure 9(a)) in a conformation that points the indole nitrogen toward the bulge in TM5 with the ring face sandwiched between Val-1605.45 and Pro-2115.50. In addition, the tryptophan’s electron deficient ring edge points toward the unpaired Val-206 carbonyl and could be envisioned to interact with the TM5 main-chain, thus helping to stabilize the bulge in TM5 initiated by Pro-2115.50. The electron rich face of the tryptophan ring is positioned proximal to the ring of Pro-2115.50. Proline residues at helix termini are often closely associated with aromatic residues that are believed to stabilize secondary structure through CH…π interactions with the aromatic π-cloud.33,34 It is possible that the break in TM5 initiated by Pro-2115.50 is stabilized by such a mechanism. Similar side-chain conformations are observed in the minimized models of the β2AR E122Y (Figure 9(b)) and E122F (Figure 9(c)) mutants. Although both rings form contacts with Val-160, neither ring is positioned to accept a CH…π bond from Pro-2115.50. The most obvious difference between the tyrosine and phenylalanine mutants lies in the interaction with the Val-2065.45 carbonyl. The tyrosine hydroxyl is positioned proximal to the unpaired Val-2065.45 carbonyl and may form a strong hydrogen bond, thus stabilizing the bulge in TM5 which may explain the four-fold difference in the thermal half-life of these mutants. In contrast, the phenylalanine ring lacks a strong hydrogen bond donor, and therefore may only weakly interact with TM5. The nature of the interactions revealed through modeling supports the differences in thermal stability between the mutants and suggests that stabilization of local unwinding of TM5 at Pro-2115.50 is responsible for the observed effect.34 Flexibility of the TM5 helix due to the Pro-2115.50 break can be envisioned to result in conformational dynamics that include rotation or tilting of the N-terminal half of TM5 (Figure 9(d)). Interaction of the tryptophan ring edge with the Val-2065.45 carbonyl and its face with Pro-2115.50 may stabilize the dynamics of TM5 at the breakpoint, and thus help to stabilize receptor secondary and tertiary structure.
Figure 9.
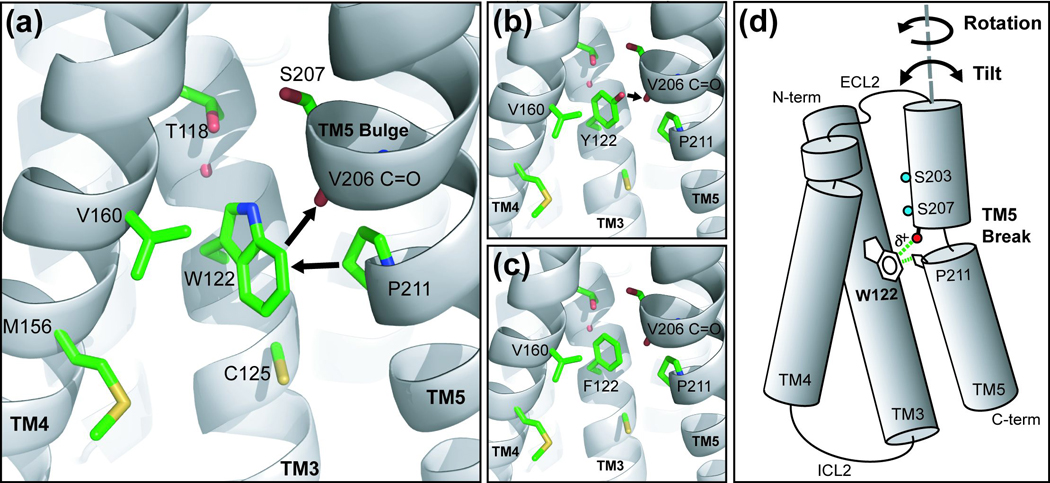
Molecular modeling of β2AR mutants and proposed TM5 movements. A previously published homology model of the human β2AR built on the structure of (inactive) bovine rhodopsin (PDB code 1F88) was used to compare structurally equivalent residues. Mutations were introduced into the β2AR structural model and amino acid side-chain bond parameters were refined by energy minimization in CNS. Side-chains are colored green and backbone atoms are represented as a gray cartoon. (a) β2AR E122W model showing the tryptophan ring proximal to Pro-211, and the carbonyl of V206. (b) β2AR E122Y model showing the tyrosine hydroxyl proximal to the carbonyl oxygen of V206. (c) β2AR E122F model. (d) Cartoon of possible TM5 dynamics and the proposed interactions with Trp-122. Structural flexibility of the TM5 helix due to the Pro-211 break can be envisioned to result in conformational dynamics that include rotation or tilting (black arrows) of the N-terminal half of TM5. Interaction of the polarized (indicated by δ+) Trp ring with the V206 carbonyl and Pro-211 could stabilize the dynamics of TM5 at the breakpoint, and thus may help to stabilize receptor secondary and tertiary structure.
Although numerous attempts have undoubtedly been made to identify thermally stable mutants of GPCRs amenable to structural study, we identified only two published reports of such mutants in the literature. One of these was a mutant of rhodopsin that contained a rationally-designed disulfide bridge between the third extracellular loop and N-terminus that dramatically enhanced thermal stability and lead to its successful crystallization for the first time from a heterologous source.5,28 The other example was the introduction of mutations at the predicted interface of TM3 and TM6 in the cannabinoid receptor (CB1R) that enhanced trafficking and thermal stability of the receptor, but dramatically altered its biochemistry.35 For the first time, we present evidence that the thermal stability of a non-rhodopsin GPCR can be significantly enhanced through engineering of the TM domain, while maintaining wild-type function. Additionally, the stability enhancement was found to beneficially affect the properties of the receptor when removed from the membrane by detergent, thus allowing high levels of functionally and structurally stable receptor to be purified in the absence of added ligand. Homology modeling revealed that the polarized tryptophan ring edge is positioned to interact with the Val-2065.45 and Pro-2115.50 and may stabilize the receptor by reducing the conformational dynamics of TM5.24 Similarly, the tyrosine hydroxyl may stabilize the bulge in TM5 by interacting with the unpaired carbonyl at the helix breakpoint. Comparison of the recently published β2AR high resolution crystal structure with the rhodopsin-based homology model used in this study revealed that the local structure predicted around Glu-1223.41 was remarkably close to that seen in the crystal structure.36,37 The newly published β2AR crystal structure will undoubtedly serve as an improved foundation for the generation of homology models for other class A receptors.
Herein, we also provide evidence that mutation of the 3.41 position in the β1AR results in the enhancement of receptor folding, and a dramatic shift in its oligomeric state (Figure 8(b)). The Trp-1223.41 substitution when translated to the β1AR (E147W) had a dramatic positive effect on its folding and stability, despite the variation in amino acids surrounding Glu-1473.41 compared to the β2AR. The rationale for the effectiveness of mutating this area may therefore be extrapolated to other members of the amine family GPCRs and is currently being pursued as a path to obtaining highly stable samples of a multitude of receptors for structural studies. We conclude that engineering of the TM4-3-5 interface provides a strategy for the functional stabilization of adrenergic receptors, and potentially other class A GPCRs.
MATERIALS AND METHODS
Synthesis of AlexaFluor532-Alprenolol (Falp)
Alprenolol-cysteamine (AlpC) was synthesized by radical addition of cysteamine to the allyl double bond of (±)-alprenolol (Sigma-Aldrich, St. Louis, MO) to form a thioether linkage. Solid cysteamine (Sigma) (8 mmol) was added with vigorous stirring to a solution of alprenolol HCl (Sigma) (1 mmol) dissolved in anhydrous degassed methanol under argon. Solid AIBN (Sigma) (0.1 mmol) was added and the mixture was refluxed under argon at 65 °C for 6 hr with a second equal addition of fresh AIBN after 3 h. AlpC was purified by reverse-phase HPLC and the product verified by MALDI-TOF mass spectroscopy.
The fluorescent β2AR antagonist (Falp) was next synthesized by coupling the free amine of AlpC to the reactive dye Alexa Fluor 532 succinimidyl ester (Invitrogen, Carlsbad, CA). Ten milligrams of AlpC (1g/ml in DMF) was added with vigorous stirring to 1 mg Alexa Fluor 532 succinimidyl ester pre-dissolved in 1 ml anhydrous DMF (Sigma) and maintained under a stream of dry nitrogen gas. A catalytic amount of N,N-diisopropylethylamine (Sigma) was added to the solution and the reaction was allowed to progress to completion (3 hr at 25 °C) as judged by reverse-phase TLC. Falp was purified from crude reaction mixture compound by reverse-phase HPLC by using a 20–50% acetonitrile gradient and monitoring the absorbance at 532 nm to detect the Alexa Fuor dye. The mass of pure Falp was verified by MALDI-TOF mass-spectrometry.
Construct Design and Mutagenesis
The coding sequence of the human β2AR in pFastBac with the third glycosylation site removed and a cleavable N terminal hemaglutinin secretion signal followed by a FLAG tag was provided by Brian Kobilka (Stanford University, Stanford, CA). PCR-based site directed mutagenesis (QuickChange II, Stratagene, CA) was used to insert an XbaI site at the C terminus of the β2AR in pFB. The coding sequence of the non-dimerizing GFP variant from Aequorea coerulescens (AcGFP1), was amplified by PCR from pIRES (Clontech, Mountain View, CA) with primers containing flanking XbaI restriction sites, restriction digested, and then ligated into XbaI-cut pFB-β2AR to create pFB-β2AR-GFP. The β2AR-GFP fragment was then subcloned by PCR and ligated into the XhoI/EagI sites of the baculovirus transfer vector pOrbigen (Orbigen, San Diego, CA). Alternatively, an 8xHis tag was fused directly to the β2AR C-terminus by PCR and cloned into the same sites in pOrbigen. For mammalian expression, β2AR-GFP was subcloned from pFB-β2AR-GFP by PCR and ligated into the XhoI/HindIII sites of pCDNA 3.1(-) (Invitrogen) to create pCDNA-β2AR-GFP. All amino acid substitution mutants were introduced by site-directed PCR mutagenesis using the QuickChange II kit and protocol and verified by DNA sequencing.
Sequence Alignment and Molecular Modeling
To facilitate the comparison of aligned residues in different GPCRs, we used the indexing method introduced by Ballesteros and Weinstein, where a residue in a particular transmembrane segment (TM) is assigned a general position indexed relative to the most conserved residue in the TM in which it is located.38 The general Ballesteros and Weinstein index position is included as a superscript to the sequence-specific residue number (e.g. Pro-2115.50 in TM5 of the β2AR). All sequence alignments were performed using Clustal W.35 The high-resolution structural model of (inactive) bovine rhodopsin (PDB code 1U19) was explored using the Pymol graphics suite (DeLano Scientific, Palo Alto, CA). A previously published and extensively validated homology model of the human β2AR built on the structure of (inactive) bovine rhodopsin (PDB code 1F88) was used to compare structurally equivalent residues.5,24 Mutations were introduced into the structural models in Pymol and conformations were manually chosen to minimize steric clashes. Amino acid side-chain conformations were further refined by energy minimization in the Crystallographic and NMR System (CNS) 32. Side-chains were positioned by chemical-based conformational sampling coupled to the usage of energy restraints derived from the force field available in the CNS. The resulting models were refined with conjugate gradient minimization with no experimental energy terms used. All backbone and Cα atoms were fixed, while all amino acid side-chain atoms were subjected to 1000 cycles of energy minimization with a continuous dielectric constant of three.
Cell Culture and Protein Expression
Sf-9 cells (Invitrogen) were seeded in suspension flasks at 1.0 × 106 cells/ml and grown at 27 °C for 24 hr with constant agitation at 115 rpm. When the cell density reached 2.0 × 106 cells/ml, (24 hr) cultures were infected with high-titer recombinant baculovirus stock at a multiplicity of infection (MOI) of three. 48–72 hr after infection, cells were harvested by centrifugation at 500 × g for 10 min and stored at −80 °C until use. HEK293T cells (Invitrogen) were cultured in Dulbecco’s modified Eagles medium (DMEM, Invitrogen) supplemented with 10 % (v/v) fetal calf serum (FCS) in a humidified incubator at 8 % (v/v) CO2 in air at 37 °C.
Flow Cytometry
Cell surface and total expression of receptors was quantified by flow cytometry using the Guava™ flow cytometer.21 Cells were stained with a Zenon™ AlexaFluor-647 R-Phycoerythrin (R-PE; Invitrogen) conjugate of the FLAG-M2 antibody (Sigma) as recommended by the manufacturer. The cultures were then assayed for fluorescence using a Guava EasyCyte microcapillary flow cytometer (Guava Technologies, Hayward, CA), utilizing laser excitation at 488 nm. GFP fluorescence and cell surface expression were analyzed simultaneously using the green and red channels, respectively. Only cells that that fluoresced above the uninfected control background were counted as expressing and were used to compute the mean fluorescence intensity (MFI). For each assay point, 2500 cellular events were collected to compute the MFI.
Radioligand Saturation Binding
Cell pellets were suspended in ice-cold binding buffer (HSG: 25 mM HEPES, 150 mM NaCl, 10% glycerol, pH 7.5), containing protease inhibitors (Complete protease inhibitor cocktail tablet, Roche Applied Science, Indianapolis, IN) and homogenized for 30 strokes with a Dounce homogenizer. Crude membranes and organelles were isolated from cytoplasmic proteins by centrifugation of the supernatants at 150,000 × g for 60 min at 4 °C. The membrane pellets were resuspended in HSG buffer and the homogenization step repeated. Resuspended membranes expressing the β2AR and truncation mutants were tested for binding with Levo-[Ring, Propyl-3H(N)]-Dihydroalprenolol Hydrochloride [3H]DHA, (81 Ci/mmol, Perkin Elmer Life Sciences, Waltham, MA). Cellular homogenates (2.5 µg of total protein per reaction) were incubated for 60 min at room temperature with serial dilutions of the radioligand (0.15–8 nM). Incubations were rapidly terminated by filtration using a Tomtec Mach III cell harvester (Tomtec, Hamden, CT) through a 96-well GF/B filter plate (MultiScreen Harvest plate, Millipore Corp., Billerica, MA), and rinsed five times with 500 µl of ice-cold wash buffer (50 mM Tris-HCl, pH 7.4). The harvest plates were dried, and 30 µl of OptiPhase “HiSafe” III scintillation liquid (Perkin Elmer Life Science) were added. The bound radioactivity was measured using a Packard's TopCounter NTX (Packard Bioscience, Meriden CT). Non-specific binding was determined in parallel reactions in the presence of an excess of alprenolol and specific binding was defined as the difference between total and non-specific binding at each point. Protein concentrations were determined with the BCA protein assay (Pierce, Rockford, IL), using bovine serum albumin as a reference. All incubations were performed in triplicates, and independent experiments were repeated five times. Equilibrium dissociation constants (Kd) and maximal receptor levels (Bmax) were calculated from the results of saturation experiments with non-linear fitting using GraphPad Prism Software.
Analytical SEC Analysis
Sf-9 whole cells or membrane fractions expressing β2AR-GFP receptor fusion proteins mutants were solubilized in HS buffer (HS: 25 mM HEPES, 150 mM NaCl, pH 7.5) containing protease inhibitors (Roche Mini Tablets). A solution of 10% n-dodecyl-β-d-maltoside (DDM; Anatrace, Maumee, OH) was added to the resuspended membranes (1% final concentration) and the mixture was agitated for 60 min at 4 °C. Crude extracts were clarified by centrifuging at 21,000 × g for 60 min and supernatants transferred to fresh tubes. Solubilized proteins were separated by analytical size-exclusion chromatography employing a Sepax™ nanofilm SEC-250 column (Sepax Technologies, Newark, DE) connected to a Dionex™ Ultimate 3000 HPLC with in-line fluorescence detection (Dionex, Bannockburn, IL). Prior to separation, DDM extracts were diluted to 0.05% DDM with HS buffer and 5 µL injected onto the column at 0.3 ml/min into a column pre-equilibrated in HS buffer containing 0.05% DDM. Receptor peaks were detected by monitoring the GFP fluorescence intensity at 505 nm (475 nm excitation). Raw chromatogram data was imported to PeakFit software (Systat Software, San Jose, CA). Initial peak detection and fitting were done by the residual method using an exponentially modified Gaussian (EMG) convolution function, and the functions were further fitted to the original peaks by using a least squares minimization algorithm. The resulting Gaussian peaks were validated by the r2 coefficient of the determinant, the degree of freedom-adjusted coefficient of determination, the fit standard error, and the F value. The area of the component peaks were computed for each sample in triplicate and the mean area used to compute the “relative monomer yield” which equals the peak area divided by the total expression defined by the mean GFP fluorescence intensity determined by flow cytometry.
Measurement of Cyclic AMP (cAMP) Accumulation in HEK293T cells
Adherent HEK293T cells were plated at a density of 40,000–60,000 cells/well and incubated overnight at 37 °C with 8% CO2. Cells were transfected with pcDNA 3.1(-) vectors containing β2AR-GFP fusion constructs using Insect Gene Juice (EMD Chemicals Inc., San Diego, CA) in 96-well plates. Cells were washed with Krebs-Ringer bicarbonate buffer and then stimulated with serial dilutions of Isoproterenol (100 µM-0.15 nM) for 20 min at 37 °C and reactions were terminated by the addition of lysis buffer. cAMP concentrations were determined by competitive ELISA using the CatchPoint assay (Molecular Devices, Sunnyvale, CA) in 384-well plates using pure cAMP as a standard. EC50 values were obtained by non-linear fitting to a one-site sigmoidal model using nonlinear regression analysis in GraphPad Prism.
Fluorescence Microscopy
HEK293T cells were transiently transfected with β2AR-GFP fusions with Gene Juice (Novagen) as outlined in the manufacturer’s protocol. Cell imaging was performed at 37 °C using a Zeiss Radiance 2100 Rainbow laser scanning microscope (Carl Zeiss, Thornwood, NY) equipped with a controlled live cell chamber (Pathology Devices, Westminster, MD). Cells were magnified with either 40×/1.3 or a 60×/1.4 numerical aperture oil immersion lens. GFP was excited with 488 nm argon/krypton laser and the emitted fluorescence was detected at 505 nM through a spectral band pass filter. Images were collected with BioRad LaserSharp 2000 software (BioRad Hercules, CA).
Measurement of Receptor Thermal Stability
For purification of the wild-type and the E122 β2AR-GFP receptor mutants, Sf-9 cells were grown in 500 ml cultures at a density of 2 × 106 cells/ml, infected with high titer virus stock at an MOI of 3 and harvested after 48 hr. Sf-9 crude membrane fractions were solubilized in HS buffer + protease inhibitors containing (Roche Mini Tablets) 1% (w/v) DDM for 60 min at 4 °C with constant agitation. Crude extracts were clarified by centrifuging at 100,000 × g for 60 min and supernatants transferred to fresh tubes. Protein was bound in batch to 1 ml FLAG-M2 immunoaffinity resin (Sigma) for 2 hr at 4 °C and washed in batch with 200 ml (4 × 50 ml) HS + 0.05% DDM. Purified receptor was eluted with 2 ml HS buffer + 0.5% DDM with 250 µg/ml FLAG peptide (Sigma) for 30 min. Stability of the FLAG purified apo-receptor was determined by incubating purified receptors at 37 °C for defined intervals (16 time points per hour) in HS buffer + 0.5% DDM and then cooled to 4 °C. 1 µM Falp was added to all samples and incubated for 120 min at 4°C. Samples were then analyzed in triplicate by F-SEC as described above. Bound Falp was detected by the fluorescence signal at 560 nm (532 nm excitation). Total receptor-GFP fusion was detected by monitoring the fluorescence signal at 505 nm. The non-specific binding of Falp and GFP signal overlap were determined in the presence of 100 µM timolol (Sigma). Chromatograms were analyzed by Gaussian fitting to determine peak areas as described above.
ACKNOWLEDGEMENTS
We thank Veli-Pekka Jaakola, Ellen Chien, Alexander Alexandrov, Peter Kuhn, Joe Ng, Mark Griffith, Jeff Velasquez, and Leila Davis for thoughtful scientific discussions, and Angela Walker for assistance with manuscript preparation. These studies were supported in part by the NIH Roadmap grant P50 GM073197.
Abbreviations used
- GPCR
G protein-coupled receptor
- βAR
β-adrenergic receptor
- TM
transmembrane domain
- AlpC
alprenolol-cysteamine
- MOI
multiplicity of infection
- [3H]DHA
Levo-[Ring, Propyl-3H(N)]-Dihydroalprenolol Hydrochloride
- DDM
n-dodecyl-β-d-maltoside
- EMG
exponentially modified Gaussian
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chelikani P, Hornak V, Eilers M, Reeves PJ, Smith SO, RajBhandary UL, Khorana HG. Role of group-conserved residues in the helical core of beta2-adrenergic receptor. Proc Natl Acad Sci U S A. 2007;104:7027–7032. doi: 10.1073/pnas.0702024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation--a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- 3.Elling CE, Frimurer TM, Gerlach LO, Jorgensen R, Holst B, Schwartz TW. Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J Biol Chem. 2006;281:17337–17346. doi: 10.1074/jbc.M512510200. [DOI] [PubMed] [Google Scholar]
- 4.Crocker E, Eilers M, Ahuja S, Hornak V, Hirshfeld A, Sheves M, Smith SO. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J Mol Biol. 2006;357:163–172. doi: 10.1016/j.jmb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 5.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 6.Beck M, Sakmar TP, Siebert F. Spectroscopic evidence for interaction between transmembrane helices 3 and 5 in rhodopsin. Biochemistry. 1998;37:7630–7639. doi: 10.1021/bi9801560. [DOI] [PubMed] [Google Scholar]
- 7.Patel AB, Crocker E, Eilers M, Hirshfeld A, Sheves M, Smith SO. Coupling of retinal isomerization to the activation of rhodopsin. Proc Natl Acad Sci U S A. 2004;101:10048–10053. doi: 10.1073/pnas.0402848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W, Eilers M, Patel AB, Smith SO. Helix packing moments reveal diversity and conservation in membrane protein structure. J Mol Biol. 2004;337:713–729. doi: 10.1016/j.jmb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Manikandan K, Ramakumar S. The occurrence of C--H…O hydrogen bonds in alpha-helices and helix termini in globular proteins. Proteins. 2004;56:768–781. doi: 10.1002/prot.20152. [DOI] [PubMed] [Google Scholar]
- 10.Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfeld R. C-H…pi-interactions in proteins. J Mol Biol. 2001;307:357–377. doi: 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- 11.Gether U, Ballesteros JA, Seifert R, Sanders-Bush E, Weinstein H, Kobilka BK. Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. J Biol Chem. 1997;272:2587–2590. doi: 10.1074/jbc.272.5.2587. [DOI] [PubMed] [Google Scholar]
- 12.Javitch JA, Fu D, Chen J. Residues in the fifth membrane-spanning segment of the dopamine D2 receptor exposed in the binding-site crevice. Biochemistry. 1995;34:16433–16439. doi: 10.1021/bi00050a026. [DOI] [PubMed] [Google Scholar]
- 13.Javitch JA, Shi L, Liapakis G. Use of the substituted cysteine accessibility method to study the structure and function of G protein-coupled receptors. Methods Enzymol. 2002;343:137–156. doi: 10.1016/s0076-6879(02)43131-x. [DOI] [PubMed] [Google Scholar]
- 14.Chaipatikul V, Erickson-Herbrandson LJ, Loh HH, Law PY. Rescuing the traffic-deficient mutants of rat mu-opioid receptors with hydrophobic ligands. Mol Pharmacol. 2003;64:32–41. doi: 10.1124/mol.64.1.32. [DOI] [PubMed] [Google Scholar]
- 15.Wuller S, Wiesner B, Loffler A, Furkert J, Krause G, Hermosilla R, Schaefer M, Schulein R, Rosenthal W, Oksche A. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279:47254–47263. doi: 10.1074/jbc.M408154200. [DOI] [PubMed] [Google Scholar]
- 16.Van Durme J, Horn F, Costagliola S, Vriend G, Vassart G. GRIS: glycoprotein-hormone receptor information system. Mol Endocrinol. 2006;20:2247–2255. doi: 10.1210/me.2006-0020. [DOI] [PubMed] [Google Scholar]
- 17.Krishna G, Chen K, Lin C, Nomeir AA. Permeability of lipophilic compounds in drug discovery using in-vitro human absorption model, Caco-2. Int J Pharm. 2001;222:77–89. doi: 10.1016/s0378-5173(01)00698-6. [DOI] [PubMed] [Google Scholar]
- 18.Cao X, Yu LX, Barbaciru C, Landowski CP, Shin HC, Gibbs S, Miller HA, Amidon GL, Sun D. Permeability dominates in vivo intestinal absorption of P-gp substrate with high solubility and high permeability. Mol Pharm. 2005;2:329–340. doi: 10.1021/mp0499104. [DOI] [PubMed] [Google Scholar]
- 19.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 20.McLean AJ, Zeng FY, Behan D, Chalmers D, Milligan G. Generation and analysis of constitutively active and physically destabilized mutants of the human beta(1)-adrenoceptor. Mol Pharmacol. 2002;62:747–755. doi: 10.1124/mol.62.3.747. [DOI] [PubMed] [Google Scholar]
- 21.Hanson MA, Brooun A, Baker KA, Jaakola VP, Roth C, Chien EY, Alexandrov A, Velasquez J, Davis L, Griffith M, Moy K, Ganser-Pornillos BK, Hua Y, Kuhn P, Ellis S, Yeager M, Stevens RC. Profiling of membrane protein variants in a baculovirus system by coupling cell-surface detection with small-scale parallel expression. Protein Expr Purif. 2007;56:85–92. doi: 10.1016/j.pep.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barak LS, Ferguson SS, Zhang J, Martenson C, Meyer T, Caron MG. Internal trafficking and surface mobility of a functionally intact beta2-adrenergic receptor-green fluorescent protein conjugate. Mol Pharmacol. 1997;51:177–184. doi: 10.1124/mol.51.2.177. [DOI] [PubMed] [Google Scholar]
- 23.Costa T, Cotecchia S. Historical review: Negative efficacy and the constitutive activity of G-protein-coupled receptors. Trends Pharmacol Sci. 2005;26:618–624. doi: 10.1016/j.tips.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Xhaard H, Rantanen VV, Nyronen T, Johnson MS. Molecular evolution of adrenoceptors and dopamine receptors: implications for the binding of catecholamines. J Med Chem. 2006;49:1706–1719. doi: 10.1021/jm0511031. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Y, Bowie JU. Building a thermostable membrane protein. J Biol Chem. 2000;275:6975–6979. doi: 10.1074/jbc.275.10.6975. [DOI] [PubMed] [Google Scholar]
- 26.Bowie JU. Stabilizing membrane proteins. Curr Opin Struct Biol. 2001;11:397–402. doi: 10.1016/s0959-440x(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 27.Chakrabarti P, Chakrabarti S. C--H…O hydrogen bond involving proline residues in alpha-helices. J Mol Biol. 1998;284:867–873. doi: 10.1006/jmbi.1998.2199. [DOI] [PubMed] [Google Scholar]
- 28.Xie G, Gross AK, Oprian DD. An opsin mutant with increased thermal stability. Biochemistry. 2003;42:1995–2001. doi: 10.1021/bi020611z. [DOI] [PubMed] [Google Scholar]
- 29.Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF. Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol. 2007;372:1179–1188. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D'Antona AM, Ahn KH, Kendall DA. Mutations of CB1 T210 produce active and inactive receptor forms: correlations with ligand affinity, receptor stability, and cellular localization. Biochemistry. 2006;45:5606–5617. doi: 10.1021/bi060067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobilka BK. Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor. Anal Biochem. 1995;231:269–271. doi: 10.1006/abio.1995.1533. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Gurskaya NG, Fradkov AF, Pounkova NI, Staroverov DB, Bulina ME, Yanushevich YG, Labas YA, Lukyanov S, Lukyanov KA. A colourless green fluorescent protein homologue from the non-fluorescent hydromedusa Aequorea coerulescens and its fluorescent mutants. Biochem J. 2003;373:403–408. doi: 10.1042/BJ20021966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steiner T, Koellner G. Hydrogen bonds with pi-acceptors in proteins: frequencies and role in stabilizing local 3D structures. J Mol Biol. 2001;305:535–557. doi: 10.1006/jmbi.2000.4301. [DOI] [PubMed] [Google Scholar]
- 35.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 38.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]