

Abstract
Combined QM/MM calculations of the active-site of cytochorme P450cam have been performed before and after the binding of P450cam to putidaredoxin. The calculations were carried out for both a 5-coordinated and a 6-coordinated active-site of cytochrome P450cam, with either a water molecule or a carbon monoxide molecule as a 6th distal ligand. An experimentally observed increase in the Fe-S stretching frequency that occurs after cytochrome P450cam binds to putidaredoxin, has been reproduced in our study. Experimentally observed changes in the Fe-C and C-O vibration frequencies that occur after binding of both proteins, have also been reproduced in our study. The computed increase of the Fe-S and Fe-C stretching frequencies is correlated with a corresponding decrease of the Fe-S and Fe-C interatomic distances. According to our calculations, for the active-site with carbon monoxide in the triplet electronic state, the binding process increases the spin densities on the iron and sulfur atoms, which changes the Fe-C and C-O stretching frequencies in opposite directions, in agreement with experimental data.
Introduction
Cytochrome P450cam (P450) is a bacterial protein, which catalyses a hydroxylation reaction of a substrate (d-camphor) at physiological conditions [1]. The catalytic activity of P450 has been extensively characterized by experimental [2; 3; 4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14; 15; 16; 17] and theoretical [18; 19; 20; 21; 22; 23; 24; 25; 26; 27; 28; 29] studies, therefore P450 serves as a model protein of the entire cytochrome family. The hydroxylation reaction in the enzyme requires two electrons to proceed [30], and both electrons are transferred into the reaction site of the enzyme after binding P450 to an iron-sulfur (2Fe-2S) protein, putidaredoxin (pdx) [31; 32; 33; 34; 35; 36]. Both experimental [37; 38] and theoretical [39; 40] studies suggest that pdx binds to P450 from the proximal side of the active-site of the heme enzyme. Although the binding of P450 to pdx has been the subject of extensive investigations, a molecular mechanism for the interaction between both proteins remains unknown. The binding process changes a spin state of P450 [2], suggesting that the interaction between both proteins perturbs the active site of the heme enzyme.
To elucidate the changes of the P450 active site after binding to pdx, we present results of a computational study that is based on a combined quantum mechanical - molecular mechanical (QM/MM) approach. In our calculations the heme active-site of P450 and axial ligands, namely a water molecule, carbon monoxide, and cysteine CYS357, were calculated at a quantum mechanical (QM) level of theory, while the rest of the protein environment was calculated at a molecular mechanical (MM) level. The calculations were performed in two steps, namely MM-based molecular dynamic (MD) simulations using the AMBER program [41], followed by combined QM/MM calculations of snapshots taken from the MD simulation using the Q-Chem [42] program. Experimental data indicate that the active site of P450 in the binding process to pdx, is a mixture of low and high spin states [2], therefore we have separately performed calculations for the doublet, quartet and sextet electronic states of the neutral QM molecule, representing the oxidized iron form. Experimental data show also that carbon monoxide binds to P450 only in the reduced form of the heme active site [4; 43]. Therefore for the protein system with this ligand, the calculations of the reduced iron form were also carried out in our study with an additional electron in the QM molecule, for the singlet and triplet electronic states. For comparison, we have also performed calculations for the oxidized iron state with the CO ligand.
Computational Details
The MD simulations of P450 were carried out before and after binding to pdx, separately for: (1) the 5-coordinated active-site center, (2) the 6-coordinated system with a water molecule, and (3) the 6-coordinated system with carbon monoxide. The initial geometry was obtained from the x-ray experimental structure of the cyanide (CN) complex of P450, as implemented in the 1O76 pdb protein file [17], and from the 1OQQ pdb file of the pdx mutation [44]. The CN ligand was removed in our calculations of the 5-coordinated system, and was replaced by a water molecule or carbon monoxide in the calculations of the 6-coordinated system. Serines 73 and 85 of the pdx mutation have been replaced in our calculations by cysteines to mimic the original wild type of this protein. Pdx has been placed close to P450 at the proximal side of the heme active-site of P450 keeping the distance around 12 A from the iron atom of P450 to one of the iron atoms of pdx, according to previous MD simulations of the P450/pdx complex [34; 39; 40]. Hydrogen atoms were added to their standard positions according to the AMBER force field. MM parameters for the heme active-site, the camphor molecule and the metal site of pdx, were taken from the literature and from simple quantum chemical calculations in the gas phase. To neutralize the entire protein system, 17 sodium counter-ions were utilized whose positions were obtained from grid electrostatic potential calculations using AMBER program. Next, the protein system was placed into a center of a 50 A (radius) water sphere, having explicit TIP3P water molecules. The MD consisted of slow 100ps heating from 0 to 300 K performed at constant volume, followed by long 10 ns equilibration dynamics performed at constant volume and constant temperature. In the simulation, an external harmonic potential outside the water sphere, and a weak harmonic potential for sodium counter-ions were applied. The similar approach has been applied to MD simulations of P450 without pdx.
After MD, 10 protein snapshots were randomly selected from the MD trajectory between 9.0ns to 10.0ns of the protein dynamics time for each MD simulation. The number of snapshots used in our calculations represents the minimum number of protein structures, which can characterize the protein dynamic process. However the number of snapshots which can be used in our QM/MM approach, depends only on computational resources, and right now we are working on similar calculations based on a larger ensemble of protein structures. Each selected protein snapshot was minimized at the MM level of theory using the AMBER program with all water molecules constrained. Then each selected snapshot was divided into a QM part and a MM part for subsequent combined QM/MM calculations. The QM part of the protein complex included the heme molecule with the ligand, and the proximal cysteine CYS357. A chemical bond between CYS357 and the protein was cut and the resulting free chemical valences were filled out by hydrogen atoms, as is routinely done in combined QM/MM simulations. The QM system was simplified by eliminating side chains of the heme molecule and side chains of CYS357, as has been done by others in similar calculations [45; 46]. Next the geometry of the QM moiety was optimized without any constraints inside the fixed MM portion of the system, separately for the doublet, quartet and sextet electronic states of the neutral QM molecule representing the oxidized iron form, and for the singlet and triplet states of the QM anion representing the reduced iron form. The geometry optimization of the QM system was carried out at the B3LYP/6-31+G* level of theory, while AMBER was used to model the MM protein environment. In the QM/MM calculations all external point charges (MM atoms of the protein and the water sphere) were included. The interaction between QM and MM atoms has been approximated by classic Lennard-Jones potentials [47]. After geometry optimization, molecular hessian calculations were performed using the same QM/MM procedure. Atomic charges and spin densities have been calculated according to the natural bond orbital (NBO) analysis [48].
Results and Discussion
Final results of our calculations are reported in Tables 1, 2, 3 and 4 as average data from a statistical ensemble of the QM/MM calculations, which are based on a series of protein snapshots obtained from the MD. Figures 1, 2 and 3 show optimal geometries of the enzyme active-site, obtained in our QM/MM calculations. Figure 4 presents the structure of the P450/pdx protein complex investigated in our study.
Table 1.
The properties of the Fe and S atoms calculated in P450cam, and in the protein complex of P450cam and pdx, for the 5-coordinated heme active-site in the oxidized form for the doublet, quartet and sextet electronic states. The “Change in protein” column represents the difference between results of calculations performed in the protein complex of P450cam/pdx, and in P450cam alone. The atomic charges and spin densities are calculated according to the NBO population [48]. The results of the calculations are average values obtained from a series of QM/MM calculations based on 10 protein snapshots taken from MM dynamics. The calculated frequencies are scaled by a factor 0.92, which reflects a common characteristic of the DFT method [50]. Experimental data are taken from reference [37].
| Heme active-site 5-coordinated | P450cam | P450cam/pdx | Change in protein |
|---|---|---|---|
| Doublet | |||
| Fe Charge (a.u.) | 1.43 | 1.41 | -0.02 |
| S Charge (a.u.) | -0.24 | -0.20 | 0.04 |
| Fe Spin Density (a.u.) | 1.16 | 1.12 | -0.04 |
| S Spin Density (a.u.) | -0.06 | -0.04 | 0.02 |
| Fe-S distance (Å) | 2.21 | 2.16 | -0.05 |
| Fe-S frequency (cm-1) | 354 | 385 | 31 |
| Quartet | |||
| Fe Charge (a.u.) | 1.62 | 1.60 | -0.02 |
| S Charge (a.u.) | -0.45 | -0.42 | 0.03 |
| Fe Spin Density (a.u.) | 2.59 | 2.55 | -0.04 |
| S Spin Density (a.u.) | 0.36 | 0.40 | 0.04 |
| Fe-S distance (Å) | 2.46 | 2.36 | -0.10 |
| Fe-S frequency (cm-1) | 282 | 314 | 32 |
| Sextet | |||
| Fe Charge (a.u.) | 1.84 | 1.82 | -0.02 |
| S Charge (a.u.) | -0.47 | -0.42 | 0.05 |
| Fe Spin Density (a.u.) | 4.16 | 4.13 | -0.03 |
| S Spin Density (a.u.) | 0.34 | 0.40 | 0.06 |
| Fe-S distance (Å) | 2.35 | 2.27 | -0.08 |
| Fe-S frequency (cm-1) | 309 | 365 | 56 |
| Experimental | |||
| Fe-S Frequency (cm-1) | 350 | 353 | 3 |
Table 2.
The properties of the Fe and S atoms calculated for the 6-coordinated heme active-site with a water molecule as the distal ligand in the oxidized form for the doublet, quartet and sextet electronic states. For computational details see the caption of Table 1.
| Heme active-site 6-coordinated (H2O ligand) | P450cam | P450cam/pdx | Change in protein |
|---|---|---|---|
| Doublet | |||
| Fe Charge (a.u.) | 1.48 | 1.47 | -0.01 |
| S Charge (a.u.) | -0.34 | -0.34 | 0.00 |
| Fe Spin Density (a.u.) | 1.06 | 1.05 | -0.01 |
| S Spin Density (a.u.) | -0.04 | -0.01 | 0.03 |
| Fe-S distance (Å) | 2.25 | 2.22 | -0.03 |
| Fe-S frequency (cm-1) | 360 | 376 | 16 |
| Quartet | |||
| Fe Charge (a.u.) | 1.67 | 1.66 | -0.01 |
| S Charge (a.u.) | -0.49 | -0.50 | -0.01 |
| Fe Spin Density (a.u.) | 2.67 | 2.64 | -0.03 |
| S Spin Density (a.u.) | 0.30 | 0.32 | 0.02 |
| Fe-S distance (Å) | 2.48 | 2.40 | -0.08 |
| Fe-S frequency (cm-1) | 291 | 291 | 0.00 |
| Sextet | |||
| Fe Charge (a.u.) | 1.88 | 1.88 | 0.00 |
| S Charge (a.u.) | -0.46 | -0.48 | -0.02 |
| Fe Spin Density (a.u.) | 4.21 | 4.20 | -0.01 |
| S Spin Density (a.u.) | 0.33 | 0.34 | 0.01 |
| Fe-S distance (Å) | 2.38 | 2.33 | -0.05 |
| Fe-S frequency (cm-1) | 281 | 303 | 22 |
| Experimental | |||
| Fe-S Frequency (cm-1) | 350 | 353 | 3 |
Table 3.
The properties of the Fe, S and C atoms calculated for the 6-coordinated heme active-site with carbon monoxide as the distal ligand in the oxidized form for the doublet, quartet and sextet electronic states. For computational details see the caption of Table 1.
| Heme active-site 6-coordinated (CO ligand) | P450cam | P450cam/pdx | Change in protein |
|---|---|---|---|
| Doublet | |||
| Fe Charge (a.u.) | 1.41 | 1.37 | -0.04 |
| S Charge (a.u.) | -0.31 | -0.28 | 0.03 |
| Fe Spin Density (a.u.) | 1.07 | 0.89 | -0.19 |
| S Spin Density (a.u.) | -0.04 | 0.14 | 0.18 |
| Fe-S Distance (Å) | 2.28 | 2.20 | -0.08 |
| Fe-S Frequency (cm-1) | 355 | 381 | 27 |
| Fe-C Distance (Å) | 1.77 | 1.77 | 0.00 |
| Fe-C Frequency (cm-1) | 473 | 473 | 0.00 |
| Quartet | |||
| Fe Charge (a.u.) | 1.45 | 1.46 | 0.01 |
| S Charge (a.u.) | -0.32 | -0.37 | -0.05 |
| Fe Spin Density (a.u.) | 1.57 | 1.63 | 0.06 |
| S Spin Density (a.u.) | 0.26 | 0.24 | -0.03 |
| Fe-S Distance (Å) | 2.29 | 2.26 | -0.03 |
| Fe-S Frequency (cm-1) | 324 | 346 | 23 |
| Fe-C Distance (Å) | 1.86 | 1.85 | -0.01 |
| Fe-C Frequency (cm-1) | 417 | 437 | 21 |
| Sextet | |||
| Fe Charge (a.u.) | 1.83 | 1.84 | 0.01 |
| S Charge (a.u.) | -0.42 | -0.45 | -0.03 |
| Fe Spin Density (a.u.) | 4.15 | 4.16 | 0.01 |
| S Spin Density (a.u.) | 0.37 | 0.36 | -0.01 |
| Fe-S Distance (Å) | 2.37 | 2.30 | -0.08 |
| Fe-S Frequency (cm-1) | 292 | 345 | 52 |
| Fe-C Distance (Å) | 3.17 | 2.93 | -0.24 |
| Fe-C Frequency (cm-1) | - | - | - |
Table 4.
The properties of the Fe, S and C atoms calculated for the 6-coordinated heme active-site with carbon monoxide in the reduced form for the singlet and triplet electronic states. For computational details see the caption of Table 1. Experimental data are taken from reference [49].
| Heme active-site 6-coordinated (CO ligand) | P450cam | P450cam/pdx | Change in protein |
|---|---|---|---|
| Singlet | |||
| Fe Charge (a.u.) | 1.21 | 1.20 | -0.01 |
| S Charge (a.u.) | -0.53 | -0.55 | -0.02 |
| Fe-S Distance (Å) | 2.45 | 2.39 | -0.06 |
| Fe-S Frequency (cm-1) | 267 | 292 | 25 |
| Fe-C Distance (Å) | 1.77 | 1.77 | 0.00 |
| Fe-C Frequency (cm-1) | 473 | 473 | 0.00 |
| Triplet | |||
| Fe Charge (a.u.) | 1.44 | 1.40 | -0.04 |
| S Charge (a.u.) | -0.37 | -0.44 | -0.07 |
| Fe Spin Density (a.u.) | 1.12 | 1.42 | 0.29 |
| S Spin Density (a.u.) | -0.06 | 0.04 | 0.10 |
| Fe-S Distance (Å) | 2.31 | 2.29 | -0.02 |
| Fe-S Frequency (cm-1) | 346 | 337 | -9 |
| Fe-C Distance (Å) | 1.94 | 1.90 | -0.04 |
| Fe-C Frequency (cm-1) | 417 | 442 | 25 |
| C-O Frequency (cm-1) | 1975 | 1955 | -20 |
| Experimental | |||
| Fe-C Frequency (cm-1) | 481 | 483 | 2 |
| C-O Frequency (cm-1) | 1940 | 1932 | -8 |
Figure 1.
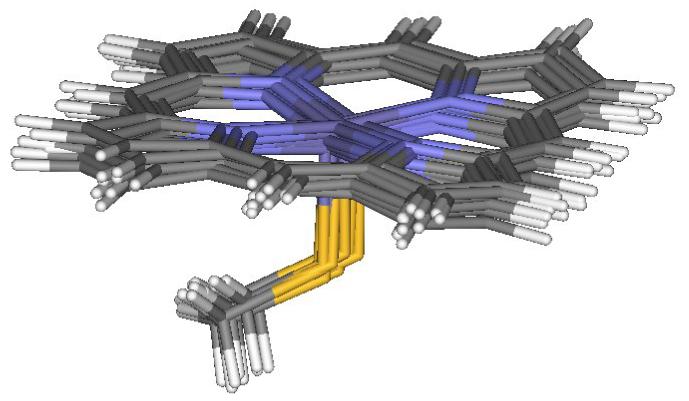
Optimal geometries of the 5-coordinated heme active site obtained in the QM/MM calculations for the doublet electronic state of the oxidized iron state, based on 10 protein snapshots after MM dynamics of cytochrome P450cam.
Figure 2.
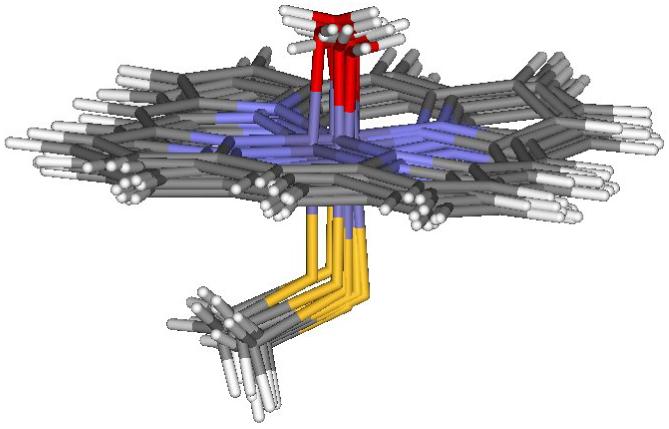
Optimal geometries of the 6-coordinated heme active site with H2O obtained in the QM/MM calculations for the doublet electronic state of the oxidized iron state, based on 10 protein snapshots after MM dynamics of cytochrome P450cam.
Figure 3.
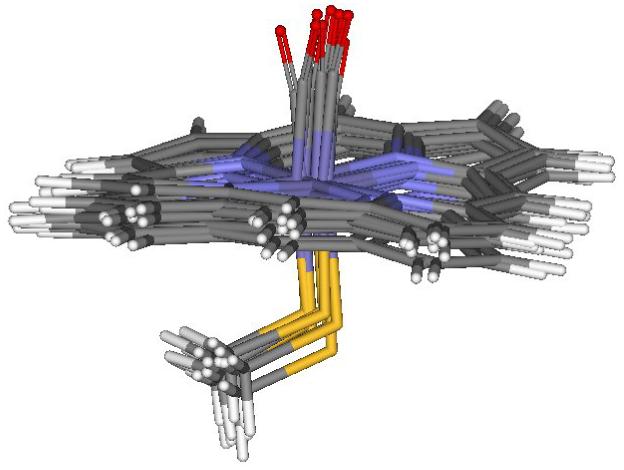
Optimal geometries of the 6-coordinated heme active site with CO obtained in the QM/MM calculations for the triplet electronic state of the reduced iron state, based on 10 protein snapshots after MM dynamics of cytochrome P450cam.
Figure 4.
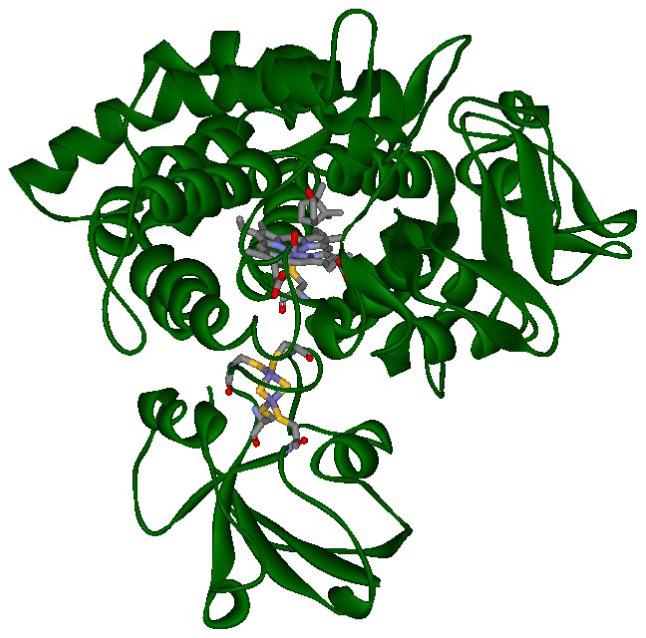
A protein snapshot of the MM dynamics of cytochrome P450cam with pdx (green ribbon), showing non-hydrogen atoms (colored sticks) of heme with CO and camphor - top, and the metal site of pdx - bottom (color code description: gray - carbon, red - oxygen, blue - nitrogen, yellow - sulfur and dark blue - iron).
Table 1 presents the results of our combined QM/MM calculations for the doublet, quartet and sextet electronic states of the system with the 5-coordinated heme active-site of the oxidized iron form. According to our results, the Fe-S stretching frequency in the doublet electronic state is increased after binding P450 to pdx. Similar increases in the Fe-S frequency are observed in our calculations for the quartet and sextet electronic states. However the absolute values of the Fe-S oscillation frequencies obtained in our study for the doublet electronic state, are closer to the experimental data than the values of these frequencies calculated for the other spin states. Our QM/MM calculations also show that the increase in the Fe-S oscillation frequency is accompanied by a decrease of the Fe-S bond length. As indicated in Table 1, substantial changes in the atomic charges and spin densities on the iron and sulfur atoms are not observed in the binding process of both proteins.
Table 2 shows the results of our calculations for the 6-coordinated heme active-site with a water molecule for the doublet, quartet and sextet electronic states of the oxidized iron form. The frequency of the Fe-S stretching vibration increases after binding P450 to pdx in the doublet and sextet electronic states, with no substantial changes of this vibration in the quartet. Analogous to the calculations of the 5-coordinated system, the absolute values of the Fe-S calculated oscillation frequencies for the doublet electronic state are closer to the experimental data than the values of these frequencies calculated in the other electronic states. Also, as was the case for the 5-coordinated system, the increase in the Fe-S frequency is accompanied by the decrease of the Fe-S interatomic distance, and there are no substantial changes in the atomic charges or the spin densities of the iron and sulfur atoms.
Results of our calculations for the 6-coordinated active-site with carbon monoxide in the doublet, quartet and sextet electronic states, representing the oxidized state of the central iron atom, are presented in Table 3. According to Table 3, the increase in the Fe-S stretching frequency is correlated with the decrease of the Fe-S distance for all three electronic states, as was the case for the previous forms of the heme active-site. However, our calculations predict a substantial change in the spin densities on the iron and sulfur atoms in the doublet electronic state. Moreover, the Fe-C interatomic distance in the sextet electronic state is much larger than in the other states, which indicates a dissociation process of carbon monoxide from the heme active site in this electronic state. According to our calculations, after dissociation the ligand stays in the docking site of the heme pocket, at a distance of about 3 A from the active-site center. The results of these calculations are consistent with experiment [4], showing that for the oxidized iron state, there is no binding process of carbon monoxide in P450.
Table 4 shows results of our calculations for the 6-coordinated system with carbon monoxide in the singlet and triplet electronic states, representing the reduced iron form. In the singlet electronic state we observe a similar increase in the Fe-S stretching frequency, as it has been calculated in our study of the other forms of the heme active-site. The frequency increase is also accompanied by a decrease in the Fe-S interatomic distance. According to Table 4, the frequency of the F-S stretching vibration in the triplet state becomes slightly smaller after binding of both proteins, as does the F-S distance, which is the opposite effect observed in our calculations of the other forms of the heme active-site. We also observe an increase in the Fe-C stretching frequency and at the same time we observe a decrease of the C-O frequency after binding P450 to pdx. The frequency increase of the Fe-C vibration is correlated in our calculations with the decrease of the Fe-C distance, however we do not observe a substantial change of the C-O distance. The computed opposite changes of the Fe-C and C-O oscillation frequencies that occurs after binding P450 to pdx, is consistent with experiment [49], however the absolute values of the calculated frequency changes are larger than experimental data. The changes of the Fe-C and C-O oscillation frequencies were the subject of many experimental investigations. For example, in the paper [5] the authors compare results of infrared and Raman spectroscopy of the P450/CO complex with their NMR results of the P450/CN protein system. According to their findings, the change of the NMR spectrum of P450 after binding to pdx indicates that the binding process alters the electronic structure of the cyanide N atom, decreasing its electron spin density. Therefore the authors assume that the binding process of both proteins transfers an electron between iron and the CN ligand The similar electron delocalization between iron and the CO ligand upon binding of both proteins, is also proposed in another paper [37]. Therefore in order to reveal the proposed electron transfer in the active site of P450, we have performed calculations of the NBO populations of the Fe, S, C and O atomic orbitals in the triplet electronic state of the reduced iron form. According to our calculations, there are no substantial changes in the atomic populations of those four atoms, however there are changes in spin densities of Fe and S, after binding P450 to pdx. Those results indicate that changes of the Fe-C and C-O vibration frequencies are because of changes of the Fe and S spin densities rather than the changes of their atomic populations. The changes of the spin densities which have been observed in our calculations are consistent with a conclusion of the NMR paper [5] indicating that the binding process of P450 to pdx changes electron spin densities on particular atoms of the P450 active-site. According to our calculations for the triplet electronic state of the reduced iron form of the P450/pdx complex with CO, the binding process of both proteins changes the proximal site of the heme pocketincreasing spin densities of the Fe and S atoms. The changes in spin densities of those two atoms perturb the distal side of the heme pocket yielding the opposite changes of the Fe-C and C-O bond force constants, increasing the Fe-C oscillation frequency and decreasing the C-O frequency. Therefore we can conclude that according to our calculations, pdx as a cofactor of the catalytic activity of P450, regulates bond force constants of a ligand after binding of both proteins.
Acknowledgments
The authors gratefully acknowledge support of NIH grants R44GM65617 and R44GM073408.
Footnotes
Corresponding author: Marek Freindorf, fax: (716) 849-6656, email: mfrein@buffalo.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mueller EJ, Loida PJ, Sligar SG. In: Cytochrome P450 Structure, Mechanism and Biochemistry. Ortiz de Montellano PR, editor. Plenum Press; New York: 1995. [Google Scholar]
- [2].Lipscomb JD. Biochemistry. 1980;19:3590–3599. doi: 10.1021/bi00556a027. [DOI] [PubMed] [Google Scholar]
- [3].Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J. J. Biol. Chem. 1985;260:16122–16130. [PubMed] [Google Scholar]
- [4].Raag R, Poulos TL. Biochemistry. 1989;28:7586–7592. doi: 10.1021/bi00445a013. [DOI] [PubMed] [Google Scholar]
- [5].Shiro Y, Iizuka T, Makino R, Ishimura Y, Morishima I. J. Am. Chem. Soc. 1989;111:7707–7711. [Google Scholar]
- [6].Simianu MC, Kincaid JR. J. Am. Chem. Soc. 1995;117:4628–4636. [Google Scholar]
- [7].Li H, Narasimhulu S, Havran LM, Winkler JD, Poulos TL. J. Am. Chem. Soc. 1995;117:6297–6299. [Google Scholar]
- [8].Mouro C, Bondon A, Simonneaux G, Jung C. FEBS Letters. 1997;414:203–208. doi: 10.1016/s0014-5793(97)00995-2. [DOI] [PubMed] [Google Scholar]
- [9].Vidakovic M, Sligar SG, Li H, Poulos TL. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]
- [10].Davydov R, Macdonald IDG, Makris TM, Sligar SG, Hoffman BM. J. Am. Chem. Soc. 1999;121:10654–10655. [Google Scholar]
- [11].Mouro C, Bondon A, Jung C, De Certaines JD, Simonneaux G. Eur. J. Biochem. 2000;267:216–221. doi: 10.1046/j.1432-1327.2000.00995.x. [DOI] [PubMed] [Google Scholar]
- [12].Schichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- [13].Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM. J. Am. Chem. Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- [14].Deng T, Macdonald IDG, Simianu MC, Sykora M, Kincaid JR, Sligar SG. J. Am. Chem. Soc. 2001;123:269–278. doi: 10.1021/ja001517d. [DOI] [PubMed] [Google Scholar]
- [15].Yoshioka S, Tosha T, Takahashi S, Ishimori K, Hori H, Morishima I. J. Am. Chem. Soc. 2002;124:14571–14579. doi: 10.1021/ja0265409. [DOI] [PubMed] [Google Scholar]
- [16].Jin S, Makris TM, Bryson TA, Sligar SG, Dawson JH. J. Am. Chem. Soc. 2003;125:3406–3407. doi: 10.1021/ja029272n. [DOI] [PubMed] [Google Scholar]
- [17].Fedorov R, Ghosh DK, Schlichting I. Arch. Biochem. Biophys. 2003;409:25–31. doi: 10.1016/s0003-9861(02)00555-6. [DOI] [PubMed] [Google Scholar]
- [18].Zakharieva O, Trautwein AX, Veeger C. Biophys. Chem. 2000;88:11–34. doi: 10.1016/s0301-4622(00)00194-0. [DOI] [PubMed] [Google Scholar]
- [19].Ogliaro F, Cohen S, de Visser SP, Shaik S. J. Am. Chem. Soc. 2000;122:12892–12893. [Google Scholar]
- [20].Yoshizawa K, Kagawa Y, Shiota Y. J. Phys. Chem. B. 2000;104:12365–12370. [Google Scholar]
- [21].Goller AH, Clark T. J. Mol. Strcut. (Theochem) 2001;541:263–281. [Google Scholar]
- [22].Yoshizawa K, Kamachi T, Shiota Y. J. Am. Chem. Soc. 2001;123:9806–9816. doi: 10.1021/ja010593t. [DOI] [PubMed] [Google Scholar]
- [23].Schoneboom JC, Lin H, Reuter N, Thiel W, Cohen S, Ogliaro F, Shaik S. J. Am. Chem. Soc. 2002;124:8142–8151. doi: 10.1021/ja026279w. [DOI] [PubMed] [Google Scholar]
- [24].Kamachi T, Yoshizawa K. J. Am. Chem. Soc. 2003;125:4652–4661. doi: 10.1021/ja0208862. [DOI] [PubMed] [Google Scholar]
- [25].Bathelt CM, Ridder L, Mulholland AJ, Harvey JN. J. Am. Chem. Soc. 2003;125:8362–8363. doi: 10.1021/ja035590q. [DOI] [PubMed] [Google Scholar]
- [26].Visser de SP, Kumar D, Cohen S, Shacham R, Shaik S. J. Am. Chem. Soc. 2004;126:8362–8363. doi: 10.1021/ja048528h. [DOI] [PubMed] [Google Scholar]
- [27].Kumar D, de Visser SP, Shaik S. J. Am. Chem. Soc. 2004;126:5072–5073. doi: 10.1021/ja0318737. [DOI] [PubMed] [Google Scholar]
- [28].Meunier B, de Visser SP, Shaik S. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- [29].Schoneboom JC, Cohen S, Lin H, Shaik S, Thiel W. J. Am. Chem. Soc. 2004;126:4017–4034. doi: 10.1021/ja039847w. [DOI] [PubMed] [Google Scholar]
- [30].Brewer CB, Peterson JA. J. Biol. Chem. 1988;263:791–798. [PubMed] [Google Scholar]
- [31].Lipscomb JD, Sligar SG, Namtvedt MJ, Gunsalus IC. J. Biol. Chem. 1976;251:1116–1124. [PubMed] [Google Scholar]
- [32].Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SG, Hoffman BM, Champion PM. Biochemistry. 2001;40:6852–6859. doi: 10.1021/bi002510b. [DOI] [PubMed] [Google Scholar]
- [33].Tosha T, Yoshioka S, Takahashi S, Ishimori K, Shimada H, Morishima I. J. Biol. Chem. 2003;278:39809–39821. doi: 10.1074/jbc.M304265200. [DOI] [PubMed] [Google Scholar]
- [34].Pochapsky SS, Pochapsky TC, Wei JW. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- [35].Nagano S, Tosha T, Ishimori K, Morishima I, Poulos TL. J. Biol. Chem. 2004;279:42844–42849. doi: 10.1074/jbc.M404217200. [DOI] [PubMed] [Google Scholar]
- [36].Tosha T, Yoshioka S, Ishimori K, Morishima I. J. Biol. Chem. 2004;279:42836–42843. doi: 10.1074/jbc.M404216200. [DOI] [PubMed] [Google Scholar]
- [37].Unno M, Christion JF, Benson DE, Gerber NC, Sligar SG, Champion PM. J. Am. Chem. Soc. 1997;119:6614–6620. [Google Scholar]
- [38].Unno M, Christian JF, Sjodin T, Benson DE, Macdonald IDG, Sligar SG, Champion PM. J. Biol. Chem. 2002;277:2547–2553. doi: 10.1074/jbc.M108917200. [DOI] [PubMed] [Google Scholar]
- [39].Pochapsky TC, Lyons TA, Kazanis S, Arkaki T, Ratnaswamy G. Biochimie. 1996;78:723–733. doi: 10.1016/s0300-9084(97)82530-8. [DOI] [PubMed] [Google Scholar]
- [40].Roitberg AE, Holden MJ, Mayhew MP, Kurnikov IV, Beratan DN, Vilker VL. J. Am. Chem. Soc. 1998;120:8927–8932. [Google Scholar]
- [41].Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Radmer RJ, Duan Y, Pitera J, Massova M, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER 8. University of California; 2004. [Google Scholar]
- [42].Kong J, White CA, Krylov AI, Sherrill D, Adamson RD, Furlani TR, Lee MS, Lee AM, Gwaltney SR, Adams TR, Ochsenfeld C, Gilbert ATB, Kedziora GS, Rassolov VA, Maurice DR, Nair N, Shao Y, Besley NA, Maslen PA, Dombrowski JP, Daschel H, Zhang W, Korambath PP, Baker J, Byrd EFC, Voorhis TV, Oumi M, Hirata S, Hsu CP, Ishikawa N, Florian J, Warshel A, Johnson BG, Gill PMW, Head-Gordon M, Pople JA. J. Comput. Chem. 2000;21:1532–1548. [Google Scholar]
- [43].Sono M, Roach MP, Coulter ED, Dawson JH. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- [44].Sevrioukova IF, Garcia C, Li H, Bhaskar B, Poulos TL. J. Molecul. Biol. 2003;333:377–392. doi: 10.1016/j.jmb.2003.08.028. [DOI] [PubMed] [Google Scholar]
- [45].Spiro TG, Zgierski MZ, Kozlowski PM. Coord. Chem. Rev. 2001;219:923–936. [Google Scholar]
- [46].Sigfridsson E, Ryde U. J. Inorg. Biochem. 2002;91:101–115. doi: 10.1016/s0162-0134(02)00426-9. [DOI] [PubMed] [Google Scholar]
- [47].Freindorf M, Shao Y, Furlani TR, Kong J. J. Comput. Chem. 2005;26:1270–1278. doi: 10.1002/jcc.20264. [DOI] [PubMed] [Google Scholar]
- [48].Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F. NBO 5.0. University of Wisconsin; Madison, WI: 2001. [Google Scholar]
- [49].Makino R, Iizuka T, Ishimura Y, Uno T, Nishimura Y, Tsuboi M. Ninth International Conference on Raman Spectroscopy; The Chemical Society of Japan: Tokyo. 1984.pp. 492–493. [Google Scholar]
- [50].Rauhut G, Pulay P. J. Phys. Chem. 1995;99:3093–3100. [Google Scholar]