

Abstract
Previous studies have suggested that coxsackievirus B (CVB) activates CD8+ T cells in vivo, but the extent of this activation and the antigen specificity of the CD8+ T cells remain uncertain. Furthermore, CVB-induced CD4+ T-cell responses have not been carefully investigated. Herein, we evaluate CD8+ and CD4+ T-cell responses both in a secondary lymphoid organ (spleen) and in peripheral tissues (heart and pancreas), using a recombinant CVB3 (rCVB3.6) that encodes well-characterized CD8+ and CD4+ T-cell epitopes. Despite reaching high levels in vivo, rCVB3.6 failed to trigger a marked expansion of CD8+ or CD4+ T cells, and T-cell activation was surprisingly limited. Furthermore, epitope-specific effector functions could not be detected using highly sensitive in vivo and ex vivo assays. Moreover, major histocompatibility complex (MHC) class I tetramer analysis indicated that our inability to detect CVB3-specific CD8+ T-cell responses could not be explained by the cells being dysfunctional. In contrast to naïve T cells, epitope-specific memory CD8+ and CD4+ T cells proliferated markedly, indicating that both of the rCVB3.6-encoded epitopes were presented by their respective MHC molecules in vivo. These data are consistent with the observation that several CVB3 proteins can limit the presentation of viral epitopes on the surface of infected cells and suggest that the level of MHC/peptide complex is sufficient to trigger memory but not naïve T cells. Finally, our findings have implications for the biological significance of cross-priming, a process thought by some to be important for the induction of antiviral CD8+ T-cell responses.
Coxsackieviruses are members of the picornavirus family and enterovirus genus, which includes type A and B coxsackieviruses, polioviruses, echoviruses, and other unclassified enteroviruses. Although the majority of type B coxsackievirus (CVB) infections in humans are subclinical or cause relatively mild disease (including rash, myalgia, or upper respiratory complications), CVB are important human pathogens, and a substantial proportion of infections can lead to severe—even lethal—acute and chronic diseases. In particular, CVB is the most common infectious cause of myocarditis, which can lead to dilated cardiomyopathy and cardiac failure (38, 44, 45). CVB also targets cells of the central nervous system and the pancreas, frequently leading to aseptic meningitis and pancreatitis (7, 12, 33, 35, 40). Overall, CVB infection can cause considerable morbidity and mortality, particularly in newborns and in young or immunocompromised individuals (35, 52).
The murine model of CVB3 infection is a valuable system for studying CVB pathogenesis and immunity, as mice infected with CVB develop diseases similar to those observed in humans (52, 53). Intraperitoneal inoculation of adult C57BL/6 mice with CVB3 results in systemic acute infection; viremia peaks on day 2 to 3 postinfection (p.i.), and infectious virus is cleared by 2 weeks p.i. (33, 34). Control of CVB3 infection depends on both cell-mediated and humoral components of the immune response. Agammaglobulinemic individuals are particularly susceptible to CVB3-associated encephalitis (15, 18), and mice lacking B cells develop a chronic infection and remain viremic for at least 2 months; viremia can be alleviated by the adoptive transfer of B cells from CVB3-immune wild-type mice (34). CD8+ T cells also play an important role in controlling virus replication. T cells are present in the inflammatory infiltrates associated with myocarditis and pancreatitis (17, 20, 41), and CD8+ T-cell depletion of CVB3-infected mice simultaneously increases viral titers and reduces myocarditis, suggesting that T-cell-mediated protection is associated with elevated immunopathology (17). This immunopathology can be uncoupled from antiviral efficacy; mice lacking perforin control cardiac infection just as well as wild-type mice but show markedly diminished myocarditis (14).
Many—probably most—acute viral infections trigger extensive CD8+ T-cell activation and division; these responses can readily be detected directly ex vivo, without any need for extensive restimulation. The convincing evidence that CD8+ T cells can contribute to control of CVB3 in mice, together with the fact that CVB3 replicates to high titers in many mouse tissues, led us to surmise that CVB3—like most other viruses—would induce readily detectable CD8+ T-cell responses in mice. Indeed, early studies had identified cytolytic T-cell activity in CVB3-infected mice, although the precise antigen specificity of the cells was unknown (16, 21, 22). Subsequent elegant work showed that synthetic peptides representing CVB3 VP1 sequences could drive in vitro T-cell proliferation, but neither the phenotype of the proliferating T cells (CD4+ or CD8+) nor the precise epitope specificity was determined (19). Therefore, we undertook a preliminary analysis of epitope-specific CD8+ T-cell responses against CVB3; contrary to our expectations, we found that CVB3-induced epitope-specific CD8+ T-cell responses were difficult to detect (42). However, those studies were incomplete: they relied on ex vivo detection methods of rather limited sensitivity, and they were limited to cells from the spleen. Furthermore, those studies focused only on CD8+ T cells, and it is clear that regulation of antiviral CD8+ T cells differs from that of CD4+ T cells. Therefore, herein we have extended our previous analysis in five ways: first, we evaluate general T-cell activation in CVB3-infected mice; second, we use more sensitive in vivo approaches to detect epitope-specific T-cell responses; third, we investigate the possibility that the virus induces the expansion of dysfunctional T cells; fourth, we extend our analyses of CVB3 epitope-specific T-cell responses to major targets of infection, such as the heart, where CD8+ T cells are present in the virus-induced infiltrate; and, fifth, we investigate CD4+ T-cell responses induced by CVB3. Our studies employ a new recombinant CVB3 (rCVB3) that encodes both a CD8 and CD4 T-cell epitope derived from lymphocytic choriomeningitis virus (LCMV). Our data are not only relevant to understanding the T-cell responses induced by coxsackievirus in particular but also have broader implications for the mechanism(s) by which CD4+ and CD8+ T cells are induced by viruses in general.
MATERIALS AND METHODS
Mice and viruses.
Male C57BL/6J mice were purchased from The Scripps Research Institute (TSRI) breeding facility or from Jackson Laboratories. All experimental procedures with mice were approved by TSRI Animal Care and Use Committee. The wild-type CVB3 (wtCVB3) used in these studies is a plaque-purified isolate (designated H3) of the myocarditic Woodruff variant of CVB3 (46). Plasmid pH3, encoding a full-length infectious clone of this virus (27), was provided by Kirk Knowlton (University of California, San Diego). The production of wtCVB3 and the development of a system to generate rCVB3 have been described previously (42). Adult C57BL/6 mice (6 to 12 weeks of age) were inoculated intraperitoneally with 1 × 107 PFU of rCVB3 or 2 × 105 PFU of LCMV Armstrong.
Recombinant coxsackievirus construction.
Plasmid pMKS1 is a modified version of plasmid pH3 and contains, immediately after the ATG start codon of the CVB3 polyprotein, a unique SfiI restriction site followed by a polyglycine linker and the CVB3 3C viral proteinase cleavage site ALFQG (42). Oligonucleotides encoding both a CD8+ and CD4+ T-cell epitope derived from LCMV (GP33-41 and GP61-80, respectively) were synthesized (Integrated DNA Technologies, Inc., Coralville, IA) as 90/91-mers with 30 bases overlapping (and each included a terminal SfiI site) (forward oligonucleotide, 5′-GAT CGA TCA TGG GGG CCG GAG GGG CCG GTA AGG CTG TCT ACA ATT TTG CCA CCT GTG GAG GGG GTG GAG GTC TTA AGG GAC CCG ACA TTT-3′; reverse oligonucleotide, 5′-GAT CGA TCG GCC CCT CCG GCC CCA TCA AAC TCC ACT GAC TTA AAT TGG TAA ACT CCT TTG TAA ATG TCG GGT CCC TTA AGA CCT CCA CCC C-3′). These oligonucleotides (50 pmol each) were annealed, extended, and amplified by PCR (denaturation, 94°C for 5 min; amplification, 10 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min; extension, 72°C for 7 min). The PCR products were cloned by the T-A method into the pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA) by using a TOPO TA cloning kit (Invitrogen) according to the manufacturer's instructions. pCR2.1-TOPO (carrying the dual epitope cassette) and pMKS1 were cut with SfiI, and the T-cell epitope insert was ligated into pMKS1 by using a rapid DNA ligation kit (Roche, Indianapolis, IN). The insert of the resulting construct (designated pCCK1) was sequenced prior to transcription (Retrogen, San Diego, CA). pCCK1 was linearized with ClaI (Invitrogen), gel purified, and used as a transcription template with a T7 mMESSAGE mMACHINE in vitro transcription kit (Ambion, Austin, TX) according to the manufacturer's instructions. Template DNA was removed by the addition of DNase, and infectious rCVB3 RNA was purified using an RNeasy mini kit (Qiagen, Valencia, CA).
HeLa cell transfection and recombinant virus isolation.
HeLa RW cells (from Rainer Wessely, formerly at University of California, San Diego) were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (complete DMEM). Infectious virus (designated rCVB3.6) was prepared by transfection of HeLa cells (at ∼80% confluence in a six-well plate) with 1 μg of pCCK1 RNA per well using Lipofectamine, Plus reagent, and Opti-MEM (all from Invitrogen) according to the manufacturer's instructions. After 3.5 h, the transfection medium was replaced with complete DMEM, and ∼50 h posttransfection the cell monolayer and culture supernatant were harvested. Cells were disrupted by three cycles of freezing and thawing, cell debris was removed by centrifugation, and the clarified medium was combined with the culture supernatant to generate a passage 1 (P1) rCVB3.6 stock. To prepare a large batch of rCVB3.6 P2 stock, HeLa cell monolayers (∼90% confluent in two T-162 flasks) were infected with the P1 stock and harvested at 8 h p.i. The culture supernatant and freeze/thaw-clarified media were pooled, the titers were determined by plaque assay, and the solutions were stored at −80°C. RNA was prepared from the rCVB3.6 stock as described below, and the T-cell epitope insert was sequenced prior to animal experiments.
One-step growth curve.
HeLa cell monolayers (∼70% confluent in six-well plates) were infected with rCVB3.6 or wtCVB3 at a multiplicity of infection of 20. After 30 min at 37°C, monolayers were washed two times with saline, and then 2 ml of complete DMEM was added to each well. Cells and supernatants were harvested at the indicated time points, the culture supernatant and freeze/thaw-clarified media were pooled, and titers were determined by plaque assay.
Tissue preparation for plaque assay and RNA isolation.
The hearts, pancreases, and spleens of infected mice were removed immediately after euthanasia and snap-frozen in liquid nitrogen. Frozen samples were weighed, homogenized in 1 ml DMEM, and stored at −80°C. In some experiments, feces were collected from rCVB3-infected mice on day 2 p.i. Fecal samples were weighed and resuspended in 250 to 500 μl DMEM. The suspensions were briefly centrifuged to pellet debris, and the supernatant was used for virus titration in a plaque assay.
Plaque assays.
Plaque assays were performed on HeLa cell monolayers as described previously (24). The titer (PFU/g) was calculated based on the weight of each organ or fecal sample.
RNA isolation and RT-PCR.
RNA was isolated from 250 μl of virus stock or organ homogenate by using Trizol LS reagent (Invitrogen) according to the manufacturer's instructions. rCVB3.6 RNA encoding the T-cell epitope insert was amplified by reverse transcription-PCR (RT-PCR) using a SuperScript one-step RT-PCR with Platinum Taq kit (Invitrogen) according to the manufacturer's instructions. The absence of plasmid or genomic DNA-derived RT-PCR products from the RNA preparations was confirmed by omitting reverse transcriptase/Platinum Taq and substituting Taq DNA polymerase (Invitrogen) in the reaction (“no RT” controls). The primers used for RT-PCR were as follows: forward primer, 5′-CTA TTG GAT TGG CCA TCC GG-3′; reverse primer, 5′-GAT TTA ATC ATG ATA TCT TTC ACT GG-3′. RT-PCR products were visualized on a 2% agarose gel.
Virus sequencing.
RT-PCR products generated using RNA template isolated from organ homogenates were cloned by the T-A method into the pCR2.1-TOPO vector (Invitrogen) by using a TOPO TA cloning kit (Invitrogen). The insert of individual clones was sequenced (Retrogen) to determine the stability and integrity of the T-cell epitopes in vivo. RT-PCR products generated using RNA isolated from P1 and P2 rCVB3.6 stocks were column purified and sequenced (Retrogen) to assess the integrity of the T-cell epitopes.
Lymphocyte isolation from organs.
A single cell suspension of splenocytes was prepared by disruption of the spleen through a 70-μm nylon cell strainer (BD Biosciences, San Jose, CA), and red blood cells were lysed with 0.83% NH4Cl. In some experiments, mice were anesthetized and perfused with cold phosphate-buffered saline, and lymphocytes/mononuclear cells from the heart and pancreas were isolated. Hearts were minced, collagenase digested for 1 h at 37°C, filtered through a cell strainer, resuspended in 44% Percoll (Amersham Biosciences, Uppsala, Sweden), underlaid with 67% Percoll, and centrifuged at ∼600 × g for 20 min. Cells were removed at the gradient interface and washed before use. Pancreases were disrupted into single cell suspensions with a Dounce homogenizer, and cells were purified on a Percoll gradient.
Flow cytometry.
Cells were stained with antibodies in phosphate-buffered saline containing 2% FBS and 0.1% sodium azide (FACS buffer) for 30 min on ice, followed by two washes in FACS buffer. Samples were immediately acquired on a FACS Calibur (BD Biosciences) or were fixed in 1% paraformaldehyde. Prior to intracellular cytokine staining (ICCS), cells were incubated for 5 h in 96-well plates in 0.2 ml/well RPMI containing 10% FBS, 50 μM 2-mercaptoethanol, penicillin-streptomycin, GolgiPlug (BD Biosciences), and synthetic peptides (1 μg/ml GP33-41 or NP396-404, 1 μM GP61-80). Cells were then stained for surface CD8 or CD4 and intracellular gamma interferon (IFN-γ) by use of a Cytofix/Cytoperm kit (BD Biosciences). CD8 (clone 53-6.7), CD4 (clone GK1.5), CD11a (clone M17/4), CD44 (clone IM7), and IFN-γ (clone XMG1.2) antibodies were purchased from eBioscience. CD4 (clone H129.19) antibody was purchased from BD Biosciences. CD62L (clone MEL-14) antibody was purchased from Caltag Laboratories. H-2Db/GP33-41 tetramers were produced by the NIH Tetramer Core Facility. Data were analyzed with FlowJo software (Tree Star, Ashland, OR).
In vivo cytotoxicity assays.
Splenocytes from naïve C57BL/6 mice were red blood cell lysed, resuspended at 10 × 106 cells/ml in complete DMEM, and labeled with peptide (LCMV GP33-41, NP396-404, NP118-126, or influenza NP366-374 at 1 μg/ml) for 1 h at 37°C. Next, the cells were resuspended at 20 × 106 cells/ml in Hanks' balanced salt solution and labeled with various concentrations of carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR) for 10 min at 37°C, i.e., 3 and 0.3 μM CFSE for two target cell populations and 4, 1, and 0.25 μM CFSE for three target cell populations. After extensive washing, equal numbers of each population were mixed, and 3 × 106 to 5 × 106 cells of each target population were injected intravenously into C57BL/6 recipients. After ∼18 h, recipient spleens were harvested and CFSE+ target cell populations were analyzed by flow cytometry. The percent specific lysis was calculated as follows: 100 − {[(% peptide coated in experimental mouse/% control peptide coated in experimental mouse)/(% peptide coated in control mouse/% control peptide coated in control mouse)] × 100}.
Plasmid DNA preparation and immunization.
A sequence spanning the N-terminal methionine to the glutamine in the artificial cleavage site (ALFQG) (Fig. 1A) was PCR amplified from the DNA plasmid encoding rCVB3.6 (pCCK1). The PCR products were cloned by the T-A method into the pCR2.1-TOPO vector (Invitrogen) by using a TOPO TA cloning kit (Invitrogen). Following a restriction digest of pCR2.1-TOPO (carrying the insertion cassette) and the pCMV expression vector (54), the insertion cassette was ligated into the multiple cloning site of pCMV by use of a rapid DNA ligation kit (Roche). The resulting construct encodes the rCVB3.6 N-terminal polypeptide sequence and was designated pCMV-Nt3.6. The insert and flanking areas of the plasmid were sequenced prior to in vivo experiments. The DNA used for immunization was prepared using an EndoFree plasmid mega kit (Qiagen) and was reconstituted in saline at a final concentration of 1 mg/ml. Mice were immunized by injection of 50 μl (50 μg) into each anterior tibial muscle (100 μg total DNA per mouse).
FIG. 1.

Insert sequence and one-step growth curve of rCVB3.6. (A) The N-terminal sequence of rCVB3.6 is shown, with the LCMV GP33-41 and GP61-80 epitopes (residues 8 to 16 and 21 to 40, respectively) underlined and separated by a polyglycine linker. The epitopes are followed by an artificial CVB3 3C viral proteinase recognition site (ALFQG, dashed underline) that, when cleaved (at position ▾), releases the N-terminal polypeptide from the CVB3 polyprotein. (B) One-step growth curves of wtCVB3 and rCVB3.6. HeLa cell monolayers were infected at a multiplicity of infection of 20, and the virus titer was determined at the indicated time points by plaque assay. Data are representative of two independent experiments.
RESULTS
Generation and characterization of an rCVB3 encoding well-characterized CD8+ and CD4+ T-cell epitopes.
The construct used to generate rCVB3 in our laboratory is a DNA plasmid named pMKS1 (42). This plasmid encodes the myocarditic Woodruff variant of CVB3 (H3) into which a unique SfiI restriction site and a linker sequence encoding the CVB3 3C viral proteinase cleavage site ALFQG have been introduced immediately downstream of the CVB3 translational initiation codon. A foreign sequence of interest can be inserted at the SfiI site, and viral translation produces a recombinant polyprotein with the foreign protein located at the N terminus; virus-driven proteolysis of the engineered cleavage site releases the foreign polypeptide from the remainder of the viral polyprotein. An oligonucleotide sequence containing well-characterized CD8+ and CD4+ T-cell epitopes derived from LCMV (GP33-41 and GP61-80, respectively) (Fig. 1A) was inserted in frame into the SfiI site, yielding infectious clones for the production of rCVB3. The sequence of the foreign epitope insertion and the region surrounding the cloning site was verified. Next, HeLa cells were transfected with infectious RNA transcribed from linearized plasmid; the resulting P1 virus (designated rCVB3.6) was used to generate a high-titer P2 stock (109 PFU/ml). Sequencing analysis of RT-PCR products amplified from RNA prepared from P1 and P2 rCVB3.6 stocks confirmed that the T-cell epitope sequence was retained after growth in tissue culture. rCVB3.6 formed small plaques on HeLa cell monolayers within 42 h (data not shown), similarly to other rCVB3 created by our laboratory (11, 42). To compare the growth rates of rCVB3.6 and wtCVB3, we performed an in vitro one-step growth curve analysis (Fig. 1B). The onset of exponential growth of rCVB3.6 was delayed by 1 h compared to that of wtCVB3, but the growth rates of both viruses were similar once viral replication had commenced. In addition, the maximum titer of rCVB3.6 was only slightly lower than that of wtCVB3 (Fig. 1B). These growth characteristics of rCVB3.6 are quite similar to those of previously described rCVB3 (42).
rCVB3.6 reaches high titers in vivo in several tissues.
The overall goal of this work was to evaluate the induction by CVB3 of CD4+ and CD8+ T-cell responses in vivo. Before analyzing T-cell responses to rCVB3.6, it was important to show that the virus could replicate and disseminate in mice and that the epitope sequences were not rapidly lost from the recombinant genome. Inoculation of C57BL/6 mice with wtCVB3 results in systemic acute infection; the wt virus achieves a peak titer of ∼1010 PFU/g in the pancreas and ∼108 PFU/g in the spleen and heart, and infectious virus is cleared from all organs between ∼7 and 14 days p.i. (33, 34). Our published studies using rCVB3 have shown that these viruses are attenuated in vivo; they achieve a peak titer similar to that of wt virus in the pancreas, but cardiac titers are moderately reduced (25, 42). To determine the in vivo replication kinetics of rCVB3.6, C57BL/6 mice were inoculated with 107 PFU of rCVB3.6, and viral titers in multiple organs were measured at several time points. High titers of rCVB3.6 were present in the spleen, pancreas, and heart on day 2 p.i. (∼107, ∼1010, and ∼3 × 106 PFU/g, respectively) (Fig. 2A). Viral titers declined in all organs by day 4 and were undetectable by day 6 in the heart and by day 7 to 8 in the spleen and pancreas. Importantly, the absence of virus from tissues harvested at the later time points reflected true virus clearance rather than a failure of the initial infection, because all of these mice had a high CVB3 titer in feces that had been harvested 2 days p.i. (∼106 PFU/g) (data not shown). Thus, rCVB3.6 reaches a high titer in vivo and is cleared moderately faster than its wt counterpart.
FIG. 2.

In vivo analyses of rCVB3.6: growth kinetics and insert stability. Mice were inoculated with 107 PFU of a P2 stock of rCVB3.6. (A) The viral titers in the spleen, pancreas, and heart were determined at the indicated time points p.i. (three mice at days 2, 6, 7, and 8 and five mice at day 4). For the day 6-, 7-, and 8-tested mice that did not harbor infectious virus, the success of the original infection was confirmed by titration of feces that had been collected at 2 days p.i.; all mice showed a high fecal titer (∼106 PFU/g) (data not shown). (B) A portion of the rCVB3.6 sequence encoding the GP33 and GP61 epitopes was amplified by RT-PCR from RNA isolated from the virus stock used to inoculate mice (P2) or from the spleen, heart, and pancreas at the indicated time points p.i., and PCR products were analyzed by gel electrophoresis. Each lane on the gel represents an individual mouse. The expected PCR product from intact rCVB3.6 is 466 bp; the dashed white line shows the expected location of a wt RNA product (310 bp). All RNA samples also were incubated in the absence of reverse transcriptase, and in all cases PCR products were undetectable (not shown). To more precisely analyze the sequence stability, samples were cloned and sequenced as described in the text. (C) To ensure that the P2 stock that was inoculated into the mice did not contain any residue of the plasmid DNA from which infectious RNA was prepared, the PCR was carried out with or without reverse transcriptase (+RT or −RT, respectively).
The inserted epitope sequences are stable for at least 7 days in vivo.
Activation of naïve T cells requires only a relatively brief (2- to 24-h) exposure to cognate antigen (26, 47), and so, in order to activate T cells, the epitopes encoded by rCVB3.6 would have to be retained and expressed in vivo for at least this time period. We thought it likely that rCVB3.6 would meet and exceed this temporal threshold, because we and others have shown that recombinant enteroviruses can retain the inserted sequences for several days in vivo (36, 42). Nevertheless, we considered it important to determine the stability of the epitopes inserted into rCVB3.6; to this end, RNA was prepared from the hearts, spleens, and pancreases at day 2, 4, 6, or 7 p.i., as indicated in Fig. 2B. Viral RNA was amplified by RT-PCR using primers flanking the cloning site, and the reaction products were evaluated by agarose gel electrophoresis. A single PCR product was visualized in all tissues at all time points examined, and, as late as day 7 p.i. in the pancreas, a faint band was visible. The PCR band was the same size as the product amplified from the P2 rCVB3.6 stock that was used to infect the mice. To more precisely determine the extent to which the epitope sequences were or were not maintained, these PCR products were cloned, and the DNA from individual transformed bacterial clones (five clones per organ) was sequenced. All clones derived from RNA from the heart (day 4 p.i.) and spleen (day 6 p.i.) were identical to the original epitope insertion sequence. This was true, too, for four of five clones isolated from the pancreas at 7 days p.i. The remaining pancreatic isolate contained a point mutation in codon 17 of the N-terminal sequence, resulting in a glycine-to-arginine substitution in the short polyglycine spacer separating the two epitopes (Fig. 1A). Thus, the native CD4+ and CD8+ T-cell epitope sequences were maintained in 100% of clones (15/15) obtained between day 4 and day 7 p.i. Taken together, these data show that the rCVB3.6 insert is remarkably stable in vivo, being detectable for up to 1 week p.i. in the pancreas.
Minimal activation of CD8+ and CD4+ T cells following rCVB3.6 infection.
When activated by virus infection, CD8+ and CD4+ T cells expand in number and also modulate their expression of cell surface molecules such as CD62L and CD44. Therefore, to broadly assess the extent of T-cell activation during CVB3 infection, we evaluated the ability of rCVB3.6 to cause these phenotypic changes, which should affect not only T cells specific for the two inserted epitopes but also T cells that are specific for “native” CVB3 epitopes (that are presumed to exist but are at present unidentified). Therefore, the CD62L and CD44 expression profiles of splenic CD8+ and CD4+ T cells (Fig. 3A, left) were compared for uninfected mice and rCVB3.6-infected mice. The changes that resulted from rCVB3.6 infection appeared minimal, suggesting that this virus—despite reaching high titers—does not drive extensive T-cell activation. This is consistent with recent work from this lab using wtCVB3 (6). It was possible that the limited activation observed in spleen might reflect the rapid departure of activated T cells to peripheral sites of virus infection, and to evaluate this, similar analyses were carried out on cells isolated from the hearts; again, minimal changes in CD62L and CD44 expression profiles were observed (Fig. 3A, center). Similar findings were observed for cells isolated from the pancreases (data not shown). The weakness of the CVB3-induced T-cell response is underscored when it is compared to the response induced by another virus, LCMV; infection with the latter virus led to dramatic down-regulation of CD62L on splenic CD8+ and CD4+ T cells as well as up-regulation of CD44 on these subsets (Fig. 3A, right). The frequencies of CD8+ and CD4+ T cells that were CD62Llow, CD44high, and CD11ahigh in the spleens or hearts of uninfected and rCVB3.6-infected mice were quantitated, and no statistically significant differences were observed (Fig. 3B). We conclude that, despite reaching high titers in several tissues, rCVB3.6 causes a very limited degree of CD8+ and CD4+ T-cell activation.
FIG. 3.

rCVB3.6 infection induces minimal changes in activation marker expression on CD8+ and CD4+ T cells. (A) CD62L and CD44 expression profiles of CD8+ and CD4+ T cells in the spleen and heart were compared for uninfected and rCVB3.6-infected mice (day 8 p.i.). Representative histograms are gated on CD8+ or CD4+ T cells, and the phenotypes of T cells from uninfected and virus-infected mice are shown by black and red lines, respectively. To show the phenotypic changes that occur during infection with a virus that induces strong T-cell activation, equivalent data are shown for splenocytes isolated from LCMV-infected mice. (B) Quantitation of changes in T-cell activation markers. The frequencies of CD62Llow, CD44high, and CD11ahigh CD8+ and CD4+ T cells in the spleens and hearts of naïve and rCVB3.6-infected mice (day 8 p.i.) are shown. Percentages for the spleen are plotted as the means ± standard deviations of three mice per group. Cells isolated from the hearts of the three mice in each group were fewer in number and so were pooled prior to phenotypic analysis. Max, maximum; Uninf, uninfected.
Primary GP33- and GP61-specific effector T cells are not detected during acute infection.
Although there was no broad activation of T cells during rCVB3.6 infection, it remained possible that weak CVB3-specific T-cell responses could be detected by more-sensitive assays based on the effector functions of virus-specific (epitope-specific) cells, e.g., ICCS or in vivo cytotoxicity; such analyses require foreknowledge of epitope specificity, and it was for this purpose that rCVB3.6 was designed and constructed. (i) ICCS assay. Splenocytes and mononuclear cells isolated from the hearts and pancreases of perfused mice were subjected to an ICCS assay (Fig. 4). The cells were stimulated with GP33-41 or GP61-80 peptides (or left untreated), and the frequency of IFN-γ-producing T cells was analyzed by flow cytometry. T-cell responses in LCMV-infected mice were used as a positive control. In contrast to the substantial GP33- and GP61-specific responses detected in LCMV-infected mice, neither CD4+ nor CD8+ T-cell epitope-specific responses were detected in the spleens, hearts, or pancreases of rCVB3.6-infected animals. (ii) In vivo cytotoxicity assay. As an additional means of detecting low-frequency CVB3-specific CD8+ T-cell responses, highly sensitive in vivo cytotoxicity assays were performed with rCVB3.6-infected mice and with appropriate negative- and positive-control mice (uninfected mice and LCMV-infected mice, respectively). The epitope specificity of any observed in vivo lysis was ensured by including not only target cells coated with the GP33-41 peptide but also cells coated with an irrelevant peptide representing another strong Db-presented epitope (NP366 from influenza virus). Following an 18-h assay, NP366- and GP33-pulsed target cells were present at almost identical frequencies in naïve and rCVB3.6-infected mice, indicating that GP33-specific lysis failed to rise above background levels in rCVB3.6-infected mice. In contrast, nearly all GP33-pulsed targets were eliminated in LCMV-infected hosts, corresponding to ∼95% specific lysis. Representative histograms from individual mice are shown in Fig. 5A, and data from five infected mice (three infected with rCVB3.6 and two infected with LCMV) are summarized in Fig. 5B.
FIG. 4.

Primary GP33- and GP61-specific T-cell responses are not detected during acute rCVB3.6 infection. Mice were infected with rCVB3.6 or with LCMV, and 8 days later, the mice were perfused and sacrificed. Splenocytes and mononuclear cells isolated from the heart and pancreas were analyzed by ICCS. The numbers indicate the proportion of cells in the quadrant, shown as the percentage of total mononuclear cells in the dot plot. Data are representative of two independent experiments. No pep, no peptide.
FIG. 5.

GP33-specific in vivo cytotoxicity is not detected in rCVB3.6-infected mice. CFSElow target cells coated with influenza A NP366 peptide were mixed with CFSEhigh targets coated with the GP33 peptide and were inoculated into uninfected (Uninf), rCVB3.6-infected, or LCMV-infected mice (day 8 p.i.). Eighteen hours later, the cells were recovered from the spleen and were enumerated by flow cytometry. (A) Representative histograms are shown; the numbers reflect the proportion of each target cell population as a percentage of the total CFSE+ splenocytes. (B) The percentages of GP33-specific lysis are shown for five individual mice, three of which were infected with rCVB3.6 and two with LCMV.
The absence of detectable CD8+ T-cell induction cannot be attributed to T-cell dysfunction.
We next considered the possibility that CVB3 infection might drive the proliferation of epitope-specific CD8+ T cells that lack effector functions such as cytokine production and cytotoxicity; such cells would not be identified using the approaches employed above. To evaluate this possibility, DbGP33 tetramers were used to enumerate GP33-specific cells in the spleens, hearts, and pancreases of rCVB3.6-infected mice. CVB3-specific CD8+ T cells could not be identified in any of the tissues, but in contrast, a sizeable DbGP33 tetramer+ CD44high CD8+ T-cell population was present in all three tissues of LCMV-infected mice (Fig. 6). Taken together, the data in Fig. 4 to 6 strongly suggest that rCVB3.6 infection directs remarkably meager expansion of epitope-specific primary CD8+ and CD4+ T cells. One possible explanation for the extremely weak induction of primary T-cell responses by rCVB3.6 is that the foreign epitope sequences encoded by the recombinant virus are in some way flawed, preventing them from being processed and/or presented by the major histocompatibility complex (MHC) class I and II antigen presentation pathways. The remaining experiments were aimed at evaluating this possibility.
FIG. 6.
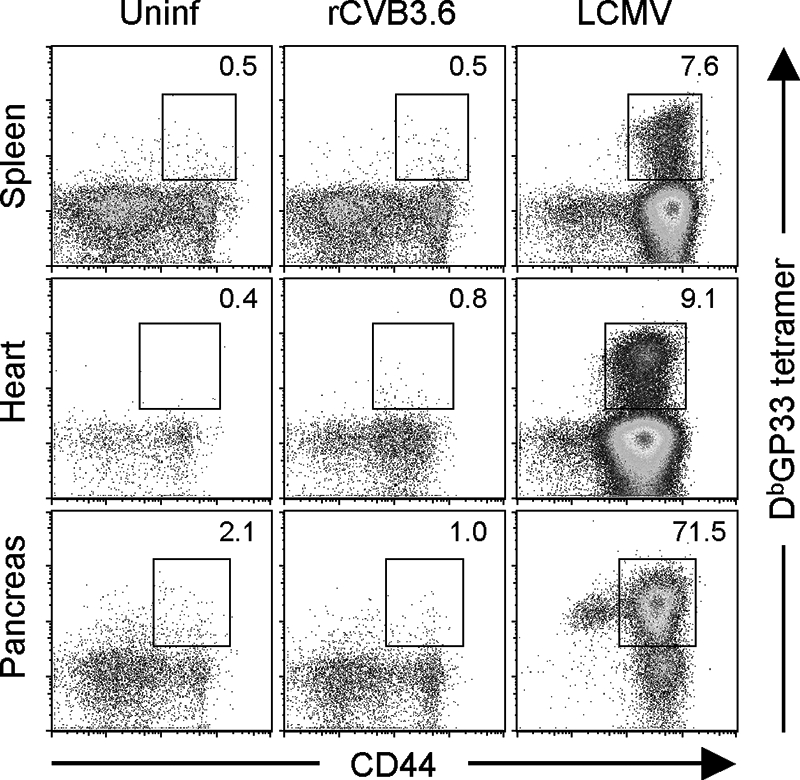
CVB3-specific CD8+ T cells cannot be readily identified in lymphoid or nonlymphoid tissues by using MHC class I peptide tetramers. Mice were infected with rCVB3.6 or with LCMV, and 8 days later, the mice were perfused and sacrificed. Splenocytes and mononuclear cells isolated from the heart and pancreas were stained with CD8 and CD44 antibodies and MHC class I H-2Db/GP33-41 tetramers and analyzed by flow cytometry. Cells isolated from the hearts and pancreases of three mice per group were pooled for analysis. The dot plots shown are gated on CD8+ T cells, and the numbers indicate the percentages of CD8+ T cells that were tetramer positive. Uninf, uninfected.
Both GP33 and GP61 T-cell epitopes are expressed in vivo during rCVB3.6 infection.
First, we sought evidence for presentation of the two epitopes during rCVB3.6 infection. To do so, we exploited a key feature of epitope-specific memory T cells; memory cells are much better able than naïve cells to respond to very low levels of cognate antigen (43), and so memory cells are more sensitive indicators of in vivo epitope presentation. C57BL/6 mice were infected with LCMV, and 71 days later (a time point at which epitope-specific memory T cells are abundant), the mice were infected with rCVB3.6. Six days thereafter, CD8+ and CD4+ T-cell responses against the GP33-41 and GP61-80 epitopes (respectively) were analyzed by ICCS. To ensure that any observed effects of rCVB3.6 were epitope specific, we also evaluated CD8+ memory cells that are specific for another LCMV epitope (NP396) that is abundant in LCMV-immune mice but that is not present in rCVB3.6. Two additional negative-control groups were included: LCMV-immune mice that were not infected with rCVB3 and LCMV-immune mice inoculated with rCVB3.3 (a virus that expresses the CD8+ T-cell epitope NP118-126, which is presented by the Ld MHC class I allele in BALB/c mice [42]). rCVB3.6 infection of LCMV-immune mice led to an ∼5-fold increase in the frequency of GP33-specific CD8+ T cells and an ∼9-fold increase in the frequency of GP61-specific CD4+ T cells (Fig. 7A). The increase was observed only for T cells specific for rCVB3.6, because no significant change in frequency occurred for NP396-specific cells. The epitope specificity of the T-cell expansion was further confirmed by the absence of any significant changes in T-cell frequency in mice that had been infected with the irrelevant virus rCVB3.3 (Fig. 7). The changes in frequency of rCVB3.6-specific T cells were also reflected in similar changes in absolute cell numbers in the mouse spleens (Fig. 7B); both GP33-specific and GP61-specific cell populations expanded at least 10-fold in mice challenged with rCVB3.6 but not in mice challenged with rCVB3.3. Taken together, these data indicate that, during rCVB3.6 infection, both the GP33 and GP61 epitopes are processed and presented by the MHC class I and class II antigen presentation pathways in vivo.
FIG. 7.

rCVB3.6 stimulates the in vivo expansion of functional epitope-specific CD8+ and CD4+ memory cells. LCMV-immune mice (day 71 p.i.) were challenged with rCVB3.6 or rCVB3.3 or were left untreated, and GP33-, NP396-, and GP61-specific T-cell responses were analyzed by ICCS 6 days later. (A) The frequency of each population of epitope-specific cells was calculated as a percentage of the total number of relevant cells (CD8+ or CD4+) in the spleen. Statistical significance was determined by an unpaired Student t test, assuming unequal variances (Microsoft Excel). (B) The absolute numbers of epitope-specific IFN-γ+ T cells were determined and are shown as the means ± standard errors of two to three mice per group. The data are representative of two independent experiments.
Immunization of mice with a DNA plasmid encoding the rCVB3.6 N-terminal polypeptide induces a GP33-specific CD8+ T-cell response.
We report above that we were unable to detect any primary CD4+ or CD8+ responses following rCVB3.6 infection of naïve mice. The difficulty in detecting CD8+ T-cell responses is particularly striking, because CD8+ T cells usually outnumber CD4+ T cells by at least 10:1. Therefore, to confirm that the CD8+ T-cell epitope could be efficiently synthesized and presented from the sequence that had been inserted into rCVB3.6 and to compare the immunogenicity of rCVB3.6 against another form of immunization, we evaluated the capacity of this sequence to induce CD8+ T cells when expressed as part of a plasmid DNA vaccine. The DNA sequence spanning the N-terminal methionine to the glutamine in the artificial cleavage site (Fig. 1A) was inserted into the pCMV vector (54) to generate a new construct encoding the N-terminal polypeptide of rCVB3.6. This plasmid (referred to as pCMV-Nt3.6) or pCMV as a negative control was used to immunize C57BL/6 mice; a control mouse was infected with LCMV. DNA-immunized mice were boosted with the same plasmids 21 days later, and in vivo cytotoxicity assays were performed 8 days later, on day 29. Histograms from individual mice and data for several individual mice are shown in Fig. 8A and B, respectively. As expected, both the GP33-pulsed targets and the NP396-pulsed targets were almost completed eliminated in LCMV-immune mice. Of greater interest, a substantial portion of the GP33-pulsed targets was eliminated in all pCMV-Nt3.6-immunized mice, indicating that the DNA vaccine had induced a GP33-specific CD8+ T-cell response. The specificity of the DNA-induced response was confirmed by the absence of lysis of target cells pulsed with the NP396 peptide. In summary, these data show that the sequence present in rCVB3.6 can induce a strong GP33-specific CD8+ T-cell response when expressed separately from the other proteins of CVB3.
FIG. 8.

The rCVB3.6 insert induces strong epitope-specific in vivo cytotoxicity when expressed from a plasmid DNA vaccine. On day 0, naïve mice were DNA immunized as described in Materials and Methods, using either pCMV-Nt3.6 or pCMV as a negative control. A positive-control mouse was infected with LCMV. Twenty-one days later, the DNA-immunized mice were boosted with the same plasmids, and an in vivo cytotoxicity assay was carried out 8 days later, on day 29. (A) Representative histograms show the CFSE low, intermediate, and high target cells pulsed with NP396, NP118, and GP33 peptides, respectively, in the spleens of mice immunized with pCMV or pCMV-Nt3.6 or an LCMV-immune mouse; the numbers reflect the proportion of each target cell population expressed as a percentage of the total CFSE+ splenocytes. (B) The percentages of GP33-specific lysis (top) and NP396-specific lysis (bottom) were determined for individual mice immunized with pCMV-Nt3.6 (n = 3) and for one LCMV-immune mouse.
DISCUSSION
In this study, we generated an rCVB3 that encodes highly immunogenic T-cell epitopes from LCMV and used it to evaluate the induction of CD4+ and CD8+ T-cell responses during CVB3 infection. Our analysis of CVB3-specific T-cell responses with highly sensitive techniques and in multiple sites of infection revealed a very limited degree of CD4+ and CD8+ T-cell activation, and primary GP33- and GP61-specific T-cell responses were not detected. Taken together, these data suggest that CVB3 infection induces minimal CD4+ and CD8+ T-cell responses. The absence of a detectable CD8+ T-cell response is particularly surprising because, as noted above, during the great majority of virus infections, virus-specific CD8+ T cells greatly outnumber their CD4+ counterparts, usually by a factor of at least 10:1.
In principle, two trivial explanations could have contributed to the poor immunogenicity of rCVB3.6: first, instability of the virus-encoded T-cell epitope insert in vivo, resulting in its rapid loss before sufficient time has passed to trigger a response in naïve mice; second, a deficit in the production, processing, or presentation of the epitopes. We show herein that the size of the rCVB3.6 RNA insert is maintained in RNA isolated from the heart, pancreas, and spleen for up to 1 week p.i. (Fig. 2B), and sequencing of 15 individual clones showed that the epitopes were intact. The reason for the unusual stability of this sequence is not clear; others have proposed that a high G/C content confers stability on poliovirus inserts (29), but our data do not allow us to draw conclusions regarding the insert in rCVB3.6. However, we can conclude that the low immunogenicity of rCVB3.6 is unlikely to be caused by the rapid loss of epitope sequence. In passing, we note that the retention of intact epitope sequences carries an implication beyond that of mere sequence stability. It is well established that CD8+ T cells, in particular, can exert selective pressures that lead to mutation within viral epitopes (3, 39, 49), and the maintenance of unaltered epitopes in rCVB3.6 therefore constitutes additional, albeit indirect, evidence reflecting the weakness of the T-cell responses that are induced by rCVB3.
The replicative fitness of rCVB3.6 in tissue culture (Fig. 1B) and in vivo (Fig. 2A) provides strong evidence that the epitopes must have been translated during the infection, because they lie in frame between the initiator ATG and the remainder of the viral polyprotein. However, this led us to the second trivial explanation for the weak T-cell responses; perhaps the amino acids flanking the epitopes prevented their proper processing and/or delivery into the antigen presentation pathways. We addressed this possibility in two ways. (i) We prepared a plasmid encoding the N-terminal sequence of rCVB3.6 and showed that this plasmid DNA can induce a strong GP33-specific CD8 T-cell response in naïve mice (Fig. 8). Thus, the polypeptide sequence can be processed sufficiently well to allow stimulation of naïve GP33-specific T cells in vivo. (ii) Both GP33-specific and GP61-specific memory T cells in LCMV-immune mice expanded in number following challenge with rCVB3.6 (but not with rCVB3.3 expressing an irrelevant epitope), indicating that the CD8+ and CD4+ T-cell epitopes in rCVB3.6 are presented to T cells during rCVB3.6 infection (Fig. 7).
What might be the explanation for the poor immunogenicity of rCVB3.6 in naïve mice? Our laboratory (4, 5) and others (9, 50, 51) have recently demonstrated that several CVB3 proteins act in concert to disrupt protein trafficking within an infected cell: 2B and 2C inhibit the movement of protein through the Golgi complex, while 3A is able to disrupt this organelle (4). Furthermore, the combined actions of 3A-mediated inhibition of anterograde transport and 2B (and/or 2BC)-mediated up-regulation of endocytosis lead to rapid down-regulation of surface MHC class I (5). Recent data from another lab have confirmed the effects of CVB3 on cell surface levels of MHC (23). Therefore, we speculate that rCVB3.6 fails to induce strong T-cell responses by severely limiting presentation of viral epitope-MHC complexes and, as a result, virus-infected antigen-presenting cells (APCs) are unable to trigger naïve T-cell differentiation. However, this powerful immune evasion mechanism is not quite perfect. The consequence of this imperfection is that some rCVB3.6-encoded epitopes do reach the cell surface, albeit in small amounts. We speculate that the level of epitope/MHC is sufficient to trigger responses in memory cells but not their naïve counterparts, which require at least 80-fold-higher levels of antigen (43). Others have studied CD8+ T-cell responses induced in mice infected with the closely related virus poliovirus and have reported detectable CD8+ T-cell responses. However, these CD8+ T cells reached detectable levels only after several days of in vitro antigen restimulation (13, 32), suggesting that the true in vivo responses were weak. Elegant studies have shown that poliovirus proteins can inhibit MHC class I antigen presentation (2, 8), and this probably contributes to the weakness of the observed responses.
Our data and the above-described interpretation (if correct) have profound implications for the processes of cross-presentation and cross-priming. In most cells, epitopes that are presented by MHC class I molecules are derived from proteins synthesized within that cell; thus, a cell will present epitopes from its own proteome and also from proteins synthesized within the cell by intracellular microbes (most often viruses, but also some intracellular bacteria). However, in recent years, it has become generally accepted that some cell types—particularly dendritic cells (DCs)—can take up proteins from the extracellular milieu or from phagocytosis of cellular debris and can direct these materials into the MHC class I antigen presentation pathway. This process, termed cross-presentation, is thought to result in cross-priming of naïve epitope-specific CD8+ T cells. Although there is no doubt that cross-presentation and cross-priming can occur both in tissue culture and in vivo, the biological significance of the processes—that is, their real importance in the induction of CD8+ T-cell responses in vivo—remains highly controversial. Some experts have proposed that this mechanism underpins the majority of CD8+ T-cell responses observed in virus infections (1), while others have questioned its biological significance (55). For the reasons outlined below, we feel that our data shed light on this controversy.
rCVB3.6 reaches very high titers in vivo in several lymphoid and nonlymphoid tissues (Fig. 2), providing abundant viral protein for cross-presentation. DCs are numerous, and only a very few (∼100 to 1,000) epitope-presenting DCs are needed to trigger a very strong CD8+ T-cell response (31). Thus, if cross-presentation and cross-priming were effective, it seems likely that this numerical threshold would quickly be reached and exceeded. One might argue that the antitrafficking attributes of several CVB3 proteins would prevent effective cross-presentation by DCs, but this would be possible only if a DC that consumed one CVB3 protein (for example, the N-terminal fragment containing the GP33 and GP61 epitopes) invariably also took up the antitrafficking proteins of CVB3. Although this is possible, we feel it to be somewhat unlikely for two reasons. First, the antitrafficking functions of the CVB3 proteins may require them to be present in a specific stoichiometry and/or in specific subcellular compartments, and these criteria might not easily be achieved via exogenous uptake. Second, given the large number of DCs present in a mouse, it seems probable that some DCs will take up the epitope-containing sequences but not the antitrafficking proteins. We conclude that cross-priming is not an efficient process for generating virus-specific CD8+ T-cell responses in the context of CVB3 infection, and we speculate that this may hold true for other virus infections, as has been suggested previously (55). It is important that this issue be resolved, because it is not merely academic; some new approaches to immunization are predicated on efficient cross-presentation, and if this pathway is much less effective than current dogma contends, then these vaccines may be doomed to failure. Studies to more directly evaluate epitope presentation by MHC class I and class II during CVB3 infection are ongoing in our laboratory.
In addition to modulating the level of viral antigen presentation, CVB3 may have other means to limit the ability of DCs to prime robust virus-specific T-cell responses. For example, much recent research has shown that the innate response that is induced by virus infection might shape the quality and quantity of the adaptive responses that subsequently develop. The innate response to CVB infections is poorly understood. Previous studies have shown that poliovirus infection inhibits proinflammatory cytokine secretion and the expression of antiviral cytokine receptors (10, 37), while echovirus infection limits the ability of DCs to up-regulate costimulatory molecules or produce cytokines in response to Toll-like receptor stimulation (28). Because some members of the picornavirus family encode proteins with conserved functions, it is worth considering that one or more CVB3 proteins may be able to alter the expression of costimulatory molecules and/or the cytokine milieu, which could interfere with DC maturation or function. In addition, plasmacytoid DC activation and type I interferon production in response to CVB3 require virus-specific antibodies (48), and therefore a delay in plasmacytoid DC activation (until the emergence of CVB3-specific antibodies at ∼day 4 p.i. [17]) could contribute to suboptimal maturation of APCs. Whether CVB3 infection affects the activation or maturation of DCs and other professional APCs in vivo is an interesting topic awaiting future study. Other factors, too, may contribute to the weak T-cell responses reported herein. We cannot exclude the possibility that regulatory T cells may play a role, perhaps suppressing the development of strong effector T-cell responses. Prior studies have identified regulatory T-cell activity during CVB3 infection (20, 30).
In summary, our investigation of the CVB3-specific immune response in both lymphoid and peripheral sites of infection reveals that virus infection induces minimal primary CD4+ and CD8+ T-cell responses. A better understanding of how CVB3 is able to restrain the adaptive immune response may open new avenues for manipulation of adaptive immunity. In particular, the induction of a more robust antiviral T-cell response could limit viral replication, lower viral persistence, and reduce chronic inflammation. It is hoped that such a strategy could be useful for the design of a CVB vaccine or for methodologies to prevent or treat chronic myocarditis.
Acknowledgments
We thank Annette Lord for excellent secretarial support.
This work was supported by National Institutes of Health grants R01 AI-42314 (J.L.W.) and T32 NS41219-06 (C.C.K.).
This is manuscript number 19255 from The Scripps Research Institute.
Footnotes
Published ahead of print on 27 February 2008.
REFERENCES
- 1.Carbone, F. R., C. Kurts, S. R. Bennett, J. F. Miller, and W. R. Heath. 1998. Cross-presentation: a general mechanism for CTL immunity and tolerance. Immunol. Today 19368-373. [DOI] [PubMed] [Google Scholar]
- 2.Choe, S. S., D. A. Dodd, and K. Kirkegaard. 2005. Inhibition of cellular protein secretion by picornaviral 3A proteins. Virology 33718-29. [DOI] [PubMed] [Google Scholar]
- 3.Ciurea, A., L. Hunziker, M. M. Martinic, A. Oxenius, H. Hengartner, and R. M. Zinkernagel. 2001. CD4+ T-cell-epitope escape mutant virus selected in vivo. Nat. Med. 7795-800. [DOI] [PubMed] [Google Scholar]
- 4.Cornell, C. T., W. B. Kiosses, S. Harkins, and J. L. Whitton. 2006. Inhibition of protein trafficking by coxsackievirus B3: multiple viral proteins target a single organelle. J. Virol. 806637-6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornell, C. T., W. B. Kiosses, S. Harkins, and J. L. Whitton. 2007. Coxsackievirus B3 proteins directionally complement each other to downregulate surface MHC class I. J. Virol. 816785-6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crocker, S. J., R. F. Frausto, J. K. Whitmire, N. Benning, and J. L. Whitton. 2007. Amelioration of coxsackievirus B3-mediated myocarditis by inhibition of TIMP-1. Am. J. Pathol. 1711762-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daley, A. J., D. Isaacs, D. E. Dwyer, and G. L. Gilbert. 1998. A cluster of cases of neonatal coxsackievirus B meningitis and myocarditis. J. Paediatr. Child Health 34196-198. [DOI] [PubMed] [Google Scholar]
- 8.Deitz, S. B., D. A. Dodd, S. Cooper, P. Parham, and K. Kirkegaard. 2000. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 9713790-13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Jong, A. S., H. J. Visch, M. F. de Mattia, M. M. van Dommelen, H. G. Swarts, T. Luyten, G. Callewaert, W. J. Melchers, P. H. Willems, and F. J. van Kuppeveld. 2006. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and Golgi, thereby inhibiting protein trafficking through the Golgi. J. Biol. Chem. 28114144-14150. [DOI] [PubMed] [Google Scholar]
- 10.Dodd, D. A., T. H. Giddings, Jr., and K. Kirkegaard. 2001. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 758158-8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feuer, R., I. Mena, R. R. Pagarigan, M. K. Slifka, and J. L. Whitton. 2002. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J. Virol. 764430-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feuer, R., R. R. Pagarigan, S. Harkins, F. Liu, I. P. Hunziker, and J. L. Whitton. 2005. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J. Neurosci. 252434-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freigang, S., D. Egger, K. Bienz, H. Hengartner, and R. M. Zinkernagel. 2003. Endogenous neosynthesis vs. cross-presentation of viral antigens for cytotoxic T cell priming. Proc. Natl. Acad. Sci. USA 10013477-13482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gebhard, J. R., C. M. Perry, S. Harkins, T. Lane, I. Mena, V. C. Asensio, I. L. Campbell, and J. L. Whitton. 1998. Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am. J. Pathol. 153417-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geller, T. J., and D. Condie. 1995. A case of protracted coxsackie virus meningoencephalitis in a marginally immunodeficient child treated successfully with intravenous immunoglobulin. J. Neurol. Sci. 129131-133. [DOI] [PubMed] [Google Scholar]
- 16.Guthrie, M., P. A. Lodge, and S. A. Huber. 1984. Cardiac injury in myocarditis induced by coxsackievirus group B, type 3 in Balb/c mice is mediated by Lyt 2+ cytolytic lymphocytes. Cell. Immunol. 88558-567. [DOI] [PubMed] [Google Scholar]
- 17.Henke, A., S. A. Huber, A. Stelzner, and J. L. Whitton. 1995. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J. Virol. 696720-6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hertel, N. T., F. K. Pedersen, and C. Heilmann. 1989. Coxsackie B3 virus encephalitis in a patient with agammaglobulinaemia. Eur. J. Pediatr. 148642-643. [DOI] [PubMed] [Google Scholar]
- 19.Huber, S., J. Polgar, A. Moraska, M. W. Cunningham, P. Schwimmbeck, and P. Schultheiss. 1993. T lymphocyte responses in CVB3-induced murine myocarditis. Scand. J. Infect. Dis. Suppl. 8867-78. [PubMed] [Google Scholar]
- 20.Huber, S. A., A. M. Feldman, and D. Sartini. 2006. Coxsackievirus B3 induces T regulatory cells, which inhibit cardiomyopathy in tumor necrosis factor-alpha transgenic mice. Circ. Res. 991109-1116. [DOI] [PubMed] [Google Scholar]
- 21.Huber, S. A., and L. P. Job. 1983. Cellular immune mechanisms in coxsackievirus group B, type 3 induced myocarditis in Balb/C mice. Adv. Exp. Med. Biol. 161491-508. [DOI] [PubMed] [Google Scholar]
- 22.Huber, S. A., L. P. Job, and J. F. Woodruff. 1980. Lysis of infected myofibers by coxsackievirus B-3-immune T lymphocytes. Am. J. Pathol. 98681-694. [PMC free article] [PubMed] [Google Scholar]
- 23.Huhn, M. H., M. Hultcrantz, K. Lind, H. G. Ljunggren, K. J. Malmberg, and M. Flodstrom-Tullberg. 2008. IFN-γ production dominates the early human natural killer cell response to coxsackievirus infection. Cell. Microbiol. 10426-436. [DOI] [PubMed] [Google Scholar]
- 24.Hunziker, I. P., C. T. Cornell, and J. L. Whitton. 2007. Deletions within the 5′UTR of coxsackievirus B3: consequences for virus translation and replication. Virology 360120-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunziker, I. P., S. Harkins, R. Feuer, C. T. Cornell, and J. L. Whitton. 2004. Generation and analysis of an RNA vaccine that protects against coxsackievirus B3 challenge. Virology 330196-208. [DOI] [PubMed] [Google Scholar]
- 26.Kaech, S. M., and R. Ahmed. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naïve cells. Nat. Immunol. 2415-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knowlton, K. U., E. S. Jeon, N. Berkley, R. Wessely, and S. A. Huber. 1996. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J. Virol. 707811-7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramer, M., B. M. Schulte, L. W. Toonen, M. A. de Bruijni, J. M. Galama, G. J. Adema, and F. J. van Kuppeveld. 2007. Echovirus infection causes rapid loss-of-function and cell death in human dendritic cells. Cell. Microbiol. 91507-1518. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S. G., D. Y. Kim, B. H. Hyun, and Y. S. Bae. 2002. Novel design architecture for genetic stability of recombinant poliovirus: the manipulation of G/C contents and their distribution patterns increases the genetic stability of inserts in a poliovirus-based RPS-Vax vector system. J. Virol. 761649-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loudon, R. P., A. F. Moraska, S. A. Huber, P. L. Schwimmbeck, and P. Schultheiss. 1991. An attenuated variant of coxsackievirus B3 preferentially induces immunoregulatory T cells in vivo. J. Virol. 655813-5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ludewig, B., S. Ehl, U. Karrer, B. Odermatt, H. Hengartner, and R. M. Zinkernagel. 1998. Dendritic cells efficiently induce protective antiviral immunity. J. Virol. 723812-3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandl, S., L. J. Sigal, K. L. Rock, and R. Andino. 1998. Poliovirus vaccine vectors elicit antigen-specific cytotoxic T cells and protect mice against lethal challenge with malignant melanoma cells expressing a model antigen. Proc. Natl. Acad. Sci. USA 958216-8221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mena, I., C. Fischer, J. R. Gebhard, C. M. Perry, S. Harkins, and J. L. Whitton. 2000. Coxsackievirus infection of the pancreas: evaluation of receptor expression, pathogenesis, and immunopathology. Virology 271276-288. [DOI] [PubMed] [Google Scholar]
- 34.Mena, I., C. M. Perry, S. Harkins, F. Rodriguez, J. R. Gebhard, and J. L. Whitton. 1999. The role of B lymphocytes in coxsackievirus B3 infection. Am. J. Pathol. 1551205-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modlin, J. F., and H. A. Rotbart. 1997. Group B coxsackie disease in children. Curr. Top. Microbiol. Immunol. 22353-80. [DOI] [PubMed] [Google Scholar]
- 36.Mueller, S., and E. Wimmer. 1998. Expression of foreign proteins by poliovirus polyprotein fusion: analysis of genetic stability reveals rapid deletions and formation of cardioviruslike open reading frames. J. Virol. 7220-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neznanov, N., A. Kondratova, K. M. Chumakov, B. Angres, B. Zhumabayeva, V. I. Agol, and A. V. Gudkov. 2001. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 7510409-10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Connell, J. B. 1987. The role of myocarditis in end-stage dilated cardiomyopathy. Tex. Heart Inst. J. 14268-275. [PMC free article] [PubMed] [Google Scholar]
- 39.Pircher, H., D. Moskophidis, U. Rohrer, K. Burki, H. Hengartner, and R. M. Zinkernagel. 1990. Viral escape by selection of cytotoxic T cell-resistant virus variants in-vivo. Nature 346629-633. [DOI] [PubMed] [Google Scholar]
- 40.Ramsingh, A. I. 1997. Coxsackieviruses and pancreatitis. Front. Biosci. 2e53-e62. [DOI] [PubMed] [Google Scholar]
- 41.Ramsingh, A. I., W. T. Lee, D. N. Collins, and L. E. Armstrong. 1997. Differential recruitment of B and T cells in coxsackievirus B4-induced pancreatitis is influenced by a capsid protein. J. Virol. 718690-8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slifka, M. K., R. R. Pagarigan, I. Mena, R. Feuer, and J. L. Whitton. 2001. Using recombinant coxsackievirus B3 to evaluate the induction and protective efficacy of CD8+ T cells in controlling picornaviral infection. J. Virol. 752377-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slifka, M. K., and J. L. Whitton. 2001. Functional avidity maturation of CD8+ T cells without selection of higher affinity TCR. Nat. Immunol. 2711-717. [DOI] [PubMed] [Google Scholar]
- 44.Sole, M. J., and P. Liu. 1993. Viral myocarditis: a paradigm for understanding the pathogenesis and treatment of dilated cardiomyopathy. J. Am. Coll. Cardiol. 2299A-105A. [DOI] [PubMed] [Google Scholar]
- 45.Tam, P. E. 2006. Coxsackievirus myocarditis: interplay between virus and host in the pathogenesis of heart disease. Viral Immunol. 19133-146. [DOI] [PubMed] [Google Scholar]
- 46.Van Houten, N., P. E. Bouchard, A. Moraska, and S. A. Huber. 1991. Selection of an attenuated coxsackievirus B3 variant, using a monoclonal antibody reactive to myocyte antigen. J. Virol. 651286-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Stipdonk, M. J., E. E. Lemmens, and S. P. Schoenberger. 2001. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2423-429. [DOI] [PubMed] [Google Scholar]
- 48.Wang, J. P., D. R. Asher, M. Chan, E. A. Kurt-Jones, and R. W. Finberg. 2007. Cutting edge: antibody-mediated TLR7-dependent recognition of viral RNA. J. Immunol. 1783363-3367. [DOI] [PubMed] [Google Scholar]
- 49.Weidt, G., W. Deppert, O. Utermohlen, J. Heukeshoven, and F. Lehmann-Grube. 1995. Emergence of virus escape mutants after immunization with epitope vaccine. J. Virol. 697147-7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wessels, E., D. Duijsings, R. A. Notebaart, W. J. Melchers, and F. J. van Kuppeveld. 2005. A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-Golgi transport. J. Virol. 795163-5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wessels, E., R. A. Notebaart, D. Duijsings, K. Lanke, B. Vergeer, W. J. Melchers, and F. J. van Kuppeveld. 2006. Structure-function analysis of the coxsackievirus protein 3A: identification of residues important for dimerization, viral RNA replication, and transport inhibition. J. Biol. Chem. 28128232-28243. [DOI] [PubMed] [Google Scholar]
- 52.Whitton, J. L. 2002. Immunopathology during coxsackievirus infection. Springer Semin. Immunopathol. 24201-213. [DOI] [PubMed] [Google Scholar]
- 53.Whitton, J. L., C. T. Cornell, and R. Feuer. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3765-776. [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama, M., J. Zhang, and J. L. Whitton. 1995. DNA immunization confers protection against lethal lymphocytic choriomeningitis virus infection. J. Virol. 692684-2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zinkernagel, R. M. 2002. On cross-priming of MHC class I-specific CTL: rule or exception? Eur. J. Immunol. 322385-2392. [DOI] [PubMed] [Google Scholar]