

Abstract
Pax3 is an essential paired- and homeodomain-containing transcription factor that is necessary for closure of the neural tube,, and morphogenesis of the migratory neural crest and myoblast lineages. Homozygous loss-of-function mutation results in mid-gestational lethality with defects in myogenesis, neural tube closure and neural crest-derived lineages including melanocytes, Schwann cells and insufficient mesenchymal cells to septate the cardiac outflow tract. To address the function of Pax3 in later fetal stages and in specific adult tissues, we generated a floxed Pax3 allele (Pax3flox). An intermediate allele (Pax3neo) was produced via creation of the floxed allele, in which the TK-neo(R) cassette is present between exons 5 and 6. It was deduced to be a hypomorph, as Pax3 protein expression is reduced by 80% and homozygote hypomorphs die postnatally. To assess the consequences of reduced Pax3 levels on the various Pax3-expressing lineages and to determine the underlying cause of lethality, we examined Pax3 spatiotemporal expression and the resultant defects. Defective limb and tongue musculature were observed and lethality was due to an inability to suckle. However, the heart, diaphragm, trunk musculature, as well as the various neural crest-derived lineages and neural tube were all unaffected by reduced Pax3 levels. Significantly, elevated levels of the related Pax7 protein were present in unaffected neural tube and epaxial somatic component. The limb and tongue myogenic defects were found to be due to a significant increase in apoptosis within the somites that leads to a paucity of migratory hypaxial myoblasts. These effects were attributed to the hypomorphic effect of the Pax3neo allele, as removal of the TK-neo(R) cassette completely relieves the hypomorphic effect, as 100% of the Pax3flox/flox mice were normal. These data demonstrate a lineage-specific response to ~80% loss of Pax3 protein expression, with myogenesis of limb and tongue being most sensitive to reduced Pax3 levels. Thus, we have established that there are different minimum threshold requirements for Pax3 within different Pax3-expressing lineages.
Keywords: mouse embryo, somites, Pax3, hypomorph/gene dosage, tongue and limb hypaxial muscle, apoptosis, Pax7
INTRODUCTION
Pax (paired-box) factors are a highly conserved family of transcription factors belonging to the helix-turn-helix class. They are characterized by the presence of a paired-domain (a bipartite DNA binding domain) and are often associated with a homeodomain which itself is able to form both homo- and hetero-dimers on DNA (Jun & Desplan, 1996). There are nine mammalian Pax genes that are expressed early during embryogenesis and guide development by promoting cell-lineage specification, as well as cell survival, proliferation and migration. Their expression is spatiotemporally restricted during development, and homozygous mutations in most of them result in specific developmental defects (Mansouri et al., 1999; Chi & Epstein, 2002). An intriguing feature of this family is that many Pax genes exhibit an unusual gene dosage requirement. Loss-of-function heterozygous mutations in Pax1, Pax2, Pax3, Pax6, Pax8, and Pax9 each cause semi-dominant phenotypes in either the mouse or human, or both (Epstein et al., 1991; Baldwin et al., 1992; Hanson et al., 1994; Keller et al., 1994; Sanyanusin et al., 1995; Macchia et al., 1998; Wilm et al., 1998; Stockton et al., 2000; van Raamsdonk & Tilghman 2000; Kist et al., 2005). Usually the affected tissue in heterozygous mutants is reduced in size, and either more severely disrupted or missing in homozygotes (van Raamsdonk & Tilghman, 2000). Similarly, sensitivity to Pax dosage is also evident from transgenic over-expression of Pax2 and Pax6 (Dressler et al., 1993; Schedl et al., 1996; Kim & Lauderdale, 2006). Remarkably, transgenic mice that over-express Pax6 exhibit a phenotype (Schedl et al. 1996) similar to that observed in heterozygous Pax6 mutants (Hill et al., 1992).
Although the molecular basis for the Pax gene dosage requirement is not known, it has generally been assumed that the protein products act within a concentration range sensitive to twofold changes. This may in turn result in prolonged times to reach required Pax factor threshold leading to developmental delays (van Raamsdonk & Tilghman, 2000). Alternatively, based on the monoallelic expression of Pax5 (Nutt et al., 1999) and the reduced size of affected tissues in the heterozygous mutants, it has been suggested that haplo-insufficiency could be explained if heterozygous cells activate either the wildtype or mutant allele, but not both (Nutt & Busslinger, 1999). Thus, a heterogeneous population of both wildtype and null cells could co-exist in Pax heterozygous animals that results in stochastic cell fate determination and ultimately smaller Pax-specified lineages.
PAX3 is expressed in the 8–9 week old human fetus (Terzic & Saraga-Babic, 1999) and from E8 onwards in mice (Goulding et al., 1991). Pax3-deficient mice embryos die in utero and have multiple defects affecting neural tube (NT) closure, myogenesis and morphogenesis of neural crest-derived cells/tissues including melanocytes, Schwann cells and a subpopulation of mesenchymal cells in outflow tract. Significantly, Pax3 is a key regulator of embryonic skeletal muscle formation, as it can directly regulate Myf5 (which plays a major role in determining myogenic cell fate at the onset of skeletal muscle formation) in the hypaxial somite and its derivatives (Bajard et al., 2006). Along with the related Pax7, it has been shown to be expressed in skeletal muscle progenitor cells (Relaix et al., 2005). However, Pax3 may be dispensable for postnatal muscle regeneration since Pax3-postive cells can not effectively regenerate muscle in the absence of Pax7 (Kuang et al., 2006). Pax3 null embryonic muscular defects are thought to result from loss of Pax3 activation of c-Met tyrosine kinase receptor (Epstein et al., 1996; Yang 1996; Relaix et al., 2003), which is essential for the delamination/migration of muscle progenitor cells (Bladt et al., 1995), while the pigmentary defects are thought to result from lack of Pax3-regulated Mitf transcription factor that controls expression of dopachrome tautomerase expression and melanin synthesis (Lang et al., 2005). Although the mechanism leading to NT defects is presently unclear, it has been shown Pax3 mutant NT defects can either be reduced by maternal folic acid supplementation (Greene & Copp, 2005) or by placing Pax3 nulls on a p53 null background to prevent apoptosis and rescue NT closure (Loeken, 2005). The Pax3 null cardiovascular defects include persistent truncus arteriosus (PTA) and obligatory interventricular septal defects (VSDs) due to a dramatic deficiency of cardiac neural crest (NC) cells (Conway et al., 1997a; Conway et al., 2000; Stoller & Epstein, 2005). Furthermore, Pax3 heterozygotes exhibit pigmentation defects of varying penetrance depending on genetic background, indicating that melanocytes are partially sensitive to a 50% reduction in Pax3 gene dosage (Conway et al., 1997b). In patients, haploinsufficient PAX3 (2q35) mutations manifest as Waardenburg syndrome I or III and cause developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair with congenital deafness (Waardenberg, 1951). Interestingly, heterozygous murine Pax3 mutations phenotypes do not include dystopia canthorum or deafness. Thus, despite the presence of multiple systemic mutant mice lines and extensive patient genotype-phenotype correlations, it is still not known how Pax3 mutations affect tissues within such a wide range of cell types.
To address these limitations, we have taken advantage of a novel, hypomorphic Pax3 allele (Pax3neo) and investigated the developmental consequences of a more drastic Pax3 gene dosage reduction. Analyses of these mice revealed myogenesis is the most sensitive developmental process affected by 80% reduction of Pax3 protein. Significantly, myogenesis is not uniformly affected in all long-range migrating myoblast precursors giving rise to muscles in different organs. The consequence of reduced gene dosage of Pax3 in hypomorphic somites is first evident as elevated apoptosis prior to myoblast migration into the limb and towards the tongue. In addition, we identified compensatory upregulation of closely-related Pax7 in Pax3neo/neo mutants, demonstrating genetic redundancy and a hitherto unappreciated role for Pax7 in NT and NC morphogenesis. We propose that the absence of myoblasts in hypomorphic embryo limbs arise due to lack of cell survival prior to migration, secondary to a delay in achieving a threshold of Pax3 required for myoblast precursor survival.
MATERIALS AND METHODS
Mice Colonies and Genotyping
Generation of the C57Bl6 Pax3 conditional allele was previously described (Koushik et al., 2002). Briefly, exon5 was flanked by a 5’ loxP and a 3’ loxP-flanked thymidine kinase (TK) promoter-driven neomycin cassette - TK-neo(R) (Fig.1A). Heterozygous Pax3neo mice are fertile, and mice and embryos resulting from intercrosses were genotyped by PCR using primers 1,2 spanning the 5’ loxP site to amplify both the wildtype (315bp) and Pax3neo (349bp) alleles (Fig.1A).
Figure 1. Characterization of Pax3neo allele.
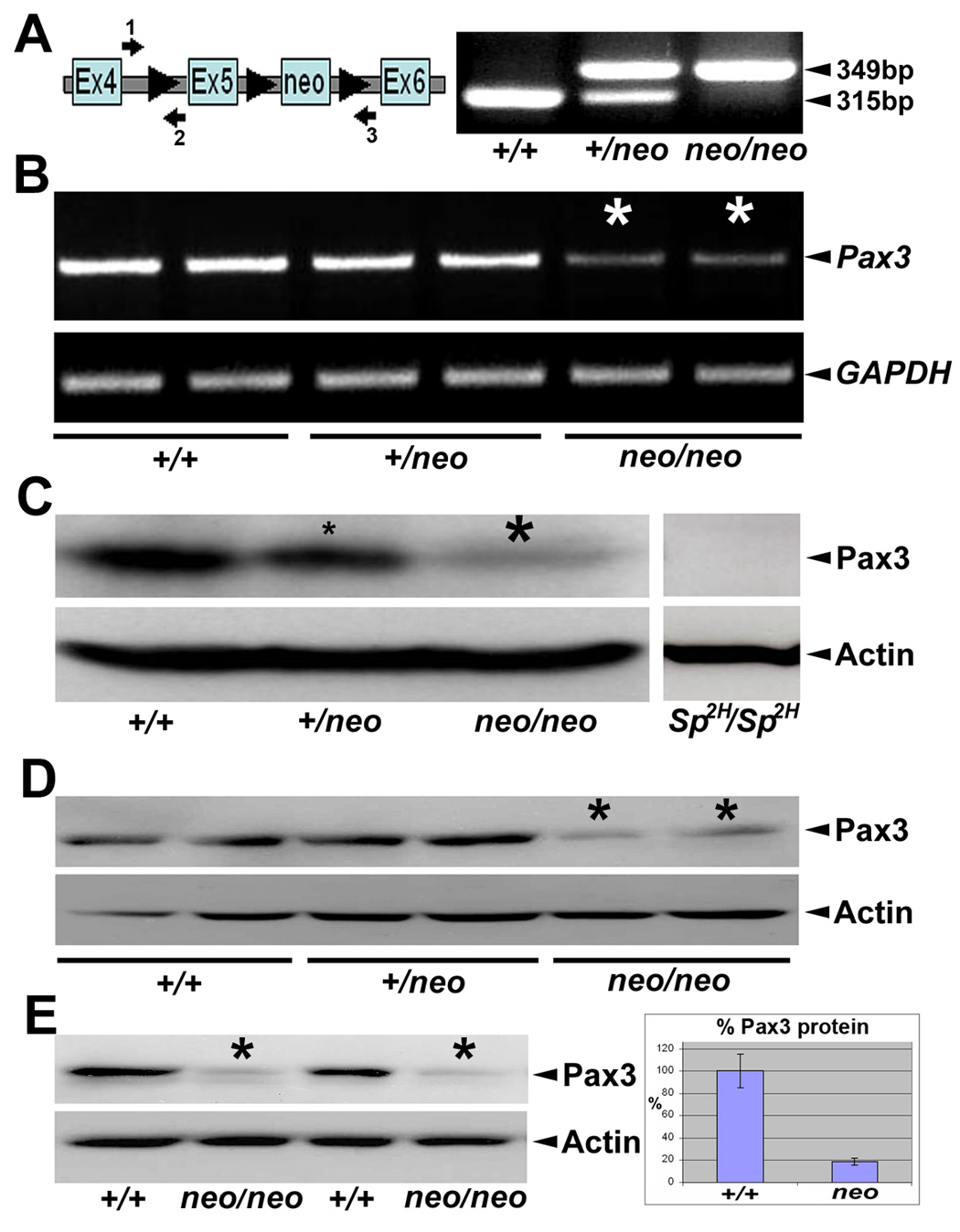
(A) Schematic representation of targeted Pax3neo allele (Koushik et al. 2002) and PCR genotyping strategy. Note loxP sites (triangles) flank exon5 of the Pax3 gene and the downstream neomycin cassette. Arrows indicate location of primers (#1–3) used for PCR genotyping. Combination of primers 1 & 2 amplifies both the wildtype (315bp) and modified (349bp) allele via the extra 34bp contributed by the 5′ loxP site. Combination of primer 1 & 3 amplifies wildtyple allele (1,861bp) and neo-removed exon5-floxed allele (1,929bp). Representative PCR with primer 1 & 2 combination is shown for wildtype (+/+), heterozygote (Pax3+/neo) and homozygous (Pax3neo/neo). (B) RT-PCR analysis of Pax3 expression levels. Note in duplicate E10.5 embryos the level of Pax3 mRNA expression is reduced in a dose-responsive manner in both the heterozygous and homozygous embryos, with the most significant decrease within Pax3neo/neo samples (indicated via *). This is in contrast to the uniform expression of the GAPDH internal control. (C-E) Western analysis of Pax3 protein expression. (C) Specificity of the anti-Pax3 antibody was verified via Sp2H/Sp2H and wildtype littermate E10.5 embryos. Note in Sp2H/Sp2H embryos, ~53kD Pax3 is absent but actin is expressed at similar levels to control littermates¨ Using the same conditions, there is a dose-responsive decrease in Pax3 levels in both the E10.5 heterozygous (~40% & small *) and homozygous (~80% & large *) Pax3neo whole embryos despite equivalent actin expression levels. (D) Similarly, there is decrease (~80%) in Pax3 levels in duplicate E10.5 homozygous Pax3neo isolated heads (*) relative to duplicate heterozygous and wildtype littermate heads (despite similar actin expression). (E) There is also reduced Pax3 levels in duplicate E9.0 homozygous Pax3neo whole embryos (*) compared to duplicate wildtype littermates. Note when the signal intensity was quantified using ImageJ software, E9.0 Pax3neo/neo embryos only contained ~20% Pax3 levels when compared to wildtype littermates (normalized to actin levels).
To selectively remove the floxed TK-neo(R) cassette in the germline, adult male Pax3neo heterozygous mice were crossed to female C57Bl6 Tie2−Cre (as Tie2−Cre is expressed in female germ cells; Koni et al., 2001). The resulting progeny were screened by PCR using primers (1,2) and primers (1,3). With this screening strategy (Fig. 1), the allele with selective removal of TK-neo (referred to flox) was identified as containing both the 349bp (1,2) and 1929bp (1,3) bands. The identity of flox allele was further confirmed as negative for neo-based PCR (data not shown). The primers sequences were as follows: #1 forward 5’-CATTTCTATCTCATTCTCTGGACC-3’; #2 reverse 5’-AAGTCGGAGTCTCTGCTGAGG-3’; #3 reverse 5’-GCAGGCAGCAATGTTAGCTTAG-3’. Resultant Pax3flox/+ males were crossed to wildtype C57Bl6 females to remove Tie2−Cre transgene, and then intercrossed to produce Pax3flox/flox mice. Genotyping and breeding of Sp2H mice was as described (Conway et al., 2000). All breeding colonies were maintained in our animal facility and studies conducted according to Institutional Animal Care and Use guidelines. For timed pregnancies, the day of observed vaginal plug was designated embryonic day 0.5 (E0.5).
Reverse Transcriptase-PCR and Western blot analysis
Total RNA isolated from individual E10.5 embryos was reverse-transcribed and RT-PCR analysis for Pax3 and GAPDH was performed as described (Conway et al., 1997a). RT-PCR was carried out twice in duplicate (n=4). For Western analysis, individual E10.5 embryos were homogenized in 300µl protein lysis buffer (10mM Tris, pH6.8, 100mM NaCl, 10mM EDTA, 1% SDS and 1x Roche proteinase inhibitor cocktail). After centrifugation (16,000rpm at 4°C), the supernatant was transferred to new tube, 60µl of 6x SDS-PAGE loading buffer added and denatured for 10 minutes at 94°C in the presence of 100mM β-mecaptoethanol. For each sample, 50µl was resolved using 10% SDS-PAGE (Bio-Rad) and transferred to nitrocellulose. Membranes were blocked for 1 hour in 5% non-fat milk (in phosphate buffered saline (PBS) with 0.5% Tween20) and probed with mouse monoclonal anti-Pax3 (1:2000 dilution) or monoclonal anti-Pax7 (1:2000 dilution) antibodies (both obtained from the Hybridoma Bank) in blocking solution. The signal was revealed via ECLPlus (Amersham) with peroxidase-conjugated goat anti-mouse secondary antibody (1:5000 dilution, Promega). To verify equal loading, all blots were subsequently stripped (0.2M NaOH for 5 minutes at room temperature), thoroughly washed, re-blocked and then probed with mouse antiactin antibody (1;5000 dilution, Sigma). X-ray films were scanned and signal intensity measured using ImageJ software (downloaded from wsr@nih.gov).
Histological, In situ hybridization and Immunohistochemical analysis
Embryos were fixed in 4% paraformaldehyde at 4°C overnight and processed for routine paraffin sections (10µm) for histology, in situ hybridization and/or immunohistochemistry using standard procedures. Embryos older than E14 were decalcified in 0.5M EDTA prior to dehydration. For histological analysis, the sections were stained with haematoxylin/eosin. In situ hybridization of Pax3, Crabp1, cMet Periostin, MyoD, Myf5, Msx2 and Sox10 was performed as previously described (Conway, 1996) using S35 or DIG-labeled cRNA probes. Hybridization with corresponding sense probe was paralleled as a negative control. Pax3, Crabp1 and Periostin probes have been previously described (Conway et al., 2000; Kruzynska-Frejtag et al., 2001), whilst MyoD, cMet and Myf5 were provided by Dr. Chen-Ming Fan (Chen et al., 2005), Msx1 and Msx2 by Dr. Robert Maxson (Kwang et al., 2002) and Sox10 was cloned via PCR amplification of E12.5 whole embryo cDNA using the following primers (5’-TCTGTCTTCACCTGGGCTTT and 3’-ATGTCAGATGGGAACCCAGA).
Immunostaining was carried out using ABC kit (Vectorstain) with DAB and hydrogen peroxide as chromogens. The endogenous peroxidase was quenched via a 20-minute incubation in 0.3% hydrogen peroxide in PBS. For detection of Pax3 and Pax7, sections were boiled in antigen retrieval buffer (Dako) for 2 minutes and then cooled to room temperature prior to blocking. The dilution of primary antibody was 1:5000 for mouse anti-alpha smooth muscle actin (αSMA; Sigma), 1:200 for goat anti-Pax3 antibody (Santa Cruz sc-7748) and mouse anti-Pax7 (Hybridoma Bank). Incubation in primary antibody was carried out at 4°C overnight, and incubation in secondary antibody and ABC was performed at room temperature for 30 minutes at 1:400 dilution. All washes were carried out in PBS with 0.5% Tween20 at room temperature. The specificity of Pax3 staining was confirmed by using age-matched Pax3 null (Sp2H/Sp2H) sections.
Skeletal Staining
E17.5 embryos (n=13) were collected in PBS and fixed in 70% ethanol, 30% acetic acid, and 0.01% Alcian blue 8GX for 3–5 days. After dehydration to 100% ethanol, embryos were stained with 0.5% KOH + 0.01% alizarine S for 10 min. Embryos were then cleared in 1% KOH/20% glycerol and then dehydrated gradually to 100% glycerol and photographed.
Detection of cell proliferation and apoptosis
Timed pregnant mice were injected with BrdU at recommended dosage (1ml/100g, ZYMED) two hours prior to sacrifice. Embryos were fixed and processed for paraffin sections. Incorporated BrdU was detected with ZYMED BrdU staining kit following the manufacturer’s instruction. TUNEL analysis was performed on 10µm paraffin sections using the ApopTag® peroxidase In Situ Apoptosis Detection Kit (S7100, Chemicon) according to manufacturer's directions. Sections were counterstained with methyl green. For both assays, serial sections were examined using at least three individual E10.5 embryos of each genotype.
RESULTS
The Pax3neo allele is hypomorphic
Mice heterozygous for the Pax3 conditional allele (Pax3neo/+)(Koushik et al., 2002) are viable and fertile (n>100), and lack the characteristic white belly spot, a feature of Pax3 loss-of-function heterozygous mutations (Koushik et al., 2002; Engleka et al., 2005). However, intercross of Pax3neo/+ failed to produce any adult homozygous offspring, suggesting the floxed allele retaining the TK-neo(R) cassette is not fully functional. Genotyping of newborns and 3-week old pups from Pax3neo/+ intercrosses reveal that Pax3neo/neo are viable and present at expected Mendelian ratios at day 1 after birth but not at day 21. In contrast, mice homozygous for loss-of-function mutation (Sp2H) alleles die ~E14 at mid-gestation (Epstein et al., 1996; Conway et al., 1997c). The postnatal death of Pax3neo/neo neonates suggests the Pax3neo allele is a hypomorph.
To determine the extent of the hypomorphic effect, we first checked Pax3 mRNA expression levels using RT-PCR. Pax3 expression was detected in all three genotypes but was significantly reduced in Pax3neo homozygote (~20%) E10.5 embryos (Fig. 1B). We chose to examine Pax3 expression at E10.5 as this was 2 days prior to any morphological alterations being evident (Fig. 2). Additionally, a larger transcript containing neomycin was amplified in Pax3neo heterozygote and homozygote samples (data not shown). Given that the TK-neo(R) cassette may interfere with gene expression (Meyers et al., 1998; Nagy et al., 1998), we sequenced RT-PCR products and confirmed neomycin insertion in the intron generates alternatively spliced Pax3 mRNA forms resulting in reduced Pax3 levels (data not shown). Next we examined Pax3 protein levels via Western analysis using a mouse anti-Pax3 monoclonal antibody that recognizes the Pax3 C-terminus which is absent in Sp2H/Sp2H embryos due to premature stop codon and predicted truncated protein (Epstein et al., 1991). The specificity of the assay was verified using E10.5 Sp2H/Sp2H embryos as a negative controls, that lack the ~53kDa Pax3 peptide (Fig. 1C). In line with RT-PCR data, a ~53kDa Pax3 band was detected in all three genotypes, with signal intensity being weakest in Pax3neo/neo embryos. Band intensity was scanned and quantified using ImageJ software. Relative to wildtype littermates, the protein level was ~60% in Pax3neo/+ and only ~20% in Pax3neo/neo (Fig. 1C). Given that Pax3 is predominantly expressed in neural tube, somites and migratory myoblast lineages at E10.5 and that subsequent analysis revealed a lack of migratory myoblasts in the Pax3neo/neo limb buds (Fig. 4 & Fig. 6), we measured Pax3 protein levels via Western analysis in isolated E10.5 heads (to examine whether reduced levels were due to tissue loss or global down-regulation within Pax3-expressing lineages). Similarly, Pax3 levels were ~60% in Pax3neo/+ and only ~20% in Pax3neo/neo heads/neural tissues when compared to actin (Fig. 1D). Finally, as there would be significantly fewer migratory limb myoblasts at E9.5 as migration is just initiating, we examined E9.5 embryos for Pax3 protein levels. Consistent with aforementioned Western data, Pax3 levels were ~20% in E9.5 Pax3neo/neo embryos relative to wildtype littermates (+/+=128,327+/−20,226 and neo/neo=23,346 +/−1,275 n= 3 Westerns of duplicate samples) (Fig. 1E,). Thus a Pax3neo hypomorphic allele encodes ~10% Pax3 protein relative to ~50% encoded by a wild-type allele, and Pax3 expressions levels are reduced in all tissues examined. These global Pax3 suppression data are consistent with later Pax3 immunohistochemistry data (Fig. 6). Further, the lack of white belly spots in Pax3neo/+ adult mice (n>100), indicates that ~60% Pax3 protein (i.e. 10% from Pax3neo allele + 50% from wildtype allele) is sufficient to initiate/maintain normal pigmentation. Taken together, these results indicate the Pax3neo allele is hypomorphic, encoding ~20% level of wildtype allele expression, and consequently is not compatible with postnatal survival.
Figure 2. Reduced Pax3 levels result in disorganization of Pax3neo/neo tongue musculature.
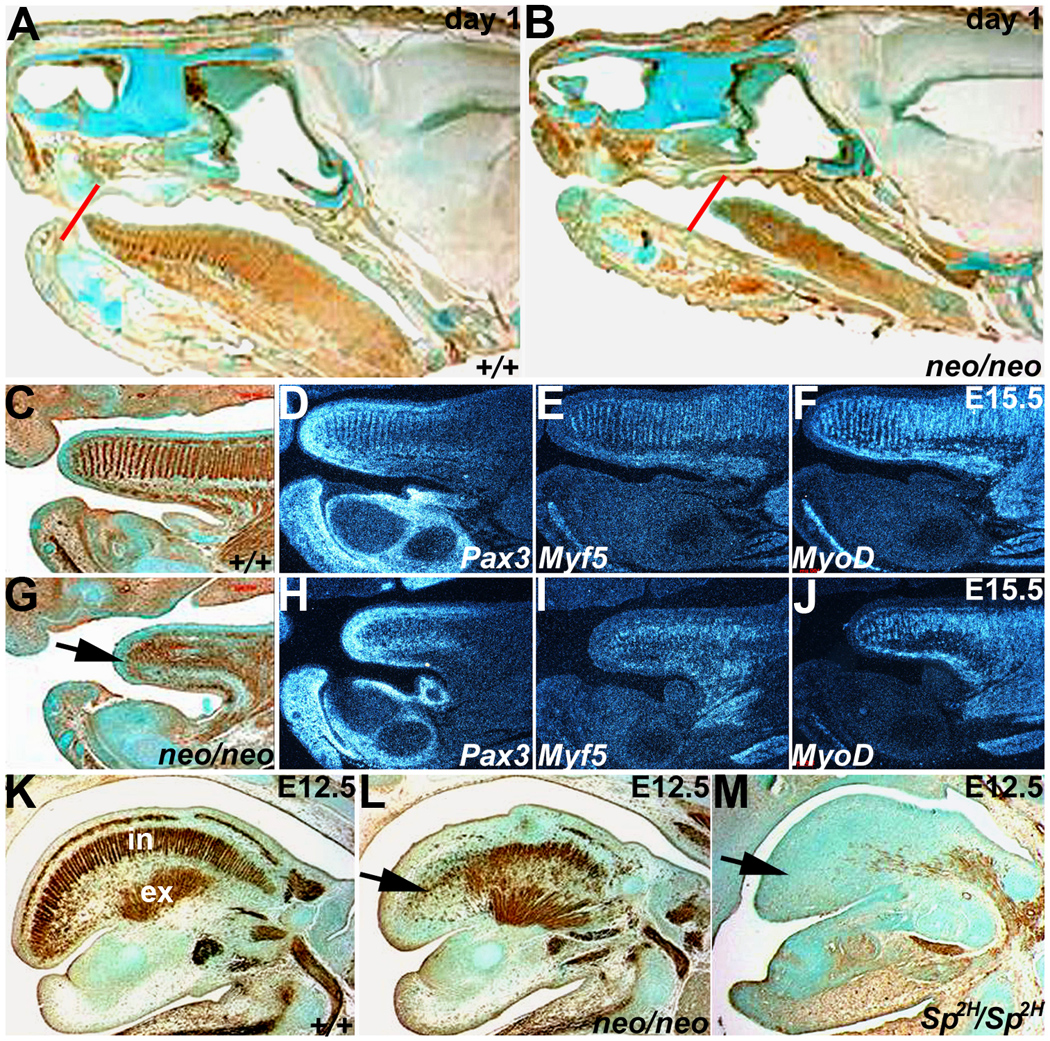
Both immunohistochemistry for alpha smooth muscle actin (αSMA) and radioactive in situ hybridization of Pax3 and early (Myf5) and late (MyoD) muscle marker expression were used to assess muscle deficiencies and pathogenesis of the anomalies at day 1 (A,B), E15.5 (C–J) and E12.5 (K–M). Day-1 sagittal sections stained for αSMA expression indicates that the Pax3neo/neo (B) tongue is shorter and thinner (red lines indicates the position of tongue tips) than in wildtype (A). (C&G) αSMA staining in E15.5 wildtype (C) and Pax3neo/neo (G) sagittal sections. Note E15.5 Pax3neo/neo tongue musculature was greatly reduced and disorganized. Pax3 (D,H), Myf5 (E,I) and MyoD (F, J) mRNA expression patterns mirror the disorganized αSMA-expressing cells on adjacent sections (C&G). The arrow in G points to location of tongue tip in Pax3neo/neo. Note the shorter tongue is already evident at this stage. (K-M) αSMA staining of sagittal E12.5 wildtype (K), Pax3neo/neo (L) and Sp2H/Sp2H (M) sections. Intrinsic (IN) muscles are partially absent whilst extrinsinc (EX) muscles are intact in Pax3neo/neo. However, both intrinsic and extrinsic Sp2H/Sp2H tongue muscles are absent. Arrow indicates intrinsic muscles affected in E12.5 Pax3 mutants.
Figure 4. Molecular marker analysis of migratory limb myoblasts and dorsal-ventral neural tube patterning.
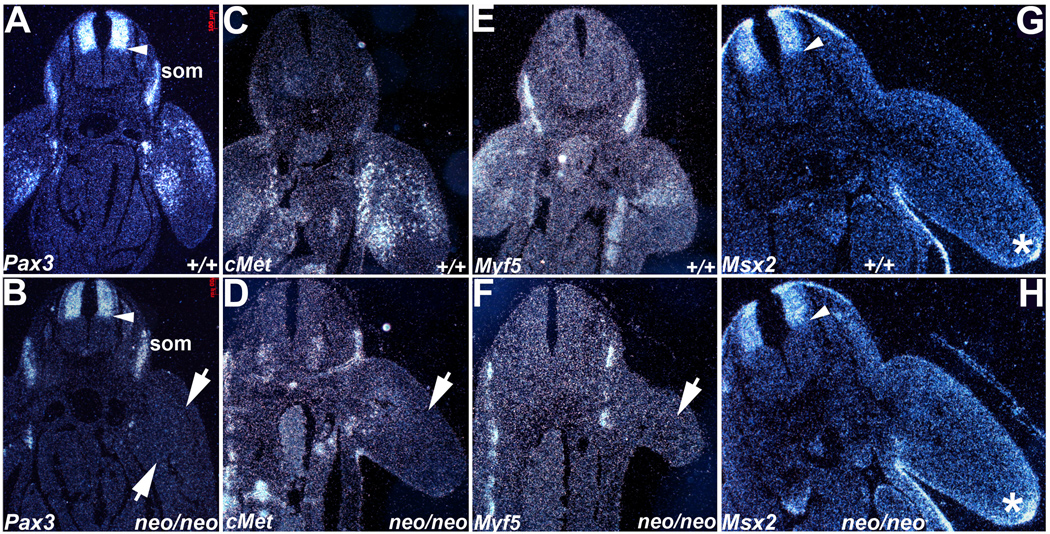
Radioactive in situ hybridization was used to assess Pax3 (A,B), cMet (C,D), Myf5 (E,F) and Msx2 (G,H) spatiotemporal expression patterns in E10 embryos. (A,B) In contrast to wildtype embryos (A), Pax3 mRNA is expressed at reduced levels within Pax3neoo/neo (neo/neo) dorsal NT (arrow head), adjacent dorsal root ganglia, somites (som) and is mostly undetectable within the hypomorphic limb buds apart from a few isolated myoblasts (B). The arrows indicate the two Pax3-expressing populations missing in hypomorphic limb buds. (C,D) cMet mRNA exhibits reduced expression in the Pax3neo/neo limb buds (arrow in D) when compared to wildtype (+/+) littermates (C). (E,F) Myf5 mRNA is also reduced in Pax3neo/neo limb buds (arrow in F) but is maintained in Pax3neo/neo somites. (G,H) Msx2 mRNA exhibits similar expression patterns in the dorsal NT, limb apical ectodermal ridge (*) and body wall within both wildtype (G) and hypomorphic embryos (H). Arrow heads points to mid-NT boundary.
Figure 6. Pax3 protein levels are uniformly reduced whilst Pax7 protein levels are upregulated in Pax3neo/neo hypomorphs.
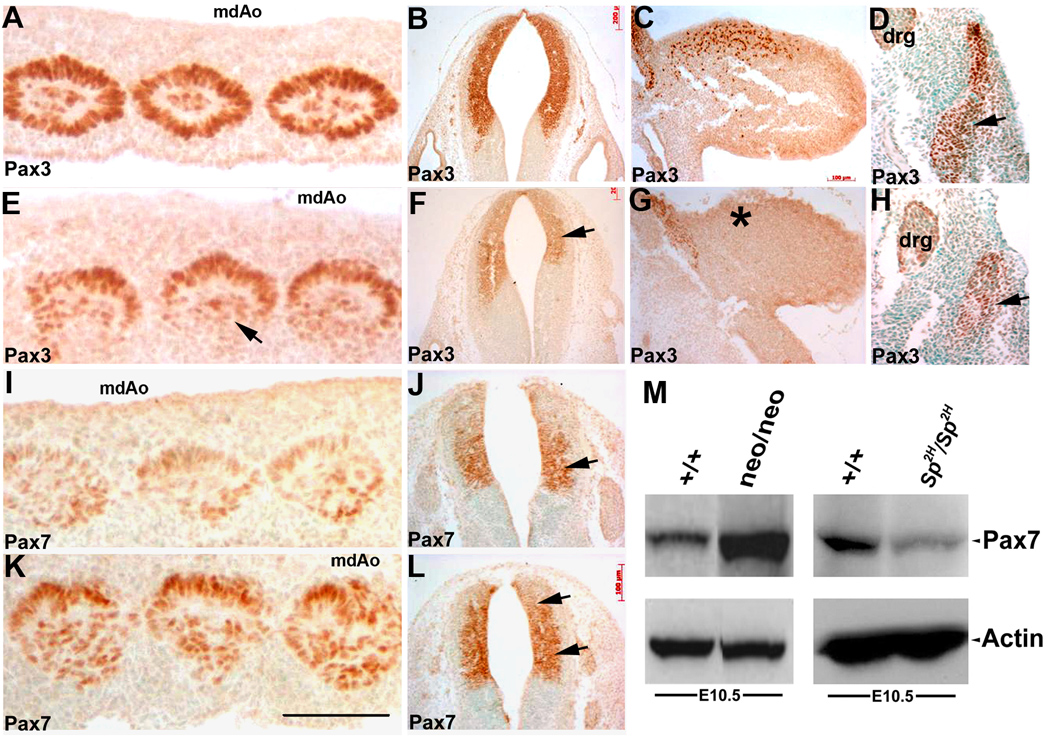
Immunodetection of Pax3 (A–H) and Pax7 (I–L) spatial expression and Western blot analysis (M) were used to assess relative Pax3 and Pax7 protein expression levels. In equivalent transverse sections through the midline dorsal aorta (mdAo) region, Pax3 protein is present in all the mesodermal cells of the concentric whorls of the E10.5 wildtytpe somites (A) but is expressed at reduced levels and is almost absent within the dorsolateral portion (arrow in E) of the Pax3neo/neo somites. Pax3 expression is also reduced in the E10.5 hypomorphic NT (F) and is absent within the E10.5 forelimb buds (G) when compared to wildtype embryos (B,C). In E11.5 Pax3neo/neo dermomyotome, the lateral region is absent and thus Pax3 expression is diminished (H) when compared to controls (D). In adjacent sections Pax7 protein is only weakly expressed in wildtype E10.5 somites (I) and restricted to the medial portion of the wildtype NT (J), but in contrast Pax7 expression is elevated in the hypomorphic somites (K) and expanded into the dorsal region of the NT in Pax3neo/neo embryos (L, indicated by arrows). (M) Western analysis of E10.5 whole embryos revealed Pax7 levels were elevated (~6 fold) in Pax3neo/neo embryos when compared to wildtype littermates, despite the actin loading control being expressed at similar levels. In contrast, Pax7 levels were reduced in Sp2H/Sp2H mutants when compared to wildtype littermates. Bar in K = 100 µm.
Neonatal lethality of Pax3neo/neo pups
Screening of 120 pups collected shortly after delivery indicates Pax3neo/neo mice were born alive in an expected Mendelian ratio (data not shown). Characteristically, all Pax3neo/neo neonatal mice had dysmorphic limbs with forelimbs being more obviously affected than hindlimbs (Fig. 3A). Having established the fully-penetrant dysmorphic phenotype, we carefully examined mice upon delivery. Newborn Pax3neo/neo pups were comparable in body size and appearance (except for the limb defects and absence of milk in their stomachs). This suggested subsequent growth retardation (noticeable by day 2) was postnatal, and that respiration and circulation were not compromised by reduced Pax3 levels. This is consistent with subsequent analysis of cardiovascular system (Supplemental Fig. 1) and lungs/diaphragm (Supplemental Fig. 2), demonstrating normal cardio-pulmonary vascular morphogenesis. The Pax3neo/neo pups all died by day 3, and progressive growth retardation is evident 24hours after birth. Importantly, we noted an absence of ingested milk and any sucking behavior in homozygous Pax3neo pups (n>30). The lack of ingested milk was further confirmed by direct histological examination (data not shown). The lumen of the stomach of wildtype pups was stretched and full of acellular protein aggregates (eosinic-positive and haematoxylin-negative), indicative of proteins from ingested milk. However, the Pax3neo/neo lumen was empty and the stomach wall thin and tightly folded due to a lack of expansion. This analysis indicates Pax3neo/neo pups were viable at birth but were unable to feed.
Figure 3. Reduced Pax3 levels result in severe limb musculature anomalies.
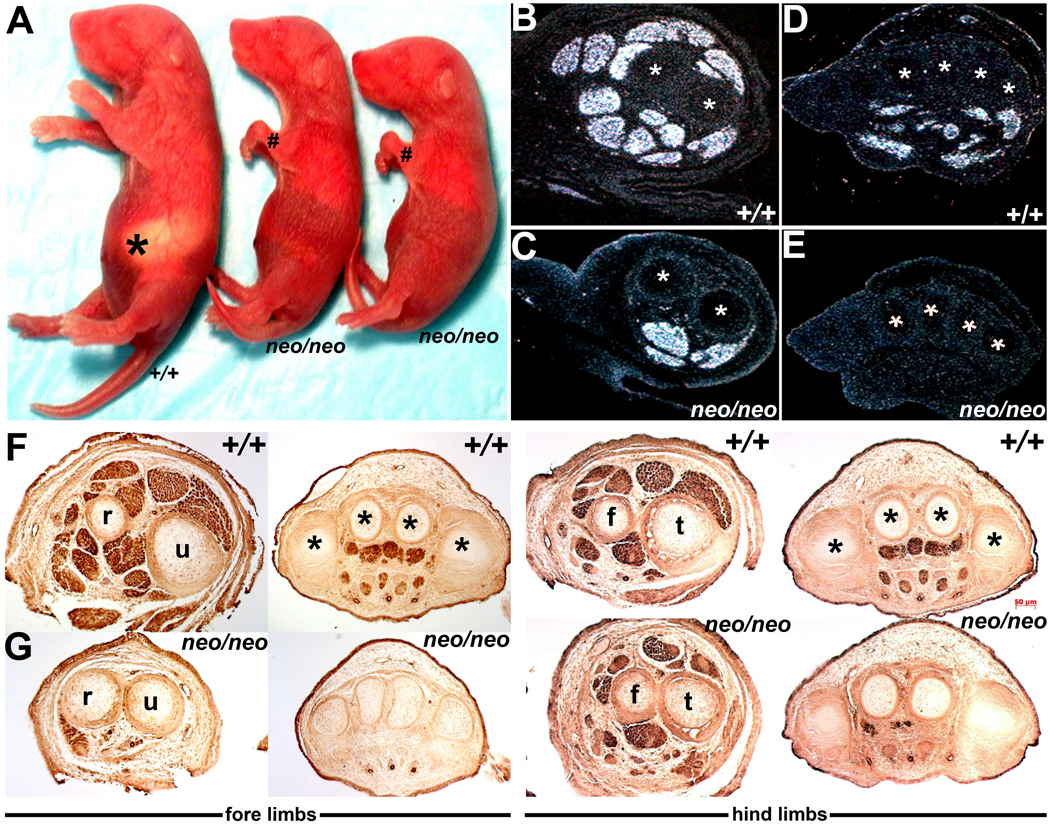
(A) Pax3neo/neo day-2 pups are readily distinguished from their wildtype (+/+) littermates via undersized and severely dysmorphic limbs (# indicate Pax3neo/neo warped forelimbs) and lack of ingested milk (asterisk). (B–E) In situ hybridization analysis of MyoD expression in the E15.5 wildtype (B,D) and Pax3neo/neo (C,E) forelimbs at the level of the ulnar (u)/radius (r) (indicated via * in B,C) and phalange (indicated via * in D,E) bones. Note most of the MyoD-positive muscle masses are absent within the hypomorphic forelimbs. The asterisks indicate the bones serving as a reference to show comparable section levels. (F,G) Immunostaining for αSMA at various medial-lateral section levels along wildtype (F) and Pax3neo/neo (G) E13.5 forelimbs and hindlimbs. Note Pax3neo/neo embryos are already abnormal at this early developmental stage and αSMA-positive muscle masses are either severely hypoplastic (hindlimbs) or completely missing (forelimbs) in hypomorphic embryos. Abbreviations: f, fibula; t, tibia.
Given the potential effects of the TK-neo(R) cassette, we next crossed heterozygous Pax3neo animals with mice expressing Cre recombinase to delete the loxP-flanked sequences. Taking advantage of known Cre expression in Tie2−Cre transgenic oocytes (Koni et al., 2001), we crossed adult male Pax3neo/+ with female Tie2−Cre mice. The Cre activity present in fertilized oocytes mediated three possible recombination events and gave rise to chimeric progeny with more than one recombined allele. Using PCR screening and intercrossing with wildtype C57Bl6 mice, we identified mice that had deleted the floxed TK-neo(R) cassette but retained the loxP flanked exon5 (termed Pax3flox). Intercross of Pax3flox/+ mice demonstrated that Pax3flox homozygotes reach adulthood (n=24/24 indicates 100% survival) with a normal Mendelian frequency (data not shown) and reproduce normally, thus deletion of TK-neo(R) cassette fully restores viability and prevents the hypomorphic effects. Further, Western analysis of E10.5 Pax3flox/flox embryos revealed same Pax3 levels when compared with wildtype littermates (data not shown), indicating that remaining two loxP sites flanking exon5 have no impact on Pax3 expression. Thus, the hypomorphic effect and lethality is solely attributed to the presence of TK-neo(R) cassette, and not any unintended potential mutational effects due to insertion of loxP sites in introns.
Reduced Pax3 levels result in lack of tongue musculature
Having observed the absence of milk in the stomachs of newborn Pax3neo/neo, we were prompted to investigate if tongue development might have been impaired due to Pax3 reduction. Following this hypothesis, we examined Pax3neo/neo tongues in day 1, E15.5 and E12.5 animals using αSMA immunohistochemistry and Pax3, Myf5 (an early myogenic determination factor) and MyoD (a later myogenic factor) in situ hybridization analysis. In newborns, αSMA expression was present in both wildtype and Pax3neo/neo tongues, however there was a striking difference in the extent of αSMA-positive cells, their organization and the size of the tongue (Fig. 2A,B). In control littermates the tongue was longer, reaching the mouth opening. In Pax3neo/neo, the newborn tongue tip merely reached halfway and was also significantly thinner. This anatomical defect would adversely impact suckling, which demands an intimate contact between tongue tip and nipple.
Although tongue thickness was comparable between wildtype and Pax3neo/neo at E15.5, it was already evident that the Pax3neo/neo tongue was shorter and αSMA-positive cells severely disorganized (Fig. 2G). The transcripts of Pax3, Myf5 and MyoD were each detectable in both E15.5 wildtype and Pax3neo/neo, tongues. However, whilst expression in wildtype was restricted to the front half of tongue and present in regularly aligned myoblasts that correspond to the intrinsic muscles (superior longitudinal, transverse and vertical components) and underlying extrinsic muscles (longitudinal genioglossus, hyoglossus and styloglossus); expression in Pax3neo/neo was diminished and exhibited an irregular organization (Fig. 2). At E12.5, while the thickness was comparable between Pax3neo/neo and control tongues, the reduced length was just becoming evident (Fig. 2L). Significantly, αSMA staining revealed the intrinsic superior longitudinal muscle layer is almost completely absent and the vertical components of the intrinsic muscles are disorganized and/or absent from the hypomorphic tongue tip. In order to compare the severity of these muscular defects with embryos that completely lack functional Pax3 protein, we assessed αSMA in Sp2H/Sp2H E12.5 embryos (Fig. 2M). As expected, the phenotype is more severe and the entire tongue was mostly devoid of αSMA-expressing cells. This more severe Sp2H/Sp2H phenotype argues in favor of the tongue defect being due to reduced Pax3 dosage rather than any non-specific effects of neomycin itself.
Collectively, these observations indicate that an ~80% loss of Pax3 protein expression results in hypoplastic tongue muscle formation and that the intrinsic musculature is especially sensitive to reduced Pax3 levels. The hypoplastic tongue is likely to result in the observed suckling defects as evidenced by lack of ingested milk, which ultimately leads to Pax3neo/neo neonatal growth retardation, wasting and lethality.
Reduced Pax3 levels also result in severe limb muscle hypoplasia
Given Pax3 nulls exhibit severe muscular hypoplasia defects (Franz et al., 1993; Epstein et al., 1996; Engleka et al., 2005), we next examined limb musculature at E13.5, E15.5 and day 2; both before and after the undersized and warped forelimb defects could be observed (Fig. 3A). As gross skeletal abnormalities are invariably present in Pax3 mutants (Tremblay et al., 1998; Dickman et al., 1999; Henderson et al., 1999), we assessed whether reduced Pax3 expression can cause skeletal malformations. Alizian red/Alizian blue staining revealed that Pax3neo/neo skeletons are unaffected, except for occasional fusion between the 1st and 2nd ribs (data not shown, n=3/10). The skeletal system was otherwise indistinguishable between Pax3neo/neo and littermates controls, and despite the subsequent muscular defects, the size, proportion and positioning of each limb bone and cartilage was normal (n=10 +/+ and Pax3neo/neo E18 littermates). Analysis of MyoD expression in E15.5 forelimbs revealed that several MyoD-expressing muscle masses present in control embryos were absent in Pax3neo/neo limbs (Fig. 2C,E). Similarly, analysis of αSMA expression at E13.5, supported the MyoD differences. αSMA staining revealed hypomorphic forelimb muscular masses were either absent or significantly reduced in size prior to any gross defects (Fig. 2F). Specifically, the more distal the muscle the more severe the hypoplasia, and dorsal extensor muscles were more affected than ventral flexor muscles. A similar phenomenon was also seen with Pax3neo/neo hindlimbs, albeit it was less grossly dysmorphic (Fig. 2G). However, musculature of dorsal, ventral and lateral body-wall appeared grossly normal in Pax3neo/neo, as evidenced via MyoD and αSMA expression (Supplemental Fig. 2). Taken together, these observations indicate that the hypomorphic allele specifically affected skeletal limb musculature derived from myoblast precursor cells that undergo medial-lateral migration from the somitic dermomyotome.
Reduced Pax3 levels results in deficient migratory limb myoblasts
In order to document if the hypomorphic reduction of Pax3 (as shown via RT-PCR and Western) globally affects all Pax3-expressing cell lineages, we initially used in situ hybridization to examine Pax3 spatiotemporal expression. In wildtype E10.5 embryos, Pax3 mRNA was robustly expressed in the dorsal NT, dorsal root ganglia, somites and limb bud in a pattern consistent with published reports (Goulding et al., 1991; Conway et al., 1997a). Specifically, Pax3-expressing cells in limb buds are distributed along the dorsal and ventral sides of limb bud, representing the migratory myoblasts (Fig. 4A). In contrast, in E10.5 Pax3neo/neo limb buds, Pax3 expression was significantly decreased, indicating a greatly reduced number of Pax3-expressing migrating myoblasts. Although reduced relative to wildtype littermates, Pax3 was detectable in Pax3neo/neo dorsal NT, dorsal root ganglia and somites in similar expression domains as control littermates (Fig. 4B). We also performed in situ hybridization targeting the 5’ and 3’ ends of Pax3 mRNA and as expected, both are appropriately co-expressed at low levels in Pax3 target sites in hypomorphic embryos (data not shown), indicating that insertion of the Neo gene does not result in ectopic expression patterns. Next we examined downstream targets such as c-Met and the myogenic target Myf5 in E10.5 limb buds. Pax3 is required for the activation of c-Met, a recepotor for HGF is required for delamination and migration of muscle progenitor cells (Bladt et al., 1995). c-Met is expressed in cells migrating from the ventral lip of the dermomyotome and present on both dorsal and ventral sides of the wildtype limb buds but is largely absent in Pax3neo/neo limb buds (Fig. 4D), and appears to be upregulated in the somite itself Similarly, expression of Myf5 (an early marker of muscle differentiation; Bajard et al., 2006) is also downregulated in Pax3neo/neo limb buds (Fig. 4F). These analyses indicate that it is an absence of colonizing myoblasts rather than normal numbers of very low Pax3-expressing myoblasts (i.e. below-detection via in situ hybridization) that is the underlying cause of hypomorphic limb muscle hypoplasia.
Reduced Pax3 levels do not affect dorsal-ventral neural tube patterning
Given the extensive published data that link altered Pax3 expression to abnormal cardiac NC and subsequent outflow tract remodeling (Conway et al., 1997a,b,c; Conway et al., 2000; Epstein et al., 2000; Chan et al., 2004); altered dorso-ventral restricted Pax3 expression in the NT and resultant abnormal limb, eye, brain and NT development (Tremblay et al., 1996); and defective closure of the NT in Pax3-deficient embryos that can result in spina bifida and/or exencephaly (Conway et al., 1997b; Pani et al., 2002) – we performed in situ hybridization with molecular markers of dorso-ventricular NT patterning and NC morphogenesis. Although expression of Crabp1 and Ap2α, both NC markers, is diminished in the migratory cardiac NC in Pax3 null embryos (Conway et al., 1997a; Conway et al., 2000), their expression is unaltered in Pax3neo/neo embryos (Supplemental Fig. 1). Specifically, Crabp1 (which is expressed in NT, migratory NC and dorsal root ganglia) exhibits similar ventral NT/floor plate exclusion (Supplemental Fig. 1E,F). Ap2α (that is only expressed in normal emigrating NC; data not shown) is appropriately expressed in dorsal root ganglia and migrating NC. Similarly, given that Msx2 is co-expressed with Pax3 in the dorsal NT and is known to be repressed by Pax3 (Kwang et al., 2002), we investigated whether Msx2 expression was altered by reduced Pax3 levels (Fig. 4G,H). Msx2 expression was normal within E10.5 Pax3neo/neo dorsal NT, neuroectoderm, and apical ectodermal ridges that control limb bud outgrowth.
Apoptotic rates are specifically increased within Pax3neo/neo somites
To determine the underlying mechanism responsible for the hypoplastic limb and tongue muscle defects, we first investigated whether reduced proliferation could account for the reduced musculature. Using BrdU in vivo labeling, we found the cell proliferation index was comparable between Pax3-expressing and non-Pax3-expressing cells in both control and Pax3neo/neo somites and limb buds at E10.5 (n=3 E10.5 embryos of each genotype; data not shown), indicating the observed 80% reduction of Pax3 expression does not affect cell proliferation. We then examined whether the hypoplastic muscle defects may be due to excessive cell death. Using TUNEL we found there was a marked increase in E10.5 Pax3neo/neo somitic cells undergoing apoptosis, when compared to wildtype littermates (Fig. 5D). Significantly, increased apoptosis was only detected within the somites and not within other Pax3neo/neo tissues, including Pax3-expressing dorsal root ganglia and NT (Fig. 5E). Similarly, increased apoptosis was also observed in the E10.5 hypomorphic occipital myotomes that give rise to the tongue muscles (data not shown), but apoptosis was not elevated in E12.5 hypomorphic tongues (Fig. 5F). Furthermore, the TUNEL labeling index was not increased in hypomorphic E10.5 limb buds (data not shown). The TUNEL assays indicate that the muscular defect in limb and tongue was not due to increased cell death during myoblasts ,migration/colonization but rather due to increased cell death of myoblast progenitors prior to myoblasts emigration/colonization of the mutant limb and tongue mesenchyme. This increased apoptosis probably also underlies the observed mis-expression of hypomorphic somitic and migratory cell population markers. Histology revealed that early E9 and E10 hypomorphic somites appear to be normal and at appropriate developmental stages (data not shown). This indicates ~20% Pax3 expression was not sufficient to maintain cell survival specifically within the somitic lineage. This analysis is consistent with previous reports that Pax3 is essential for somitic survival and it is also required for delamination and migration of myoblast progenitor cells (Epstein et al., 2000; Buckingham et al., 2003).
Figure 5. Apoptotic rates are specifically increased within Pax3neo/neo somites.
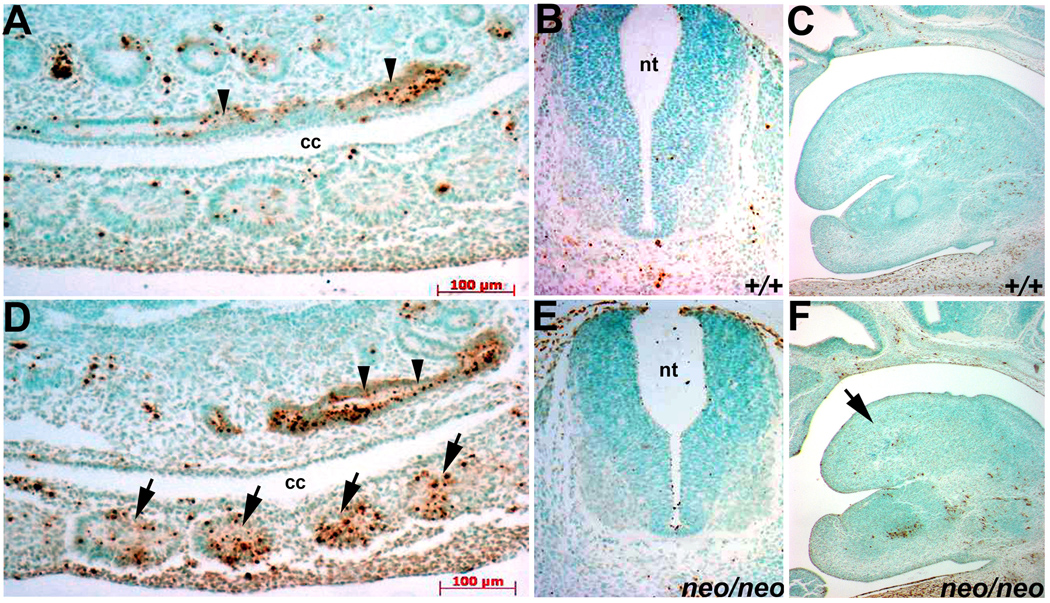
Apoptotic cells were detected via TUNEL in wildtype (A–C) and Pax3neo/neo (D–F) serial transverse sections. Note a significant proportion of the mesodermal cells of the E10.5 hypomorphic somites are undergoing apoptosis (D), specifically within the lateral edge of the dermomyotome that gives rise to the migratory limb muscle precursors. In contrast, only a few E10.5 wildtype lateral somitic cells are TUNEL-positive (A). Each arrow indicates a somite. However, the apoptotic index is comparable in wildtype and Pax3neo/neo NT (B&E) and hindgut (indicated by arrowheads in A&D). Similarly, the apoptotic index is comparable in wildtype and Pax3neo/neo (arrow) E12.5 tongues (C&F).
Pax3 protein levels are uniformly reduced whilst Pax7 protein levels are upregulated in Pax3neo/neo embryos
As Pax3 reduction is a global phenomenon in Pax3neo/neo embryos but the defects are not evident in all Pax3-expressing lineages, we investigated whether Pax3 protein was globally reduced and/or if specific Pax3-expressing lineages were absent (Fig. 6A–H). Consistent with in situ hybridization analysis (Fig. 4A,B), Pax3 was detected in E10.5 NT, somites and proximal dorsal-lateral limb bud in wildtypes. Specifically, Pax3 was evenly distributed throughout the entire somite. However, in Pax3neo/neo embryos, Pax3 protein is largely absent in the dorsolateral somitic (hypaxial myotome) cells that are destined to migrate into the limb bud to form muscles (Fig. 6E) and from the occipital myotomes (data not shown). In E11.5 Pax3neo/neo dermomyotome, the lateral region is absent and thus Pax3 expression is diminished (Fig. 6H). However, Pax3 is spatiotemporally normally expressed in hypomorphic NT but is down-regulated (Fig. 6F). Few, if any, Pax3-positive cells were detected in E10.5 Pax3neo/neo limb bud (Fig. 6G).
Given Pax3 is most closely related to Pax7 and they represent a Pax subfamily (Buckingham & Relaix, 2006) and during somitogenesis Pax7 is activated in the dermomyotome after Pax3, but before the onset of expression of muscle regulatory factors (Jostes et al., 1990), we investigated whether Pax7 protein is up-regulated in a cell lineage-specific manner that could partially compensate for reduced Pax3 expression. We checked Pax7 expression at E10.5 via immunohistochemistry and Western analysis using a Pax7-specific antibody (Fig. 6I–M). In wildtypes, both somitic and dorsal NT expression was observed, but Pax7 expression was mainly restricted to the medial NT with the dorsal-most region being negative (Fig. 6J). However in Pax3neo/neo embryos, Pax7 was elevated in both the somites and the NT (Fig. 6K,L). Additionally, Pax7-expressing zone was dorsally expanded in the NT. This up-regulation of Pax7 was confirmed by Western analysis of E10.5 whole embryo lysates showing a ~6-fold increase of Pax7 (densitometry analysis via ImageJ) in Pax3neo/neo embryos (Fig. 6M). In order to test whether upregulation of Pax7 levels is invariably a consequence of reduced Pax3 levels, Pax7 expression levels were analyzed via Western analysis of E10.5 Sp2H/Sp2H embryos (Fig. 6M). Pax7 levels are not upregulated but rather reduced in Pax3 null embryos, indicating that Pax7 levels are differentially modulated via reduced Pax3 levels as opposed to absent Pax3.
Reduced Pax3 levels do not affect heart development
Given Pax3 null mutation results in PTA and associated VSD (Auerbach 1954; Franz et al., 1989; Conway et al, 1997a & b; Conway et al., 2003; Engleka et al., 2005), the reduction of Pax3 expression and neonatal death prompted us to investigate whether Pax3neo/neo pups had heart defects. Both wholemount and histological examination of Pax3neo/neo hearts at E14.5, a stage at which both ventricular septation and outflow tract division are normally complete, reveals that Pax3neo/neo hearts are normal (n=10). Specifically, separate aortic and pulmonary outlet vessels are present, correctly aligned and exit the appropriate ventricle, whilst the muscular and membranous interventricular septum is closed. Furthermore, the thickness of ventricular and atrial walls and ventricular septum also all appear unaffected by reduced Pax3 levels (Supplemental Fig. 1A–D). Given that 100% of the Pax3neo/neo neonates die shortly after birth and the presence of patent ductus arteriosus in partially-rescued transgenic Sp homozygous newborns (Li et al., 1999), we also analyzed the ability of the in utero Pax3neo/neo cardiovascular system to adapt to postnatal requirements. Histology revealed the Pax3neo/neo ductus arteriosus and foramen ovale were both closed within 6 hours of birth (not shown), and thus the Pax3neo/neo newborn pups were able to increase arterial oxygen tension upon delivery, a process known to trigger closure of the ductus arteriosus (Conway et al., 2003). Collectively, these analyses demonstrated that Pax3neo/neo hearts were structurally (and probably functionally) normal, given the healthy appearance of the newborns and intact circulation (Fig. 3A).
The cardiac NC is known to be dependent upon Pax3 and be required for normal outflow tract morphogenesis. Indeed, failure of migration, proliferation, colonization and/or differentiation can lead to pathogenesis of PTA and associated VSD (Conway et al., 1997a & b; Snider et al., 2007). The normal outflow tract morphogenesis observed in Pax3neo/neo suggests that either cardiac NC cells are affected but are present in sufficient numbers to support outflow tract septation or that the cardiac NC lineage is unaffected by reduced Pax3 expression. To distinguish which of these possibilities is present in Pax3neo/neo embryos, we performed in situ hybridization to examine molecular markers of NC morphogenesis (Supplemental Fig. 1E-J). We have previously shown that Crabp1 expression is unaltered in Pax3neo/neo NT, and wholemount analysis reveals there are also an equivalent number and appropriate spatiotemporal migration route taken by Pax3neo/neo cardiac NC (data not shown). Similarly, expression of Periostin (Supplemental Fig. 1G,H) and Sox10 (Supplemental Fig. 1I,J), markers of differentiated NC-derived outflow tract mesenchyme and Schwann cells respectively (Britsch et al., 2001; Lindsley et al., 2007), are comparably expressed in Pax3neo/neo and wildtype dorsal root ganglion and outflow tract. Thus, molecular marker analysis indicates the cardiac NC lineage is unaffected by reduction of Pax3 in Pax3neo/neo embryos.
Diaphragmatic muscle morphogenesis tolerates reduced Pax3 levels
In vertebrates, all skeletal muscles are derived from the paraxial mesoderm. Some muscle groups develop in situ, such as the epaxial muscles of the trunk, which form the deep muscles of the back. In contrast, several muscle groups derive from cells that migrate over long distances as undifferentiated precursor myoblasts before undergoing myogenesis and fusing. Hypaxial muscles including those of the limb, the ventral body wall, the tongue and the diaphragm fall into this category (Epstein et al., 1996; Schäfer & Braun 1999; Buckingham et al., 2003). Given the diaphragm is severely affected by various Pax3 null mutations and its function is vital for postnatal survival, we also analyzed potential effects of reduced Pax3 expression upon the Pax3neo/neo diaphragm using both histology and immunohistochemistry. In contrast to the hypoplastic diaphragm reported in Pax3 null mutants (Li et al., 1999; Engleka et al., 2005), the Pax3neo/neo diaphragm is indistinguishable from that of littermate controls with respect to its appearance and thickness. Furthermore, immunostaining of E15.5 differentiated myofibers using αSMA revealed the Pax3neo/neo diaphragm and ventral body wall were also indistinguishable from that of controls, indicating they had appropriately undergone muscularization (Supplemental Fig. 2). Similarly, the newborn diaphragm was normal (not shown).
To further document that the diaphragm was functionally normal in Pax3neo/neo animals, we analyzed neonatal lungs collected 6 hours after delivery. The Pax3neo/neo and control littermate lungs were also indistinguishable, with both genotypes showing inflated and similar-sized alveoli (Supplemental Fig. 2C,F) indicating respiration had been initiated in Pax3neo/neo animals. We next examined MyoD expression at E15.5 by in situ hybridization to assess whether whether MyoD is normally switched on. Consistent with αSMA staining, MyoD is robustly expressed in Pax3neo/neo intercostal muscles of the ventral body wall and diaphragm with a similar intensity as wildtypes (Supplemental Fig. 2G,H). These analyses of the heart and diaphragm were consistent with gross observation of Pax3neo/neo pups, which exhibited a healthy coloration and a smooth rhythm of respiration similar to their control littermates. Given Pax3neo/neo pups did not appear cyanotic due to respiration failure or pale due to compromised circulation. This suggests both their diaphragm and cardiopulmonary systems are intact and functionally normal. Taken together, these analyses demonstrate that ~20% Pax3 protein was sufficient to initiate/maintain normal organogenesis of heart and diaphragm.
DISCUSSION
Our analysis of the Pax3neo/neo hypomorphic defects clearly indicates that long-range myoblast migration into the limb and tongue are particularly sensitive to reduced Pax3 levels, but that long-range myoblast migration to diaphragm, intercostal, body wall and trunk are able to tolerate an 80% reduction in Pax3. Furthermore, NT closure and NC-derived tissues and organs are all also unaffected by reduced Pax3 expression, indicating that ~20% Pax3 protein levels are sufficient to initiate/maintain all non-myogenic biological processes. Indeed, Pax3 dosage-dependent anomalies have been observed for decades, as heterozygous loss-of-function PAX3 mutation results in pigmentary disturbances in both mouse and human and cochlear deafness in man (Tassabehji et al., 1992). These Pax3 mutant data clearly indicate a ~50% loss of Pax3 protein can affect melanocytes and ear pigment cells. Given we can now further titrate Pax3 expression levels, it is interesting to note that adult Pax3neo/+ (~60% Pax3 levels) mice do not exhibit any pigmentation defects, however, further studies will be required to test whether a ~10% difference in Pax3 levels or potential genetic background effects account for this intriguing result. Although it is critically dependent upon Pax3 and the initial site of Pax3 expression in the embryo (Goudling et al., 1991), the dorsal neural folds/NT and NC lineages derived thereof, appear to be unaffected by ~80% reduction in Pax3 levels. This clearly establishes that there are different minimum threshold level requirements for Pax3 amongst the various Pax3-expressing lineages. Significantly, characterization of a Pax3–engrailed fusion hypomorphic allele by Bajard et al. found a similar attenuated mutant phenotype, with partial conservation of the hypaxial somite and its myogenic derivatives, including some hindlimb muscles and NT closure defects (Bajard et al., 2006). However, the Pax3–engrailed fusion hypomorphic allele does not lead to extensive loss of myogenic progenitor cells in the hypaxial somite, although the migration of these cells to the limb buds was affected. This indicates that neomycin (acts via suppression of Pax3 levels) and Pax3–engrailed fusion (acts via interference of Pax3 function) alleles each result in overlapping but distinct anomalies.
In terms of the myogenic defects, the titration of Pax3 expression levels down to ~20% has revealed that Pax3-expressing epaxial muscles are unaffected, and that only some hypaxial-derived muscles are affected. As the hypaxial musculature (i.e., limb muscles, tongue, diaphragm, intercostal and abdominal wall muscles) are all derived from ventrolateral regions of the dermomyotome (Ordahl & Le Dourain, 1992; Christ & Ordahl, 1995; Ordahl & Williams, 1998; Kablar & Rudnicki, 2000), it is surprising that only the limb and tongue muscles were hypoplastic. Furthermore, Pax3neo/neo forelimb muscle hypoplasia is significantly more severe than that observed in hindlimbs, and that dorsal limb muscle hypoplasia is more severe than ventral limb muscle hypoplasia. Significantly, localized muscle deficiencies in Pax3neo/neo can be observed as early as E10.5 in the limb and E12.5 in craniofacial region. Underlying this muscle hypoplasia is an elevation of apoptosis within E9.5–10.5 hypomorphic somites, that results in a drastic reduction of long-range migratory progenitor hypaxial myoblasts and consequently reduced colonization of the limbs and tongue. Although we detected a significant increase in apoptotic cells in Pax3neo/neo somites, we are unable to demonstrate whether apoptosis is confined to distinct populations that would normally give rise to the specific affected muscular masses in tongue and limbs. Further detailed molecular marker analysis and lineage mapping studies will be required to address this possibility. As Pax3 is normally expressed in both dorsal and ventral colonizing myoblasts in both the fore- and hindlimbs and the remaining hypomorphic tongue and forelimb flexor muscle precursors switch on normal Myf5 and MyoD expression, the Pax3neo/neo hypomorphic partial hypaxial muscle defects may be due to: 1) hypaxial muscles existing as distinct muscle precursor cells, specified via different Pax3 expression levels; 2) hypaxial precursors depending on Pax3-regulated lineage-specific environmental cues that require different Pax3 thresholds; 3) dependence upon lineage-specific positional signals; or 4) partial genetic compensation via other Pax family members.
Similar to the Pax3neo/neo hypomorphic limb anomalies, compound Eya1/2 mouse mutants exhibit more severe muscle hypoplasia of the forelimbs than the hindlimbs (Grifone et al., 2007). Eya1, Eya2 and Eya4 are all co-expressed with Pax3 during mouse somitogenesis at the limb level and are thought to lie genetically upstream of Pax3 in the formation of ventrolateral dermomyotome hypaxial region. Additionally, a similar spectrum of severe extensor muscle hypoplasia but normal flexor muscles observed in Pax3neo/neo was reported in Lbx1h null embryos that ectopically express Pax3 in migratory myoblasts, but not in malformed Lbx1h null limbs (Schäfer & Braun, 1999). Indeed, both Lbx1 and Lbx1h mutants have hypoplastic musculature in limbs, but the tongue and diaphragm appear unaffected (Schafer & Braun, 1999; Gross et al., 2000). Lbx1h is specifically expressed in migrating muscle cells and is thought to be a key regulator of muscle precursor cell migration required for the acquisition of dorsal identities of forelimb muscles. Combined, these data indicate that specification of the various hypaxial muscle groups is probably dependent upon coordinated and lineage-specific pathways that are differentially sensitive to alterations in gene dosage.
As Pax3 is required for the activation of c-Met (Epstein et al., 1996; Yang et al., 1996; Relaix et al., 2003), which is important for delamination and migration of muscle progenitor cells (Bladt et al., 1995), it is not surprising that c-Met mRNA expression is diminished in the Pax3neo/neo limb buds. In fact, c-Met nulls phenocopy the Sp2H/Sp2H mutant muscle defects and exhibit a generalized loss of musculature in limb, tongue, diaphragm and body wall (Bladt et al., 1995; Prunotto et al., 2004). Although it is not known whether different Pax3 levels can differentially activate c-Met and reduced levels of c-Met are still detectable by RT-PCR in Pax3 mutant limb buds (Yang et al., 1996), future lineage-restricted and temporally-regulated Pax3 deletion and assessment of c-Met expression is required to test whether different Pax3 thresholds specify different Pax3-dependent hypaxial subpopulations and/or whether c-Met is able to respond differently to various Pax3 levels. Similarly, although there are no data to suggest any differences in Pax3 expression between fore- and hindlimbs, it is known that limb-specific expression of Pitx1, Tbx4, and Tbx5 regulates the determination of limb identity (Margulies et al., 2001). Thus, the finding that forelimb muscles are more severely affected than the hindlimb hypaxial muscles, may similarly reflect lineage-specific positional signaling and/or that the forelimbs develop earlier than hindlimbs and the existence of compensatory pathways in developmentally older embryos.
In terms of genetic compensation via other Pax family members, of particular interest is the observation that Pax3 can be partially replaced by Pax7 in the mouse (Relaix et al., 2004). Specifically, Pax7 can substitute for Pax3 function in dorsal NT, NC cell, and somite development, but not in the formation of muscles involving long-range migration of muscle progenitor cells as these mutants die perinatally. Similar to the Pax3neo/neo hypomorph, the severity of the limb muscle phenotype increases as the number of Pax7 replacement alleles is reduced, with the hypomorphic forelimb more affected than the hindlimb (Relaix et al., 2004). Expression studies have shown that Pax7 is not expressed in myoblasts migrating from the occipital trunk somites to the tongue primordium and is not expressed in limb myoblasts until E11.5 as the limb muscles develop (Jostes et al., 1990; Relaix et al., 2004; Horst et al., 2006). Thus, elevated Pax7 would not be able to compensate for reduced Pax3 levels in these specific hypaxial lineages. However, in the myoblasts that migrate to the septum transversum (which partly gives rise to the diaphragm), Pax3 and Pax7 are co-expressed during myoblast migration (Gross et al., 2000). Thus, the defects observed in Pax3neo/neo intrinsic tongue muscles and limb musculature, but not in the diaphragm and trunk, most likely reflect the fact that Pax7 co-expression can compensate for 80% reduction in Pax3 levels and rescue the myoblasts that colonize the diaphragm. Given that Pax7 can prevent apoptosis (Borycki et al., 1999; Kassar-Duchossoy et al., 2005), these data could suggest that compensatory Pax7 upregulation in Pax3neo/neo may be sufficient to overcome commitment to the apoptotic pathway. The lack of Pax3neo/neo NT and NC-associated anomlies most likely reflects that these Pax3-expressing lineages are “rescued” via the compensatory upregulation of Pax7 within the dorsal NT. Thus, the limited lineage-restricted hypomorphic defects observed in Pax3neo/neo embryos, reflect those tissues in which spatiotemporally coordinated Pax7 upregulation is not able to compensate for 80% reduced Pax3 dosage levels.
Taken together, the similarities in Pax3 and Pax7 expression, structure, and function suggest that, while the two gene products perform distinct functions, there is overlap. Future conditional Pax3 and Pax7 deletion and compound mutational studies will be required to dissect their relative roles during myogenesis.
Supplementary Material
Acknowledgements
We are grateful to Dr. C-M Fan, Carnegie Institution of Washington for providing the Pax7 antibody and MyoD, Myf5 and c-Met probes, and Dr. Robert Maxson, USC for the Msx1/2 probes. The Pax3 and Pax7 monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. These studies were supported, in part, by the Riley Children’s Foundation (H.Z.), the IU Department of Pediatrics/Cardiology and National Institutes of Health grant HL60714 (S.J.C).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Auerbach C. After-effects of radiation and chemicals on chromosomes and genes. Br J Radiol. 1954;314:122–124. doi: 10.1259/0007-1285-27-314-122. [DOI] [PubMed] [Google Scholar]
- Bajard L, Relaix F, Lagha M, Rocancourt D, Daubas P, Buckingham ME. A novel genetic hierarchy functions during hypaxial myogenesis: Pax3 directly activates Myf5 in muscle progenitor cells in the limb. Genes Dev. 2006;20:2450–2464. doi: 10.1101/gad.382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin CT, Hoth CF, Amos JA, da-Silva EO, Milunsky A. An exonic mutation in the HuP2 paired domain gene causes Waardenburg's syndrome. Nature. 1992;355:637–638. doi: 10.1038/355637a0. [DOI] [PubMed] [Google Scholar]
- Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- Bober E, Brand-Saberi B, Ebensperger C, Wilting J, Balling R, Paterson BM, Arnold HH, Christ B. Initial steps of myogenesis in somites are independent of influence from axial structures. Development. 1994;120:3073–3082. doi: 10.1242/dev.120.11.3073. [DOI] [PubMed] [Google Scholar]
- Borycki AG, Li J, Jin F, Emerson CP, Epstein JA. Pax3 functions in cell survival and in pax7 regulation. Development. 1999;126:1665–1674. doi: 10.1242/dev.126.8.1665. [DOI] [PubMed] [Google Scholar]
- Brand-Saberi B. Genetic and epigenetic control of skeletal muscle development. Ann Anat. 2005;187:199–207. doi: 10.1016/j.aanat.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Daubas P, Esner M, Lagha M, Relaix F, Rocancourt D. Myogenic progenitor cells in the mouse embryo are marked by the expression of Pax3/7 genes that regulate their survival and myogenic potential. Anat Embryol (Berl) 2006;211 Suppl 1:51–56. doi: 10.1007/s00429-006-0122-0. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Relaix F. The Role of Pax Genes in the Development of Tissues and Organs: Pax3 and Pax7 Regulate Muscle Progenitor Cell Functions. Annu Rev Cell Dev Biol. 2006 doi: 10.1146/annurev.cellbio.23.090506.123438. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Chan WY, Cheung CS, Yung KM, Copp AJ. Cardiac neural crest of the mouse embryo: axial level of origin, migratory pathway and cell autonomy of the splotch (Sp2H) mutant effect. Development. 2004;131:3367–3379. doi: 10.1242/dev.01197. [DOI] [PubMed] [Google Scholar]
- Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature. 2005;433:317–322. doi: 10.1038/nature03126. [DOI] [PubMed] [Google Scholar]
- Chi N, Epstein JA. Getting your Pax straight: Pax proteins in development and disease. Trends Genet. 2002;18:41–47. doi: 10.1016/s0168-9525(01)02594-x. [DOI] [PubMed] [Google Scholar]
- Christ B, Ordahl CP. Early stages of chick somite development. Anat Embryol (Berl) 1995;191:381–396. doi: 10.1007/BF00304424. [DOI] [PubMed] [Google Scholar]
- Conway SJ. In Situ Hybridization of Cell and Tissue Sections, Chapter 15. In: Cotter FE, editor. Methods in Molecular Medicine, Molecular Diagnosis of Cancer. Totowa, NJ, USA: Humana Press Inc.; 1996. pp. 193–206. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997a;124:505–514. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Kirby ML, Anderson RH, Copp AJ. Development of a lethal congenital heart defect in the splotch (Pax3) mutant mouse. Cardiovasc Res. 1997b;36:163–173. doi: 10.1016/s0008-6363(97)00172-7. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Godt RE, Hatcher CJ, Leatherbury L, Zolotouchnikov VV, Brotto MA, Copp AJ, Kirby ML, Creazzo TL. Neural crest is involved in development of abnormal myocardial function. J Mol Cell Cardiol. 1997c;29:2675–2685. doi: 10.1006/jmcc.1997.0499. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc Res. 2000;47:314–328. doi: 10.1016/s0008-6363(00)00098-5. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35:1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- Dickman ED, Rogers R, Conway SJ. Abnormal skeletogenesis occurs coincident with increased apoptosis in the Splotch (Sp2H) mutant: putative roles for Pax3 and PDGFRalpha in rib patterning. Anat Rec. 1999;255:353–361. doi: 10.1002/(SICI)1097-0185(19990701)255:3<353::AID-AR11>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Dressler GR, Wilkinson JE, Rothenpieler UW, Patterson LT, Williams-Simons L, Westphal H. Deregulation of Pax-2 expression in transgenic mice generates severe kidney abnormalities. Nature. 1993;362:65–67. doi: 10.1038/362065a0. [DOI] [PubMed] [Google Scholar]
- Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA, Epstein JA. Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Dev Biol. 2005;280:396–406. doi: 10.1016/j.ydbio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, Lo CW. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127:1869–1878. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Shapiro DN, Cheng J, Lam PY, Maas RL. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci U S A. 1996;93:4213–4218. doi: 10.1073/pnas.93.9.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax3. Cell. 1991;67:767–774. doi: 10.1016/0092-8674(91)90071-6. [DOI] [PubMed] [Google Scholar]
- Franz T. Persistent truncus arteriosus in the Splotch mutant mouse. Anat Embryol (Berl) 1989;180:457–464. doi: 10.1007/BF00305120. [DOI] [PubMed] [Google Scholar]
- Franz T, Kothary R, Surani MA, Halata Z, Grim M. The Splotch mutation interferes with muscle development in the limbs‥. Anat Embryol (Berl) 1993;187:153–160. doi: 10.1007/BF00171747. [DOI] [PubMed] [Google Scholar]
- Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10:1135–1147. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding M, Lumsden A, Paquette AJ. Regulation of Pax-3 expression in the dermomyotome and its role in muscle development. Development. 1994;120:957–971. doi: 10.1242/dev.120.4.957. [DOI] [PubMed] [Google Scholar]
- Greene ND, Copp AJ. Mouse models of neural tube defects: investigating preventive mechanisms. Am J Med Genet C Semin Med Genet. 2005;135:31–41. doi: 10.1002/ajmg.c.30051. [DOI] [PubMed] [Google Scholar]
- Grifone R, Demignon J, Giordani J, Niro C, Souil E, Bertin F, Laclef C, Xu PX, Maire P. 1 and Eya2 proteins are required for hypaxial somitic myogenesis in the mouse embryo. Dev Biol. 2007;302:602–616. doi: 10.1016/j.ydbio.2006.08.059. [DOI] [PubMed] [Google Scholar]
- Gross MK, Moran-Rivard L, Velasquez T, Nakatsu MN, Jagla K, Goulding M. Lbx1 is required for muscle precursor migration along a lateral pathway into the limb. Development. 2000;127:413–424. doi: 10.1242/dev.127.2.413. [DOI] [PubMed] [Google Scholar]
- Hammond CL, Hinits Y, Osborn DP, Minchin JE, Tettamanti G, Hughes SM. Signals and myogenic regulatory factors restrict pax3 and pax7 expression to dermomyotome-like tissue in zebrafish. Dev Biol. 2007;302:504–521. doi: 10.1016/j.ydbio.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson IM, Fletcher JM, Jordan T, Brown A, Taylor D, Adams RJ, Punnett HH, van Heyningen V. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters' anomaly. Nat Genet. 1994;6:168–173. doi: 10.1038/ng0294-168. [DOI] [PubMed] [Google Scholar]
- Henderson DJ, Conway SJ, Copp AJ. Rib truncations and fusions in the Sp2H mouse reveal a role for Pax3 in specification of the ventro-lateral and posterior parts of the somite. Dev Biol. 1999;209:143–158. doi: 10.1006/dbio.1999.9215. [DOI] [PubMed] [Google Scholar]
- Hill R, Van Heyningen V. Mouse mutations and human disorders are paired. Trends Genet. 1992;8:119–120. doi: 10.1016/0168-9525(92)90359-C. [DOI] [PubMed] [Google Scholar]
- Horst D, Ustanina S, Sergi C, Mikuz G, Juergens H, Braun T, Vorobyov E. Comparative expression analysis of Pax3 and Pax7 during mouse myogenesis. Int J Dev Biol. 2006;50:47–54. doi: 10.1387/ijdb.052111dh. [DOI] [PubMed] [Google Scholar]
- Jostes B, Walther C, Gruss P. The murine paired box gene, Pax7, is expressed specifically during the development of the nervous and muscular system. Mech Dev. 1990;33:27–37. doi: 10.1016/0925-4773(90)90132-6. [DOI] [PubMed] [Google Scholar]
- Jun S, Desplan C. Cooperative interactions between paired domain and homeodomain. Development. 1996;122:2639–2650. doi: 10.1242/dev.122.9.2639. [DOI] [PubMed] [Google Scholar]
- Kablar B, Rudnicki MA. Skeletal muscle development in the mouse embryo. Histol Histopathol. 2000;15:649–656. doi: 10.14670/HH-15.649. [DOI] [PubMed] [Google Scholar]
- Kassar-Duchossoy L, Giacone E, Gayraud-Morel B, Jory A, Gomès D, Tajbakhsh S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev. 2005;19:1426–1431. doi: 10.1101/gad.345505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SA, Jones JM, Boyle A, Barrow LL, Killen PD, Green DG, Kapousta NV, Hitchcock PF, Swank RT, Meisler MH. Kidney and retinal defects (Krd), a transgene-induced mutation with a deletion of mouse chromosome 19 that includes the Pax2 locus. Genomics. 1994;23:309–320. doi: 10.1006/geno.1994.1506. [DOI] [PubMed] [Google Scholar]
- Kim J, Lauderdale JD. Analysis of Pax6 expression using a BAC transgene reveals the presence of a paired-less isoform of Pax6 in the eye and olfactory bulb. Dev Biol. 2006;292:486–505. doi: 10.1016/j.ydbio.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Kist R, Watson M, Wang X, Cairns P, Miles C, Reid DJ, Peters H. Reduction of Pax9 gene dosage in an allelic series of mouse mutants causes hypodontia and oligodontia. Hum Mol Genet. 2005;14:3605–3617. doi: 10.1093/hmg/ddi388. [DOI] [PubMed] [Google Scholar]
- Koushik SV, Chen H, Wang J, Conway SJ. Generation of a conditional loxP allele of the Pax3 transcription factor that enables selective deletion of the homeodomain. Genesis. 2002;32:114–117. doi: 10.1002/gene.10051. [DOI] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–758. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- Kuang S, Chargé SB, Seale P, Huh M, Rudnicki MA. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol. 2006;172:103–113. doi: 10.1083/jcb.200508001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe R, Kuratani S. Evolutionary perspectives from development of mesodermal components in the lamprey. Dev Dyn. 2007 doi: 10.1002/dvdy.21177. [DOI] [PubMed] [Google Scholar]
- Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, Ishii M, Sangiorgi FO, Rauchman M, Sucov HM, Maas RL, Maxson RE. Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129:527–538. doi: 10.1242/dev.129.2.527. [DOI] [PubMed] [Google Scholar]
- Lang D, Lu MM, Huang L, Engleka KA, Zhang M, Chu EY, Lipner S, Skoultchi A, Millar SE, Epstein JA. Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature. 2005;433:884–887. doi: 10.1038/nature03292. [DOI] [PubMed] [Google Scholar]
- Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126:2495–2503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- Lindsley A, Snider P, Zhou H, Rogers R, Wang J, Olaopa M, Kruzynska-Frejtag A, Koushik SV, Lilly B, Burch JB, Firulli AB, Conway SJ. Identification and characterization of a novel Schwann and outflow tract endocardial cushion lineage-restricted periostin enhancer. Dev Biol. 2007 doi: 10.1016/j.ydbio.2007.04.041. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeken MR. Current perspectives on the causes of neural tube defects resulting from diabetic pregnancy. Am J Med Genet C Semin Med Genet. 2005;135:77–87. doi: 10.1002/ajmg.c.30056. [DOI] [PubMed] [Google Scholar]
- Macchia PE, Lapi P, Krude H, Pirro MT, Missero C, Chiovato L, Souabni A, Baserga M, Tassi V, Pinchera A, Fenzi G, Grüters A, Busslinger M, Di Lauro R. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet. 1998;19:83–86. doi: 10.1038/ng0598-83. [DOI] [PubMed] [Google Scholar]
- Machado AF, Martin LJ, Collins MD. Pax3 and the splotch mutations: structure, function, and relationship to teratogenesis, including gene-chemical interactions. Curr Pharm Des. 2001;7:751–785. doi: 10.2174/1381612013397726. [DOI] [PubMed] [Google Scholar]
- Mansouri A, Goudreau G, Gruss P. Pax genes and their role in organogenesis. Cancer Res. 1999;59:1707–1709. [PubMed] [Google Scholar]
- Mansouri A, Stoykova A, Torres M, Gruss P. Dysgenesis of cephalic neural crest derivatives in Pax7−/− mutant mice. Development. 1996;122:831–838. doi: 10.1242/dev.122.3.831. [DOI] [PubMed] [Google Scholar]
- Margulies EH, Kardia SL, Innis JW. A comparative molecular analysis of developing mouse forelimbs and hindlimbs using serial analysis of gene expression (SAGE) Genome Res. 2001;10:1686–1698. doi: 10.1101/gr.192601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Nagy A, Moens C, Ivanyi E, Pawling J, Gertsenstein M, Hadjantonakis AK, Pirity M, Rossant J. Dissecting the role of N-myc in development using a single targeting vector to generate a series of alleles. Curr Biol. 1998;8:661–664. doi: 10.1016/s0960-9822(98)70254-4. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Busslinger M. Monoallelic expression of Pax5: a paradigm for the haploinsufficiency of mammalian Pax genes? Biol Chem. 1999;380:601–611. doi: 10.1515/BC.1999.077. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Vambrie S, Steinlein P, Kozmik Z, Rolink A, Weith A, Busslinger M. Independent regulation of the two Pax5 alleles during B-cell development. Nat Genet. 1999;21:390–395. doi: 10.1038/7720. [DOI] [PubMed] [Google Scholar]
- Ordahl CP, Le Douarin NM. Two myogenic lineages within the developing somite. Development. 1992;114:339–353. doi: 10.1242/dev.114.2.339. [DOI] [PubMed] [Google Scholar]
- Ordahl CP, Williams BA. Knowing chops from chuck: roasting myoD redundancy. Bioessays. 1998;20:357–362. doi: 10.1002/(SICI)1521-1878(199805)20:5<357::AID-BIES1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Otto A, Schmidt C, Patel K. Pax3 and Pax7 expression and regulation in the avian embryo. Anat Embryol (Berl) 2006;211:293–310. doi: 10.1007/s00429-006-0083-3. [DOI] [PubMed] [Google Scholar]
- Oustanina S, Hause G, Braun T. Pax7 directs postnatal renewal and propagation of myogenic satellite cells but not their specification. EMBO J. 2004;23:3430–3439. doi: 10.1038/sj.emboj.7600346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3- dependent development and tumorigenesis. Genes Dev. 2002;16:676–680. doi: 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunotto C, Crepaldi T, Forni PE, Ieraci A, Kelly RG, Tajbakhsh S, Buckingham M, Ponzetto C. Analysis of Mlc-lacZ Met mutants highlights the essential function of Met for migratory precursors of hypaxial muscles and reveals a role for Met in the development of hyoid arch-derived facial muscles. Dev Dyn. 2004;231:582–591. doi: 10.1002/dvdy.20177. [DOI] [PubMed] [Google Scholar]
- Relaix F, Polimeni M, Rocancourt D, Ponzetto C, Schäfer BW, Buckingham M. The transcriptional activator PAX3-FKHR rescues the defects of Pax3 mutant mice but induces a myogenic gain-of-function phenotype with ligand-independent activation of Met signaling in vivo. Genes Dev. 2003;17:2950–2965. doi: 10.1101/gad.281203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F, Rocancourt D, Mansouri A, Buckingham M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004;18:1088–1105. doi: 10.1101/gad.301004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435:948–953. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- Sanyanusin P, McNoe LA, Sullivan MJ, Weaver RG, Eccles MR. Mutation of PAX2 in two siblings with renal-coloboma syndrome. Hum Mol Genet. 1995;4:2183–2184. doi: 10.1093/hmg/4.11.2183. [DOI] [PubMed] [Google Scholar]
- Schäfer K, Braun T. Early specification of limb muscle precursor cells by the homeobox gene Lbx1h. Nat Genet. 1999;23:213–216. doi: 10.1038/13843. [DOI] [PubMed] [Google Scholar]
- Schedl A, Ross A, Lee M, Engelkamp D, Rashbass P, van Heyningen V, Hastie ND. Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell. 1996;86:71–82. doi: 10.1016/s0092-8674(00)80078-1. [DOI] [PubMed] [Google Scholar]
- Schubert FR, Tremblay P, Mansouri A, Faisst AM, Kammandel B, Lumsden A, Gruss P, Dietrich S. Early mesodermal phenotypes in splotch suggest a role for Pax3 in the formation of epithelial somites. Dev Dyn. 2001;222:506–521. doi: 10.1002/dvdy.1211. [DOI] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Snider P, Olaopa M, Firulli AB, Conway SJ. Cardiovascular development and the colonizing cardiac neural crest lineage. TSW Development & Embryology. 2007;2 doi: 10.1100/tsw.2007.189. ePub ahead of press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soejima H, Fujimoto M, Tsukamoto K, Matsumoto N, Yoshiura KI, Fukushima Y, Jinno Y, Niikawa N. Three novel PAX3 mutations observed in patients with Waardenburg syndrome type 1. Hum Mutat. 1997;9:177–180. doi: 10.1002/(SICI)1098-1004(1997)9:2<177::AID-HUMU11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Stockton DW, Das P, Goldenberg M, D'Souza RN, Patel PI. Mutation of PAX9 is associated with oligodontia. Nat Genet. 2000;24:18–19. doi: 10.1038/71634. [DOI] [PubMed] [Google Scholar]
- Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–715. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh S, Buckingham M. The birth of muscle progenitor cells in the mouse: spatiotemporal considerations. Curr Top Dev Biol. 2000;48:225–268. doi: 10.1016/s0070-2153(08)60758-9. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Read AP, Newton VE, Harris R, Balling R, Gruss P, Strachan T. Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature. 1992;355:635–636. doi: 10.1038/355635a0. [DOI] [PubMed] [Google Scholar]
- Terzić J, Saraga-Babić M. Expression pattern of PAX3 and PAX6 genes during human embryogenesis. Int J Dev Biol. 1999;43:501–508. [PubMed] [Google Scholar]
- Tremblay P, Dietrich S, Mericskay M, Schubert FR, Li Z, Paulin D. A crucial role for Pax3 in the development of the hypaxial musculature and the long-range migration of muscle precursors. Dev Biol. 1998;203:49–61. doi: 10.1006/dbio.1998.9041. [DOI] [PubMed] [Google Scholar]
- Tremblay P, Pituello F, Gruss P. Inhibition of floor plate differentiation by Pax3: evidence from ectopic expression in transgenic mice. Development. 1996;122:2555–2567. doi: 10.1242/dev.122.8.2555. [DOI] [PubMed] [Google Scholar]
- Underhill DA. Genetic and biochemical diversity in the Pax gene family. Biochem Cell Biol. 2000;78:629–638. [PubMed] [Google Scholar]
- van Raamsdonk CD, Tilghman SM. Dosage requirement and allelic expression of PAX6 during lens placode formation. Development. 2000;127:5439–5448. doi: 10.1242/dev.127.24.5439. [DOI] [PubMed] [Google Scholar]
- Venters SJ, Argent RE, Deegan FM, Perez-Baron G, Wong TS, Tidyman WE, Denetclaw WF, Marcelle C, Bronner-Fraser M, Ordahl CP. Precocious terminal differentiation of premigratory limb muscle precursor cells requires positive signalling. Dev Dyn. 2004;229:591–599. doi: 10.1002/dvdy.20016. [DOI] [PubMed] [Google Scholar]
- Waardenberg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet. 1951;3:195–253. [PMC free article] [PubMed] [Google Scholar]
- Wilm B, Dahl E, Peters H, Balling R, Imai K. Targeted disruption of Pax1 defines its null phenotype and proves haploinsufficiency. Proc Natl Acad Sci U S A. 1998;95:8692–8697. doi: 10.1073/pnas.95.15.8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XM, Vogan K, Gros P, Park M. Expression of the met receptor tyrosine kinase in muscle progenitor cells in somites and limbs is absent in Splotch mice. Development. 1996;122:2163–2171. doi: 10.1242/dev.122.7.2163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.