

Abstract
Although the catalytic (C) subunit of cAMP-dependent protein kinase is N-myristylated, it is a soluble protein, and no physiological role has been identified for its myristyl moiety. To determine whether the interaction of the two regulatory (R) subunit isoforms (RI and RII) with the N-myristylated C subunit affects its ability to target membranes, the effect of N-myristylation and the RI and RII subunit isoforms on C subunit binding to phosphatidylcholine/phosphatidylserine liposomes was examined. Only the combination of N-myristylation and RII subunit interaction produced a dramatic increase in the rate of liposomal binding. To assess whether the RII subunit also increased the conformational flexibility of the C subunit N terminus, the effect of N-myristylation and the RI and RII subunits on the rotational freedom of the C subunit N terminus was measured. Specifically, fluorescein maleimide was conjugated to Cys-16 in the N-terminal domain of a K16C mutant of the C subunit, and the time-resolved emission anisotropy was determined. The interaction of the RII subunit, but not the RI subunit, significantly increased the backbone flexibility around the site of mutation and labeling, strongly suggesting that RII subunit binding to the myristylated C subunit induced a unique conformation of the C subunit that is associated with an increase in both the N-terminal flexibility and the exposure of the N-myristate. RII subunit thus appears to serve as an intermolecular switch that disrupts of the link between the N-terminal and core catalytic domains of the C subunit to expose the N-myristate and poise the holoenzyme for interaction with membranes.
Cyclic AMP-dependent protein kinase (cAPK) exists in nearly all eukaryotic cells and plays a critical role in the regulation of cellular growth, metabolism, and homeostasis by catalyzing the phosphorylation of a variety of proteins (1–3). The holoenzyme configuration of cAPK comprises two catalytic (C) subunits and a cAMP-binding regulatory (R) subunit homodimer. Three mammalian forms of the C subunit have been identified (Cα, Cβ1,2,3, and Cγ) (1–3). With the exception of some of the novel Cβ splice variants, all the mammalian isoforms are myristylated (4, 5). Two pharmacologically and structurally distinct types of the R subunits, RI and RII, are known, along with α and β isoforms of each (1–3). Tissue-specific membrane targeting of each type has been observed (1–3) and appears to be mediated by specific membrane anchoring proteins (A-kinase anchoring proteins) that bind to the dimerization domain of the R subunit but not to the C subunit (6).
Because protein N-myristylation is often part of a membrane targeting signal that permanently or reversibly steers proteins to membranes (7), it is perplexing that the C subunit is thought to lack a membrane targeting signal and that membrane binding of the holoenzyme results solely from A-kinase anchoring protein interaction with the nonmyristylated R subunits. The x-ray structure of the mammalian Cα subunit suggests why the free C subunit is not necessarily membrane-bound. The conserved catalytic core of the C subunit is preceded by myristylated Gly-1 and a short unstructured segment (residues 1–9) followed by a long amphipathic helix (residues 10–31). The myristyl moiety is tucked into a hydrophobic pocket formed by the N-terminal and core catalytic domains (Fig. 1) (8). Although N-myristylation contributes to the thermostability of the phosphotransferase activity of the C subunit (9), it is unclear whether this is the only role of C subunit myristylation (10). Two observations suggest that the cAPK myristate could participate in membrane targeting. The 2H-NMR spectrum of myr-d27-GNAAAKKGSEQES, a myristylated 14-mer encompassing the first 14 residues of the C subunit, shows that the myristate inserts into the hydrocarbon interior of bicelles (11). Also, the replacement of the 14 N-terminal amino acids of the myristylated oncoprotein pp60v-src with the comparable N-terminal residues from the C subunit does not block normal pp60v-src membrane binding, indicating that the myristylated N terminus of cAPK is capable of directing membrane association of an intact protein (12).
Figure 1.

Backbone structure of the C subunit of cAPK with the sites of X-to-Cys substitution (K16C and C199A), wild-type cysteines (Cys-199 and Cys-343), phosphorylation (Ser-10, Thr-197, and Ser-338), and myristylation (at Gly-1) highlighted.
To determine whether the myristyl moiety of the C subunit participates in membrane targeting when the C subunit is in a holoenzyme configuration with specific R subunit isoforms, we developed an assay to monitor C subunit binding to liposomes. Specifically, the binding-dependent changes in the fluorescence resonance energy transfer (FRET) between fluorescein maleimide (FM) attached to Cys-343 in a C subunit mutant (C199A) and C12-Texas Red incorporated into l-α-phosphatidylcholine (PC)/l-α-phosphatidylserine (PS) liposomes were monitored. Surprisingly, myristylation alone and each of the R subunits had only a small effect on the rate of liposomal binding. However, myristylation plus the RII subunit synergistically increased the rate of liposomal binding. To determine whether the RII subunit also induced greater conformational flexibility of the N-terminal segment to which the myristate is attached, the backbone flexibility of the N terminus was examined by measuring the rotational diffusion with time-resolved fluorescence anisotropy of FM attached to Cys-16 in the N-terminal domain of another mutant C subunit (K16C). Whereas myristylation of the N terminus of the C subunit alone significantly reduces the N-terminal backbone flexibility, as suggested by the x-ray structure of C subunit, the RII subunit, but not the RI subunit, reversed the immobilizing effects of myristylation. Together, the results suggest that RII subunit binding selectively acts as an intermolecular switch that releases the N-terminal segment from the core catalytic domain of the C subunit to activate a portion of the type II cAPK membrane targeting signal.
Experimental Procedures
Materials.
The methods for the preparation and characterization of the mutant C subunits (13), the different phosphoisoforms of the C subunit (13), the myristylated C subunit (8), and the regulatory subunits (RIα and RIIβ) (14) have been described elsewhere. C12-Texas Red was synthesized following the procedures described by Johnson and Nuss (15).
Fluorescein Maleimide Labeling.
The mutant C subunit (0.5–1 mg) was labeled with FM by using the procedures described in ref. 16 with the exception that the C199A and the myr-K16C mutants were labeled with a 7-fold excess of FM. Aliquots of the concentrated sample were subjected to gel electrophoresis under denaturing conditions (12% SDS/PAGE) (17) and the fluorescent bands were visualized with a mineral lamp to assess whether any unconjugated fluorescein was associated with the labeled-mutant C subunit. Stoichiometry of labeling was determined spectroscopically (16), and the phosphotransferase activity of the labeled C subunits was quantified by the method of Cook et al. (18) with kemptide as a substrate.
Formation of Holoenzyme.
The cAMP was removed from the R subunits by urea denaturation as described elsewhere (9). FM-labeled K16C or myristylated K16C was complexed with the R subunits following the procedures described in ref. 9. After this step, the holoenzyme was eluted through a Sephacryl S-200 (2.5 × 20 cm) column equilibrated with buffer B (50 mM MOPS/100 mM KCl/5 mM β-mercaptoethanol, pH 7.1) plus 5 mM MgCl2 and 1 mM ATP for all sample purifications involving the type I holoenzyme.
Steady State Emission Spectra.
Steady state emission spectra were measured by using an Instruments SA (Edison Park, NJ) Jobin Yvon/Spex FluoroMax II spectrofluorometer with the excitation and emission bandwidths set at 5 nm.
Time-Resolved Fluorescence Anisotropy.
The time-resolved emission anisotropy was determined by time-correlated single photon counting with an EEY scientific nanosecond spectrofluorometer (La Jolla, CA) (19) equipped with an IBH System 5000 coaxial hydrogen flash lamp (Edinburgh). A detailed description of the data collection procedures can be found in ref. 16.
For vertically polarized exciting light, time-resolved fluorescence anisotropy, r(t), is defined as
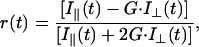 |
1 |
where I∥(t) and I⊥(t) are the vertically and orthogonally polarized emission decays, respectively, and G is polarization bias of the detection instrumentation. The fluorescence lifetimes and rotational correlation times were obtained by simultaneously deconvoluting the vertical and horizontal emission anisotropy decay curves for each sample by using the Globals Unlimited (Laboratory for Fluorescence Dynamics, Urbana, IL) software package (20). Goodness of fit was evaluated from the value of χ2 and visual inspection of the difference between experimental and an empirical nonassociative anisotropy decay model. This model can be expressed as a sum of two exponential expressions,
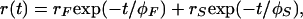 |
2 |
where r is the amplitude of the decay and φ is the rotational correlation time. The subscripts F and S denote the “fast” and “slow” decay processes, respectively. The deconvolution analysis utilized data points of the emission decay profiles the values of which were at least 1% of the maximum peak emission counts and consequently included data points before the flash peak.
Liposome Preparation.
Liposomes were prepared by dissolving l-α-phosphatidylcholine (Calbiochem, catalogue no. 524616) and l-α-phosphatidylserine (Avanti Polar Lipids, catalogue no. 840032) in a 4:1 molar ratio in 2:1 (vol/vol) methanol/chloroform. The phospholipids were dried by rotary evaporation for 5 min followed by placement under vacuum (<1 mtorr) for at least 1 h. The thin film was then hydrated in buffer A (50 mM MOPS/100 mM KCl, pH 7.1) to a final concentration of 400 μM in total phospholipids immediately before use.
Liposomal Binding.
The liposomes were diluted with buffer B to a final concentration of 10 μM in phospholipid, and C12-Texas Red was added to this suspension to a final concentration of 80 nM. This suspension was then incubated at room temperature for 20 min. The fluorescence emission (excitation at 450 nm) from fluorescein (emission at 525 nm) and Texas Red (emission at 610 nm) was monitored for 45 s before the addition of the various labeled proteins (final concentration, 60 nM in C subunit). The data were fit by using Scientist for Windows (Micromath, Salt Lake City) to a single exponential equation:
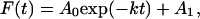 |
3 |
where F(t) is the fluorescence at time t, A1 is the fluorescence at equilibrium, A0 is the maximum change in fluorescence, and k represents the apparent rate of binding of the labeled cAPK configurations to the liposomes.
Results
Characterization of the Labeled Mutants.
The various FM-labeled mutants were prepared as described in Experimental Procedures. SDS/PAGE analysis and UV illumination of the gels revealed no detectable amounts of unconjugated probe. Based on spectroscopic analysis, the stoichiometry of labeling ranged between 0.4 and 0.6 of probe/protein (data not shown). Analysis of the catalytic activity revealed no detectable effects of labeling (data not shown). Finally, the excitation and emission spectra of the myr-FM-K16C and myr-FM-C199A mutants were essentially identical with those of the previously published FM-K16C and FM-C199A mutants (data not shown) (16).
Effect of Myristylation and R Subunit Isoforms on Liposomal Binding.
Because enhanced exposure of the myristate would increase the rate of C subunit membrane binding, a simple FRET binding assay was developed to monitor the time course of the increase in FRET associated with the interaction of the FM-labeled C subunit mutants (C199A or myr-C199A) and PC/PS liposomes into which C12-Texas Red was incorporated. The negatively charged PS was added to the liposomes to approximate the surface charge of the typical mammalian membrane. Labeling near the C terminus (at Cys-343), at a maximal distance from the N terminus (Fig. 1) minimizes the possibility that the charge and/or hydrophobicity of the probe would influence liposomal binding.
The mixing of the various configurations of FM-labeled mutants with suspensions of C12-Texas-Red-PC/PS liposomes showed the expected time-dependent decrease in fluorescein emission (donor quenching) and parallel increase in Texas Red emission (acceptor sensitization), that is indicative of FRET. Fig. 2 illustrates typical time tracings of the changes in Texas-Red emission after the mixing of the various FM-labeled C subunit complexes with the PC/PS liposomes. Because the donor quenching and acceptor sensitization data yielded essentially identical results, only the acceptor sensitization, Texas Red emission, results are presented here. Each tracing was well fit to a single exponential equation (Eq. 3), and the best fit parameters are summarized in Table 1. An assumption is necessary to interpret the changes in the “maximum fluorescence” parameter, namely, that when the various configurations of cAPK bind to the liposomes, the distance between the fluorophore and the surface of the bilayer is constant.
Figure 2.

Effect of myristylation and R subunit isoforms on the time course of FM-labeled C subunit binding to PC/PS (4:1) liposomes. Each tracing was fit to Eq. 3, and the best fit parameters are summarized in Table 1. Liposomes (10 μM in phospholipids) and C12-Texas Red (80 nM) were allowed to incubate for at least 20 min before the addition of labeled proteins (60 nM in C subunit). Texas Red (610 nm) emission was monitored.
Table 1.
Effect of myristylation and R subunits on C subunit binding to PC/PS liposomes
| FM-labeled configurations | Relative rate,* s−1 | Relative maximum fluorescence† |
|---|---|---|
| FM-C199A‡ alone | 1 | 1 |
| myr-FM-C 199A§ alone | 1.64 ± 0.17 | 2.0 ± 0.2 |
| RI:(FM-C199A)2‡ | 1.21 ± 0.14 | 5.0 ± 1.8 |
| RI:(myr FM-C199A§)2 | 1.60 ± 0.32 | 4.4 ± 1.2 |
| RII:(FM-C199A)2‡ | 1.56 ± 0.08 | 6.4 ± 1.1 |
| RII:(myr FM-C199A§)2 | 3.41 ± 0.29 | 5.1 ± 0.6 |
The data tracings (Fig. 2) were fit to a single exponential equation (Eq. 3). Shown are the mean ± SD of three determinations of the relative best, fit parameters. The concentration of C12-Texas Red and labeled protein were 80 and 60 nM, respectively. PC/PS (4:1) liposomes were present at a total concentration of 10 μM.
The relative rates were calculated by dividing the observed rate by that of FM-C199A (0.0017 = 0.0001 s−1).
The relative maximum fluorescence was calculated by dividing the observed maximum by that of FM-C199A (19,000 ± 13,000, arbitrary fluorescence units).
Phosphoisoform II of C subunit where residues Ser-10, Thr-197, and Ser-338 are phosphorylated.
Phosphorylation at Thr-197 and Ser-338.
During the period examined (10 min), the apparent rate of nonmyristylated C subunit binding was slow (0.0017 ± 0.0001 s−1), and the time tracings suggest little or no liposomal binding (Fig. 2). Myristylation of the C subunit was associated with a 60% increase in the apparent rate of binding and a 200% increase in the maximum amplitude, both suggesting some bilayer interaction and some exposure of the myristate with the bilayer. Interestingly, in the absence of the myristate, the interaction of the RI or the RII subunits with the labeled C subunit was associated with a 21 and 56% increase in the apparent rate of FM-K16C binding, respectively, and ≈570% increase in the maximum amplitude change of fluorescence. This suggests that the two R subunit isoforms have an intrinsic ability to interact with the liposomes.
The combination of both myristylation and R subunit binding produced differential effects on C subunit binding. The addition of the RI subunit had no effect on the apparent rate of the myristylated C subunit binding (≈60%) but increased the maximum change of fluorescence (≈440%) to that observed for all samples containing either R subunit isoform. The RII subunit interaction plus C subunit myristylation, on the other hand, yielded a dramatic synergistic effect, increasing the apparent rate of binding by ≈340% and the maximum change of fluorescence increased by ≈510%. This later result is consistent with the RII subunit interaction, significantly increasing the myristate involvement in bilayer. This would occur if the myristate is partially exposed when the C subunit is free in solution, and the RII subunit dramatically increases the myristate exposure.
The maximum fluorescence amplitudes of all the holoenzyme configurations were essentially the same, lying within a standard deviation of each other, whereas the liposomal binding rates differed from one another (Table 1). This presumably occurs because the Kd values for the various holoenzyme configurations to bind the PC/PS liposomes are significantly less than the lipid concentration (10 μM), so that at equilibrium essentially all the holoenzyme is liposome-bound. The differential rates are observed because the association rate constants differ and are reflected in the Kd values if these could be measured.
Because the myristylated FM-C199A used in the above experiments is not phosphorylated at Ser-10 (21) and the myristylated C199A is, the possibility that the effects of myristylation were secondary to the absence of the phosphate at Ser-10 was examined. The less abundant C199A isoform III (phosphorylated at Thr-197 and Ser-338) was isolated and labeled with FM, and the relative rate of binding to PC/PS liposomes was determined. The rate of binding of the less negatively charged diphosphorylated FM-C199A isoform III to the PC/PS liposomes was essentially identical with the triphosphorylated FM-C199A isoform II (0.0017 versus 0.0020 s−1, respectively), indicating that Ser-10 phosphorylation has little or no effect on the rate of liposome binding (data not shown).
N-terminal Flexibility of C Subunit.
If interaction with the R subunit isoforms differentially affects the exposure of the myristyl moiety in the C subunit, then the strength of the linkage between the N-terminal myristate and the core catalytic domain is conformation- or isoform-dependent. This may be reflected in the rotational freedom and, therefore, the flexibility of the N-terminal segment. To examine this possibility, myristylated and nonmyristylated forms of the K16C mutant were labeled with FM, and their time-resolved emission anisotropy was measured in the presence and absence of the two R subunit isoforms. Neither myristylation nor interaction with either RI or RII subunits had a significant effect on the previously reported FM-K16C emission spectrum (data not shown) (9).
Table 2 summarizes the emission and anisotropy decay parameters of the FM-K16C and myr-FM-K16C alone and complexed to the two R subunit isoforms. The emission decays of the myristylated samples are illustrated in Fig. 3, A–C, and all were reasonably well fit by a single exponential decay equation. Myristylation was associated with small increase (0.2 ns) in the emission lifetime (from 3.9 to 4.1 ns). RII subunit binding, but not RI subunit binding, was associated with a further slight increase (0.1 ns) in the fluorescence lifetime of both FM-K16C and myr-FM-K16C. The anisotropy decays were well fit by a biexponential equation (Eq. 2) and were consistent with our previous observations of fast and slow rotational diffusional processes (12). As previously reported, the slow rotational correlation time, φS, corresponds to the whole body rotational diffusion, so both the myristylated or nonmyristylated C subunit free in solution displayed essentially the same φS (≈25 ns), which is close to that predicted by the Stokes–Einstein equation [20.6 ns by using hydrodynamic radius of 27.1 A (10)]. Similarly, the binding of two C subunits to either the 92.5-kDa RI subunit (homodimer) or the 108.6-kDa RII subunit (homodimer) was associated with 133 ± 21 ns and 142 ± 19 ns φS values, respectively. (The Stokes–Einstein equation predicts rotational correlation time of 160 ns and 190 ns for spheres with the hydrodynamic radius of the RI:C2 (53.8 Å) and RII:C2 (56.8 Å) complexes (22).
Table 2.
Effect of myristylation and R subunits on the best fit parameters for the anisotropy decay of FM-K16C
| FM-labeled configurations | rVF* | rF† | φF,‡ ns | φ,§ ns | τ,¶ ns |
|---|---|---|---|---|---|
| Nonmyristylated FM-K16C isoform II | |||||
| FM-K16C‖ alone | 0.10 | 0.08 | 1.9 ± 0.1 | 26 ± 1 | 3.9 |
| RI:(FM-K16C)2 | 0.10 | 0.09 | 4.6 ± 0.3 | 133 ± 21 | 3.9 |
| RII:(FM-K16C)2 | 0.07 | 0.14 | 1.8 ± 0.1 | 142 ± 19 | 4.0 |
| Nonmyristylated FM-K16C isoform III | |||||
| FM-K16C** alone | 0.06 | 0.12 | 1.9 ± 0.4 | 28 ± 1 | 4.2 |
| Myristylated FM-K16C | |||||
| myr-FM-K16C** alone | 0.11 | 0.13 | 2.9 ± 0.1 | 25 ± 1 | 4.1 |
| RI:(myr-FM-K16C)2 | 0.11 | 0.08 | 3.1 ± 0.2 | 117 ± 18 | 4.1 |
| RII:(myr-FM-K16C)2 | 0.08 | 0.12 | 1.8 ± 0.1 | 134 ± 21 | 4.2 |
Anisotropy decays were fit to an empirical nonassociative biexponential anisotropy decay model (Eq. 2) with the Globals Unlimited computer program, and the experimental details are described in Experimental Procedures. Each value represents the mean of at least three determinations, and the average χ2 for fitting the data from the various samples ranged between 1.3 and 3.3.
The amplitude of the very fast decay process which equals anisotropy of immobilized fluorescein (0.34) minus r0, and all the SDs were 0.01.
The amplitude of the very fast decay process, and all the SDs were 0.01.
Fast rotational correlation time.
Slow rotational correlation time.
Fluorescein lifetime.
Phosphorylated at Ser-10, Thr-197, and Ser-338.
Phosphorylation at Thr-197 and Ser-338.
Figure 3.

Emission and anisotropy decays of myr-FM-K16C alone and bound to the RI and RII subunits. A–C illustrate the parallel (∥) and perpendicular (⊥) emission decays (single datum points), a smooth line through these points generated with the best fit parameters for a single exponential decay equation, and a flash lamp profile (wavy solid line). D–F illustrate the time-resolved anisotropy decay (single datum points) and a smooth line through these points generated with the best fit parameters (Table 2) for a double exponential decay equation (Eq. 2). (A, D) Myristylated FM-K16C alone. (B, E) Myristylated type II holoenzyme [RII:(FM-K16C)2]. (C, F) Myristylated type I holoenzyme [RI:(FM-K16C)2].
The fast rotational correlation time, φF, is inversely correlated to the average main chain atom B (or temperature) factors around the site of FM labeling and thus provides a measure of backbone flexibility (16). In the absence of the myristate, the φF of FM-K16C is relatively fast (1.9 ns; Table 2), indicating a significant level of flexibility around Cys-16 in the N-terminal domain, which is consistent with the crystal structure where residues 1–14 are rarely resolved in the nonmyristylated protein. Myristylation significantly increases φF to 2.9 ns, which is typical for a surface residue of the C subunit (16), and suggests that the myristate plays a major role in anchoring the N-terminal domain to the core catalytic domain. For the nonmyristylated FM-K16C, the binding of the RI subunit dramatically increased the φF (from 1.9 to 4.6 ns; Table 2), indicating a reduction in the N-terminal flexibility of the C subunit, whereas RII subunit interaction had no measurable effect. RI binding to the myristylated FM-K16C slightly increased φF (2.9 to 3.1 ns). In contrast, RII subunit binding significantly reduced φF (from 2.9 to 1.8 ns; Table 2) and therefore increased the flexibility of the N-terminus of the C subunit. These effects cannot be explained by the differences in the phosphorylation state between the myristylated and nonmyristylated FM-K16C, because diphosphorylated K16C (usually denoted isoform III) and the tri-phosphorylated K16C mutants were associated with identical values of φF (1.9 ns) (Table 2). We thus observe an isoform-specific intermolecular switch that controls the conformational flexibility of the myristylated N-terminal segment and contributes to the membrane targeting of the holoenzyme.
Discussion
Molecules that are involved in signal transduction pathways tend to be highly dynamic proteins at many levels. Typically, they are only transiently activated in response to some signal, and they also frequently move between subcellular sites. This dynamic behavior whether it be activation or subcellular translocation is often mediated by specific cotranslational or posttranslational modifications. In contrast to posttranslational modifications such as phosphorylation, isoprenylation/methylation, and palmitylation, myristylation occurs cotranslationally as the polypeptide chain is being synthesized (23). The acyl group does not subsequently turn over; it remains as a stable part of the polypeptide chain. Unlike palmitylation and isoprenylation/methylation, myristylation on its own is a weak membrane anchor that is insufficient to anchor a protein to a membrane. Resh and McLaughlin and their coworkers (24) have shown that for many myristylated proteins such as src there is also a requirement for basic amino acids that adds an electrostatic component to the interaction energy. Both the electrostatic component and the myristyl group are required for membrane anchoring. In the case of src, this electrostatic component lies immediately adjacent to the acyl group.
The results described here provide new insights into the role of the myristyl moiety and the dynamic properties of the N-terminal segment that interacts with both lobes of the conserved catalytic core. The results indicate that this region is considerably more mobile than had been previously thought and that the mobility of this region of the C subunit is strongly influenced by other proteins. By using association with liposomes as an assay, we showed that myristylation alone has about the same effect as either the RI or the RII complexed to the nonmyristylated C subunit to bind to liposomes. Holoenzyme formed with the RI subunit and either the myristylated or nonmyristylated C subunit displays about the same tendency to bind to liposomes. However, when the myristylated C subunit is complexed with the RII subunit, there is a synergistic enhancement of the rate of liposome binding. This is the first direct example of a myristyl group being positioned for membrane association by its interaction with another protein.
To probe the intrinsic mobility of the N-terminal segment of the C subunit, time-resolved emission anisotropy was used by taking advantage of an engineered C subunit where Lys-16 was replaced with cysteine and then modified with a fluorescent tag, FM. Lys-16 is located in the long A helix that joins the myristylated N terminus to the N-terminal lobe of the conserved core. In the absence of myristylation, this site is rather mobile with a relatively fast anisotropy decay rate (φF = 1.9 ns). This is consistent with the crystal structures that show the first 12–15 residues as disordered (8, 25). Addition of the myristyl moiety significantly reduces the fast decay rate (φF = 2.9 ns) so that Lys-16 → Cys now behaves as a typical surface residue in a structured globular protein. Addition of the RI subunit has no effect on the fast anisotropy decay rate of the myristylated C subunit, although for the nonmyristylated C subunit the N terminus is much more rigid in the presence of RI. The anisotropy decay rate goes from 1.9 ns to 4.6 ns, indicating that the N-terminal segment is now tightly anchored. In the absence of myristylation, RII has no effect on the local mobility around Lys-16. When the myristylated C subunit is complexed with the RII subunit, however, the fast anisotropy decay rate is accelerated significantly indicating a considerable degree of mobility. This is also the complex that preferentially binds to liposomes.
The ability of the N-myristate to anchor the N terminus to the catalytic core of the C subunit is actually consistent with the crystal structures (25). Not only is this region disordered when there is no acyl group, but also the interactions with the core even when it is myristylated are mediated primarily by hydrophobic contacts (8). Furthermore, the temperature factors for the N-terminal residues are high. Thus, even though the acyl group is folded into a hydrophobic pocket, the interactions are not strong. Whether a portion of the RII subunit physically displaces the N-terminal segment or whether it induces a conformational change that radiates to the acyl pocket where the acyl group binds is unclear. To answer this will require a crystal structure of an R:C complex. Nevertheless, the results demonstrate clearly that the RI and RII subunit differentially interact with this region of the C subunit. This is also the first biophysical evidence of a clear functional difference between these two isoforms.
Association of myristylated proteins with membranes typically requires a patch of basic amino acids. Like src, the C subunit has a patch of such residues that lies C terminal to the acyl group. In addition, the surface of the C subunit in that region is basic. Thus, the requirements are present for membrane association. Other myristylated proteins are also followed by an amphipathic helix. One example is the Gtα subunit of transducin. This N-terminal helical region also was not seen in the original structure of free Gtα and was actually physically missing from the free transducin, a structure that was solved (26). In the Gtαβγ complex, however, the helix was clearly seen even though the myristyl group was absent. The helix is actually juxtapositioned against the Gtβ subunit and is poised for synergistic association with the membrane in conjunction with the farnesyl group at the end of the Gtγ subunit (27). A similar positioning could be induced by binding of the RII subunit to the myristylated C subunit. Recoverin is another example of a myristylated protein that shows conformational flexibility. In this case, the conformational change is intramolecular and induced by calcium binding (28).
The catalytic subunit thus appears to be capable of contributing to its own localization when it is associated with the RII subunit. Localization of this complex to membranes is further mediated by specific A-kinase anchoring proteins. It remains to be determined to what extent myristylation of the C subunit contributes to the phosphorylation of membrane proteins that are associated with the A-kinase anchoring protein complexes.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (GM 19301) to S.S.T. and a grant from the National Science Foundation (IBN-9515330) to D.A.J.
Abbreviations
- cAPK
cAMP-dependent protein kinase
- C subunit
the α isoform of the catalytic subunit of cAPK
- R subunit
regulatory subunit homodimer of cAPK
- RI subunit
type Iα regulatory subunit homodimer
- RII subunit
the type IIβ regulatory subunit homodimer
- r0
anisotropy at time zero
- rVF
difference between the r0 of immobilized fluorescein (0.34) and the observed r0
- rF
amplitude of the “fast” anisotropy decay process
- rS
amplitude of the slow anisotropy decay process
- FRET
fluorescence resonance energy transfer
- FM
fluorescein maleimide
- φF
fast rotational correlation time
- φS
slow rotational correlational time
- C subunit isoform II
the C subunit with residues Ser-10, Thr-197, and Ser-338 phosphorylated
- C subunit isoform III
C subunit with residues Thr-197 and Ser-338 phosphorylated
- PC
phosphatidylcholine
- PS
phosphatidylserine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.McKnight G S. Curr Opin Cell Biol. 1991;3:213–217. doi: 10.1016/0955-0674(91)90141-k. [DOI] [PubMed] [Google Scholar]
- 2.Krebs E G. In: The Enzymes: Control by Phosphorylation. Boyer P D, Krebs E G, editors. New York: Academic; 1986. pp. 3–20. [Google Scholar]
- 3.Taylor S S, Buechler J A, Yonemoto W. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 4.Carr S, Biemann K, Shozo S, Parmalee D C, Titani K. Proc Natl Acad Sci USA. 1982;79:6128–6131. doi: 10.1073/pnas.79.20.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guthrie C R, Skalhegg B S, McKnight G S. J Biol Chem. 1997;272:29560–29565. doi: 10.1074/jbc.272.47.29560. [DOI] [PubMed] [Google Scholar]
- 6.Gray P C, Scott J D, Catterall W A. Curr Opin Neurobiol. 1998;8:330–334. doi: 10.1016/s0959-4388(98)80057-3. [DOI] [PubMed] [Google Scholar]
- 7.Resh M D. Cell. 1994;76:411–413. doi: 10.1016/0092-8674(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 8.Zheng J, Knighton D R, Nguyen-Huu X, Taylor S S, Sowadski J M, Ten Eyck L F. Protein Sci. 1993;2:1559–1573. doi: 10.1002/pro.5560021003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herberg F W, Zimmerman B, McGlone M, Taylor S S. Protein Sci. 1997;6:569–579. doi: 10.1002/pro.5560060306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yonemoto W, McGlone M L, Taylor S S. J Biol Chem. 1993;268:2348–2352. [PubMed] [Google Scholar]
- 11.Struppe J, Komives E A, Taylor S S, Vold R R. Biochemistry. 1998;37:15523–15527. doi: 10.1021/bi981326b. [DOI] [PubMed] [Google Scholar]
- 12.Silverman L, Resh M D. J Cell Biol. 1992;119:415–425. doi: 10.1083/jcb.119.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herberg F W, Bell S M, Taylor S S. Protein Eng. 1993;6:771–777. doi: 10.1093/protein/6.7.771. [DOI] [PubMed] [Google Scholar]
- 14.Buechler Y J, Taylor S S. J Biol Chem. 1991;266:3491–3497. [PubMed] [Google Scholar]
- 15.Johnson D A, Nuss J M. Biochemistry. 1994;33:9070–9077. doi: 10.1021/bi00197a007. [DOI] [PubMed] [Google Scholar]
- 16.Gangal M, Cox S, Lew J, Clifford T, Garrod S M, Aschbaher M, Taylor S S, Johnson D A. Biochemistry. 1998;37:13728–13735. doi: 10.1021/bi980560z. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Cook P F, Neville M E, Jr, Varana K E, Hartl F T, Roskoski R., Jr Biochemistry. 1982;21:5794–5799. doi: 10.1021/bi00266a011. [DOI] [PubMed] [Google Scholar]
- 19.Hanson D C, Yguerabide J, Schumaker V N. Biochemistry. 1981;20:6842–6852. doi: 10.1021/bi00527a016. [DOI] [PubMed] [Google Scholar]
- 20.Beechem J M, Gratton E, Ameloot M, Knutson J R, Brand L. In: Topics in Fluorescence Spectroscopy. Lakowicz J R, editor. Vol. 2. New York: Plenum; 1991. pp. 241–305. [Google Scholar]
- 21.Yonemoto W, McGlone M L, Grant B, Taylor S S. Protein Eng. 1997;10:915–925. doi: 10.1093/protein/10.8.915. [DOI] [PubMed] [Google Scholar]
- 22.Zoller M J, Kerlavage A R, Taylor S S. J Biol Chem. 1979;254:2408–2412. [PubMed] [Google Scholar]
- 23.Casey P J. Science. 1995;268:221–225. doi: 10.1126/science.7716512. [DOI] [PubMed] [Google Scholar]
- 24.Murray D, Hermida-Matsumoto L, Buser C A, Tsang J, Sigal C T, Ben-Tal N, Honig B, Resh M D, McLaughlin S. Biochemistry. 1998;37:2145–2159. doi: 10.1021/bi972012b. [DOI] [PubMed] [Google Scholar]
- 25.Karlsson R, Zheng J, Xuong N-H, Taylor S S, Sowadski J M. Acta Crystallogr D. 1993;49:381–388. doi: 10.1107/S0907444993002306. [DOI] [PubMed] [Google Scholar]
- 26.Lambright D G, Noel J P, Hamm H E, Sigler P B. Nature (London) 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- 27.Lambright D G, Sondek J, Bohm A, Skiba N P, Hamm H E, Sigler P B. Nature (London) 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 28.Dizhoor A M, Chen C-K, Olshevskaya E, Sinelnikova V V, Phillipov P, Hurley J B. Science. 1993;259:829–832. doi: 10.1126/science.8430337. [DOI] [PubMed] [Google Scholar]