

Abstract
The nuclear hormone receptors hepatocyte nuclear factor 4 (HNF4) and retinoid X receptor α (RXRα) plus peroxisome proliferator-activated receptor α (PPARα) heterodimer support hepatitis B virus (HBV) pregenomic RNA synthesis and viral replication in nonhepatoma cells. Small heterodimer partner (SHP), an orphan nuclear hormone receptor lacking a DNA binding domain, inhibits nuclear hormone receptor-mediated viral transcription and replication. The inhibition of HBV replication by SHP is dependent on the presence of nuclear hormone receptors. HBV replication that is dependent on HNF4 is considerably more sensitive to SHP-mediated inhibition than RXRα/PPARα-directed viral biosynthesis. SHP inhibition of HBV biosynthesis in HepG2 cells suggests that multiple nuclear hormone receptors mediate viral replication in this human hepatoma cell line. These observations suggest that the physiological regulation of HBV biosynthesis by SHP in the liver will depend on both the level of SHP expression and the relative contribution of HNF4 and RXRα/PPARα, plus potentially additional nuclear hormone receptors, to HBV RNA synthesis and replication.
The level of hepatitis B virus (HBV) replication observed in the livers of infected individuals is probably dependent on a variety of factors. As the template for HBV reverse transcription and, consequently, viral DNA synthesis is the viral 3.5-kb pregenomic RNA, regulation of the expression of this transcript is a critical determinant of virus production (43). The regulation of the expression of HBV 3.5-kb RNA synthesis has been investigated, and a variety of ubiquitous and liver-enriched transcription factors that modulate the activity of the nucleocapsid promoter and, hence, the abundance of this transcript have been identified (42). However, only the liver-enriched nuclear hormone receptors hepatocyte nuclear factor 4 (HNF4) and retinoid X receptor α (RXRα)/ peroxisome proliferator-activated receptor α (PPARα) have been shown to have the capacity to support HBV pregenomic RNA synthesis and viral biosynthesis in nonhepatoma cells (43). These observations suggest that the liver-specific tropism of this hepadnavirus is determined, in part, by these particular transcription factors (43). In addition, the physiological regulation of the activities of these nuclear hormone receptors is likely to have an important role in determining the level of HBV pregenomic RNA and subsequent replication observed in the infected liver (38).
In this study, the potential role of the small heterodimer partner (SHP), a known physiological regulator of several nuclear hormone receptors (36), in the control of HBV transcription and replication was examined. In an attempt to establish the potential physiological relevance of SHP to the regulation of HBV transcription and replication, the effect of expressing SHP in the human hepatoma cell line HepG2, which supports HBV biosynthesis independently of the expression of additional transcription factors, was examined (41). HBV transcription and replication were inhibited in a dose-dependent manner by SHP in HepG2 cells, suggesting that SHP might be a relevant regulator of HBV biosynthesis in the livers of infected individuals under certain physiological circumstances. To address the transcription factor activities that might be susceptible to modulation by SHP, the effect of SHP on HNF4- and RXRα/PPARα-mediated HBV transcription and replication was analyzed in nonhepatoma cells. This analysis demonstrated that HBV replication directed by HNF4 was highly sensitive to SHP modulation, whereas HBV replication directed by RXRα/PPARα was relatively insensitive to SHP modulation. In addition, it was apparent that the inhibition of HBV replication by SHP in HepG2 cells was intermediate between HNF4- and RXRα/PPARα-mediated HBV biosynthesis in nonhepatoma cells. This suggests that either HNF4 and RXRα/PPARα contribute approximately equally to supporting HBV replication in HepG2 cells or additional nuclear hormone receptors that are sensitive to modulation by SHP govern HBV biosynthesis in these cells. Interestingly, the level of SHP in the liver can be modulated by the levels of cytokines such as tumor necrosis alpha (TNF-α) and bile acids, suggesting that multiple physiological stimuli modulate HBV transcription and replication by activating SHP gene expression and thereby modulating the activities of the nuclear hormone receptors that govern HBV biosynthesis (8, 12, 17, 21).
MATERIALS AND METHODS
Plasmid constructions.
The steps in the cloning of the plasmid constructs used in the transfection experiments were performed by use of standard techniques (34). HBV DNA sequences in these constructions were derived from plasmid pCP10, which contains two copies of the HBV genome (subtype ayw) cloned into the EcoRI site of pBR322 (10). The HBV DNA (4.1 kbp) construct that contains 1.3 copies of the HBV genome includes the viral sequence from nucleotide positions 1072 to 3182 and 1 to 1990 (Fig. 1A). This plasmid was constructed by cloning the NsiI/BglII HBV DNA fragment (nucleotide positions 1072 to 1990) into pUC13, generating pHBV(1072-1990). Subsequently, a complete copy of the 3.2-kbp viral genome linearized at the NcoI site (nucleotide positions 1375 to 3182 plus 1 to 1374) was cloned into the unique NcoI site (HBV nucleotide position 1374) of pHBV(1072-1990), generating the HBV DNA (4.1-kbp) construct. The pCMVHBVayw construct contains the cytomegalovirus (CMV) immediate-early promoter (region at positions −522 to −1) (4) located directly upstream of the HBV sequence from nucleotide positions 1821 to 3182 plus 1 to 1990 (Fig. 1B). In this construct, the expression of the HBV pregenomic 3.5-kb RNA is controlled by the CMV immediately-early promoter.
FIG. 1.

Structure of the HBV constructs supporting viral transcription and replication in human hepatoblastoma HepG2 and embryonic kidney 293T cells. (A) Structure of the HBV DNA (4.1-kbp) construct used in transient transfection analysis. The 4.1-kbp greater-than-genome-length HBV DNA sequence in this construct spans positions 1072 to 3182 and 1 to 1990 of the HBV genome (subtype ayw). The locations of the HBV 3.5-kb, 2.4-kb, 2.1-k, and 0.7-kb transcripts are indicated. EnhI/Xp, enhancer I/X gene promoter region; Cp, nucleocapsid or core promoter; pA, polyadenylation site; PS1p, presurface antigen promoter; Sp, major surface antigen promoter; X, X gene; S, surface antigen gene; C, core gene; P, polymerase gene; ORF, open reading frame. (B) Structure of the pCMVHBV DNA construct used in transient transfection analyses. The CMV immediate-early promoter (CMVp) (region at positions −522 to −1) directs the expression of the HBV pregenomic 3.5-kb RNA from the greater-than-genome-length HBV DNA sequence in this construct that spans positions 1821 to 3182 and 1 to 1990 of the HBV genome (subtype ayw). The locations of the HBV 3.5-kb, 2.4-kb, 2.1-kb, and 0.7-kb transcripts are the same as those indicated for the HBV DNA (4.1-kbp) construct.
The pCMXSHP, pCMVHNF4, pRS-hRXRα, and pCMVPPARα-G vectors express SHP, HNF4, RXRα, and PPARα-G polypeptides from mouse SHP, rat HNF4, human RXRα, and mouse PPARα-G cDNAs, respectively, using the CMV immediate-early promoter (pCMX and pCMV) or the Rous sarcoma virus long terminal repeat (pRS) (6, 23, 25, 30, 32, 33). The PPARα-G polypeptide contains a mutation in PPARα cDNA, changing Glu282 to Gly, that may decrease the affinity of the receptor for the endogenous ligand. Consequently, this mutation increases the peroxisome proliferator-dependent (i.e., clofibric acid-dependent) activation of transcription from a peroxisome proliferator response element-containing promoter (25) and was used in this study to demonstrate the peroxisome proliferator-dependent transcriptional transactivation of the nucleocapsid promoter.
Cells and transfections.
The human embryonic kidney 293T and hepatoblastoma HepG2 cell lines were grown in RPMI 1640 medium and 10% fetal bovine serum at 37°C in 5% CO2-air. Transfections for viral RNA and DNA analysis were performed as previously described (24) by using 10-cm plates containing approximately 1 × 106 cells. Isolation of DNA and RNA was performed 3 days posttransfection. The transfected DNA mixture was composed of 5 μg of HBV DNA (4.1 kbp) plus 1.5 μg of the liver-enriched transcription factor expression vectors pCMVHNF4, pRS-hRXRα, and pCMVPPARα-G and various amounts of the pCMXSHP expression vector (6, 23, 25, 30, 32, 33, 43). Controls were derived from cells transfected with HBV DNA and the pCMV expression vector lacking a liver-enriched transcription factor cDNA insert (32). All-trans retinoic acid and clofibric acid at 1 μM and 1 mM, respectively, were used to activate the nuclear hormone receptors RXRα and PPARα (43).
Characterization of HBV transcripts and viral replication intermediates.
Transfected cells from a single plate were divided equally and used for the preparation of total cellular RNA and viral DNA replication intermediates as described previously (40), with minor modifications. For RNA isolation (9), the cells were lysed in a solution containing 1.8 ml of 25 mM sodium citrate (pH 7.0), 4 M guanidinium isothiocyanate, 0.5% (vol/vol) sarcosyl, and 0.1 M 2-mercaptoethanol. After the addition of 0.18 ml of 2 M sodium acetate (pH 4.0), the lysate was extracted with 1.8 ml of water-saturated phenol plus 0.36 ml of chloroform-isoamyl alcohol (49:1). After centrifugation for 30 min at 3,000 rpm in a Sorval RT6000 centrifuge, the aqueous layer was precipitated with 1.8 ml of isopropanol. The precipitate was resuspended in 0.3 ml of a solution containing 25 mM sodium citrate (pH 7.0), 4 M guanidinium isothiocyanate, 0.5% (vol/vol) sarcosyl, and 0.1 M 2-mercaptoethanol and precipitated with 0.6 ml of ethanol. After centrifugation for 20 min at 14,000 rpm in an Eppendorf 5417C microcentrifuge, the precipitate was resuspended in 0.3 ml of a solution containing 10 mM Tris hydrochloride (pH 8.0), 5 mM EDTA, and 0.1% (wt/vol) sodium lauryl sulfate and precipitated with 45 μl of 2 M sodium acetate plus 0.7 ml of ethanol.
For the isolation of viral DNA replication intermediates, the cells were lysed in a solution containing 0.4 ml of 100 mM Tris hydrochloride (pH 8.0) plus 0.2% (vol/vol) NP-40. The lysate was centrifuged for 1 min at 14,000 rpm in an Eppendorf 5417C microcentrifuge to pellet the nuclei. The supernatant was adjusted to 6.75 mM magnesium acetate plus 200 μg/ml DNase I and incubated for 1 h at 37°C to remove transfected plasmid DNA. The supernatant was readjusted to contain 100 mM NaCl, 10 mM EDTA, 0.8% (wt/vol) sodium lauryl sulfate, and 1.6 mg/ml pronase and incubated for an additional 1 h at 37°C. The supernatant was extracted twice with phenol, precipitated with 2 volumes of ethanol, and resuspended in 100 μl of a solution containing 10 mM Tris hydrochloride (pH 8.0) and 1 mM EDTA. RNA (Northern) and DNA (Southern) filter hybridization analyses were performed using 10 μg of total cellular RNA and 30 μl of viral DNA replication intermediates, respectively, as described previously (34).
RNase protection assays were performed using the Pharmingen Riboquant kit, and riboprobes were synthesized using the Ambion Maxiscript kit according to instructions provided by the manufacturers. Transcription initiation sites for the HBV 3.5-kb transcripts were examined using 20 μg of total cellular RNA and a 333 (HBV positions 1990 to 1658)-nucleotide-long 32P-labeled HBV riboprobe. This riboprobe contains additional flanking vector sequences of 46 nucleotides that are not protected by HBV RNA.
RESULTS
Inhibition of HBV replication by SHP in human hepatoma HepG2 cells.
Nuclear hormone receptors have been shown to support HBV pregenomic 3.5-kb RNA synthesis and viral replication in nonhepatoma cells (43). In addition, the transcriptional activities of several nuclear hormone receptors have been shown to be sensitive to inhibition by the corepressor properties of the liver-enriched orphan nuclear receptor SHP (20, 20, 35, 36). Consequently, it was of interest to determine if HBV transcription and replication might be subject to regulation by the SHP nuclear hormone receptor in human hepatoma cells. Transfection of the HBV DNA (4.1-kbp) construct into HepG2 cells supports HBV transcription and replication (Fig. 2, lane 1). The expression of increasing levels of SHP inhibits both HBV 3.5-kb RNA synthesis and viral replication in a dose-dependent manner (Fig. 2). Interestingly, the degree to which SHP inhibits HBV 3.5-kb RNA synthesis is less than the observed reduction in the level of viral DNA replication (Fig. 2C). This observation is similar to the effect of HNF3 on the inhibition of HBV 3.5-kb RNA and DNA synthesis in nonhepatoma cells (43). In the case of HNF3-mediated inhibition, the discrepancy between RNA and DNA syntheses is due to the greater inhibition of HBV pregenomic 3.5-kb RNA synthesis than the precore 3.5-kb RNA (44).
FIG. 2.

Modulation of HBV transcription and replication by SHP. Human hepatoblastoma HepG2 cells were transiently transfected with the HBV DNA (4.1-kbp) construct plus 0, 1, 2, 5, and 10 μg of the SHP expression vector (lanes 1 to 5, respectively). (A) RNA (Northern) filter hybridization analysis of HBV transcripts. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (B) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA. (C) Quantitative analysis of the 3.5-kb HBV RNA and HBV DNA replication intermediates. The levels of the 3.5-kb HBV RNA and total HBV DNA replication intermediates are reported relative to the HBV DNA (4.1-kbp) construct in the absence of SHP expression (lane 1), which are designated as having a relative activity of 1.0. The mean RNA and DNA levels plus standard deviations from three independent analyses are shown.
Limited inhibition of RXRα/PPARα-mediated HBV transcription and replication by SHP in human embryonic kidney 293T cells.
To investigate how SHP inhibits HBV transcription and replication in hepatoma cells, the effect of SHP on RXRα/PPARα-mediated viral biosynthesis was examined in the nonhepatoma 293T cell line (Fig. 3). Increasing the level of SHP expression had only a very modest effect on HBV transcription and replication. HBV transcription was not obviously modulated by SHP expression (Fig. 3A and C). At the highest levels of SHP expression, viral replication was reduced approximately twofold (Fig. 3B and C). These observations suggest that HBV transcription and replication mediated by RXRα/PPARα are largely resistant to inhibition by SHP. This indicates that HBV transcription and replication in HepG2 cells are not mediated primarily by RXRα/PPARα.
FIG. 3.

Modulation of RXRα-PPARα-dependent HBV transcription and replication by SHP. Human embryonic kidney 293T cells were transiently transfected with the HBV DNA (4.1kbp) construct (lanes 1 to 6) plus the RXRα-PPARα expression vector and 0, 1, 2, 5, and 10 μg of the SHP expression vectors (lanes 2 to 6, respectively). (A) RNA (Northern) filter hybridization analysis of HBV transcripts. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (B) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA. (C) Quantitative analysis of the 3.5-kb HBV RNA and total HBV DNA replication intermediates. The levels of the 3.5-kb HBV RNA and HBV DNA replication intermediates are reported relative to those of the HBV DNA (4.1-kbp) construct in the presence of RXRα-PPARα expression (lane 2), which are designated as having a relative activity of 1.0. All-trans retinoic acid and clofibric acid at 1 μM and 1 mM, respectively, were used to activate the nuclear hormone receptors RXRα and PPARα. The mean RNA and DNA levels plus standard deviations from three independent analyses are shown.
Inhibition of HNF4-mediated HBV transcription and replication by SHP in human embryonic kidney 293T cells.
In contrast to RXRα/PPARα-mediated viral biosynthesis, SHP inhibited HNF4-mediated HBV transcription and replication in the nonhepatoma 293T cell line (Fig. 4). HBV transcription was reduced approximately threefold at the highest level of SHP expression (Fig. 4A and C). However, at the highest levels of SHP expression, viral replication was almost undetectable (Fig. 4B and C). These observations indicate that HBV transcription and replication mediated by HNF4 are highly sensitive to inhibition by SHP. This finding is consistent with the suggestion that HNF4 may contribute to HBV transcription and replication in HepG2 cells (Fig. 2 and 4). In addition, the observation that HNF4-mediated HBV replication is more sensitive to SHP inhibition than HBV 3.5-kb RNA synthesis, as also seen in HepG2 cells, suggests that SHP may be preferentially inhibiting HBV 3.5-kb pregenomic RNA compared to HBV 3.5-kb precore RNA under both conditions.
FIG. 4.

Modulation of HNF4-dependent HBV transcription and replication by SHP. Human embryonic kidney 293T cells were transiently transfected with the HBV DNA (4.1kbp) construct plus the HNF4 expression vector and 0, 1, 2, 5, and 10 μg of the SHP expression vectors (lanes 1 to 5, respectively). (A) RNA (Northern) filter hybridization analysis of HBV transcripts. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (B) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA. (C) Quantitative analysis of the 3.5-kb HBV RNA and total HBV DNA replication intermediates. The levels of the 3.5-kb HBV RNA and HBV DNA replication intermediates are reported relative to the HBV DNA (4.1-kbp) construct in the presence of HNF4 expression (lane 1), which are designated as having a relative activity of 1.0. The mean RNA and DNA levels plus standard deviations from three independent analyses are shown.
The expression of SHP does result in the preferential decrease of the level of HBV 3.5-kb pregenomic RNA compared with that of HBV 3.5-kb precore RNA (Fig. 5). At the highest level of SHP expression, the relative level of precore RNA synthesis is approximately fivefold greater than that seen in the absence of SHP expression (Fig. 5B). This observation can account for the greater decrease in viral replication compared with the decrease in HBV 3.5-kb RNA synthesis, as the majority of the decrease in the HBV 3.5-kb RNA was due to the reduced synthesis of pregenomic RNA.
FIG. 5.
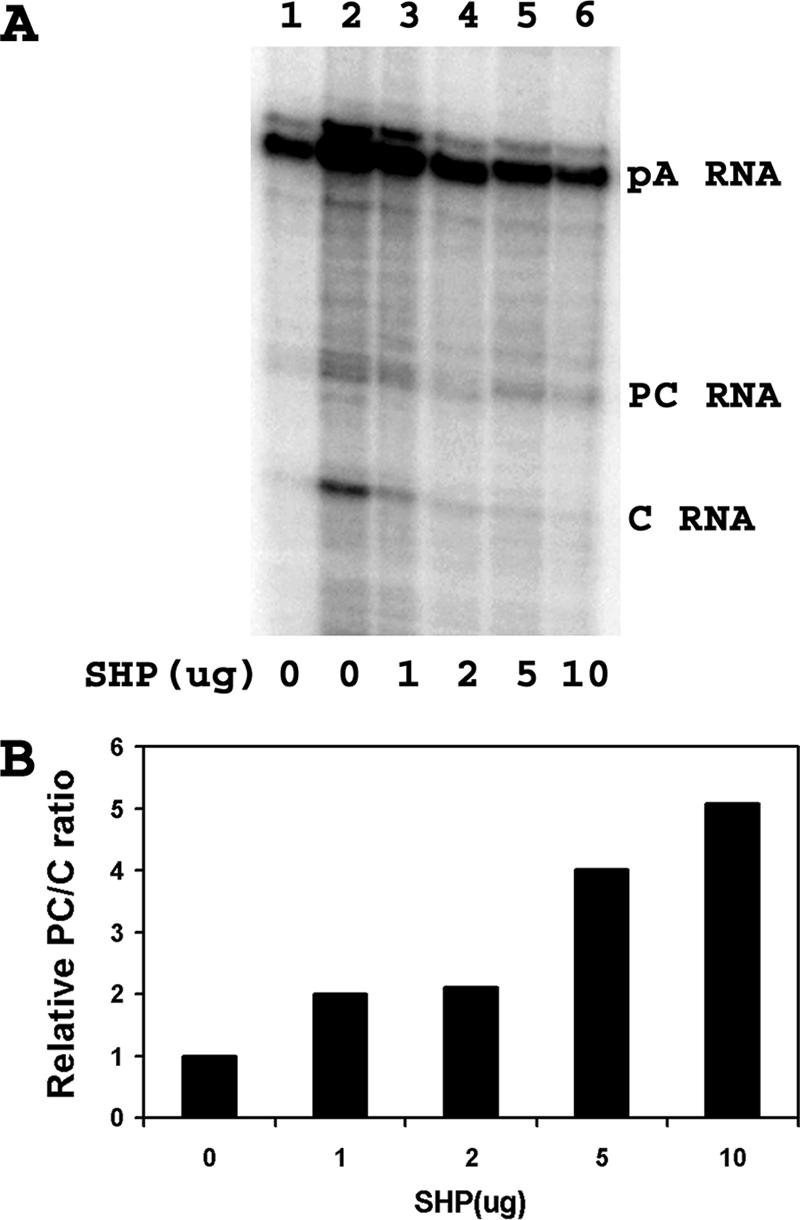
Effect of SHP on the relative levels of precore and pregenomic RNA syntheses. Human embryonic kidney 293T cells were transiently transfected with the HBV DNA (4.1-kbp) construct, the HNF4 expression vector (lanes 2 to 6), and 0, 1, 2, 5, and 10 μg of the SHP expression vector (lanes 1 to 6) as indicated. (A) RNase protection analysis was performed to map the transcription initiation sites of the HBV precore (PC) and pregenomic or core (C) transcripts. The HBV probe also protected a fragment (pA) derived from the 3′ ends of all the HBV RNAs that terminated at the HBV polyadenylation site. (B) Quantitative analysis of the 3.5-kb HBV precore and core RNA levels. The ratios of the 3.5-kb HBV precore to core RNA levels are reported relative to the precore/core RNA ratio transcribed from the HBV DNA (4.1-kbp) construct in the presence of HNF4 expression but in the absence of SHP expression (lane 2), which is designated as having a relative precore-to-core ratio of 1.0. Quantitative analyses of lanes 2 to 6 (A) are shown.
SHP fails to inhibit HBV pregenomic RNA synthesis from a heterologous promoter in human embryonic kidney 293T cells.
The mode of SHP inhibition of HBV transcription and replication appears to be similar to that previously described for HNF3 inhibition of HBV biosynthesis (44). A notable exception to this similarity is the finding that SHP appears to efficiently inhibit only HNF4-mediated viral biosynthesis and not RXRα/PPARα-mediated HBV transcription and replication (Fig. 3 and 4). In an attempt to determine if SHP is inhibiting HBV transcription and replication by interacting directly with HNF4 or by an alternative mechanism involving the indirect inhibition of pregenomic RNA synthesis, the effect of SHP expression on viral biosynthesis from the pCMVHBVayw construct was investigated. The construct, pCMVHBVayw, directs the expression of the pregenomic RNA from the CMV immediate-early promoter (44). Viral replication occurs from the pregenomic RNA synthesized from this construct in the absence of nuclear hormone receptors in human embryonic kidney 293T cells (Fig. 6, lane 1). The expression of SHP in 293T cells does not inhibit viral transcription or replication (Fig. 6). These observations suggest that SHP inhibits HNF4-mediated HBV 3.5-kb pregenomic RNA synthesis by acting as a corepressor that binds directly to HNF4, as previously suggested for several other HNF4-regulated genes (11, 20, 27, 37, 46). In contrast to HNF3, SHP does not appear to greatly modulate HBV transcription and replication directed by RXRα/PPARα from the nucleocapsid promoter or from a heterologous promoter such as the CMV immediate-early promoter. Therefore, SHP appears to preferentially modulate HBV transcription that is controlled by HNF4, suggesting that HBV biosynthesis may be sensitive or insensitive to SHP expression depending on the physiological state of the hepatocytes and the nuclear hormone receptors occupying the nucleocapsid promoter.
FIG. 6.

SHP requires the nucleocapsid promoter to inhibit HBV pregenomic RNA synthesis and viral replication in human embryonic kidney 293T cells. Cells were transiently transfected with the pCMVHBV DNA construct and 0, 1, 2, 5, and 10 μg of the SHP expression vector (lanes 1 to 5, respectively), as indicated. (A) RNA (Northern) filter hybridization analysis of HBV transcripts. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (B) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA. (C) Quantitative analysis of the 3.5-kb HBV RNA and total HBV DNA replication intermediates. The levels of the 3.5-kb HBV RNA and HBV DNA replication intermediates are reported relative to those of the pCMVHBV DNA construct in the absence of SHP expression (lane 1), which are designated as having a relative activity of 1.0. The mean RNA and DNA levels plus standard deviations from two independent analyses are shown.
DISCUSSION
HBV transcription and replication are supported in human hepatoma cells, and in nonhepatoma cells, HBV biosynthesis can be observed when these cells are complemented with the nuclear hormone receptor HNF4 or RXRα/PPARα (43). These nuclear hormone receptors are restricted to the liver, kidney, pancreas, stomach, and intestine (19, 39) and may therefore contribute to a significant degree to the liver-specific tropism of HBV biosynthesis (43). In addition, the activities of nuclear hormone receptors are modulated by their ligands and a variety of signal transduction pathways so that they can support the necessary physiological demands faced by the various tissues as part of the overall homeostatic regulation of the whole organism (3). In this context, SHP has been shown to modulate the activities of a number of nuclear hormone receptors as part of the network of interactions that self-limit the response to changing physiological conditions mediated by these transcription factors (2). In particular, SHP has been reported to modulate the activities of both HNF4 and RXR/PPAR (11, 26, 37, 46).
As it was previously demonstrated that SHP could modulate the activities of HNF4 and RXR/PPAR, it was of interest to establish if the orphan nuclear hormone receptor might influence the ability of HNF4 and RXRα/PPARα to mediate HBV transcription and replication. Initially, the effect of SHP on HBV biosynthesis in the human hepatoma HepG2 cell line was investigated (Fig. 2). SHP inhibited both HBV transcription and replication in a dose-dependent manner in HepG2 cells. As HBV transcription in HepG2 cells is mediated by endogenous transcription factors, this observation suggested that the activities of these factors were sensitive to SHP inhibition and indicated that the endogenous nuclear hormone receptors present in HepG2 cells were directing HBV RNA synthesis.
The nuclear hormone receptors known to support HBV transcription and replication in nonhepatoma cells are HNF4 and RXRα/PPARα (43). Consequently, the susceptibility of RXRα/PPARα- and HNF4-dependent viral replication to SHP-mediated inhibition was examined in human embryonic kidney 293T cells (Fig. 3 and 4). SHP inhibited RXRα/PPARα-dependent viral replication to a very modest extent, which suggests that this mode of HBV transcription and replication is relatively insensitive to the presence of SHP. In contrast, HNF4-dependent viral replication was highly sensitive to SHP-mediated inhibition in human embryonic kidney 293T cells (Fig. 4). Importantly, the inhibition of nuclear hormone receptor-dependent viral replication appears to be occurring at the level of transcription initiation rather that at a latter stage of the RNA synthesis process, as pregenomic RNA production and viral biosynthesis directed by the immediate-early CMV promoter were insensitive to SHP expression (Fig. 6). This distinguished the mechanism of SHP inhibition of HBV RNA synthesis from that of HNF3, which appears to inhibit viral transcription at the stage of RNA elongation (44). This is consistent with previous suggestions that SHP binds to nuclear hormone receptors directly and inhibits their activities by acting as a corepressor (20, 28, 35, 36). Even though the mechanisms of action of SHP and HNF3 in reducing HBV transcription appear to be different, both decrease HBV 3.5-kb RNA synthesis to a greater extent than viral DNA synthesis (Fig. 2 to 4) (1, 44). It appears that in both cases, the synthesis of HBV 3.5-kb pregenomic RNA is inhibited to a greater extent than is HBV 3.5-kb precore RNA, accounting for the greater effect on HBV DNA synthesis than on HBV 3.5-kb RNA synthesis (Fig. 5) (1, 44).
Quantitative analysis of the degree to which SHP inhibits viral replication in HepG2 and 293T cells indicates that the observed inhibition in human hepatoma cells cannot be explained by the inhibition of either HNF4 or RXRα/PPARα alone (Fig. 7A). This suggests that either HNF4 or RXRα/PPARα must contribute approximately equally to the level of HBV biosynthesis in HepG2 cells or additional transcription factors, possibly nuclear hormone receptors because they must be sensitive to SHP inhibition, contribute to viral pregenomic RNA transcription. Under normal physiological conditions in the HBV transgenic mouse, RXRα/PPARα contributes very little to the level of HBV biosynthesis (14). In addition, the level and activity of RXRα/PPARα in HepG2 cells are believed to be extremely low (18). Together, these observations suggest that RXRα/PPARα probably does not contribute to a large extent to the level of HBV transcription and replication in HepG2 cells. Consequently, it appears possible that additional nuclear hormone receptors contribute to the level of HBV biosynthesis in HepG2 cells. Further analysis will be required to validate this suggestion.
FIG. 7.

Multiple transcription factors are targets of SHP inhibition in the human hepatoma HepG2 cell line. (A) Effect of SHP on the relative levels of total HBV DNA replication intermediates in 293T cells in the presence of RXRα/PPARα (RXR/PPAR), 293T cells in the presence of HNF4 (HNF4), and HepG2 cells (HepG2). The levels of the total HBV DNA replication intermediates are reported relative to those of the HBV DNA (4.1-kbp) construct in the absence of SHP expression, which are designated as having a relative activity of 1.0. The mean DNA levels plus standard deviations from Fig. 2 to 4 are shown. A theoretical curve showing the effect of SHP inhibition on total HBV DNA replication intermediates expected if 44% and 56% of the replication were mediated by RXRα/PPARα and HNF4, respectively, is shown. (B) Potential pathways regulating SHP expression in the liver. Cytokines such as TNF-α can activate c-Jun (AP1) through JNK to increase the level of SHP expression, and bile acids can activate FXR to activate the level of SHP expression. The modulation of SHP expression could differentially affect HNF4α- and RXRα/PPARα-mediated HBV transcription and replication in hepatocytes.
The observation that SHP can modulate HBV transcription and replication suggests possible physiologically relevant processes that might modulate HBV biogenesis. Proinflammatory cytokines such as TNF-α have been shown to modulate HBV biosynthesis in vivo (5, 13, 15, 16). The mechanism of action of theses cytokines, however, has not been clearly established. It is possible that TNF-α might modulate HBV biosynthesis by activating the JNK signaling cascade, leading to the activation of c-Jun (AP1), which subsequently increases SHP gene expression (Fig. 7B) (8, 17). In this manner, the level of HBV pregenomic RNA and viral biosynthesis might be down-regulated in response to TNF-α signaling. As very limited alterations in HBV transcription were previously shown to have large effects on HBV replication in the transgenic mouse model of chronic HBV infection (1, 14, 31), it is possible that even a small increase in SHP expression might significantly affect nuclear hormone receptor-dependent viral transcription and, subsequently, HBV replication. Additional studies will be required to establish the importance of this TNF-α signal transduction pathway to HBV biosynthesis.
Alternatively, the farnesoid X receptor (FXR) can directly activate SHP expression in the liver (Fig. 7B) (12, 21). FXR is activated by its ligands, bile acids (22, 29, 45). Conditions associated with elevated bile acid levels within the liver include choleostatic liver diseases or increased dietary cholesterol uptake (7). Under these conditions, the levels of SHP may be elevated, and consequently, HBV pregenomic RNA expression directed by nuclear hormone receptors might be decreased. This suggests that the therapeutic activation of FXR with appropriate ligands might represent a potential approach to inhibiting HBV replication in chronic carriers, although the effects of FXR activation on lipid homeostasis would need to be addressed.
Acknowledgments
We are grateful to Amy Brideau and Stefan Wieland (The Scripps Research Institute, La Jolla, CA) for plasmid pCMVHBVayw, Eric F. Johnson (The Scripps Research Institute, La Jolla, CA) for plasmids pCMVHNF4 and pCMVPPARα-G, Ronald M. Evans (The Salk Institute, La Jolla, CA) for plasmid pRS-hRXRα, and David Mangelsdorf (Southwestern Medical Center, Dallas, TX) for plasmid pCMX-mSHP.
This work was supported by Public Health Service grant AI30070 from the National Institutes of Health.
Footnotes
Published ahead of print on 30 January 2008.
REFERENCES
- 1.Banks, K. E., A. L. Anderson, H. Tang, D. E. Hughes, R. H. Costa, and A. McLachlan. 2002. Hepatocyte nuclear factor 3β inhibits hepatitis B virus replication in vivo. J. Virol. 7612974-12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bavner, A., S. Sanyal, J. A. Gustafsson, and E. Treuter. 2005. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol. Metab. 16478-488. [DOI] [PubMed] [Google Scholar]
- 3.Bookout, A. L., Y. Jeong, M. Downes, R. T. Yu, R. M. Evans, and D. J. Mangelsdorf. 2006. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 126789-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boshart, M., F. Weber, G. Jahn, K. Dorsch-Hasler, B. Fleckenstein, and W. Schaffner. 1985. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 41521-530. [DOI] [PubMed] [Google Scholar]
- 5.Cavanaugh, V. J., L. G. Guidotti, and F. V. Chisari. 1998. Inhibition of hepatitis B virus replication during adenovirus and cytomegalovirus infections in transgenic mice. J. Virol. 722630-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, D., G. Lepar, and B. Kemper. 1994. A transcriptional regulatory element common to a large family of hepatic cytochrome P450 genes is a functional binding site of the orphan receptor HNF-4. J. Biol. Chem. 2695420-5427. [PubMed] [Google Scholar]
- 7.Chiang, J. Y. L. 2002. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr. Rev. 23443-463. [DOI] [PubMed] [Google Scholar]
- 8.Choi, Y. H., M. J. Park, K. W. Kim, H. C. Lee, Y. H. Choi, and J. Cheong. 2004. The orphan nuclear receptor SHP is involved in monocytic differentiation, and its expression is increased by c-Jun. J. Leukoc. Biol. 761082-1088. [DOI] [PubMed] [Google Scholar]
- 9.Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162156-159. [DOI] [PubMed] [Google Scholar]
- 10.Dubois, M. F., C. Pourcel, S. Rousset, C. Chany, and P. Tiollais. 1980. Excretion of hepatitis B surface antigen particles from mouse cells transformed with cloned viral DNA. Proc. Natl. Acad. Sci. USA 774549-4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furihata, T., M. Hosokawa, M. Masuda, T. Satoh, and K. Chiba. 2006. Hepatocyte nuclear factor-4α plays pivotal roles in the regulation of mouse carboxylesterase 2 gene transcription in mouse liver. Arch. Biochem. Biophys. 447107-117. [DOI] [PubMed] [Google Scholar]
- 12.Goodwin, B., S. A. Jones, R. R. Price, M. A. Watson, D. D. McKee, L. B. Moore, C. Galardi, J. G. Wilson, M. C. Lewis, M. E. Roth, P. R. Maloney, T. M. Willson, and S. A. Kliewer. 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 6517-526. [DOI] [PubMed] [Google Scholar]
- 13.Guidotti, L. G., P. Borrow, M. V. Hobbs, B. Matzke, I. Gresser, M. B. A. Oldstone, and F. V. Chisari. 1996. Viral cross talk: intracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver. Proc. Natl. Acad. Sci. USA 934589-4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guidotti, L. G., C. M. Eggers, A. K. Raney, S. Y. Chi, J. M. Peters, F. J. Gonzalez, and A. McLachlan. 1999. In vivo regulation of hepatitis B virus replication by peroxisome proliferators. J. Virol. 7310377-10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guidotti, L. G., T. Ishikawa, M. V. Hobbs, B. Matzke, R. Schreiber, and F. V. Chisari. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 425-36. [DOI] [PubMed] [Google Scholar]
- 16.Guidotti, L. G., B. Matzke, and F. V. Chisari. 1997. Hepatitis B virus replication is cell cycle independent during liver regeneration in transgenic mice. J. Virol. 714804-4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta, S., R. T. Stravitz, P. Dent, and P. B. Hylemon. 2001. Down-regulation of cholesterol 7α-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J. Biol. Chem. 27615816-15822. [DOI] [PubMed] [Google Scholar]
- 18.Hsu, M. H., U. Savas, K. J. Griffin, and E. F. Johnson. 2001. Identification of peroxisome proliferator-responsive human genes by elevated expression of the peroxisome proliferator-activated receptor α in HepG2 cells. J. Biol. Chem. 27627950-27958. [DOI] [PubMed] [Google Scholar]
- 19.Issemann, I., and S. Green. 1990. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347645-650. [DOI] [PubMed] [Google Scholar]
- 20.Lee, Y. K., H. Dell, D. H. Dowhan, M. Hadzopoulou-Cladaras, and D. D. Moore. 2000. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol. Cell. Biol. 20187-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu, T. T., M. Makishima, J. J. Repa, K. Schoonjans, T. A. Kerr, J. Auwerx, and D. J. Mangelsdorf. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 6507-515. [DOI] [PubMed] [Google Scholar]
- 22.Makishima, M., A. Y. Okamoto, J. J. Repa, H. Tu, R. M. Learned, A. Luk, M. V. Hull, K. D. Lustig, D. J. Mangelsdorf, and B. Shan. 1999. Identification of a nuclear receptor for bile acids. Science (New York) 2841362-1365. [DOI] [PubMed] [Google Scholar]
- 23.Mangelsdorf, D. J., E. S. Ong, J. A. Dyck, and R. M. Evans. 1990. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 345224-229. [DOI] [PubMed] [Google Scholar]
- 24.McLachlan, A., D. R. Milich, A. K. Raney, M. G. Riggs, J. L. Hughes, J. Sorge, and F. V. Chisari. 1987. Expression of hepatitis B virus surface and core antigens: influences of pre-S and precore sequences. J. Virol. 61683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muerhoff, A. S., K. J. Griffin, and E. F. Johnson. 1992. The peroxisome proliferator-activated receptor mediates the induction of CYP4A6, a cytochrome P450 fatty acid omega-hydroxylase, by clofibric acid. J. Biol. Chem. 26719051-19053. [PubMed] [Google Scholar]
- 26.Nishizawa, H., K. Yamagata, I. Shimomura, M. Takahashi, H. Kuriyama, K. Kishida, K. Hotta, H. Nagaretani, N. Maeda, M. Matsuda, S. Kihara, T. Nakamura, H. Nishigori, H. Tomura, D. D. Moore, J. Takeda, T. Funahashi, and Y. Matsuzawa. 2002. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor γ transactivation. J. Biol. Chem. 2771586-1592. [DOI] [PubMed] [Google Scholar]
- 27.Ogata, M., T. Awaji, N. Iwasaki, S. Miyazaki, G. I. Bell, and Y. Iwamoto. 2002. Nuclear translocation of SHP and visualization of interaction with HNF-4α in living cells. Biochem. Biophys. Res. Commun. 2928-12. [DOI] [PubMed] [Google Scholar]
- 28.Ortlund, E. A., Y. Lee, I. H. Solomon, J. M. Hager, R. Safi, Y. Choi, Z. Guan, A. Tripathy, C. R. H. Raetz, D. P. McDonnell, D. D. Moore, and M. R. Redinbo. 2005. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat. Struct. Mol. Biol. 12357-363. [DOI] [PubMed] [Google Scholar]
- 29.Parks, D. J., S. G. Blanchard, R. K. Bledsoe, G. Chandra, T. G. Consler, S. A. Kliewer, J. B. Stimmel, T. M. Willson, A. M. Zavacki, D. D. Moore, and J. Lehmann. 1999. Bile acids: natural ligands for an orphan nuclear receptor. Science (New York) 2841365-1368. [DOI] [PubMed] [Google Scholar]
- 30.Raney, A. K., A. J. Easton, D. R. Milich, and A. McLachlan. 1991. Promoter-specific transactivation of hepatitis B virus transcription by a glutamine- and proline-rich domain of hepatocyte nuclear factor 1. J. Virol. 655774-5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raney, A. K., C. M. Eggers, E. F. Kline, L. G. Guidotti, M. Pontoglio, M. Yaniv, and A. McLachlan. 2001. Nuclear covalently closed circular viral genomic DNA in the liver of hepatocyte nuclear factor 1α-null hepatitis B virus transgenic mice. J. Virol. 752900-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raney, A. K., J. L. Johnson, C. N. A. Palmer, and A. McLachlan. 1997. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J. Virol. 711058-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raney, A. K., P. Zhang, and A. McLachlan. 1995. Regulation of transcription from the hepatitis B virus large surface antigen promoter by hepatocyte nuclear factor 3. J. Virol. 693265-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 35.Seol, W., M. Chung, and D. D. Moore. 1997. Novel receptor interaction and repression domains in the orphan receptor SHP. Mol. Cell. Biol. 177126-7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seol, W., H. S. Choi, and D. D. Moore. 1996. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science (New York) 2721336-1339. [DOI] [PubMed] [Google Scholar]
- 37.Shimamoto, Y., J. Ishida, K. Yamagata, T. Saito, H. Kato, T. Matsuoka, K. Hirota, H. Daitoku, M. Nangaku, K. Yamagata, H. Fujii, J. Takeda, and A. Fukamizu. 2004. Inhibitory effect of the small heterodimer partner on hepatocyte nuclear factor-4 mediates bile acid-induced repression of the human angiotensinogen gene. J. Biol. Chem. 2797770-7776. [DOI] [PubMed] [Google Scholar]
- 38.Shlomai, A., N. Paran, and Y. Shaul. 2006. PGC-1α controls hepatitis B virus through nutritional signals. Proc. Natl. Acad. Sci. USA 10316003-16008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sladek, F. M., W. Zhong, E. Lai, and J. E. Darnell, Jr. 1990. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 42353-2365. [DOI] [PubMed] [Google Scholar]
- 40.Summers, J., P. M. Smith, M. Huang, and M. Yu. 1991. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J. Virol. 651310-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sureau, C., J.-L. Romet-Lemonne, J. I. Mullins, and M. Essex. 1986. Production of hepatitis B virus by a differentiated human hepatoma cell line after transfection with cloned circular HBV DNA. Cell 4737-47. [DOI] [PubMed] [Google Scholar]
- 42.Tang, H., K. E. Banks, A. L. Anderson, and A. McLachlan. 2001. Hepatitis B virus transcription and replication. Drug News Perspect. 14325-334. [PubMed] [Google Scholar]
- 43.Tang, H., and A. McLachlan. 2001. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc. Natl. Acad. Sci. USA 981841-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang, H., and A. McLachlan. 2002. Mechanisms of inhibition of nuclear hormone receptor dependent hepatitis B virus replication by hepatocyte nuclear factor 3β. J. Virol. 768572-8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang, H., J. Chen, K. Hollister, L. C. Sowers, and B. M. Forman. 1999. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3543-553. [DOI] [PubMed] [Google Scholar]
- 46.Yamagata, K., H. Daitoku, Y. Shimamoto, H. Matsuzaki, K. Hirota, J. Ishida, and A. Fukamizu. 2004. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J. Biol. Chem. 27923158-23165. [DOI] [PubMed] [Google Scholar]