

Abstract
Dynorphins, glutamate, and glutamate-sensitive N-Methyl-d-Aspartate (NMDA) receptors exist in the mammalian cochlea. Dynorphins produce neural excitation and excitotoxic effects in the spinal cord through a κ-opioid facilitation of NMDA receptor sensitivity to glutamate. The κ-opioid receptor drug agonists N-dimethylallyl-normetazocine [(-)-pentazocine (50 mmol)] and trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]-benzeneacetamide [U-50488H (100 mmol)] were administered across the cochlear round window membrane in the chinchilla. Each drug produced significant post-baseline amplitude changes in the click-evoked auditory nerve compound action potential. Amplitude changes at threshold amounted to increases in sensitivity that ranged from 4-8 decibels, measured in sound pressure level (dB SPL). The large neural amplitude increases at threshold were accompanied by progressively smaller amplitude changes at 5 and 10 dB above threshold (dB SL). However, at stimulus intensities ≥ 20dB SL, post-baseline neural amplitudes were suppressed to levels below baseline and control values. These bi-phasic intensity-dependent neural amplitude changes have never before been observed following i.v. administered (-)-pentazocine in this species. Finally, the bi-phasic neural amplitude changes in U-50488H-treated (100 mmol) animals were partially blocked (except at 20dB SL), following a round window pretreatment with the NMDA receptor drug antagonist, dizocilpine hydrogen maleate [(+)-MK-801 (8 mmol)]. Our data suggests that endogenous dynorphins within lateral efferent olivocochlear neurons differentially modulate auditory neural excitation, possibly through cochlear NMDA receptors and glutamate. The role played by lateral efferent opioid neuromodulation at cochlear NMDA receptors, is discussed.
Keywords: (+)-MK-801 (dizocilpine hydrogen maleate), U-50488H, (-)-pentazocine, N-Methyl-d-Aspartate (NMDA) receptors, lateral efferent olivocochlear system, κ-opioid receptors, dynorphins, glutamate, auditory nerve, compound action potential
1.Introduction
Comparatively little is known regarding the function of the lateral efferent olivocochlear (LEOC) system. This system is recognized as originating from discrete nuclear regions near or surrounding the brainstem lateral superior olive (Azeredo et al., 1999; Pujol, 2001; Warr 1992). Lateral superior olivary nuclei give rise to a system of descending axons that terminate in the cochlea, in close proximity to spiral ganglion Type I auditory dendrites. Type I auditory dendrites in turn, innervate the cochlear inner hair cells (Guinan, 1996; Simmons, 2002; Warr et al., 1997). Recent evidence indicates that the LEOC system modulates afferent auditory nerve output by inducing both a suppression and an enhancement of cochlear neural responses independent of the organ of Corti, in some as yet, unknown and presumably complex manner (Groff and Liberman, 2003; Le Prell et al., 2003). Such evidence, taken together with anatomical investigations of lateral efferent axon termination patterns on Type I auditory dendrites (Liberman, 1990; Merchan-Perez and Liberman, 1996), as well as pharmacologic (Le Prell et al., 2004) and developmental evidence (Walsh et al., 1998) has suggested that the LEOC system participates in modulating auditory neural sensitivity (Liberman, 1988; 1990; McMahon et al., 2004).
Several LEOC neurotransmitter-modulators have been identified within the mammalian brainstem and cochlea, including endogenous neuroactive opioid dynorphins (Eybalin, 1993; Le Prell et al., 2001; Maison et al., 2003; Puel, 1995; Sewell, 1996; Simmons, 2002). Deca-, trideca- and heptadeca- peptide derivatives of prodynorphin synthesis coexist within LEOC brainstem nuclei of the guinea pig and rat (Abou-Madi et al., 1987; Altschuler et al., 1988). Dynorphin- and enkephalin-related opioid peptides are also co-distributed within descending LEOC fiber bundles and in the terminal varicosities that innervate Type I auditory dendrites in the cochlea (Abou-Madi et al., 1987; Altschuler et al., 1988; Eybalin, 1993; Puel, 1995; Safieddine et al., 1997). In the chinchilla brainstem, prodynorphin derivatives are also found within auditory nuclei and fiber pathways. For instance, moderate to intense immunoreactivity to α-neoendorphin and dynorphin-B, respectively, has been observed pericellularly in fibers and within cell bodies of neurons scattered throughout the lateral nucleus of the trapezoid body (Sahley et al., 1995). This brainstem nucleus contributes descending fibers as part of the LEOC system, and these fibers terminate in the cochlea (Sahley et al., 1997; Warr, 1992). Brainstem medial efferent olivocochlear neurons that innervate the cochlear outer hair cells are void of dynorphin-like activity in all species investigated (Eybalin, 1993; Puel, 1995), including the chinchilla (Sahley et al., 1995).
Neuroactive products of prodynorphin synthesis bind selectively to the ketocyclazocine (κ)-opioid receptor (Basbaum and Jessell, 2000; Gutstein and Akil, 2001). There is direct evidence that κ-opioid receptors exist in the cochlea of the rat (Jongkamonwiwat et al., 2003) and guinea pig (Jongkamonwiwat et al., 2006). In both species, κ-opioid receptor immunoreactivity was detected on the sensory Type I auditory dendrites that are anatomically positioned at the bases of the cochlear inner hair cells. While the presence of κ-opioid receptors has yet to be investigated in the chinchilla cochlea, pharmacological investigations in this species have demonstrated neural amplitude changes following i.v. administrations of κ-opioid receptor drug agonists. For instance, the mixed non-opioid/opioid receptor drug agonist (±)-pentazocine (16mg/kg), or the κ-opioid receptor enantiomer (-)-pentazocine (8mg/kg) (Sahley and Nodar, 1994; Sahley et al., 1991) have produced post-baseline intensity-dependent amplitude facilitations in the first negative peak of the auditory whole nerve compound action potential (CAP - N1). The neural amplitude facilitation was most prominent at threshold stimulus intensities, and was progressively less prominent as the stimulus was raised to 5 and 10 decibels above threshold (dB SL). Neural amplitude changes occurred in the absence of corresponding changes in the CAP - N1 peak latency, or in the amplitude of the auditory hair cell-generated cochlear microphonic (CM) potential. The intensity-dependent facilitation in the CAP - N1 amplitude following (-)-pentazocine was partially but significantly suppressed by an i.v. administration of the pure, non-specific opioid receptor-drug antagonist, naloxone HCl (5mg/kg) (Sahley et al., 1996a), or by a cochlear round window administration of the potent and specific κ-opioid receptor-drug antagonist, nor-binaltorphimine (4 mmol) (Sahley et al., 1996b). The non-opioid, sigma (σ)-receptor drug agonist (+)-pentazocine, and the powerful μ-opioid receptor drug agonists fentanyl and morphine produce no measurable changes in the same dependent measures (Sahley and Nodar, 1994; Sahley et al., 1991; Samra et al., 1984; 1985). While generally consistent with the excitatory properties reported for κ-opioid-receptor ligands (Arcaya et al., 1999; Crain and Shen, 1998; Laughlin et al., 1997), the intensity-dependent neural amplitude changes following i.v. (-)-pentazocine (Sahley and Nodar, 1994; Sahley et al., 1991; 1996a; 1996b) had suggested a more complex role for endogenous dynorphins in the peripheral auditory system.
Considerable evidence indicates that κ-opioid receptor ligands can directly interact with the N-Methyl-d-Aspartate (NMDA) receptor (Hauser et al., 1999; Shukla and Lemaire, 1994; Tang et al., 1999), quite possibly at multiple binding sites (Caudle and Dubner, 1998; Chen et al., 1995; Laughlin et al., 2001; Zhang et al., 1997). While many, but certainly not all of the reported properties of dynorphins at the NMDA receptor are excitatory, a distinct, high affinity dynorphin binding site has been detected on the NMDA receptor in rat cortex (Tang et al., 1999). The excitatory and often neurotoxic actions of dynorphins at NMDA receptors is well documented in dorsal horn neurons of the spinal cord (Hauser et al., 2001; Lai et al., 2001; Laughlin et al., 1997; 2001; Shukla and Lemaire, 1994). Dynorphins also produce anti-analgesic effects by way of interactions at the NMDA receptor (Arcaya et al., 1999). High dynorphin concentrations also result in a significant release of glutamate into the extracellular fluid in rat spinal cord (Skilling et al., 1992), and produces a significant dose-dependent increase in extracellular glutamate and aspartate in the rat hippocampus (Faden, 1992). Finally, dynorphins are reported to produce excitation in dorsal horn neurons by potentiating NMDA receptor-sensitivity to glutamate (Dubner and Ruda, 1992; Shukla and Lemaire, 1994; Skilling et al., 1992). From these investigations, it appears that high concentrations of κ-opioid receptor ligands can increase extracellular levels of glutamate and/or potentiate NMDA receptor-sensitivity to glutamate. Hence, the NMDA receptor appears to be a likely mediator of many non-analgesic actions of dynorphins (Lai et al., 2001; Laughlin et al., 1997; Vanderah et al., 1996).
It is also generally accepted that glutamate is the excitatory neurotransmitter responsible for neural signaling in the mammalian cochlea and in brainstem auditory pathways (Ehrenberger and Felix, 1995; Puel, 1995; Suneja et al., 1995a; 1995b; Usami et al., 2000). Ionotropic glutamate receptors are classified according to their pharmacology, molecular structure and biophysical properties. They consist of the NMDA receptor, as well as the non-NMDA subtypes; the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and kainate receptors (Dingledine et al., 1999; Kandel and Siegelbaum, 2000; Waxham, 2003). Cochlear NMDA receptors have been detected in a number of mammalian species. Their presence in the mammalian cochlea is supported by a body of evidence ranging from in situ hybridization histochemistry (Kuriyama et al., 1993; Niedzielski et al., 1997; Safieddine and Eybalin, 1992) to pharmacological (Chen et al., 2001; Ehrenberger and Felix, 1991; 1995; Felix and Ehrenberger, 1989; 1990; 1991; Ostreicher et al., 2002; Pujol et al., 1992; 1993), physiological (Kleinlogel et al., 1999; Puel et al., 1997) and electrophysiological investigations (d’Aldin et al., 1997; Puel et al., 1991). NMDA receptors have also been detected in the human cochlea (Nordang et al., 2000).
The present set of experiments was designed to examine amplitude and latency changes in the auditory whole nerve CAP- N1, as well as amplitude changes in the CM potential following the administration of κ-opioid receptor drug agonists into the cochlea. The two negative peaks (N1; N2) of the auditory whole nerve CAP is generated almost entirely within the cochlea by Type I auditory neurons (Brown et al., 2004; McMahon et al., 2004). The CAP-N1 peak amplitude represents neural activity and synchronization coincident with the onset discharges produced by single units within the peripheral auditory nerve (Brown et al., 2004; Miller et al., 1993). The CM potential is an extracellular, frequency- and intensity-dependent manifestation of a response to sound. Lacking a true threshold and exhibiting no measurable onset latency, the CM potential is accepted as a useful index of outer hair cell transduction (Sahley et al.,1997; Trautwein et al., 1996). All auditory potentials were generated at six stimulus intensities and were obtained repetitiously over a series of baseline and post-baseline (post-administration) recording periods.
Post-baseline recording was preceded by the administration of the potent and selective κ-opioid receptor drug agonists (-)-pentazocine (experiment 1) or U-50488H (experiments 2 and 3) delivered directly to the cochlear round window membrane. (-)-Pentazocine [N-dimethylallyl-normetazocine (TALWIN) MW = 403.5] is a highly lipophilic κ-opioid receptor drug agonist (Gutstein and Akil, 2001; Lahti et al., 1985; Tam, 1985; Walker et al., 1990) and U-50488H {trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]-benzeneacetamide; MW = 465.4} is a potent and specific κ-opioid receptor drug agonist (Lahti et al., 1985; North, 1986; Tam, 1985; Von Voigtlander 1983; Zukin and Zukin, 1984). Both (-)-pentazocine and U-50488H possess pharmacological properties that mimic the actions of endogenous dynorphins at κ-opioid receptors (Gutstein and Akil, 2001). Both κ-receptor drug agonists were administered at relatively high concentrations since NMDA receptors require a significant level of pre-synaptic activity before they can function maximally (Stricker and Huganir, 2002; Wisden et al., 2000). We also investigated the possibility that κ-opioid-induced changes in auditory neural potentials may be mediated by NMDA receptor activation. Therefore, the non-competitive open-channel (voltage-dependent) NMDA receptor antagonist (+)-MK-801 [dizocilpine hydrogen maleate {5R,10S-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclo-hepten-5,10-imine hydrogen maleate; MW = 337.4; pH = 7.4}] (Dingledine et al., 1999; Hollmann and Heinemann, 1994; McBain and Mayer, 1994) was delivered to the cochlear round window membrane 30 min prior to the post-baseline round window administration of U-50488H, in experiment 3.
2. Materials and methods
2.1 Animals
Adult male (N = 28) pigmented chinchillas (Chinchilla laniger) 0.5 to 1.5 years in age, weighing <550g (Mean = 487.5g; ± 59.3, S.D.) were obtained from a licensed vendor, for use in experiments 1 (n = 6), 2 (n = 12) and 3 (n = 10).
2.2 Auditory threshold verification
Normal hearing sensitivity was confirmed in each animal by obtaining auditory thresholds as indexed by far-field brainstem auditory evoked responses. Animals were anesthetized with tiletamine/zolazepam (TELAZOL; 20mg/kg i.m.) and tested for auditory thresholds in an Industrial Acoustics Corporation sound-attenuating chamber (<30dB) located within an electrically isolated laboratory. Bioelectric activity was differentially recorded with subdermal platinum-alloy needle electrodes (Grass E-2) using standard cephalic positions: vertex- (+), nasion- (-) and neck musculature- (common ground). Electrode impedances for both active electrodes were within 2-5kΩ. Bioelectric activity was amplified ×104 (Nicolet HGA-200A), band-pass filtered (Nicolet 501A) from 150 to 1500 Hz (6dB/octave roll off), and averaged on-line over a 10ms recording window with a Nicolet Clinical Averaging System (CA-1000). Rectangular electrical pulses (100μs duration) were delivered from a click generator (Nicolet 1007A) and negative-polarity clicks were presented at a high stimulus rate (68.3/s) from a ceramic microphone (Etymotic ER-3A), mounted through a form-fitting foam earplug that sealed the left external ear canal. The ER-3A microphone delivers a nearly flat frequency response from 250Hz to 3kHz (Beauchaine et al., 1987; Musiek and Baran, 1990). A signal attenuator (Grason Stadler #1292), calibrated in 1.3 (±0.1) dB steps additionally modified the 22 to 135dB range peak sound pressure level output (SPL; re 20 μPa) from the Nicolet system. Frequency-specific whole nerve CAP thresholds in chinchillas are reported to be 35-40dB SPL at 1kHz-2kHz, and within 10-20 dB SPL for the frequencies 3kHz-8kHz (Spagnoli and Saunders, 1987). Therefore, identifiable brainstem auditory evoked responses (wave V peak) to clicks presented at ≤35dB SPL were accepted as evidence for normal threshold sensitivity in this species (Sahley and Nodar, 1994; Sahley et al., 1996a; 1996b).
2.3 Acute surgical procedures
The procedures employed in all three experiments were identical to those used previously (Sahley and Nodar, 1994; Sahley et al., 1991; 1996a; 1996b). Chinchillas were food-and water-deprived (15 h) prior to pre-anesthesia with ketamine HCl (KETASET; 50 mg/kg; i.m.). A polyethylene tracheal tube (Intramedic PE #205) was introduced into the exposed trachea and secured in place using standard tracheotomy procedures. Animal core temperature was maintained (34.5-36°C) by an auto-regulating heating blanket system (YSI-73A) and was digitally monitored throughout surgery and testing. A surgical operating microscope (Zeiss) was securely suspended at all times within the test chamber, above the surgical field.
A cannula (Intramedic PE #10) was placed in the right internal jugular vein to a depth of 2cm using standard microsurgical procedures. A zero dead-volume three-way stopcock valve (Hamilton) was mounted on the opposite end of the venous cannula tubing. Additional tubing extended from the stopcock to a syringe that was mounted on a multispeed perfusion pump system, also located within the test chamber. Surgical anesthesia, consisting of sodium pentobarbital diluted in sterile lactated Ringer’s to a concentration of 5mg/ml, was delivered at a rate of 10μl/min) and was maintained through the venous cannula throughout the surgical procedures and testing. To prepare each animal for testing, the head was secured at a fixed and predetermined ventrolateral angle using a non-invasive, custom designed table-mounted head-appliance located within the test chamber. The left pinna was retracted anteriorward. Post auricular vessels were surgically ligated or cauterized, and an opening into the osseous auditory canal was created for the insertion of an Etymotic ER-3A (insert) microphone foam earplug. The ER-3A earplug reduces ambient noise by 30dB (Beauchaine et al., 1987). The tissue overlying the left auditory bulla was surgically excised, the bulla was breached, and the round window membrane at the base of the cochlea was exposed. At the end of each experiment, chinchillas were euthanized with 1ml sodium pentobarbital delivered through the venous cannula. The care and use of the animals reported in these investigations conformed to NIH guidelines, and was approved of by the Institutional Animal Care and Use Committee of Cleveland State University. The Animal Research Facility at Cleveland State University has a full-time DVM.
2.4 Near-field electrophysiological measurements
2.41 Equipment And Stimuli
Auditory potentials were simultaneously generated in response to negative-polarity clicks and differentially recorded from a location near the round window membrane. Rectangular electrical pulses, 100 μs in duration, were delivered (18.3/s) from the Nicolet click generator and negative-polarity clicks were produced with the ER-3A insert microphone. Subdermal platinum-alloy needle electrodes (Grass E2) were inserted at the vertex- (+) and neck musculature-(common ground). The inverting (-)-electrode consisted of a Grass E2 wrapped with fine diameter (30G) silver wire, heated at the tip to form a silver ball (≤ 0.5 mm in diameter). The spherical tip was positioned ventral to the round window and secured in place, permitting fine adjustment, effortless removal and reliable repositioning of the electrode when required. The round window electrode impedance was restricted to 8-10kΩ by the minute application of a hypoallergenic conductive gel. A non-patent form-fitting foam earplug was inserted into the contralateral ear to eliminate any possibility of unwanted stimulation by ambient noise within the closed test chamber. Responses to clicks, at each of six stimulus intensities, were amplified (×104), band-pass filtered (150-1500 Hz; 6dB/octave roll-off), and averaged on-line over a 5ms recording epoch with the Nicolet CA-1000 system. All waveforms were stored for subsequent off-line analyses.
2.42 Threshold Acquisition
During the initial baseline period, a neural ”threshold” intensity was determined for each animal. This established a reference for all subsequent suprathreshold (i.e. dB SL) testing. Threshold was defined as the weakest click intensity producing visually identifiable and repeatable CAP - N1 / N2 wave peaks. Click intensity was systematically reduced in 2dB steps from about 30 to 40dB SPL until no measurable peaks were observed (zero amplitude). The intensity level was then increased in 2dB steps until reliable peak responses were measured. The intensity was reduced once again to a zero amplitude, and again increased. With this descending/ascending 2dB-step tracking procedure, neural peak threshold estimates obtained on descending trials invariably matched those obtained on the ascent (Sahley and Nodar, 1994; Sahley et al., 1996a; 1996b). During baseline testing, the 28 animals exhibited mean CAP- N1 / N2 click thresholds of 19.38 dB SPL (± 7.15dB S.D.).
2.43 Stimulus Presentation and Data Acquisition
Auditory neural and CM potentials were systematically recorded in all 28 animals across a series of six stimulus intensities: threshold, and 5, 10, 20, 30 and 40dB SL. Click stimuli were always presented in ascending order within each series, beginning at the lowest intensity (threshold) and ending at the highest (40dB SL). To assure on-line within series (intra-series) reliability, two samples (an original and replication) were always obtained at each of the six intensities, yielding twelve waveforms per series. In each animal, baseline amplitude stability in the CAP- N1 was initially established and was defined as the absence of consistent intra-series or between series (inter-series) variance in threshold peak amplitudes across 5 complete and consecutive testing series.
Following the affirmation of stable thresholds, the neural and CM amplitudes and latencies were repetitiously recorded across the stimulus intensities, for a total recording duration of 180 min in each animal. The total recording duration of 180 min consisted of three consecutive 30 min baseline and three consecutive 30 min post-baseline recording intervals. This resulted in a 6 (30 min recording period) x 6 (stimulus intensity) data matrix for each of the 28 animals. Between six to fifteen complete intensity series were recorded during each of the six, 30 min recording intervals, resulting in the acquisition of 12 to 30 CAP - N1 and CM data entries within each of the 36 recording blocks, per animal.
2.5 Round window pharmacologic manipulations
2.51 Baseline treatment
At the conclusion of the second 30 min baseline recording period, all animals received 1μl of artificial perilymph [130 mmol NaCl; 4 mmol KCl; 4 mmol CaCl2; 2 mmol MgCl2; 12.5 mmol HEPES buffer; 10 mmol glucose; osmol = 309 mosmol/l; pH = 7.40] (APS) in combination with the universal solvent, dimethyl sulfoxide (DMSO). APS/DMSO solutions were applied directly to the cochlear round window membrane and were allowed to infuse into the cochlear fluids. The pH of all APS/DMSO solutions was maintained at 7.40(±0.02) with a Microelectrodes Inc. (MI-410) micro pH probe and Accumet 825 MP pH meter. APS/DMSO solutions were non-invasively administered at room temperature using a 33G needle and microliter syringe assembly, mounted within a mini-micromanipulator system. The blunt microliter syringe tip was guided to the round window membrane surface under the Zeiss operating microscope. The procedure required temporarily displacing the (-)-electrode from the basal surface of the cochlea. The position and impedance of the (-)-electrode were immediately restored following this procedure, and the repetitious recording procedure was resumed over the final, 30 min baseline period.
In experiment 1, the APS-to-DMSO ratio of the 1μl baseline solution was APS (80%)/DMSO (20%). For the animals in experiments 2 and 3, the APS-to-DMSO ratio of the 1μl baseline solution was APS (50%)/DMSO (50%). In addition, for the animals of experiment 3, the APS/DMSO contained an 8 mmol (2.69 μg/μl) concentration of the NMDA-receptor antagonist (+)-MK-801.
2.52 Post-Baseline Treatment
Following completion of the third and final 30 min baseline recording period, animals received a second (post-baseline) round window administration of APS combined with DMSO. All post-baseline APS/DMSO solutions were administered at a 2μl volume at room temperature and at a pH of 7.4. The repetitious recording procedure was then resumed over the three remaining 30 min post-baseline recording periods.
Experiment 1
(-)-Pentazocine succinate was dissolved in APS/DMSO at a high concentration of 10.08μg/μl, and post-baseline administered to the round window in the treatment animals (n = 3), at a 2μl volume (50 mmol). To maintain the required 7.4 pH, (-)-pentazocine was made water-soluble by employing the lowest workable APS-to-DMSO ratio, of APS (80%)/DMSO (20%). Control animals (n = 3) received a 2μl post-baseline administration of APS (80%)/DMSO (20%).
Experiment 2
U-50488H was dissolved in APS/DMSO at a high concentration of 23.27μg/μl, and post-baseline administered to the round window in the treatment animals (n = 5), at a 2μl volume (100 mmol). To maintain the required 7.4 pH, U-50488H was made water-soluble by employing the lowest workable APS-to-DMSO ratio, of APS (50%)/DMSO (50%). Control animals (n = 7) received a 2μl post-baseline administration of APS (50%)/DMSO (50%).
Experiment 3
The baseline round window administration of (+)-MK-801 (8 mmol) in the treatment animals (n = 5) was followed by a 2 μl post-baseline round window administration of U-50488H (100 mmol) in APS (50%)/DMSO (50%). The baseline round window administration of (+)-MK-801 (8 mmol) in the control animals (n = 5) was followed by a 2 μl post-baseline administration of APS (50%) / DMSO (50%).
2.6 Data analyses
Post-baseline changes in the amplitude and latency of the CAP - N1, and in the amplitude of the CM were analyzed for all experiments. CAP - N1 amplitudes were measured from the negative (N1) peak to the following positive trough (P1). The latency of each CAP - N1 peak of interest was measured in ms at the point of maximum amplitude. Click-evoked CM potentials were measured from maximum peak to the 0-baseline and were consistently recorded at the same onset latency, independent of stimulus intensity and drug administration (Sahley and Nodar, 1994; Sahley et al., 1991; 1996a; 1996b).
A 6 (30 min recording period) X 6 (stimulus intensity) data matrix of means was generated from the 12 to 30 CAP - N1 amplitudes obtained at all six intensities, and from the CM amplitudes and CAP - N1 latencies obtained at threshold and at 5dB SL, for each animal. This resulted in a 6 X 6 matrix of mean CAP - N1 amplitudes and mean latencies, and mean CM amplitudes. Mean data from each animal were then used to create several 6 X 6 summary data matrices of mean dependent variables for the three treatment, and three control groups of animals.
2.61 CAP - N1 amplitudes
Experiments 1 and 2
Additional summary matrices were created in which the three baseline and post-baseline CAP - N1 amplitudes were collapsed, creating a mean of means baseline and a mean of means post-baseline CAP - N1 amplitude per animal at each intensity. The mean of means summary matrices provided an unambiguous differentiation of baseline vs. post-baseline under the factor ”time (treatment)” for the overall multifactor ANOVAs. CAP - N1 amplitude data were submitted to separate multifactor ANOVAs [2 (group) X 2 (time: baseline vs. post-baseline) X 6 (intensity)] with repeated measures on factors, time and intensity. Statistical significance was defined as α ≤ 0.05. In all cases, additional planned comparisons were performed only when warranted by the results from either of the two, 2X2X6 multifactor ANOVAs. Planned comparisons consisted of repeated measures one-way ANOVAs and/or a set of correlated t-tests, with α adjusted for the number of comparisons.
Experiment 3
The two 6 × 6 summary data matrices of mean CAP - N1 amplitudes for the treatment (n = 5) and control animals (n = 5) of experiment 3 were analyzed using two separate repeated measures one-way ANOVAs. Each ANOVA was performed exclusively on the three, 30 min baseline recorded CAP - N1 amplitude values obtained at each of the six intensity levels in each group. A set of planned comparisons (adjusted correlated t-tests) were then performed when warranted to compare the computed average of the three, 30 min baseline amplitudes with each of the three, 30 min post-baseline amplitude values at each of the six intensities in each group of animals. Statistical significance for all analyses was defined by α ≤ 0.05.
2.62 CM amplitudes and CAP - N1 latencies
Experiments 1, 2 and 3
Summary data matrices of mean CM amplitudes and mean CAP - N1 latencies at the two of the six stimulus intensities (threshold and 5dB SL) in each of the three treatment and three control groups were analyzed using twelve separate one-way repeated measures ANOVAs. In all six groups, repeated measures ANOVAs were performed on mean CM amplitudes and CAP - N1 latencies across each of the six (3 baseline; 3 post-baseline) 30 min recording periods at the two intensities indicated. Statistical significance was defined by α ≤ 0.05. Planned comparisons were conducted only if warranted by the repeated measures one-way ANOVAs. All analyses were performed with the Statistical Package for the Social Sciences (SPSS) program, version 13.0.
3. Results
Results of the multifactor (2X2X6) ANOVA performed on the six animals of experiment 1 (F [1,4] = 46.14, P < 0.005) and the twelve animals of experiment 2 (F [1,10] = 59.50, P < 0.001) indicated significant baseline vs. post-baseline neural amplitude changes overall (main effect of time). Significant changes in overall CAP - N1 amplitude were also observed as a main effect of intensity level in experiments 1 (F [1,4] = 250.88, P < 0.001) and 2 (F [1,10] = 92.55, P < 0.001). The significant time × group/treatment interactions in either experiment further indicated that post-baseline neural amplitudes varied significantly as a function of group membership [i.e. (-)-pentazocine vs. control (F [1,4] = 31.04, P < 0.01), or U-50488H vs. control (F [1,10] = 32.14, P < 0.001)] Post-baseline neural amplitudes also varied significantly as a function of overall intensity level, as indicated by the significant time × intensity interaction in experiments 1 (F [1,4] = 34.19, P < 0.005) and 2 (F [1,10] = 64.39, P < 0.001). Furthermore, the interaction of the two factors, time and intensity varied as a function of group membership, as indicated by the significant interaction between the three factors of time, intensity and group/treatment in experiments 1 (F [1,4] = 24.88, P < 0.01) and 2 (F [1,10] = 35.81, P < 0.001). The overall neural amplitude changes observed in experiments 1 and 2 are illustrated in Figs. 1 and 2, respectively.
Fig. 1.
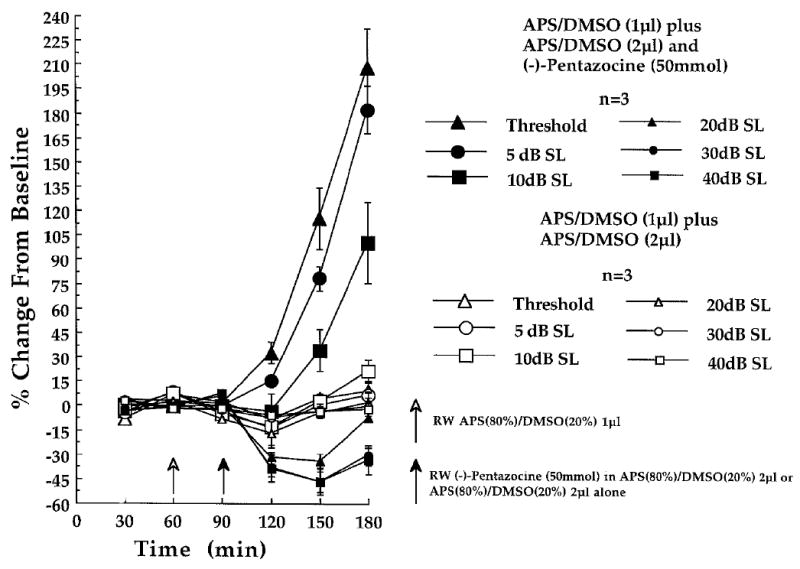
Effect of cochlear round window (RW)-administered (-)-pentazocine (50 mmol) on CAP - N1 amplitudes recorded at the six indicated stimulus intensities in the (-)-pentazocine-treated (n = 3) and control animals (n = 3), of experiment 1. At each of the three baseline (time 30, 60 and 90 min) and three post-baseline recording periods (time 120, 150 and 180 min) each point represents a mean percent change in neural amplitude relative to the grand baseline mean (±S.E.M.). Open arrow at time 60 min indicates the 1μl administration of APS(80%) / DMSO(20%), delivered to the cochlear round window membrane in all animals, prior to the final 30 min baseline recording period. Filled arrow at time 90 min indicates the 2μl post-baseline administration of (-)-pentazocine (50 mmol) dissolved in APS(80%) / DMSO(20%) (n = 3), or the APS(80%) / DMSO(20%) solution delivered alone (n = 3).
Fig. 2.
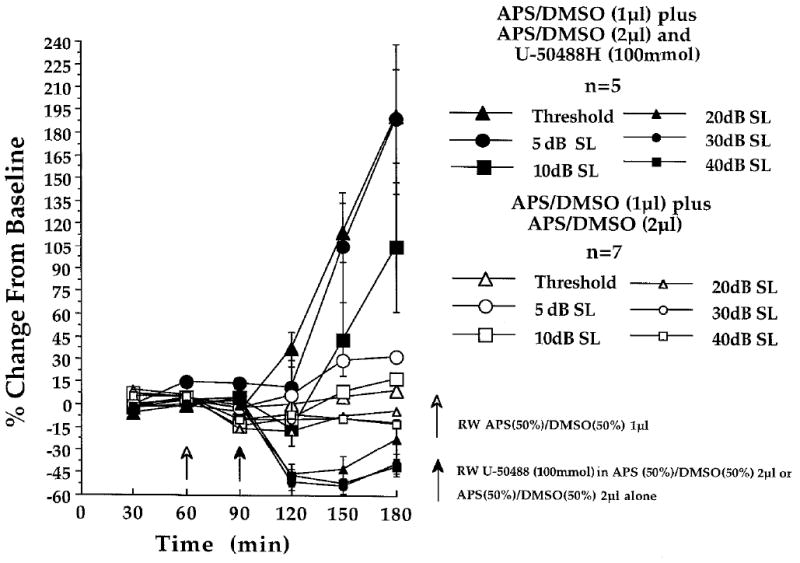
Effect of cochlear round window (RW)-administered U-50488H (100 mmol) on CAP - N1 amplitudes, recorded at the six indicated stimulus intensities in the U-50488H-treated (n = 5) and control animals (n = 7) of experiment 2. At each of the three baseline (time 30, 60 and 90 min) and three post-baseline recording periods (time 120, 150 and 180 min) each point represents a mean percent change in neural amplitude relative to the grand baseline mean (±S.E.M.). Open arrow at time 60 min indicates the 1μl administration of APS(50%) / DMSO(50%), delivered to the cochlear round window membrane in all animals, prior to the final 30 min baseline recording period. Filled arrow at time 90 min indicates the 2μl post-baseline administration of U-50488H (100 mmol) dissolved in APS(50%) / DMSO(50%) (n = 5), or the APS(50%) / DMSO(50%) solution delivered alone (n = 7).
3.1 Baseline CAP - N1 amplitude analyses: experiments 1, 2 and 3
The significant main effects of time, as well as the time x group/treatment interactions in experiments 1 and 2 justified an overall evaluation of the baseline neural amplitude stability of each group of animals in both experiments. A set of within subjects planned comparisons were conducted on baseline CAP - N1 values at each of the six intensity levels for the (-)-pentazocine-treated and control animals in experiment 1 (Fig. 1; time 30, 60 and 90 min), as well as the U-50488H-treated and control animals in experiment 2 (Fig. 2; time 30, 60 and 90 min). Planned comparisons consisted of separate repeated measures one-way ANOVAs performed on the baseline neural amplitudes recorded over the three, 30 min periods at each stimulus intensity. Baseline CAP - N1 amplitudes were found to be stable at five of the six stimulus intensities in the (-)-pentazocine-treated animals of experiment 1 (n = 3) following baseline APS (80%)/DMSO (20%) administration. At threshold, a slight though significant baseline instability was observed (F [1,2] = 20.89, P < 0.05) in this group of animals. In the control animals, APS (80%)/DMSO (20%) produced no significant baseline instabilities in the CAP - N1 amplitude at any stimulus intensity. In experiment 2, APS (50%)/DMSO (50%) produced no measurable baseline neural amplitude instabilities in the control or U-50488H-treated animals, at any stimulus intensity.
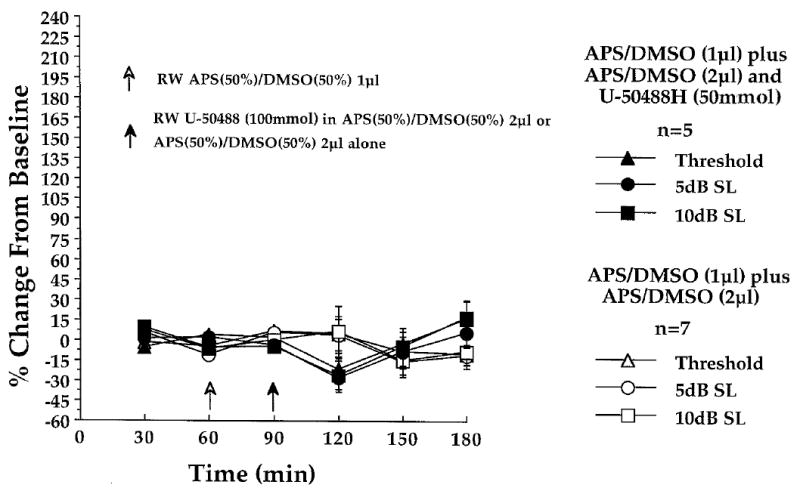
In the [(+)-MK-801 + APS (50%)/DMSO (50%)] controls of experiment 3, neural amplitude stability during the three, 30 min baseline periods (Fig. 3; time 30, 60 and 90 min) was evaluated at each of the six stimulus intensities using repeated measures one-way ANOVAs. Outlying data from one chinchilla produced an apparent though non-significant degree of neural baseline instability at threshold (F [1,4] = 1.12), 5dB SL (F [1,4] = 0.97) and 10dB SL (F [1,4] = 0.92). Fig. 3 contrasts the CAP - N1 baseline (and post-baseline) amplitude changes at each stimulus intensity in the [(+)-MK-801 + APS (50%)/DMSO (50%)]-treated control animals of experiment 3, to those of the APS (50%)/DMSO (50%) control animals of experiment 2, as percent changes from baseline.
Fig. 3.
Lack of effect of cochlear round window (RW)-administered U-50488H (100 mmol) on the CM amplitudes, recorded at the three indicated stimulus intensities in the U-50488H-treated (n = 5) and control animals (n = 7), of experiment 2. At each of the three baseline (time 30, 60 and 90 min) and three post-baseline recording periods (time 120, 150 and 180 min) each point represents a mean percent change in the microphonic amplitude relative to the grand baseline mean (±S.E.M.). Open arrow at time 60 min indicates the 1μl administration of APS(50%) / DMSO(50%), delivered to the cochlear round window membrane in all animals, prior to the final 30 min baseline recording period. Filled arrow at time 90 min indicates the 2μl post-baseline round window administration of U-50488H (100 mmol) dissolved in APS(50%) / DMSO(50%) (n = 5), or the APS(50%) / DMSO(50%) solution delivered alone (n = 7).
Fig. 4 contrasts baseline (and post-baseline) neural amplitude changes in the [(+)-MK-801 + U-50488H]-treated animals of experiment 3 with baseline data obtained in the U-50488H-treated animals of experiment 2, as percent changes from baseline at each stimulus intensity. In the [(+)-MK-801 + U-50488H]-treated animals, the 1μl round window administration of (+)-MK-801 in APS (50%)/DMSO (50%) prior to the administration of U-50488H (Fig. 4 time 30, 60 and 90 min) produced a significant baseline neural instability at 10dB SL (F [1,4] = 10.46, P < 0.05) consisting of a slight μV increase during the third of three baseline recording periods. No other CAP - N1 baseline instabilities were observed at the remaining five stimulus intensities.
Fig. 4.
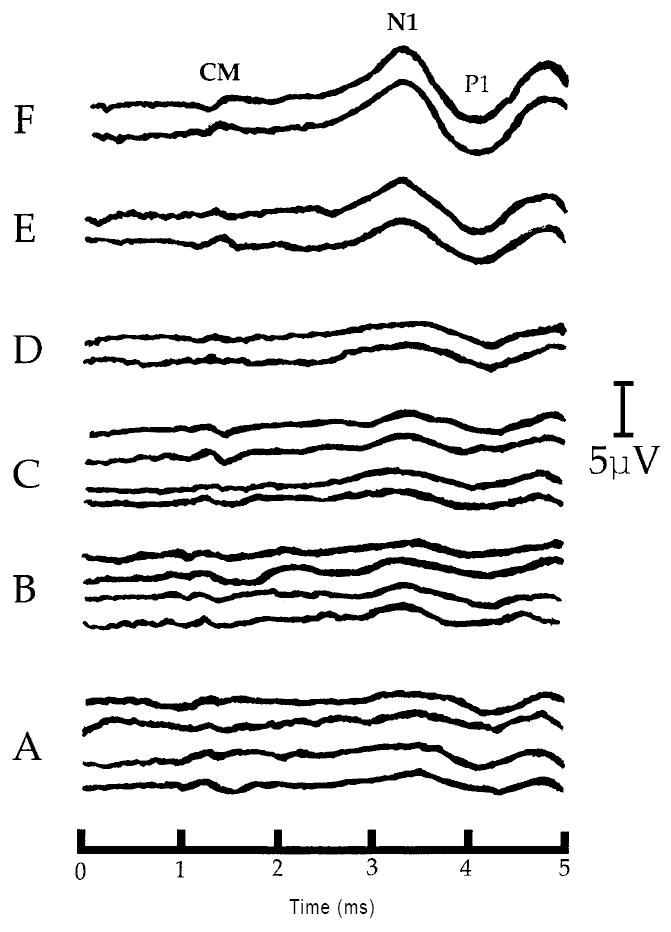
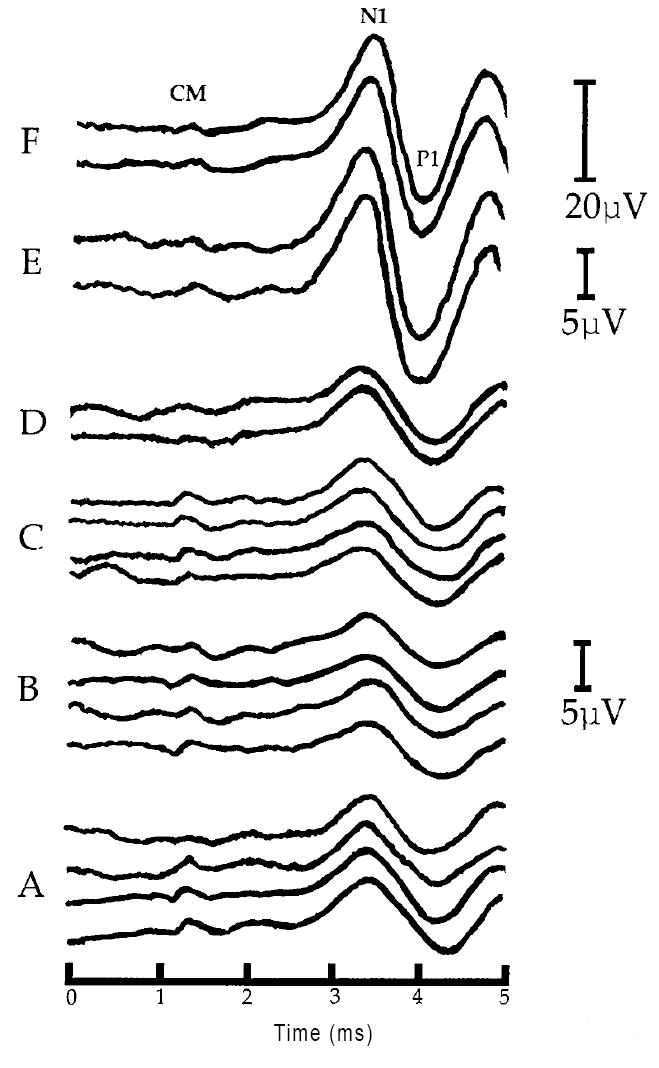
A: Positive post-baseline effect of U-50488H on the CAP - N1 amplitude at threshold in one representative chinchilla (#U5-01) from experiment 2. Each waveform was obtained at the same threshold intensity level (22dB SPL). Shown are sets of waveforms obtained during each of the six, 30 min recording periods labeled A-F. Four responses are shown at each of the three baseline recording intervals (A-C) corresponding to time 30, 60 and 90 min, respectively. Two responses are shown at each of the three post-baseline recording intervals (D-F) corresponding to time 120, 150 and 180 min, respectively. Positive post-baseline amplitude changes at threshold are clearly visible during the post-baseline periods E and F (time 150 and 180 min, respectively). All waveforms are displayed with negative-polarity up. Labeled are the cochlear microphonic (CM), and the first negative peak (N1) and first positive trough (P1) of the auditory whole nerve compound action potential. All latencies are uncorrected for the 0.9 ms acoustic delay produced by the 28cm in length sound tube assembly of the ER-3A microphone (e.g. Beauchaine et al., 1987). Note: lack of a corresponding change in the CM amplitude and CAP - N1 latency following U-50488H.
B: Positive post-baseline effect of U-50488H on the CAP - N1 amplitude at 5dB above threshold (SL) in one representative chinchilla (#U5-01) from experiment 2. Shown are sets of waveforms obtained during each of the six, 30 min recording periods labeled A-F. Four responses are shown at each of the three baseline recording intervals (A-C) corresponding to time 30, 60 and 90 min, respectively. Two responses are shown at each of the three post-baseline recording intervals (D-F) corresponding to time 120, 150 and 180 min, respectively. Positive post-baseline amplitude changes at 5dB SL are clearly visible during the post-baseline periods E and F (time 150 and 180 min, respectively). All waveforms are displayed with negative-polarity up. Labeled are the cochlear microphonic (CM), the first negative peak (N1) and the first positive trough (P1) of the auditory whole nerve compound action potential. All latencies are uncorrected for the 0.9 ms acoustic delay produced by the 28cm in length sound tube assembly of the ER-3A microphone (e.g. Beauchaine et al., 1987). Note: lack of a corresponding change in the CM amplitude and CAP - N1 latency following U-50488H, as well as the corresponding scale adjustments (μV) as amplitude continues to increase from the post-baseline recording periods E to F (time 150 to 180 min, respectively).
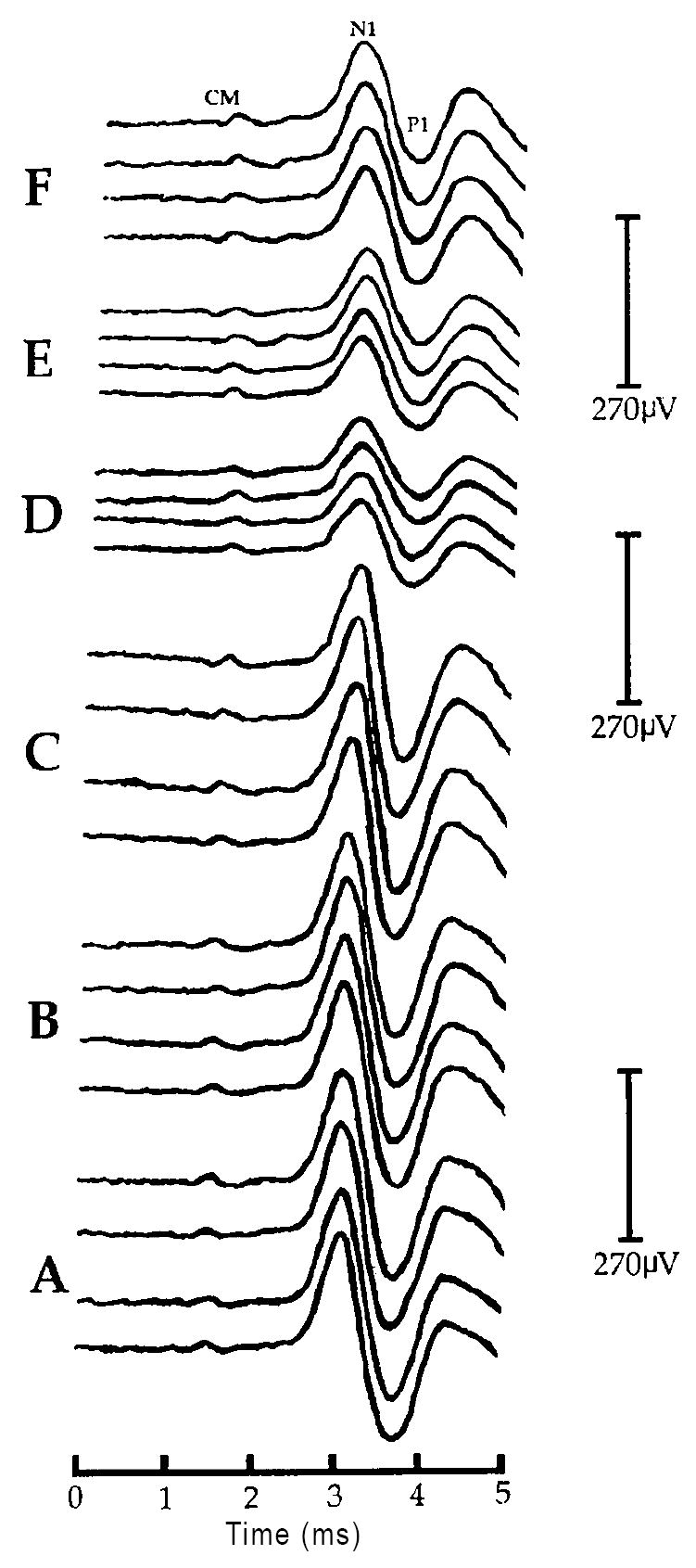
C: Negative post-baseline effect of U-50488H on the CAP - N1 amplitude at 30dB above threshold (SL) in one representative chinchilla (#U5-01) from experiment 2. Shown are sets of waveforms obtained during each of the six, 30 min recording periods labeled A-F. Four responses are shown at each of the three baseline recording intervals (A-C) corresponding to time 30, 60 and 90 min, respectively. Two responses are shown at each of the three post-baseline recording intervals (D-F) corresponding to time 120, 150 and 180 min, respectively. Negative post-baseline amplitude changes at 30dB SL are clearly visible during post-baseline periods D-F (time 120, 150 and 180 min, respectively). All waveforms are displayed with negative-polarity up. Labeled are the hair cell-generated cochlear microphonic (CM), the first negative peak (N1) and the first positive trough (P1) of the auditory whole nerve action potential. All latencies are uncorrected for the 0.9 ms acoustic delay produced by the 28cm in length sound tube assembly of the ER-3A microphone (e.g. Beauchaine et al., 1987). Note: lack of a corresponding change in the CM amplitude and CAP - N1 latency following U-50488H.
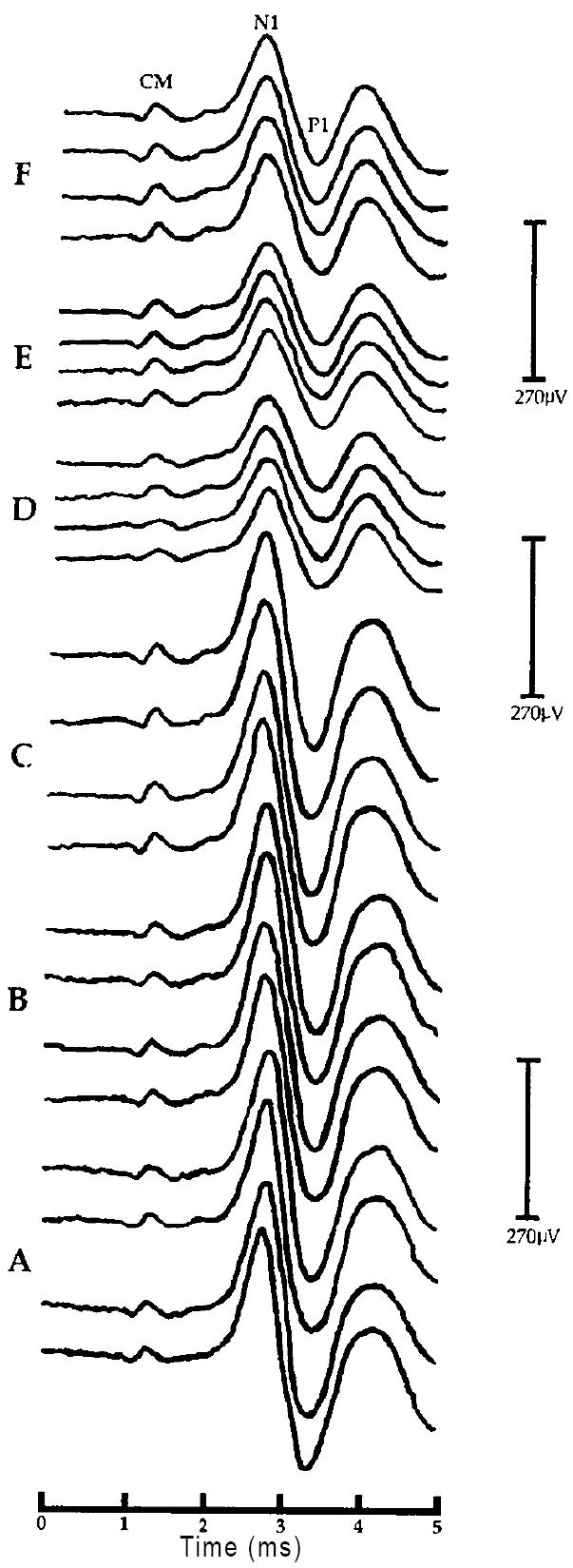
D: Negative post-baseline effect of U-50488H on the CAP - N1 amplitude at 40dB above threshold (SL) in one representative chinchilla (#U5-01) from experiment 2. Shown are sets of waveforms obtained during each of the six, 30 min recording periods labeled A-F. Four responses are shown at each of the three baseline recording intervals (A-C) corresponding to time 30, 60 and 90 min, respectively. Two responses are shown at each of the three post-baseline recording intervals (D-F) corresponding to time 120, 150 and 180 min, respectively. Negative post-baseline amplitude changes at 40dB SL are clearly visible during post-baseline periods D-F (time 120, 150 and 180 min, respectively). All waveforms are displayed with negative-polarity up. Labeled are the hair cell-generated cochlear microphonic (CM), the first negative peak (N1) and the first positive trough (P1) of the auditory whole nerve compound action potential. All latencies are uncorrected for the 0.9 ms acoustic delay produced by the 28cm in length sound tube assembly of the ER-3A microphone (e.g. Beauchaine et al., 1987). Note: lack of a corresponding change in the CM amplitude and CAP - N1 latency following U-50488H.
3.2 Post-baseline analyses: experiments 1 and 2
To directly examine the significant time x intensity x group interactions in experiments 1 and 2, and to statistically verify the observed post-baseline neural amplitude changes following 50 mmol of (-)-pentazocine or 100 mmol of U-50488H (Figs. 1 and 2, respectively), additional within subjects planned comparisons were conducted. Correlated t-tests compared the computed average of the three, 30 min baseline CAP - N1 amplitudes with each of the three, 30 min post-baseline CAP - N1 amplitudes at each intensity level in each group of animals. These planned comparisons employed the very conservative Bonferroni adjustment in which alpha (α ≤ 0.05) was divided by the number of comparisons (three), yielding a corrected α of ≤ 0.0167 (George and Mallery, 1999). The Bonferroni adjustment avoids experiment-wise error by shrinking the power of each test, consequently decreasing the probability of committing Type I errors. Comparisons were either one or two-tailed, as statistically appropriate.
3.21 Post-baseline CAP - N1 amplitude facilitation
(-)-Pentazocine-treated animals
Consistent with previous investigations in which (-)-pentazocine was administered by a systemic route, were the significant post-baseline amplitude increases in the CAP - N1 at threshold, in experiment 1. Relative to a mean (of means) neural baseline at a threshold of 2.29 μV (±0.19 S.D.), post-baseline mean of means neural amplitudes of 3.02 μV (±0.13 S.D.), 4.91 μV (±0.49 S.D.) and 6.99 μV (±0.33 S.D.) were obtained during the three post-baseline recording periods (Fig. 1; time 120, 150 and 180 min), respectively. By 60 min post-baseline (Fig. 1; time 150), the neural amplitude changes at threshold corresponded to a 6 to 8dB increase in auditory sensitivity. The average growth in neural amplitudes for the 60 and 90 min post-baseline periods (Fig. 1; time 150 and 180 min) corresponded to a mean change from baseline of +115.64% and +207.68%, respectively, as illustrated in Fig. 1 . Finally, one-tailed planned comparisons indicated that the neural amplitude increases at threshold were significant by 30 (t [2] = 6.44, P < 0.0167), 60 (t [2] = 8.06, P < 0.0167) and 90 min post-baseline (t [2] = 15.36, P < 0.0167) in the (-)-pentazocine-treated animals.
At 5dB SL, neural baselines of 6.29 μV (±2.58 S.D.) contrasted with post-baseline neural amplitudes of 7.27 μV (±3.61 S.D.), 11.47 μV (±6.25 S.D.) and 17.88 μV (±9.22 S.D.) by 30, 60 and 90 min respectively. In addition, mean percent amplitude changes from baseline were +78.38% and +182.17% by 60 and 90 min post-baseline (Fig. 3). However, the small sample size probably contributed to greater variance as these differences failed to reach statistical significance.
Lastly at 10dB SL, neural baseline values of 27.40 μV (±10.27 S.D.) contrasted with post-baseline neural amplitudes of 25.22 μV (±5.87 S.D.), 35.26 μV (±8.89 S.D.) and 51.70 μV (±9.18 S.D.) by 30, 60 and 90 min post-baseline (Fig. 1; time 120, 150 and 180 min) respectively. One-tailed comparisons indicated that these differences were only significant by 90 min post-baseline (t [2] = 13.422, P < 0.0167) (Fig. 1; time 180 min).
U-50488H-treated animals
Consistent with the results of experiment 1 were the large post-baseline neural amplitude increases at threshold and at 5dB SL in the U-50488H-treated animals of experiment 2 (Fig. 2). At threshold, mean amplitudes changed from 1.75 μV (±0.27 S.D.) during baseline, to 2.55 μV (±0.74 S.D.), 3.93 μV (±0.96 S.D.) and 5.27 μV (±0.97 S.D.) by 30, 60, and 90 min post-baseline (Fig. 2; time 120, 150 and 180 min), respectively. The neural amplitude changes at threshold corresponded to a 4 to 6dB increase in auditory sensitivity by 60 and 90 min post-baseline. One-tailed planned comparisons also indicated that the differences were statistically significant by 60 (t [4] = 5.67, P < 0.0167) as well as by 90 min post-baseline (t [4] = 8.18, P < 0.0167), as illustrated in Fig. 2; time 150 and 180 min.
At 5dB SL, neural amplitudes changed from a baseline of 8.39 μV (±1.73 S.D.), to 8.38 μV (±2.70 S.D.), 15.21 μV (±4.79 S.D.) and 21.43 μV (±5.90 S.D.) by 30, 60, and 90 min post-baseline respectively. One-tailed planned comparisons indicated that U-50488H produced significant neural amplitude increases by 60 (t [4] = 3.23, P = 0.0167) and 90 min post-baseline (t [4] = 4.69, P < 0.0167) (Fig. 2; time 150 and 180 min).
Lastly, at 10dB SL, less dramatic changes in neural amplitude were observed relative to baselines. These were 17.07 μV (±5.20 S.D.), 28.69 μV (±8.95 S.D.) and 40.29 μV (±12.88 S.D.) by the 30, 60, and the 90 min post-baseline recording periods, respectively, relative to baseline values of 20.94 μV (±4.37 S.D. One-tailed planned comparisons indicated that amplitude increases in the CAP - N1 only approached a level of significance, and only within the 90 min post-baseline period (t [4] = 3.13, P = 0.0175) at 10dB S (Fig. 2; time 180 min). The overall neural amplitude increases in the U-50488H-treated animals are presented in Fig. 2 as percent changes from baseline.
3.22 Post-baseline CAP - N1 amplitude suppression
(-)-Pentazocine-treated animals
As illustrated in Fig. 3, (-)-pentazocine (50 mmol) administered across the round window membrane produced an unpredicted post-baseline suppression of the CAP- N1 amplitude relative to baseline values. Neural suppression occurred at stimulus intensities (i.e. ≥ 20dB SL) that were greater than those associated with neural amplitude facilitations. Relative to CAP- N1 baselines of 140.75 μV (±18.21 S.D.) at 20dB SL, neural amplitudes of 95.94 μV (±18.56 S.D.), 94.09 μV (±25.02 S.D.) and 129.95 μV (±19.08 S.D.) were obtained by 30, 60 and 90 min post-baseline, respectively at this stimulus intensity. Two-tailed planned comparisons indicated that the neural suppression at 20dB SL was statistically significant at 30 (t [2] = -9.03, P < 0.0167), 60 (t [2] = -17.32, P < 0.0167) and at 90 min post-baseline (t [2] = -3.57, P = 0.0167) (Fig. 1; time 120, 150 and 180 min).
At 30dB SL, baselines of 264.86 μV (±19.36 S.D.) contrasted with post-baseline neural amplitudes of 163.14 μV (±27.16 S.D.), 140.51 μV (±36.64 S.D.) and 184.69 μV (±37.72 S.D.) by 30, 60 and 90 min post-baseline, respectively. Two-tailed planned comparisons indicated that neural suppression was significantly different from baseline at the 30 (t [2] = -7.95, P < 0.0167) (Fig. 1; time 120) and 60 min (Fig. 1; time 150) post-baseline periods (t [2] = -8.86, P < 0.0167), but not at 90 min post-baseline (t [2] = -5.18).
Neural amplitude suppression was also observed at 40dB SL following (-)-pentazocine. CAP- N1 baseline values of 289.77 μV (±25.86 S.D.) were reduced to 176.61 (±43.25 S.D.), 154.65 (±48.30 S.D.), and 191.91 μV (±49.02 S.D.) by 30, 60 and 90 min post-baseline. This amounted to a -38.52%, a -46.55% and a -33.50% mean change from baseline, respectively (Fig. 3). However, the suppression failed to reach levels of statistical significance at this intensity. The overall neural amplitude suppression at intensities ≥ 20dB SL following round window administered (-)-pentazocine (50 mmol) is presented in Fig. 1 as percent changes from baseline.
U-50488H-treated animals
U-50488H (100 mmol) delivered across the cochlear round window membrane also produced an unpredicted post-baseline suppression of CAP - N1 amplitudes at stimulus intensities ≥ 20dB SL (Fig. 4), similar to the neural suppression observed in the (-)-pentazocine-treated animals of experiment 1. At 20dB SL, baseline neural amplitudes of 121.97 μV (±27.96 S.D.) were reduced to post-baseline amplitudes of 62.75 μV (±30.51 S.D.), 69.18 μV (±27.89 S.D.) and 97.15 μV (±41.75 S.D.) by 30, 60 and 90 min post-baseline (Fig. 2; time 120, 150 and 180), respectively. Two-tailed planned comparisons indicated that neural suppression was statistically significant at 30 (t [4] = -7.48, P < 0.0167) and at 60 min post-baseline (t [4] = -5.46, P < 0.0167) (Fig. 2; time 120 and 150), but not significantly different from baseline at 90 min (Fig. 2; time 180), post-baseline (t [4] = -1.73).
At 30 and at 40dB SL, two-tailed planned comparisons indicated that U-50488H administration resulted in a significant neural amplitude suppression during all three post-baseline recording periods (Fig. 2; time 120, 150 and 180). At 30dB SL, baselines of 231.15 μV (±64.61 S.D.) were reduced to 116.95 μV (±57.90 S.D.), 106.76 μV (±38.87 S.D) and 140.31 μV (±59.46 S.D.) by 30 (t [4] = -15.39, P < 0.0167), 60 (t [4] = -7.05 P < 0.0167) and 90 min post-baseline (t [4] = -5.70, P < 0.0167), respectively. At 40dB SL, neural baseline amplitudes of 257.68 (±81.43 S.D.) were reduced to post-baseline amplitudes of 142.60 μV (±69.65 S.D., t [4] = -18.32, P < 0.0167), 122.27 μV (±42.78 S.D., t [4] = -8.41 P < 0.0167) and 153.28 μV (±63.29 S.D., t [4] = -6.72, P < 0.0167) by 30, 60 and 90 min post-baseline, respectively.
3.23 CAP - N1 amplitude controls: experiments 1 and 2
One-tailed planned comparisons indicated that the 2μl post-baseline round window APS (80%)/DMSO (20%) administered to the control animals of experiment 1 (n = 3) failed to result in significant neural amplitude changes relative to baseline, at any stimulus intensity (Fig. 1; time 120, 150 and 180). The higher concentration of DMSO [i.e. APS (50%)/DMSO (50%)] administered to control animals of experiment 2 (n = 7) did result in a significant amplitude suppression at 20dB SL, and only during the initial 30 min (Fig. 2; time 120), post-baseline period (t [2] = -3.50, P < 0.0167). By 60 (t [2] = -1.80) and 90 min post-baseline (t [2] = -0.96), amplitudes returned to values closely approximating baseline (Fig. 2; time 150 and 180). No other post-baseline variability was observed at any other stimulus intensity in this group. Overall post-baseline neural amplitude changes observed in the (-)-pentazocine and U-50488H control animals are presented in Figs. 1 and 2, respectively, at each stimulus intensity as percent changes from baseline.
3.24 Post-baseline CAP - N1 latencies
(-)-Pentazocine and U-50488H-treated animals
CAP - N1 latencies obtained during the six, 30 min recording periods were submitted to repeated-measures one-way ANOVAs. Despite the large post-baseline neural amplitude changes observed at threshold and 5dB SL in the (-)-pentazocine and U-50488H-treated animals of experiments 1 and 2, respectively, no corresponding neural latency changes were ever observed in the same animals across the six recording periods at either intensity. Hence, neural latencies remained consistent at threshold (F [1,2] = 0.030) and at 5dB SL (F [1,2] = 0.000) following (-)-pentazocine (50 mmol). Neural latencies also remained stable at threshold (F [1,4] = 0.65) and 5dB SL (F [1,4] = 0.41) following U-50488H (100 mmol).
3.25 Post-baseline cochlear microphonic (CM) amplitudes
(-)-Pentazocine and U-50488H-treated animals
Repeated measures one-way ANOVAs were performed on the recorded amplitudes of the hair cell-generated CM potentials, obtained at threshold and at 5dB SL across the six, 30 min recording periods in the (-)-pentazocine (experiment 1) and U-50488H-treated animals (experiment 2). Following (-)-pentazocine (experiment 1), CM amplitude variance at 5dB SL was not significant across the six, 30 min recording periods (F [1,2] = 15.52). At threshold however, overall variance was significant (F [1,2] = 18.69, P = 0.05). To examine this variance, two-tailed planned comparisons were performed with correlated t-tests that compared the computed average of the three, 30 min baseline amplitudes with each of the three (mean) post-baseline amplitudes at threshold. In contrast to the large positive post-baseline neural amplitude changes obtained at threshold, round window-administration of (-)-pentazocine resulted in a significant and paradoxical reduction in the amplitude of the CM during the 30 (t [2] = -14.46, P < 0.0167) and the 90 min post-baseline periods (t [2] = -9.85, P < 0.0167). In experiment 2, U-50488H (100 mmol) administration resulted in no significant CM amplitude changes. Post-baseline amplitude changes in the CM were absent at threshold (F [1,4] = 0.004) and at 5dB SL (F [1,4] = 0.34) in spite of the large positive post-baseline neural amplitude changes observed at either intensity, in the same animals. Baseline and post-baseline amplitude variability in the CM potentials of experiment 2 are illustrated in Fig. 3.
Fig. 4(A-D) illustrates pre- and post-U-50488H changes in CAP - N1 amplitudes, CAP - N1 latencies, and CM amplitudes recorded simultaneously at the separate stimulus intensities of threshold (Panel A), 5dB SL (Panel B), 30dB SL (Panel C) and at 40dB SL (Panel D) in one representative chinchilla (#U5-01). In each of the four panels, waveforms are also labeled with an A, B or C corresponding to the 30, 60 or 90 min baseline recording periods, respectively. Waveforms labeled D, E or F correspond to post-baseline recording periods 30 (time 120), 60 (time 150), or 90 (time 180) min, respectively. In each of the four panels, note the large bi-phasic intensity-dependent post-baseline amplitude changes in the CAP - N1 (Panels A and B vs. Panels C and D), concomitant with unchanging CAP - N1 latencies and unchanging CM amplitudes, recorded within the same animal, following a round window administration of U-50488H (100 mmol).
3.3 Post-baseline analyses: experiment 3
3.31 Antagonism of CAP-N1 amplitude facilitation
As illustrated in Fig. 5, the round window administration of U-50488H (100 mmol) in experiment 2 produced a large and significant post-baseline neural amplitude facilitation at threshold, of +113.84% and +191.19% by 60 and 90 min (Fig. 5; time 150 and 180) post-baseline, respectively. In experiment 3, introduction of the NMDA receptor antagonist (+)-MK-801 (8 mmol) to the round window membrane 30 min prior (Fig. 5; time 60) to the same round window administration of U-50488H (100 mmol) resulted in a post-baseline neural amplitude facilitation of only +37.42% and +47.10% by 60 and 90 min post-baseline (Fig. 5; time 150 and 180). One-tailed planned comparisons consisting of α-adjusted correlated t-tests, compared the computed average of the three, 30 min baseline amplitudes with each of the three, 30 min post-baseline amplitude values recorded at each intensity, in each group of animals.
Fig. 5.
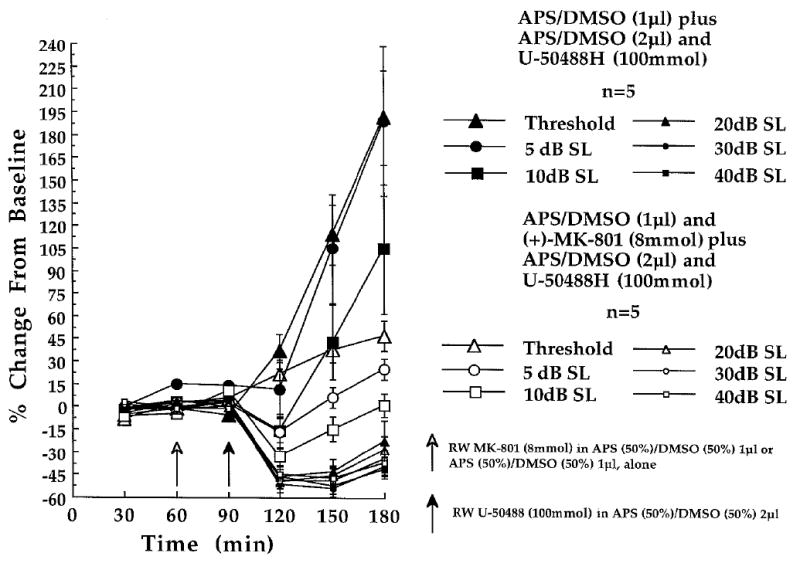
Partial antagonism of U-50488H (100 mmol)-induced neural amplitude changes by a cochlear round window (RW) pre-treatment with (+)-MK-801 (8 mmol), recorded at the six indicated stimulus intensities in experiment 3. Shown are the amplitude changes obtained in the [(+)-MK-801 + U-50488H (n = 5)]-treated animals of experiment 3 (open characters) and the amplitude changes obtained in the [APS / DMSO + U-50488H]-treated animals (n = 5) of experiment 2 (solid characters). At each of the three baseline (time 30, 60 and 90 min) and three post-baseline recording periods (time 120, 150 and 180 min) each point represents a mean percent change in neural amplitude relative to the grand baseline mean (±S.E.M.). Open arrow at time 60 min indicates the 1μl administration of APS(50%) / DMSO(50%) delivered to the round window membrane in the presence (n = 5; experiment 3) or absence of (+)-MK-801 (n = 5; experiment 2), prior to the final 30 min baseline recording period. Filled arrow at time 90 min indicates the 2μl post-baseline administration of U-50488H (100 mmol) dissolved in the APS(50%) / DMSO(50%) solution delivered to all animals.
In the [(+)-MK-801 + U-50488H]-treated animals, increases in CAP - N1 amplitude at threshold were diminished, but were still significantly elevated from baselines by the 60 (t [4] = 3.96, P = 0.0167) and 90 min (t [4] = 5.13, P < 0.0167) post-baseline periods (Fig. 5; time 150 and 180). This suggested that (+)-MK-801 (8 mmol) may have produced only a partial and non-significant amplitude suppression of the U-50488H-induced neural amplitude facilitation, at this intensity.
At 5dB SL, (+)-MK-801 (8 mmol) introduced 30 min prior to U-50488H (100 mmol) resulted in mean amplitude changes of +6.28% (60 min post-baseline, Fig. 5; time 150) and +24.87% (90 min post-baseline, Fig. 5; time 180) relative to baseline. These relatively smaller neural amplitude changes may be contrasted with much larger and significant mean neural amplitude increases of +104.54% (60 min post-baseline) and +189.33% (90 min post-baseline) obtained in the U-50488H-treated animals of experiment 2 (Fig. 5; time 150 and 180). Indeed, post-baseline CAP - N1 amplitude facilitations in the [(+)-MK-801 + U-50488H]-treated animals of experiment 3 failed to reach levels of statistical significance during the 60 (t [4] = 0.54) and 90 min (t [4] = 3.64) post-baseline periods, suggesting that (+)-MK-801 afforded a nearly complete suppression of U-50488H effects on neural amplitudes, at the stimulus intensity of 5dB SL.
At 10dB SL, CAP - N1 amplitude changes of +1.45% were obtained by 90 min post-baseline in the [(+)-MK-801 + U-50488H]-treated animals (Fig. 5; time 180). These negligible changes may be contrasted with a much larger (+104.47%) mean increase in neural amplitude during the same post-baseline period in the U-50488H-treated animals of experiment 2. CAP - N1 amplitudes recorded 90 min post-baseline at 10dB SL failed to differ from baseline amplitudes (t [4] = -0.06) in the [(+)-MK-801 + U-50488H]-treated animals of experiment 3. This suggested that (+)-MK-801 may have afforded an antagonism of the U-50488H-induced effect on CAP - N1 amplitudes at 10dB SL. The overall antagonistic effects of (+)-MK-801 (8 mmol) on U-50488H (100 mmol)-induced neural amplitude increases at threshold, 5dB SL and 10dB SL are presented in Fig. 5 as percent changes from baseline.
3.32 Antagonism of CAP - N1 amplitude suppression
In experiment 2, U-50488H administration resulted in a mean CAP - N1 amplitude suppression of -46.38%, -42.93% and -22.85% by 30 , 60 , and 90 min post-baseline, respectively, at the stimulus intensity of 20dB SL (Fig. 5; time 120, 150 and 180). Because the amplitude suppression following the round window administration of the κ-opioid receptor drug agonist U-50488H was unpredicted, planned comparisons consisted of two-tailed, α-adjusted correlated t-tests. (+)-MK-801 (8 mmol), delivered 30 min prior to U-50488H (100 mmol) resulted in a post-baseline CAP - N1 amplitude suppression that was indistinguishable from the amplitude suppression observed in experiment 2, following U-50488H administered alone. As in experiment 2, the neural amplitude suppression was significant at 30 (t [4] = -6.71, P < 0.0167) and 60 min (t [4] = -3.92, P = 0.0167), but not by 90 min, post-baseline (t [4] = -2.51) in the [(+)-MK-801 + U-50488H]-treated animals of experiment 3. Therefore, the results suggest that (+)-MK-801 (8 mmol) failed to antagonize the post-baseline neural amplitude suppression induced by U-50488H, at 20dB SL (Fig. 5).
Round window administration of U-50488H (100 mmol) in experiment 2 resulted in a significant post-baseline neural amplitude suppression across the three post-baseline periods, at stimulus intensities of 30dB and 40dB SL (Fig. 5; time 120, 150 and 180). In experiment 3, (+)-MK-801 (8 mmol) delivered 30 min prior to U-50488H (100 mmol) also resulted in a significant post-baseline CAP - N1 amplitude suppression by 30 min post-baseline (Fig. 5; time 120) at the stimulus intensities of 30dB (t [4] = -4.51, P < 0.0167), and at 40dB SL (t [4] = -4.39, P < 0.0167). However, by 60 min post-baseline, CAP - N1 amplitudes failed to differ significantly from baseline amplitudes, at 30dB (t [4] = -3.59) and 40dB SL (t [4] = -3.62). A similar antagonism of U-50488H-induced amplitude suppression by MK-801 was observed 90 min post-baseline at 30dB (t [4] = -2.63) and at 40dB SL (t [4] = -2.98) in these animals. The antagonism of U-50488H-induced CAP - N1 amplitude suppression afforded by the administration of (+)-MK-801 at 30 and 40dB SL is presented in Fig. 5.
3.33 CAP N1 amplitude controls
One-tailed planned comparisons indicated that the NMDA receptor antagonist (+)-MK-801 (8 mmol), round window-administered at 1μl, 30 min prior to the high concentration of APS (50%)/DMSO (50%) at 2μl, produced no significant post-baseline changes in CAP - N1 amplitudes relative to baseline, at any intensity. Baseline and post-baseline amplitude variability in the (+)-MK-801 plus APS (50%)/ DMSO (50%)–treated control animals of experiment 3 are contrasted with the APS (50%)/DMSO (50%) plus APS (50%)/DMSO (50%)–treated control animals of experiment 2, in Fig. 6.
Fig. 6.
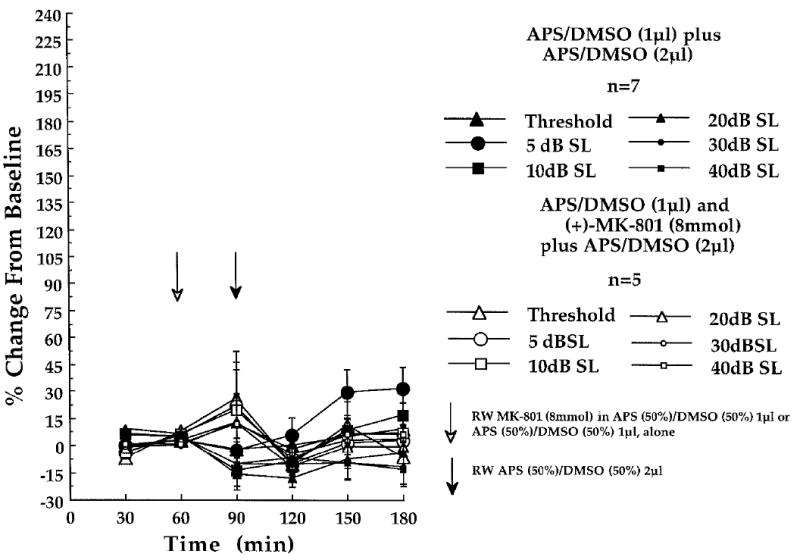
Lack of effect of cochlear round window (RW)-administered (+)-MK-801 (8 mmol) on the CAP - N1 amplitudes, recorded at the six indicated stimulus intensities in experiment 3. Shown are the amplitude changes obtained in the [(+)-MK-801 + APS / DMSO (n = 5)]-treated control animals of experiment 3 (open characters), and the amplitude changes obtained in the APS / DMSO + APS / DMSO-treated control animals (n = 7) of experiment 2 (solid characters). At each of the three baseline (time 30, 60 and 90 min) and three post-baseline recording periods (time 120, 150 and 180 min) each point represents a mean percent change in neural amplitude relative to the grand baseline mean (±S.E.M.). Open arrow at time 60 min indicates the 1μl administration of APS(50%) / DMSO(50%), delivered to the cochlear round window membrane in the presence (n = 5; experiment 3) or absence of (+)-MK-801 (n = 7; experiment 2) prior to the final 30 min baseline recording period. Filled arrow at time 90 min indicates the 2μl post-baseline administration of the APS(50%) / DMSO(50%) solution delivered to all animals.
3.34 CAP - N1 latencies
CAP - N1 latencies obtained during the six, 30 min recording periods in both the control [(+)-MK-801 + APS (50%)/DMSO (50%)] and [(+)-MK-801 + U-50488H]-treated animals of experiment 3 were submitted to repeated-measures one-way ANOVAs. CAP - N1 latency changes were analyzed at threshold and at 5dB SL. At threshold (F [1,4] = 0.000) and at 5dB SL (F [1,4] = 0.13), no significant post-baseline neural latency changes were observed in the control animals across the six recording periods. Similarly, no significant post-baseline neural latency changes were observed across the six recording periods at threshold (F [1,4] = 2.23) or at 5dB SL (F [1,4] = 1.91) in the [(+)-MK-801 + U-50488H]-treated animals.
3.35 Cochlear microphonic (CM) amplitudes
Repeated measures one-way ANOVAs were performed on the CM amplitudes obtained across the six, 30 min recording periods at threshold and 5dB SL, in the control [(+)-MK-801 + APS (50%)/DMSO (50%)] and [(+)-MK-801 + U-50488H]-treated animals of experiment 3. No significant post-baseline changes were observed in the hair cell-generated CM potentials across the six recording periods at threshold (F [ 1,4] = 1.10) or 5dB SL (F [1,4] = 1.29) in the control animals. Furthermore, no significant post-baseline CM amplitude changes were observed at threshold (F [1,4] = 0.24) or at 5dB SL (F [1,4] = 1.75) in the [(+)-MK-801 + U-50488H]-treated animals.
4. Discussion
The robust, intensity-dependent increase in the CAP - N1 amplitudes at low stimulus intensities following the administration of the opioid κ-receptor drug agonist (-)-pentazocine or the structurally divergent U-50488H delivered to the round window at high concentrations, was consistent with results of previous investigations that employed i.v. (-)-pentazocine (Sahley and Nodar, 1994; Sahley et al., 1991; 1996a; 1996b). The large post-baseline growth in neural amplitudes at threshold and 5dB SL occurred with no corresponding or consistent decrease in CAP - N1 latency, or increase in the amplitude of the simultaneously recorded, hair cell-generated CM potential, as illustrated in Fig. 6 (panels A-C). Therefore, as previously reported, the intensity-dependent post-baseline amplitude effects of (-)-pentazocine and U-50488H at low stimulus intensities are likely to be the result of neural as opposed to hair cell-receptor modulation in the cochlea (Sahley and Nodar, 1994; Sahley et al., 1991; 1996a; 1996b). The relatively high concentrations of DMSO used in the present set of investigations may be ruled out as a contributing factor in the observed electrophysiological changes, since any likely baseline or post-baseline effects of DMSO would also have been observed in the controls. In addition, DMSO is not associated with membrane damage or toxicity (Brayton, 1986) and is often used with APS in pharmacologic studies of inner ear function (e.g., Kujawa et al., 1996; Searchfield et al., 2004). Indeed, DMSO has high water solubility and significantly enhances the absorption rate of chemicals applied to tissue surfaces (Brayton, 1986; Mallory et al., 1993). Confounding barbiturate or ketamine effects may also be ruled out as contributing to the amplitude changes observed, since electrophysiological indices of peripheral auditory neural function (latency and peak amplitude) recorded within 10 ms of the delivery of a stimulus, are extremely resistant to high levels of barbiturates (Bobbin et al., 1979; Hall, 1992; Hall et al., 1985). These ‘early’ responses are also resistant to ketamine administered at the low, 50mg/kg dose employed in these investigations (Bobbin et al., 1979; Smith and Mills, 1989). Moreover, any effect of anesthesia on the recorded potentials in these investigations would have been observed in the control animals.
Quite unexpected was the post-baseline neural suppression obtained in the same animals at intensities ≥ 20dB SL, following the high concentrations of (-)-pentazocine (50 mmol) and U-50488H. (100 mmol) applied to the cochlear round window, in experiments 1 and 2. Neural amplitude suppression, and therefore, bi-phasic neural amplitude changes have never before been observed following an i.v. route of (-)-pentazocine administration (Sahley and Nodar, 1994; Sahley et al., 1991; 1996a; 1996b). The semi-permeable round window membrane is recognized as a non-invasive delivery route for administering substances directly into the intact cochlea prior to electrophysiological measurements (Dolan et al., 1990; Henry, 1995; Ohlsén et al., 1992; 1993; Searchfield et al., 2004). Substances having low molecular weights (<1000 Da) are generally incorporated into the cochlea through the round window membrane in less than 20 min (Juhn et al., 1988; Ohlsén et al., 1992). However, the 10-14μm-thick round window membrane in the chinchilla has been observed to pass macromolecules as large as 70kDa (Goycoolea and Lundman, 1997; Goycoolea et al., 1988). While (-)-pentazocine’s relatively high tissue solubility (Walker et al., 1990) and relatively low molecular weight additionally argue for an easy passage across the blood-labyrinthine barrier (Juhn et al., 1981), additional studies are required to determine the precise amount of recoverable perilymphatic (-)-pentazocine following i.v. administrations in this species.
To summarize, in the U-50488H-treated animals (Experiment 2), the positive neural amplitude changes observed at threshold and 5dB SL were statistically significant by 60 and 90 min post-baseline, and only approached significance by 90 min post-baseline at 10dB SL. In the (-)-pentazocine-treated animals (experiment 1), positive-amplitude changes were statistically significant during all three post-baseline periods at threshold, and only reached significance by 90 min post-baseline at 10dB SL. Due perhaps to the small sample sizes employed in experiment 1, positive-amplitude changes following (-)-pentazocine failed to reach statistical significance at 5dB SL. In the U-50488H-treated animals (experiment 2), neural amplitude suppression at 20dB SL was statistically significant by 30 and 60, but not by 90 min post-baseline, and was significant during all three post-baseline periods at 30 and 40dB SL. In the (-)-pentazocine-treated animals (experiment 1), neural suppression was statistically significant during all three post-baseline periods at 20dB SL, but was only significant by 30 and 60 min post-baseline at 30dB SL. At 40dB SL, neural suppression following (-)-pentazocine failed to reach levels of significance.
(-)-Pentazocine and U-50488H are virtually void of opioid δ-opioid receptor activity (Lahti et al., 1985; North, 1986; Tam, 1985). Hence, the observed bi-phasic changes in the CAP - N1 amplitude are likely to be mediated through κ-opioid receptors in the chinchilla cochlea. This conclusion is supported by a previous investigation in chinchillas that demonstrated a significant blockade of i.v. (-)-pentazocine-induced amplitude changes by a round window administration of the specific, κ-opioid receptor drug antagonist, norbinaltorphimine (Sahley et al., 1996b). A recent set of investigations conducted in guinea pigs also reported similar results (Le Prell et al., 2006; submitted). (-)-Pentazocine and U-50488H delivered to the cochlear round window membrane at a 6μl volume, and over a range of concentrations (0.1 to 5.0 mmol), produced a concentration and intensity-dependent bi-phasic effect on CAP- N1 amplitudes, very similar to the effects reported in the present investigation. In addition, the bi-phasic amplitude changes occurred independently of the organ of Corti, and were antagonized by norbinaltorphimine. Such evidence corroborates a κ-opioid-receptor neural modulatory role for endogenous dynorphins in the mammalian cochlea.
It is possible that the observed opioid-induced neural modulation in the present set of investigations was mediated by receptors located near the LEOC-Type I auditory synapse. There is no evidence for endogenous opioid-like activity within mammalian medial efferent olivocochlear neurons (Abou-Madi et al., 1987; Eybalin and Altschuler, 1990) and there is currently no evidence that endogenous dynorphin-like activity exists within medial efferent olivocochlear neurons in the chinchilla (Sahley et al., 1995). Therefore, it is unlikely that opioid receptors exist at or near the large medial efferent olivocochlear terminals located near the bases of the outer hair cells. It is more plausible that opioid κ-receptors in the chinchilla are anatomically positioned within Type I auditory dendrites found near the bases of the inner hair cells, similar to that reported in the rat (Jongkamonwiwat et al., 2003) and guinea pig cochlea (Jongkamonwiwat et al., 2006). Consequently, the available evidence suggests a κ-opioid-induced neural modulatory role for endogenous LEOC dynorphins in the mammalian cochlea.
Opioid peptides and their unique receptors are generally found within neural systems that regulate the body’s overall biologic response to physical and emotional stress (Basbaum and Jessell, 2000; Gutstein and Akil, 2001). They exhibit widespread distribution in central and peripheral nervous system structures involved in vision, audition, olfaction, and somatic sensation (Basbaum and Jessel, 2000; Stein, 1996). This fact has underscored their neurotransmitter-neuromodulatory role in the filtering of sensory information, and in the enhancement and fine-tuning of regulatory functions exerted by other neurotransmitters (Gutstein and Akil, 2001; Mansour et al., 1996). Pioneering investigations of cochlear neurotransmitter candidates in guinea pig perilymph have indicated that exposure to intense (80-115 dB SPL) and presumably stressful wide-band noise, significantly elevated levels of glutamate and [Met5]-enkephalin-like opioid peptides, relative to control values obtained in quiet (Drescher and Drescher, 1985). An intensity-dependent, in vitro release of aspartate and glutamate has also been demonstrated in guinea pig perilymph following exposure to wideband noise, presented at 101dB SPL (Jäger et al., 1998). Unfortunately none of these investigations included a perilymphatic analysis of dynorphin-like opioid neuropeptides. Oddly enough, intense stress was shown to temporarily lower CAP thresholds in guinea pigs (Muchnik et al., 1992). Considered paradoxical, no adequate explanation for the increase in auditory sensitivity was offered by these investigators.
In experiment 3, round window administered (+)-MK-801 (8 mmol) introduced 30 min prior to the round window application of U-50488H (100 mmol) produced a partial and often significant antagonism of the bi-phasic intensity-dependent neural amplitude changes subsequent to U-50488H administration. To summarize, (+)-MK-801 lead to a partial yet non-significant antagonism of U-50488H-effects at threshold and 10dB SL, and completely failed to antagonize U-50488H at 20dB SL. However, the NMDA receptor antagonism of U-50488H-induced neural changes was statistically significant by 60 and 90 min post-baseline, at 5dB, 30dB and 40dB SL. Consistent with previous investigations (Puel et al., 2002a; 2002b), was the lack of post-baseline CAP - N1 amplitude changes following (+)-MK-801 administered alone in the controls. Inasmuch as (+)-MK-801 binds only to the activated state of the NMDA receptor (Dingledine et al., 1999; Hollmann and Heinemann, 1994; Nowak et al., 1984), the absence of neural amplitude changes in the (+)-MK-801 control animals (Experiment 3) is consistent with the view that NMDA receptors are probably inactive at the relatively low range of stimulus intensities used in the present set of investigations. Indeed, using much higher stimulus intensities (100dB SPL) and long exposure durations, NMDA receptor antagonism by (+)-MK-801 afforded a significant degree of neural protection from a noise-induced permanent shift in auditory thresholds (Chen et al., 2001). Finally, dynorphin-binding affinity is reported to be greater when the NMDA receptor is inactive, whereas site-specific binding of dynorphins activates NMDA receptors (Lai et al., 2001; Shukla and Lemaire, 1994; Tang et al., 1999). Therefore, the high concentrations of κ-opioid receptor drug agonists introduced to the cochlea in the present investigation were likely to have promoted an active state in cochlear NMDA receptors.
It is tempting to speculate that the bi-phasic intensity-dependent neural amplitude changes observed in these investigations were the result of two factors. First, is the expected increased availability of cochlear glutamate as stimulus levels are increased, since it is logical to assume that the release of glutamate by cochlear inner hair cells would be proportional to the intensity of the stimulus (Jäger et al., 1998). Second, there may be a κ-opioid receptor-mediated potentiation of glutamate sensitivity at cochlear NMDA receptors resulting from the high concentrations of (-)-pentazocine or U-50488H administered to the round window membrane. The prediction from such an interaction would be an increase in auditory neural sensitivity at low stimulus intensities, and neural over-stimulation at higher intensities as pre-synaptic levels of glutamate increase (Sahley and Nodar, 2001a; 2001b; Sahley et al., 1999). Indeed, intra-cochlear perfusion of glutamate at high concentrations has been reported to reduce the discharge of auditory units during the presentation of sound (Comis and Leng, 1979). Such effects have been attributed to auditory neural over stimulation (Eybalin, 1993).
Overall, the results of the present set of investigations demonstrate κ-opioid neural modulation in the cochlea and strongly suggest an interaction between κ-opioid receptor ligands and cochlear NMDA receptors. The observation that (+)-MK-801 blocks κ-opioid-induced changes in neural amplitudes in an intensity-dependent manner suggests a complex interaction between glutamate, κ-opioid receptor ligands, and the (+)-MK-801 binding site at NMDA receptors in the chinchilla cochlea. The interaction between κ-opioid receptor ligands at cochlear NMDA receptors may be complex, and may involve elaborate allosteric interactions between opioid and/or non-opioid receptors or binding sites (Chen et al., 1995; Lai et al., 2001; Skilling et al., 1992; Zhang et al., 1997). This investigation also provides indirect evidence that the LEOC system differentially modulates auditory neural sensitivity in the cochlea by inducing both suppression and enhancement of neural responses, independent of the organ of Corti (Groff and Liberman, 2003). The differential modulation of auditory neural sensitivity by the LEOC system may incorporate responses from the modiolar-oriented Type I afferents. These cochlear afferent fibers are characterized by lower rates of spontaneous discharge and higher thresholds (Merchan-Perez and Liberman, 1996). The modiolar-oriented Type I cochlear afferents also express NMDA receptors that are postsynaptic to the inner hair cells (Ostreicher et al., 2002; Pujol et al., 1992).
Acknowledgments
Supported by NIH grant #R03-DC03360 from the National Institute On Deafness and Other Communication Disorders (NIDCD). Very special thanks to Dr. Lida C. Allen for invaluable graphics support. Special thanks also to Cleveland State University staff members Pat Reid, Elizabeth Lana Conley, and Joanne Cornelius as well as to Cleveland State University faculty members Dr. Steven Slane for valuable statistical consultation, and to Drs. Steven Slane and Benjamin Wallace for the very helpful comments on earlier drafts of this manuscript. Finally, thanks to Z. Richman for helpful edits on earlier drafts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abou-Madi L, Pontarotti P, Tramu G, Cupo A, Eybalin M. Coexistence of putative neuroactive substances in lateral olivocochlear neurons in guinea pig and rat. Hear Res. 1987;30:135–146. doi: 10.1016/0378-5955(87)90131-6. [DOI] [PubMed] [Google Scholar]
- Altschuler RA, Reeks KA, Fex J, Hoffman DW. Lateral olivocochlear neurons contain both enkephalin and dynorphin immunoreactivities. J Histochem Cytochem. 1988;36:797–801. doi: 10.1177/36.7.2898496. [DOI] [PubMed] [Google Scholar]
- Arcaya J-L, Cano G, Gomez G, Maixner W, Suarez-Roca H. Dynorphin A increases substance P release from trigeminal primary afferent C-fibers. Eur J Pharmacol. 1999;366:27–34. doi: 10.1016/s0014-2999(98)00897-8. [DOI] [PubMed] [Google Scholar]
- Azeredo WJ, Kliment MJ, Morley BJ, Relkin E, Slepecky NB, Sterns A, Warr WB, Weekly JM, Woods CI. Olivocochlear neurons in the chinchilla: a retrograde fluorescent labelling study. Hear Res. 1999;134:57–70. doi: 10.1016/s0378-5955(99)00069-6. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Jessell TM. The perception of pain. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. fourth. McGraw-Hill; New York, NY: 2000. pp. 472–491. [Google Scholar]
- Beauchaine KA, Kaminski JR, Gorga P. Comparison of Beyer DT48 and Etymotic insert earphones: auditory brain stem response measurements. Ear Hear. 1987;8:292–297. doi: 10.1097/00003446-198710000-00007. [DOI] [PubMed] [Google Scholar]
- Bobbin RP, May JG, Lemoine RL. Effects of pentobarbital and ketamine on brain stem auditory potentials. Arch Otolaryngol. 1979;105:467–470. doi: 10.1001/archotol.1979.00790200029006. [DOI] [PubMed] [Google Scholar]
- Brayton CF. Dimethyl sulfoxide (DMSO): a review. Cornell Vet. 1986;76:61–90. [PubMed] [Google Scholar]
- Brown DJ, McMahon CM, Patuzzi RB. K+ currents produce P1 in the round window CAP: evidence from DC current bias, K+ channel blockade and recordings from cochlea and brainstem. Hear Res. 2004;190:60–74. doi: 10.1016/S0378-5955(03)00404-0. [DOI] [PubMed] [Google Scholar]
- Caudle RM, Dubner R. Ifenprodil blocks the excitatory effects of the opioid peptide dynorphin 1-17 on NMDA receptor-mediated currents in the CA3 region of the guinea pig hippocampus. Neuropeptides. 1998;32:87–95. doi: 10.1016/s0143-4179(98)90022-1. [DOI] [PubMed] [Google Scholar]
- Chen G-D, Kong J, Reinhard K, Fechter LD. NMDA receptor blockage protects against permanent noise-induced hearing loss but not its potentiation by carbon monoxide. Hear Res. 2001;154:108–115. doi: 10.1016/s0378-5955(01)00228-3. [DOI] [PubMed] [Google Scholar]
- Chen L, Gu Y, Huang L-YM. The mechanism of action for the block of NMDA receptor channels by the opioid peptide dynorphin. J Neurosci. 1995;15:4602–4611. doi: 10.1523/JNEUROSCI.15-06-04602.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comis SD, Leng G. Action of putative neurotransmitters in the guinea pig cochlea. Exp Brain Res. 1979;36:119–128. doi: 10.1007/BF00238472. [DOI] [PubMed] [Google Scholar]
- Crain SM, Shen K-F. GM1 ganglioside-induced modulation of opioid receptor-mediated functions. Ann NY Acad Sci. 1998;845:106–125. doi: 10.1111/j.1749-6632.1998.tb09665.x. [DOI] [PubMed] [Google Scholar]
- d’Aldin CG, Ruel J, Assié R, Pujol R, Puel JL. Implication of NMDA type glutamate receptors in neural regeneration and neoformation of synapses after excitotoxic injury in the guinea pig cochlea. Int J Dev Neurosci. 1997;15:619–629. doi: 10.1016/s0736-5748(96)00116-5. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Dolan DF, Nuttall AL, Avinash G. Asynchronous neural activity recorded from the round window. J Acoust Soc Am. 1990;87:2621–2627. doi: 10.1121/1.399054. [DOI] [PubMed] [Google Scholar]
- Drescher DG, Drescher MJ. HPLC analysis of presumptive neurotransmitters in perilymph. In: Drescher DG, editor. Auditory Biochemestry. C.C. Thomas; Springfield, Il: 1985. pp. 50–67. [Google Scholar]
- Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- Ehrenberger K, Felix D. Glutamate receptors in afferent cochlear neurotransmission in guinea pigs. Hear Res. 1991;52:73–80. doi: 10.1016/0378-5955(91)90188-f. [DOI] [PubMed] [Google Scholar]
- Ehrenberger K, Felix D. Receptor pharmacological models for inner ear therapies with emphasis on glutamate receptors: a survey. Acta Otolaryngol. 1995;115:236–240. doi: 10.3109/00016489509139299. [DOI] [PubMed] [Google Scholar]
- Eybalin M. Neurotransmitters and neuromodulators of the mammalian cochlea. Physiol Rev. 1993;73:309–373. doi: 10.1152/physrev.1993.73.2.309. [DOI] [PubMed] [Google Scholar]
- Faden AI. Dynorphin increases extracellular levels of excitatory amino acids in the brain through a non-opioid mechanism. J Neurosci. 1992;12:425–429. doi: 10.1523/JNEUROSCI.12-02-00425.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix D, Ehrenberger K. The role of glutamate at the afferent synapses of mammalian inner hair cells: a microiontophoretic study. Inner Ear Biol. 1989;26:19. doi: 10.1007/BF00634769. [DOI] [PubMed] [Google Scholar]
- Felix D, Ehrenberger KA. Microiontophoretic study of the role of excitatory amino acids at the afferent synapses of mammalian inner hair cells. Eur Arch Otorhinolaryngol. 1990;248:1–3. doi: 10.1007/BF00634769. [DOI] [PubMed] [Google Scholar]
- Felix D, Ehrenberger K. N-methyl-d-aspartate-induced oscillations in excitatory afferent neurotransmission in the guinea pig cochlea. Eur Arch Otorhinolaryngol. 1991;248:429–431. doi: 10.1007/BF00627627. [DOI] [PubMed] [Google Scholar]
- George D, Mallery P. SPSS for Windows. Allyn & Bacon; Needham Heights, MA: 1999. [Google Scholar]
- Goycoolea MV, Lundman L. Round window membrane structure function and permeability: a review. Microsc Res Tech. 1997;36:201–211. doi: 10.1002/(SICI)1097-0029(19970201)36:3<201::AID-JEMT8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Goycoolea MV, Muchow D, Schachem PA. Experimental studies on round window membrane membrane structure function and permeability. Laryngoscope. 1988;98:1–20. doi: 10.1288/00005537-198806001-00002. [DOI] [PubMed] [Google Scholar]
- Groff JA, Liberman MC. Modulation of cochlear afferent responses by the lateral olivocochlear system: activation via electrical stimulation of the inferior colliculus. J Neurophysiol. 2003;90:3178–3200. doi: 10.1152/jn.00537.2003. [DOI] [PubMed] [Google Scholar]
- Guinan JJ. Physiology of olivocochlear efferents. In: Dallos P, Popper AN, Fay RR, editors. The Cochlea: Springer Handbook of Auditory Research. Springer-Verlag; New York, N.Y.: 1996. pp. 435–502. [Google Scholar]
- Gutstein HB, Akil A. Opioid analgesics. In: Hardman JG, Limbird LE, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. tenth. McGraw-Hill; New York, N.Y.: 2001. pp. 569–619. [Google Scholar]
- Hall JW. Handbook of Auditory Evoked Responses. Allyn & Bacon; Needham Heights, MA: 1992. Effect of nonpathologic subject characteristics; pp. 70–103. [Google Scholar]
- Hall JW, Mackey-Hargadine J, Allen SJ. Monitoring neurologic status of comatose patients in the intensive care unit. In: Jacobson JT, editor. The Auditory Brainstem Response. College-Hill Press; San Diego, CA: 1985. pp. 253–283. [Google Scholar]
- Hauser KF, Foldes JK, Turbek CS. Dynorphin A (1-13) neurotoxicity in vitro: opioid and non-opioid mechanisms in mouse spinal cord neurons. Exp Neurol. 1999;160:361–375. doi: 10.1006/exnr.1999.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, Knapp PE, Turbek CS. Structure-activity analysis of dynorphin-A toxicity in spinal cord neurons: intrinsic neurotoxicity of dynorphin-A and its carboxyl-terminal, nonopioid metabolites. Exp Neurol. 2001;168:78–87. doi: 10.1006/exnr.2000.7580. [DOI] [PubMed] [Google Scholar]
- Henry KR. Auditory nerve neurophonic recorded from the round window of the Mongolian gerbil. Hear Res. 1995;90:176–184. doi: 10.1016/0378-5955(95)00162-6. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann SF. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Jäger W, Goiny M, Herrera-Marschitz M, Flock Å, Hökfelt T, Brundin L. Sound-evoked efflux of excitatory amino acids in the guinea-pig cochlea in vitro. Exp Brain Res. 1998;121:425–432. doi: 10.1007/s002210050477. [DOI] [PubMed] [Google Scholar]
- Jongkamonwiwat N, Phansuwan-Pujito P, Sarapoke P, Chetsawang B, Casalotti SO, Forge A, Dodson H, Govitrapong P. The presence of opioid receptors in rat inner ear. Hear Res. 2003;181:85–93. doi: 10.1016/s0378-5955(03)00175-8. [DOI] [PubMed] [Google Scholar]
- Jongkamonwiwat N, Phansuwan-Pujito P, Casalotti SO, Forge A, Dodson H, Govitrapong P. The existence of opioid receptors in the cochlea of guinea pigs. Eur J Neurosci. 2006;23:2701–2711. doi: 10.1111/j.1460-9568.2006.04810.x. [DOI] [PubMed] [Google Scholar]
- Juhn SK, Hamaguchi Y, Goycoolea MV. Review of round window membrane permeability. Acta Otolaryngol Suppl. 1988;457:43–48. doi: 10.3109/00016488809138883. [DOI] [PubMed] [Google Scholar]
- Juhn SK, Rybak LP, Prado S. Nature of blood-labyrinth barrier in experimental conditions. Ann Otol. 1981;90:135–141. doi: 10.1177/000348948109000208. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Siegelbaum SA. Synaptic integration. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. fourth. McGraw-Hill; New York, N.Y.: 2000. pp. 207–228. [Google Scholar]
- Kleinlogel S, Oestreicher E, Arnold T, Ehrehberger K, Felix D. Metabotropic glutamate receptors group I are involved in cochlear neurotransmission. Neuroreport. 1999;10:1979–1882. doi: 10.1097/00001756-199906230-00015. [DOI] [PubMed] [Google Scholar]
- Kujawa SG, Fallon M, Skellett RA, Bobbin RP. Time-varying alterations in the f2-f1 DPOAE response to continuous primary stimulation: II. influence of local calcium-dependent mechanisms. Hear Res. 1996;97:153–164. [PubMed] [Google Scholar]
- Kuriyama H, Albin RL, Altschuler RA. Expression of NMDA-receptor mRNA in the rat cochlea. Hear Res. 1993;69:215–220. doi: 10.1016/0378-5955(93)90110-m. [DOI] [PubMed] [Google Scholar]
- Lahti RA, Mickelson MM, McCall JM, Von Voigtlander PF. [3H] U-69593, a highly selective ligand for the opioid K receptor. Eur J Pharmacol. 1985;109:281–284. doi: 10.1016/0014-2999(85)90431-5. [DOI] [PubMed] [Google Scholar]
- Lai J, Ossipov MH, Vanderah TW, Malan TP, Porreca F. Neuropathic pain: the paradox of dynorphin. Pharmacol Rev. 2001;1:160–167. [PubMed] [Google Scholar]
- Laughlin TM, Larson AA, Wilcox GL. Mechanisms of induction of persistent nociception by dynorphin. J Pharmacol Exp Ther. 2001;299:6–11. [PubMed] [Google Scholar]
- Laughlin TM, Vanderah TW, Lashbrook J, Nichols ML, Ossipov M, Porreca F, Wilcox GL. Spinally administered dynorphin A produces long-lasting allodynia: involvement of NMDA but not opioid receptors. Pain. 1997;72:253–260. doi: 10.1016/s0304-3959(97)00046-8. [DOI] [PubMed] [Google Scholar]
- Le Prell CG, Bledsoe SC, Bobbin RP, Puel J-L. Neurotransmission in the inner ear: functional and molecular analyses. In: Jahn AF, Santos-Sacchi J, editors. Physiology of The Ear. second. Singular-Thomson Learning; San Diego, CA.: 2001. pp. 575–611. [Google Scholar]
- Le Prell CG, Halsey KI, Dolan DF, Bledsoe SC., JR Kappa opioid-receptor agonists depress compound action potential amplitude. Abs Assoc Res Otolaryngol. 2004;27:329–330. [Google Scholar]
- Le Prell CG, Shore SE, Hughes LF, Bledsoe SC. Disruption of lateral efferent pathways: functional changes in auditory evoked responses. J Assoc Res Otolaryngol. 2003;4:276–290. doi: 10.1007/s10162-002-3018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC. Physiology of cochlear efferent and afferent neurons: direct comparisons in the same animal. Hear Res. 1988;34:179–192. doi: 10.1016/0378-5955(88)90105-0. [DOI] [PubMed] [Google Scholar]
- Liberman MC. Effects of chronic cochlear de-efferentation on auditory-nerve response. Hear Res. 1990;49:209–224. doi: 10.1016/0378-5955(90)90105-x. [DOI] [PubMed] [Google Scholar]
- Maison SF, Adams JC, Liberman MC. Olivocochlear innervations in the mouse: immunocytochemical maps, crossed versus uncrossed contributions, and transmitter colocalization. J Comp Neurol. 2003;455:406–416. doi: 10.1002/cne.10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory SB, Lehman PA, Vanderpool DR, Franz TJ. Topical lidocaine for anesthesia in patients undergoing pulsed dye laser treatment for vascular malformations. Pediatr Dermatol. 1993;10:370–375. doi: 10.1111/j.1525-1470.1993.tb00403.x. [DOI] [PubMed] [Google Scholar]
- Mansour A, Burke S, Pavlic RJ, Akil H, Watson SJ. Immunohistochemical localization of the cloned K1 receptor in the rat CNS and pituitary. Neurosci. 1996;71:671–690. doi: 10.1016/0306-4522(95)00464-5. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- McMahon CM, Brown DJ, Patuzzi RB. Transient focal cooling at the round window and cochlear nucleus shows round window CAP originates from cochlear neurones alone. Hear Res. 2004;190:75–86. doi: 10.1016/S0378-5955(03)00403-9. [DOI] [PubMed] [Google Scholar]
- Merchan-Perez A, Liberman MC. Ultrastructural differences among afferent synapses on cochlear hair cells: correlations with spontaneous discharge rate. J Comp Neurol. 1996;371:208–221. doi: 10.1002/(SICI)1096-9861(19960722)371:2<208::AID-CNE2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Miller CA, Abbas PJ, Robinson BK. Characterization of wave I of the electrically evoked auditory brainstem response in the guinea pig. Hear Res. 1993;69:35–44. doi: 10.1016/0378-5955(93)90091-e. [DOI] [PubMed] [Google Scholar]
- Muchnik C, Sahartov E, Peleg E, Hildesheimer M. Temporary threshold shift due to noise exposure in guinea pigs under emotional stress. Hear Res. 1992;58:101–106. doi: 10.1016/0378-5955(92)90013-d. [DOI] [PubMed] [Google Scholar]
- Musiek FE, Baran JA. Canal electrode electrocochleography in patients with absent wave I ABRs. Otolaryngol Head Neck Surg. 1990;103:25–31. doi: 10.1177/019459989010300104. [DOI] [PubMed] [Google Scholar]
- Niedzielski AS, Safieddine S, Wenthold RJ. Molecular analysis of excitatory amino acid receptor expression in the cochlea. Audiol Neur-Otol. 1997;2:79–91. doi: 10.1159/000259232. [DOI] [PubMed] [Google Scholar]
- Nordang L, Oestreicher E, Arnold W, Anniko M. Glutamate is the afferent neurotransmitter in the human cochlea. Acta Otolaryngol. 2000;120:359–362. doi: 10.1080/000164800750000568. [DOI] [PubMed] [Google Scholar]
- North RA. Opioid receptor types and membrane ion channels. Trends Neurosci. 1986;9:114–117. [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochaintz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Oestreicher E, Arnold W, Felix D. Neurotransmission of the cochlear inner hair cell synapse--implications for inner ear therapy. Adv Oto-Rhino-Laryngol. 2002;59:131–139. doi: 10.1159/000059245. [DOI] [PubMed] [Google Scholar]
- Ohlsén KA, Didier A, Baldwin D, Miller JM, Nuttall AL, Hultcrantz E. Cochlear blood flow in response to dilating agents. Hear Res. 1992;58:19–25. doi: 10.1016/0378-5955(92)90004-7. [DOI] [PubMed] [Google Scholar]
- Ohlsén KA, Hultcrantz E, Engström B. The effect of topical application of vasodilating agents on cochlear electrophysiology. Acta Otolaryngol. 1993;113:55–61. doi: 10.3109/00016489309135767. [DOI] [PubMed] [Google Scholar]
- Puel J-L, d’Aldin C, Ruel J, Ladrech S, Pujol R. Synaptic repair mechanisms responsible for functional recovery in various cochlear pathologies. Acta Otolaryngol. 1997;117:214–218. doi: 10.3109/00016489709117773. [DOI] [PubMed] [Google Scholar]
- Puel J-L, Ladrech S, Chabert R, Pujol R, Eybalin M. Electrophysiological evidence for the presence of NMDA receptors in the guinea pig cochlea. Hear Res. 1991;51:255–264. doi: 10.1016/0378-5955(91)90042-8. [DOI] [PubMed] [Google Scholar]
- Puel JL. Chemical synaptic transmission in the cochlea. Prog Neurobiol. 1995;47:449–476. doi: 10.1016/0301-0082(95)00028-3. [DOI] [PubMed] [Google Scholar]
- Pujol R. Neural anatomy of the cochlea: development and plasticity. In: Jahn AF, Santos-Sacchi J, editors. Physiology of The Ear. second. Singular-Thomson Learning; San Diego, CA: 2001. pp. 515–528. [Google Scholar]
- Pujol R, Puel J-L, Gervais-d’Aldin C, Eybalin M. Pathophysiology of the glutamate synapses in the cochlea. Acta Otolaryngol. 1993;113:330–334. doi: 10.3109/00016489309135819. [DOI] [PubMed] [Google Scholar]
- Pujol R, Puel J-L, Eybalin M. Implication of non-NMDA and NMDA receptors in cochlear ischemia. Neuroreport. 1992;3:299–302. doi: 10.1097/00001756-199204000-00002. [DOI] [PubMed] [Google Scholar]
- Safieddine S, Eybalin M. Co-expession of NMDA and AMPA/kainate receptor mRNAs in cochlear neurones. Neuroreport. 1992;3:1145–1148. doi: 10.1097/00001756-199212000-00029. [DOI] [PubMed] [Google Scholar]
- Safieddine S, Prior AMS, Eybalin M. Choline acetyltransferase, glutamate decarboxylase, tyrosine hydroxylase, calcitonin gene-related peptide and opioid peptides coexist in lateral efferent neurons of rat and guinea-pig. Eur J Neurosci. 1997;9:356–367. doi: 10.1111/j.1460-9568.1997.tb01405.x. [DOI] [PubMed] [Google Scholar]
- Sahley TL, Chernicky CL, Nodar RH, Musiek FE. Pro-dynorphin peptides are co-localized within auditory structures of the chinchilla brainstem. Abs Assoc Res Otolaryngol. 1995;18:170. [Google Scholar]
- Sahley TL, Kalish RB, Musiek FE, Hoffman DW. Effects of opioid drugs on auditory evoked potentials suggest a role of lateral olivocochlear dynorphins in auditory function. Hear Res. 1991;55:133–142. doi: 10.1016/0378-5955(91)90099-u. [DOI] [PubMed] [Google Scholar]
- Sahley TL, Musiek FE, Nodar RH. Naloxone blockade of (-)-pentazocine-induced changes in auditory function. Ear Hear. 1996a;17:341–353. doi: 10.1097/00003446-199608000-00006. [DOI] [PubMed] [Google Scholar]
- Sahley TL, Nodar RH, Musiek FE. Blockade of opioid-induced changes in auditory function at the level of the cochlea. Ear Hear. 1996b;17:552–558. doi: 10.1097/00003446-199612000-00011. [DOI] [PubMed] [Google Scholar]
- Sahley TL, Nodar RH, Musiek FE. Efferent auditory system: structure and function. Singular Publishing; San Diego, CA: 1997. [Google Scholar]
- Sahley TL, Nodar RH, Musiek FE. Endogenous dynorphins: possible role in peripheral tinnitus. Int Tinnitus J. 1999;5:76–91. [PubMed] [Google Scholar]
- Sahley TL, Nodar RH. A biochemical model of peripheral tinnitus. Hear Res. 2001a;152:43–54. doi: 10.1016/s0378-5955(00)00235-5. [DOI] [PubMed] [Google Scholar]
- Sahley TL, Nodar RH. Improvement in auditory function following pentazocine suggests a role for dynorphins in auditory sensitivity. Ear Hear. 1994;15:422–431. doi: 10.1097/00003446-199412000-00003. [DOI] [PubMed] [Google Scholar]
- Sahley TL, Nodar RH. Tinnitus: present and future. In: Donald PJ, Gluckman JL, editors. Current opinion: Otolaryngol Head Neck Surg. Vol. 9. 2001b. pp. 323–328. [Google Scholar]
- Samra SK, Krutak-Krol H, Pohorecki R, Domino EF. Scopolamine, morphine, and brain-stem auditory evoked potentials in awake monkeys. Anesthesiol. 1985;62:437–441. doi: 10.1097/00000542-198504000-00011. [DOI] [PubMed] [Google Scholar]
- Samra SK, Lilly DJ, Rush NL, Kirsh MM. Fentanyl anesthesia and human brainstem auditory evoked potentials. Anesthesiol. 1984;61:261–265. doi: 10.1097/00000542-198409000-00005. [DOI] [PubMed] [Google Scholar]
- Searchfield GD, Muñoz DJB, Thorne PR. Ensemble spontaneous activity in the guinea-pig cochlear nerve. Hear Res. 2004;192:23–35. doi: 10.1016/j.heares.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Sewell WF. Neurotransmitters and synaptic transmission. In: Dallos P, Popper AN, Fay RR, editors. The Cochlea. Springer Verlag; New York, N.Y.: 1996. pp. 503–533. [Google Scholar]
- Shukla VK, Lemaire S. Non-opioid effects of dynorphins: possible role of the NMDA receptor. Trends Pharmacol Sci. 1994;15:420–424. doi: 10.1016/0165-6147(94)90091-4. [DOI] [PubMed] [Google Scholar]
- Simmons DD. Development of the inner ear efferent system across vertebrate species. J Neurobiol. 2002;53:228–250. doi: 10.1002/neu.10130. [DOI] [PubMed] [Google Scholar]
- Skilling SR, Sun X, Kurtz HJ, Larson AA. Selective potentiation of NMDA-induced activity and release of excitatory amino acids by dynorphin: possible roles in paralysis and neurotoxicity. Brain Res. 1992;575:272–278. doi: 10.1016/0006-8993(92)90090-v. [DOI] [PubMed] [Google Scholar]
- Smith DI, Mills JH. Anesthesia effects: auditory brain-stem response. Electroencephalogr Clin Neurophysiol. 1989;72:422–428. doi: 10.1016/0013-4694(89)90047-3. [DOI] [PubMed] [Google Scholar]
- Spagnoli SD, Saunders JC. Threshold sensitivity and frequency selectivity measured with round window whole nerve action potentials in the awake, restrained chinchilla. Otolaryngol Head Neck Surg. 1987;96:99–105. doi: 10.1177/019459988709600117. [DOI] [PubMed] [Google Scholar]
- Stein C. Nociceptors and neuroimmune interactions. In: Belmonte C, Cervero F, editors. Neurobiology of Nociceptors. Oxford University Press; New York, N.Y.: 1996. pp. 439–452. [Google Scholar]
- Stricker NL, Huganir RL. AMPA/kainate receptors. In: Moss SJ, Henley J, editors. Receptor and Ion-Channel Trafficking: cell biology of ligand-gated and voltage-sensitive ion channels. Oxford University Press; New York, N.Y.: 2002. pp. 131–155. [Google Scholar]
- Suneja SK, Benson CG, Gross J, Potashner SJ. Uptake and release of d-aspartate, GABA and glycine in guinea pig brainstem auditory nuclei. J Neurochem. 1995a;64:147–160. doi: 10.1046/j.1471-4159.1995.64010147.x. [DOI] [PubMed] [Google Scholar]
- Suneja SK, Benson CG, Gross J, Potashner SJ. Evidence for glutamatergic projections from the cochlear nucleus to the superior olive and ventral nucleus of the lateral lemniscus. J Neurochem. 1995b;64:161–171. doi: 10.1046/j.1471-4159.1995.64010161.x. [DOI] [PubMed] [Google Scholar]
- Tam SW. [3H]-(+)-SKF-10,047, (+)-[3H]-ethylketocyclazocine, μ, K, ä and phencyclidine binding sites in guinea pig brain membranes. Eur J Pharmacol. 1985;109:33–41. doi: 10.1016/0014-2999(85)90536-9. [DOI] [PubMed] [Google Scholar]
- Tang Q, Gandhoke R, Burritt A, Hruby VJ, Porreca F, Lai J. High-affinity interaction oF [des-Tyrosyl) dynorphin A (2-17) with NMDA receptors. J Pharmacol Exp Ther. 1999;291:760–765. [PubMed] [Google Scholar]
- Trautwein P, Hofstetter P, Wang J, Salvi R, Nostrant A. Selective inner hair cell loss does not alter distortion product otoacoustic emissions. Hear Res. 1996;96:71–82. doi: 10.1016/0378-5955(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Usami S, Matsubara A, Fujita S, Takumi Y, Otterson OP. Glutamate neurotransmission in the mammalian inner ear. In: Ottersen OP, Storm-Mathison J, editors. Handbook of Chemical Anatomy, Vol 18: glutamate. Elsevier Publishers; New York, N.Y.: 2000. pp. 225–271. [Google Scholar]
- Vanderah TW, Laughlin T, Lashbrook JM, Nichols ML, Wilcox GL, Ossipov MH, Malan TP, Porreca F. Single intrathecal injections of dynorphin A or des-Tyr-dynorphins produce long lasting allodynia in rats: blockade by MK-801 but not by naloxone. Pain. 1996;68:275–281. doi: 10.1016/s0304-3959(96)03225-3. [DOI] [PubMed] [Google Scholar]
- Von Voigtlander PF, Lahti RA, Ludens JH. U-50488: a selective and structurally novel non-mu kappa opioid agonist. J Pharmacol Exp Ther. 1983;224:7–12. [PubMed] [Google Scholar]
- Walker JM, Bowen WD, Walker FO, Matsumoto RR, de Costa B, Rice KC. Sigma receptors: biology and function. Pharmacol Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- Walsh EJ, McGee J, McFadden SL, Liberman MC. Long-term effects of sectioning the olivocochlear bundle in neonatal cats. J Neurosci. 1998;18:3859–3869. doi: 10.1523/JNEUROSCI.18-10-03859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr WB, Boche JB, Neely ST. Efferent innervation of the inner hair cell region: origins and terminations of two lateral olivocochlear systems. Hear Res. 1997;108:89–111. doi: 10.1016/s0378-5955(97)00044-0. [DOI] [PubMed] [Google Scholar]
- Warr WB. Organization of olivocochlear efferent systems in mammals. In: Webster DB, Popper AN, Fay RR, editors. The Mammalian Auditory Pathway: neuroanatomy. Springer-Verlag; New York, N.Y.: 1992. pp. 410–448. [Google Scholar]
- Waxham MN. Neurotransmitter receptors. In: Squire LR, Bloom FE, McConnell SK, Roberts JL, et al., editors. Fundamental Neuroscience. secnd. Academic Press; New York, N.Y.: 2003. pp. 225–258. [Google Scholar]
- Wisden W, Seeburg PH, Monyer H. AMPA, kainate and NMDA ionotropic glutamate receptor expression-an in situ hybridization atlas. In: Ottersen OP, Storm-Mathison J, editors. Handbook of Chemical Anatomy, Vol 18: glutamate. Elsevier Publishers; New York, N.Y.: 2000. pp. 99–143. [Google Scholar]
- Zhang L, Peoples RW, Oz M, Harvey-White J, Weight FF, Brauneis U. Potentiation of NMDA receptor-mediated responses by dynorphin at low extracellular glycine concentrations. J Neurophysiol. 1997;78:582–590. doi: 10.1152/jn.1997.78.2.582. [DOI] [PubMed] [Google Scholar]
- Zukin RS, Zukin SR. The case for multiple opiate receptors. Trends Neurosci. 1984;7:160–164. [Google Scholar]