

Abstract
During virus assembly, the capsid proteins of RNA viruses bind to genomic RNA to form nucleocapsids. However, it is now evident that capsid proteins have additional functions that are unrelated to nucleocapsid formation. Specifically, their interactions with cellular proteins may influence signaling pathways or other events that affect virus replication. Here we report that the rubella virus (RV) capsid protein binds to poly(A)-binding protein (PABP), a host cell protein that enhances translational efficiency by circularizing mRNAs. Infection of cells with RV resulted in marked increases in the levels of PABP, much of which colocalized with capsid in the cytoplasm. Mapping studies revealed that capsid binds to the C-terminal half of PABP, which interestingly is the region that interacts with other translation regulators, including PABP-interacting protein 1 (Paip1) and Paip2. The addition of capsid to in vitro translation reaction mixtures inhibited protein synthesis in a dose-dependent manner; however, the capsid block was alleviated by excess PABP, indicating that inhibition of translation occurs through a stoichiometric mechanism. To our knowledge, this is the first report of a viral protein that inhibits protein translation by sequestration of PABP. We hypothesize that capsid-dependent inhibition of translation may facilitate the switch from viral translation to packaging RNA into nucleocapsids.
The rubella virus (RV) capsid is an RNA-binding phosphoprotein (40). During virus assembly, the capsid engages in homotypic and heterotypic binding interactions to package the viral genome into a compact nucleocapsid structure (reviewed in reference 18). Assembly and disassembly of the nucleocapsid appear to be regulated by dynamic phosphorylation of serine/threonine residues in the RNA-binding motif of the capsid (34, 35). Nucleocapsid assembly occurs on membranes of the Golgi complex, and association of the capsid with this organelle presumably reflects its role in virus budding (5, 20). Similar to alphavirus budding (53), interactions between the capsid and viral glycoproteins E2 and E1 are thought to drive virus assembly. As well as being targeting to the virus budding site, the RV capsid associates with other intracellular membranes, including endocytic vacuoles (13) and mitochondria (7, 37). These organelles have no obvious link to virus assembly, and the presence of the capsid at these sites is indicative of its nonstructural roles.
Recent studies revealed the unexpected finding that capsid modulates the synthesis of viral RNAs (8, 55-57). It is not clear how the capsid protein affects viral transcription, but the fact that it binds to the nonstructural protein p150 (57) may indicate that it regulates the activity of the replicase complex. Interactions between capsid and host proteins may also influence viral transcription. For example, capsid binds to the mitochondrial matrix protein p32 (7, 44), and indirect evidence indicates that this interaction is important for virus replication (6). Specifically, ablation of the p32-binding site within the capsid results in decreased levels of subgenomic RNA and structural proteins in infected cells.
In addition to p32, the RV capsid protein interacts with a number of other host cell-encoded proteins. Affinity purification revealed that capsid binds to the poly(A)-binding protein (PABP), a translation initiation factor that forms a complex with other initiation factors, including eIF4E and eIF4G (23, 25, 36, 54). The PABP-containing complex promotes protein synthesis by circularization of cellular mRNAs. Through sequestration of PABP, it is possible that capsid inhibits translation, which in addition to inhibiting cellular defense systems, may serve to increase the efficiency of nucleocapsid formation by blocking translation of the viral genome.
MATERIALS AND METHODS
Reagents and cDNA clones.
Protein A- and protein G-Sepharose were purchased from Pharmacia Biotech (Alameda, CA). Bovine serum albumin and general lab chemicals were purchased from Sigma Chemical Co. (St. Louis, MO). TNT quick coupled transcription/translation systems were purchased from Promega (Madison, WI). 14C-labeled protein standards and Redivue l-[35S]methionine aqueous solution were purchased from GE Healthcare (Princeton, NJ). Media and sera for cell culture were purchased from Life Technologies-Invitrogen, Inc. (Carlsbad, CA). PerFectin transfection reagent was purchased from Gene Therapy Systems, Inc. (San Diego, CA). Vero, A549, COS-1, and HEK293T cells were obtained from the American Type Culture Collection (Manassas, VA.). The M33 strain of RV and pBRM33, the infectious RV cDNA clone (60), were kindly provided by S. Gillam (University of British Columbia). The infectious Sindbis virus plasmid clone (pBR TOTO 1101) was a gift from C. Rice (Rockefeller University).
Mammalian cell culture.
HEK293T and A549 cells were cultured in Dulbecco's minimal essential medium (high glucose) containing 10% fetal bovine serum, 2 mM glutamine, 1 mM HEPES, and antibiotics. COS-1 and Vero cells were cultured in Dulbecco's minimal essential medium (high glucose) containing 5% fetal bovine serum, 2 mM glutamine, 1 mM HEPES, and antibiotics. Cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Plasmid construction.
A cDNA encoding histidine-tagged capsid lacking the E2 signal peptide was constructed by PCR using a forward primer with an EcoRI site (shown underlined) (5′-CGCGAATTCAGGAGGACAGCTATGGCTTCCACTACCC-3′) and a reverse primer with a HindIII site (shown underlined) (5′-CCCAAGCTTGCGGATGCGCCAAGGATG-3′). Pfx polymerase (Invitrogen, Carlsbad, CA) was used to amplify product from the template pCMV5-Capsid (19). Prior to digestion with restriction endonucleases, the PCR product was separated from proteins and deoxynucleoside triphosphates using QIAquick PCR purification kit (Qiagen, Mississauga, Ontario, Canada). The digestion product was separated by agarose electrophoresis, purified with a QIAEX II gel extraction kit (Qiagen, Mississauga, Ontario, Canada) and then ligated into the bacterial expression vector pET-23 (+) (EMD Biosciences, San Diego, CA) such that the capsid protein was in frame with a carboxyl-terminal six-histidine tag. The resulting plasmid was named pET-23:CapsidΔSP-His.
Expression plasmids encoding the N- and C-terminal regions of human PABP were constructed as follows. The coding region for the N-terminal region of PABP (amino acid residues 1 to 368) was amplified by PCR using a forward primer containing a BamHI site (shown underlined) (5′-TATGGATCCATGAACCCCAGTGCCCC-3′) and a reverse primer containing a NotI site (shown underlined) (5′-TATGCGGCCGCTCATTTGCGCTGAGCTAAAGCTAC-3′). The coding region for the C-terminal region of PABP (amino acid residues 369 to 636) was amplified by PCR using a forward primer containing a BamHI site (shown underlined) (5′-TATGCGGCCGCTCATTTGCGCTGAGCTAAAGCTAC-3′) and a reverse primer containing a NotI site (shown underlined) (5′-TATGCGGCCGCTTTAAACAGTTGGAACACCGGTG-3′). In both cases, the template DNA was pCDNA 3.1-hPABP (from N. Sonenberg, McGill University). The resulting products were digested with BamHI and NotI and then ligated in frame with the 3′ end of the glutathione S-transferase (GST) cassette in the bacterial expression vector pGEX-6P1 (Amersham Biosciences). The resulting constructs were named pGEX-6P1-PABP NT and pGEX-6P1-PABP CT. The authenticities of all plasmid constructs were verified by DNA sequencing at the Molecular Biology Facility (Department of Biological Sciences, University of Alberta).
Identification of capsid-interacting proteins by GST pulldown.
COS or HEK293T cells (9 × 105/100-mm dish) were transiently transfected with 6 μg of either pEBG or pEBG-Capsid (35). Forty-eight hours posttransfection, cells were lysed in 1% Nonidet P-40 (NP-40) lysis buffer (1% NP-40, 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 2 mM EDTA) containing Complete protease inhibitors (EDTA free) (Roche Applied Sciences, Indianapolis) on ice for 5 min. Lysates were cleared by centrifugation (10,000 × g), and the supernatants were incubated with glutathione Sepharose beads overnight at 4°C. Beads containing protein complexes were washed three times with lysis buffer after which the complexes were eluted by boiling for 5 min in 2× gel sample buffer and then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Capsid-associated proteins were visualized by silver staining.
Briefly, gels were fixed in 50% (vol/vol) methanol and 10% (vol/vol) acetic acid for 30 min at room temperature, sensitized in 0.02% (vol/vol) sodium thiosulfate for 1 min at room temperature, and then treated with 0.1% (wt/vol) silver nitrate for 25 min at 4°C. Gels were developed in 2% (wt/vol) anhydrous sodium carbonate and 0.02% (vol/vol) formaldehyde for 5 to 10 min at room temperature. Development was stopped by immersing gels in 1.4% (wt/vol) EDTA.
Proteins that copurified with GST-tagged capsid (GST-capsid) were subjected to mass spectrometry at the Institute for Bimolecular Design (University of Alberta). The data were analyzed using the Mascot search engine (Matrix Science).
Infection of cells with rubella virus, Sindbis virus, and vesicular stomatitis virus.
Rubella virus stocks were diluted with cell culture medium and then added to cells that had been washed with phosphate-buffered saline (PBS). Cells were incubated with the virus inoculum (1 ml/35-mm dish) for 4 h at 35°C after which time the inoculum was replaced with normal growth medium. Infected cultures were kept at 35°C until experimental analyses.
For experiments involving infection with vesicular stomatitis virus (Indiana strain) or Sindbis virus, Vero cells in 35-mm dishes were grown to 80% confluence and then infected with at a multiplicity of infection (MOI) of 1 for 1 hour at 37°C after which time the inoculum was replaced with normal growth medium. Sindbis virus stocks were generated by electroporating BHK-21 cells with the infectious plasmid clone pBR TOTO 1101 (48).
Infected cells were lysed in 1% NP-40 lysis buffer containing Complete protease inhibitors at the indicated time points. Lysates were clarified by centrifugation at 10,000 × g for 10 min at 4°C, and protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL). Equivalent amounts of proteins from each lysate were resolved on 10% SDS-polyacrylamide gels and blotted onto polyvinylidene difluoride membranes.
Expression and purification of recombinant proteins.
To purify the RV capsid from bacteria, Escherichia coli BL21(DE3) (Invitrogen) cells were transformed with pET-23:CapsidΔSP-His, and Luria broth cultures (500 ml) containing ampicillin (100 μg/ml) were grown at 37°C to an A590 of 0.6. Expression of the capsid protein was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 16 h at 23°C. Bacterial pellets were resuspended in 300 mM NaCl, 0.1 mM CaCl2, 10 mM MgCl2, 1% Triton X-100, and 50 mM NaH2PO4 (pH 8.0) containing Complete protease inhibitors (EDTA free), frozen at −80°C, and then thawed in ice-water. Thawed cell suspensions were incubated with lysozyme (1 mg/ml), DNase I (0.1 mg/ml), and RNase A (1 mg/ml) for 30 min on ice before sonication with a Branson sonifier 450 (six pulses of 10 seconds each at maximum output). Sonicated samples were then incubated for 15 min at 37°C and then put on ice for 2 min before centrifugation at 10,000 × g for 30 min at 4°C.
The soluble fraction containing CapsidΔSP-His was subjected to chromatography on Ni Sepharose 6 Fast Flow resin (GE Healthcare, Princeton, NJ). Columns were washed with a buffer containing 20 mM NaH2PO4 (pH 7.4), 500 mM NaCl, 10 mM imidazole, and protease inhibitors. Capsid was eluted with 20 mM NaH2PO4 (pH 7.4), 500 mM NaCl, and 500 mM imidazole. Fractions were analyzed by immunoblotting to determine the presence of CapsidΔSP-His. Positive fractions were then pooled and further purified by fast-performance liquid chromatography using a HiTrap SP FF cation-exchange chromatography column (GE Healthcare, Princeton, NJ). Columns were washed extensively with 50 mM bicine (pH 8.5), and then capsid was eluted with 50 mM bicine (pH 8.5) buffer containing 1 M NaCl. Fractions containing capsid were pooled and then concentrated using Amicon centrifugation cartridges (10-kDa cutoff) (Millipore, Bedford, MA) before dialysis against buffer containing 20 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 7.4) and 10 mM KCl.
For purification of GST-PABP fusion proteins, E. coli BL21(DE3) cells were transformed with pGEX-6P1-PABP NT and pGEX-6P1-PABP CT constructs. Expression of the proteins was induced by the addition of IPTG (1 mM) at 37°C for 3 h. The cells were harvested and lysed with 100 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 1 mM EDTA containing Complete protease EDTA-free inhibitors. The lysates were clarified by centrifugation at 10,000 × g for 10 min. GST fusion proteins in the resulting supernatants were bound to glutathione Sepharose 4B resin (GE Healthcare, Princeton, NJ) with rotation for 1 h at 4°C.
Biologically active PABP was purified as described previously (27) with slight modifications. The bacterial strain E. coli BL21(DE3) carrying pLys was transformed with pETb-PABP-His, and 2-liter cultures (A590 of 0.3 to 0.8) were induced with 0.1 mM IPTG at 20 to 30°C for 12 to 16 h. Bacterial pellets were washed in buffer containing 20 mM Tris-HCl (pH 7.5) and 0.15 mM NaCl and then frozen at −20°C. Frozen pellets were resuspended in 50 ml of buffer containing 2 M KCl, 20 mM HEPES (pH 7.5), 10% glycerol, 0.1% Triton X-100, 5 mM 2-mercaptoethanol, and EDTA-free protease inhibitors. The bacterial cells were disrupted by sonication and then incubated on ice for 30 min. Lysates were clarified by centrifugation at 30,000 rpm in a Ti-70 rotor (Beckman) after which the supernatants were supplemented with imidazole (20 mM) before mixing with 2 ml of Ni-nitrilotriacetic acid agarose (Qiagen). The resin was washed with 20 ml of buffer containing 2 M KCl, 20 mM HEPES (pH 7.5), 10% glycerol, 0.1% Triton X-100, and 5 mM 2-mercaptoethanol. Bound proteins were eluted with 8 to 10 ml of buffer containing 0.25 M imidazole, 0.1 M KCl, 20 mM HEPES (pH 7.5), 10% glycerol, 0.1% Triton X-100, and 5 mM 2-mercaptoethanol. Samples were dialyzed against buffer containing 20 mM PIPES (pH 7.4) and 10 mM KCl and then concentrated using Centricon cartridges with a 30-kDa-molecular-size cutoff (Millipore).
In vitro binding assays.
Five micrograms of glutathione Sepharose-immobilized GST-PABP NT or GST-PABP CT fragments were incubated in PBS containing 1% casein enzymatic hydrolysate, 0.1% NP-40, and protease inhibitors with rotation for 1 h at 4°C. Next, 10 μg of purified CapsidΔSP-His was added, and the mixture was incubated with rotation at 4°C overnight. The resin was washed three times with 500 μl of PBS containing 0.1% NP-40. Proteins were eluted from the beads by boiling in sample buffer, resolved by SDS-PAGE, and detected by Coomassie blue staining or immunoblotting.
Immunoprecipitation.
COS cells (1.5 × 105) in 35-mm culture dishes were transfected with 2 μg of each plasmid combined with 7 μl of PerFectin transfection reagent as described by the manufacturer. HEK293T cells (3 × 105) in 60-mm-diameter culture dishes were transfected with 6 μg of each plasmid combined with 21 μl of PerFectin transfection reagent according to the manufacturer's protocol. Cells were incubated in culture medium for 48 h prior to lysis in 1% NP-40 lysis buffer supplemented with 2 mM dithiothreitol and protease inhibitors. Cell lysates were clarified by centrifugation at 10,000 × g at 4°C for 10 min. Immunoprecipitation was performed with clarified lysates, and 0.5 μg of His-probe (H-15) rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C with rotation. Twenty-five microliters of 50% suspension protein A-Sepharose was added and incubated for 1 h at 4°C.
Interactions between PABP and capsid in RV-infected cells were detected as follows. Vero cells were infected with the M33 strain of RV, and 40 h postinfection, cells were lysed in NP-40 lysis buffer as described above. Lysates were subjected to immunoprecipitations with mouse monoclonal antibodies to PABP (Sigma Chemical Co., St. Louis, MO) or capsid (H15C22; Abbott Laboratories, Chicago, IL). Samples were analyzed by immunoblotting with rabbit antibodies to PABP and capsid as described below.
Where indicated, micrococcal nuclease treatments were performed after immunoprecipitation. The Sepharose beads were incubated for 1 h at 4°C in 1% NP-40 lysis buffer containing 5 mM CaCl2 and 30 units/ml of micrococcal nuclease (obtained from M. Schultz, University of Alberta). After centrifugation, the beads were washed three times with lysis buffer containing 2 mM dithiothreitol and protease inhibitors. Proteins were eluted by heating at 95°C for 5 min in sample buffer and subjected to SDS-PAGE.
Immunoblotting.
Polyvinylidene difluoride membranes (Immobilon-P; Millipore, Bedford, MA) were incubated for 1 h at room temperature with one of the following antibodies and dilutions: rabbit anti-RV capsid (7W7 (7), 1:2,000; rabbit anti-Sindbis virus capsid (from C. Rice, Rockefeller University), 1:1,000; rabbit anti-PABP C terminus (from N. Sonenberg, McGill University), 1:2,000; rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (anti-GAPDH) (Abcam, Cambridge, MA), 1:2,000; rabbit anti-GST (Abcam, Cambridge, MA), 1:2,000; mouse monoclonal anti-penta-His (Qiagen), 1:1,000; goat anti-eIF4G and anti-eIF4E (Santa Cruz Biotechnology, Santa Cruz CA) and mouse anti-vesicular stomatitis virus protein G (P5D4 [32]), 1:2,000.
After three washes with Tris-buffered saline containing Tween, the membranes were incubated with either goat anti-mouse or goat anti-rabbit immunoglobulin G conjugated to horseradish peroxidase (Bio-Rad Hercules, CA) for 1 h. For detection of goat primary antibodies, rabbit anti-immunoglobulin G conjugated to horseradish peroxidase (Abcam) was used. Membranes were washed with Tris-buffered saline containing Tween four times, and immunoreactive proteins were detected using Supersignal West Pico chemiluminescent substrate (Pierce Biotechnology, Rockford, IL) and exposure to X-ray film (Fuji Photo Film Co., Ltd., Tokyo, Japan).
Indirect immunofluorescence microscopy.
Vero cells cultured on glass coverslips were either infected with RV (MOI of 1) or transiently transfected with pCMV5-Capsid or pCMV5-E2E1 (19). Cells were processed for indirect immunofluorescence 48 h after infection or 24 h after transfection by fixing in 4% paraformaldehyde for 30 min, followed by quenching with PBS containing 50 mM ammonium chloride. Cell membranes were permeabilized by incubating with PBS containing 0.2% Triton X-100 for 5 min before incubation with primary and secondary antibodies. All the washes were done in PBS supplemented with 0.1 mM CaCl2 and 1 mM MgCl2.
RV proteins were detected with rabbit anti-capsid or hyperimmune human anti-RV serum (19). PABP was detected with clone 10E10 mouse monoclonal antibody (Sigma Chemical Co., St. Louis, MO). The secondary antibodies utilized in these studies were Alexa Fluor 594-conjugated chicken anti-mouse, Alexa Fluor 488-conjugated donkey anti-rabbit, Alexa Fluor 488-conjugated donkey anti-mouse, Alexa Fluor 647-conjugated donkey anti-mouse (Molecular Probes, Invitrogen, Carlsbad, CA), and tetramethyl rhodamine isothiocyanate (TRIC)-conjugated goat anti-human antibody (Cappel Laboratories, Cochranville, PA). Coverslips were mounted onto microscope slides using ProLong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI) (Molecular Probes, Invitrogen), and samples were examined using a Zeiss 510 confocal microscope.
In vitro transcription-translation assays.
Radiolabeled luciferase and green fluorescent protein (GFP) were synthesized using the TNT quick coupled transcription/translation system. The reactions were carried out per the manufacturer's instructions, with the following modifications: instead of using nuclease-free water in the reaction mixtures, buffer containing 20 mM PIPES (pH 7.4) and 10 mM KCl was included in all the reaction mixtures, and when indicated CapsidΔSP-His, PABP-HisHis, and/or Sec17 in 20 mM PIPES (pH 7.4) and 10 mM KCl were added to the reaction mixtures at the indicated concentrations.
Samples were resolved by 8% SDS-PAGE, and the gels were fixed in 50% (vol/vol) methanol and 10% (vol/vol) acetic acid solution for 45 min at room temperature. Next, the gels were treated with 1 M sodium salicylate and 0.01% (vol/vol) β-mercaptoethanol for 45 min to intensify the 35S signal before drying onto filter paper. Radiolabeled proteins were detected by using X-ray film (Kodak Eastman Co., Rochester, NY) or a phosphorimager.
Where indicated, capped luciferase RNA was first synthesized with T7 RNA polymerase to synthesize using mMessage mMachine from Ambion (Austin, TX), according to the manufacturer's protocol. The capped RNA was then used to program the rabbit reticulocyte lysates.
Northern blot analyses.
Total RNA was extracted from mock- and RV-infected cells, and Northern blot analyses were performed as described previously (6). 32P-labeled probes specific for RV capsid and PABP were used for hybridization.
RESULTS
Affinity purification of capsid-binding proteins.
To identify novel capsid-binding proteins, we used an affinity purification protocol that employed expression of GST-tagged capsid in mammalian cells. The capsid cDNA was subcloned in frame and downstream from the GST cassette in the mammalian expression vector pEBG (43). Transcription of the GST fusion proteins is driven by the powerful EF-1α promoter. GST-capsid was expressed in transiently transfected COS cells and then isolated together with associated proteins on glutathione-agarose beads. Proteins bound to the GST-capsid fusion were visualized after SDS-PAGE and silver staining. As expected, GST-capsid bound to a number of cellular proteins that did not bind to GST alone (Fig. 1A). The most prominent capsid-binding proteins were identified by mass spectrometry. The most abundant protein that copurified with GST-capsid was the previously identified capsid-binding protein p32 (7, 44). The second major capsid-binding protein was PABP. This protein functions in translational initiation by circularizing mRNAs through heterotypic interactions with the 3′ ends of mRNA and other translation initiation factors (15).
FIG. 1.

(A) COS cells were transiently transfected with plasmids encoding GST or GST-capsid. Forty-eight hours posttransfection, lysates were prepared and mixed with glutathione Sepharose beads. Beads were washed, and bound proteins were eluted and subjected to SDS-PAGE and silver staining. Proteins that copurified with GST-capsid but not GST were identified by mass spectrometry. The positions of the poly(A)-binding protein (PABP) and the previously identified capsid-binding protein p32 are indicated. Asterisks denote partially degraded GST-capsid products. The locations of molecular mass markers (in kilodaltons) are indicated to the left of the gel. (B) GST-capsid or GST alone was expressed in HEK293T cells and then immobilized on glutathione Sepharose beads as described above. Eluates were subjected to SDS-PAGE and immunoblotting with antibodies to PABP. (C) Capsid together with histidine-tagged PABP or vector alone was expressed in HEK293T cells. Lysates were subjected to immunoprecipitation with rabbit antihistidine antibodies, followed by SDS-PAGE and immunoblotting. Where indicated, samples were treated with micrococcal nuclease (+) to destroy RNA and DNA. The membranes were probed with rabbit anticapsid or mouse antihistidine antibody, followed by enhanced chemiluminescence detection. (D) Vero cells were infected with RV, and lysates were prepared at 48 h postinfection. Lysates were immunoprecipitated (IP) with monoclonal antibodies to capsid or PABP and then subjected to immunoblot (IB) analyses with rabbit antibodies to capsid and PABP.
We confirmed that capsid-PABP interactions were not limited to COS cells by demonstrating that endogenous PABP from HEK293T cells copurified with GST-capsid but not GST alone (Fig. 1B). Reciprocal immunoprecipitation and immunoblotting were used to verify the authenticity of the interaction. Specifically, immunoprecipitation of PABP followed by immunoblotting with anticapsid antibodies showed that capsid copurified with PABP (Fig. 1C). Because capsid and PABP are both RNA-binding proteins, we were concerned that the capsid-PABP interaction may be nonspecific and simply the result of binding to an RNA intermediate. Accordingly, we next determined whether the capsid-PABP interaction was sensitive to nuclease treatment. Treatment of samples with micrococcal nuclease (Fig. 1C) or RNase A (data not shown) did not significantly alter the amount of capsid that coimmunoprecipitated with PABP. These results indicate that intact RNA is not required for the interaction between capsid and PABP. Finally, reciprocal coimmunoprecipitation and immunoblot analyses were used to demonstrate that capsid forms stable complexes with PABP in RV-infected Vero cells (Fig. 1D, lanes 4 and 6).
RV infection induces upregulation of PABP.
Binding of virus proteins to cellular proteins can have a number of effects, including relocalization, blocking their function, or inducing degradation of the cellular protein. With respect to PABP, it is the target of proteases encoded by picornaviruses, caliciviruses, and retroviruses (3, 24, 26, 33). We first examined the relative localizations of PABP in mock-infected and RV-infected cells. Vero cells were infected with the M33 strain of RV and then processed for indirect immunofluorescence microscopy. In mock-infected cells, PABP was localized throughout the cytoplasm in regularly distributed small puncta (Fig. 2). Very little PABP was detected in the nuclei. By contrast, PABP distribution was not homogenous in RV-infected cells but rather was concentrated in the perinuclear region. Extensive colocalization between PABP and capsid was evident in the perinuclear structures (Fig. 2). The capsid/PABP-positive perinuclear structures were closely associated with mitochondria as evidenced by partial colocalization with the mitochondrial protein p32 (Fig. 2). In contrast, there was no discernible overlap between PABP and the Golgi resident protein giantin in RV-infected or mock-infected samples (Fig. 2). These data suggest that PABP is not recruited to the site of virus assembly but rather is associated with a pool of capsid that localizes to mitochondria.
FIG. 2.

PABP colocalizes with capsid in infected cells. Vero cells were infected with RV or mock infected. Samples were then processed for indirect immunofluorescence microscopy after 48 h. Capsid, PABP, and the mitochondrial p32 protein were detected with rabbit, mouse, and goat antibodies, respectively. The Golgi resident giantin was stained with rabbit primary antibodies followed by a goat anti-rabbit Fab bridge. Primary and bridging antibodies were detected with secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647. Bar, 10 μm.
The PABP staining was consistently brighter in the infected cells, prompting us to wonder whether RV infection resulted in upregulation of PABP expression. To investigate this further, we harvested infected-cell lysates at various times postinfection and then processed them for immunoblot analyses. The relative levels of PABP and the housekeeping enzyme GAPDH were determined at each time point, and the ratios were calculated. The PABP/GAPDH ratio was arbitrarily set at 1.0 at the beginning of the time course. Between 0 and 24 h, the PABP levels were relatively constant (Fig. 3A). However, between 36 and 48 h when viral protein levels approach peak levels, relative PABP levels increased dramatically. To verify that the effect was not an artifact of the particular MOI that was employed, we repeated the experiment using a lower MOI, and similar results were observed (data not shown). Intriguingly, the levels of PABP mRNA were not affected by RV infection (Fig. 3B), suggesting that upregulation of PABP protein was occurring through a posttranscriptional mechanism. We also verified that the RV-induced PABP upregulation was not specific to A549 cells, as similar results were observed in Vero cells that were infected with RV (Fig. 3C). In both cell lines, by 48 h postinfection, the levels of PABP increased by more than 100%.
FIG. 3.

PABP expression is upregulated by RV infection. (A) A549 cells were infected with RV (MOI of 1), and at various time points, cell lysates were harvested and immunoblotted for capsid, PABP, and GAPDH (loading control). (B) Total RNA was extracted from mock- or RV-infected A549 cells at 48 h postinfection and then subjected to Northern blot analyses. The levels of genomic RV RNA (40S viral RNA [40S vRNA]) and PABP mRNA are shown. Equivalent amounts of RNA based on 28S rRNA were loaded in each lane. (C) Vero and A549 cells were infected with RV and processed for immunoblotting as described above for panel A. The average ratios of PABP to GAPDH at each time point (from two independent experiments) are plotted as a function of time. Infection results in a >100% increase in PABP levels between 24 and 48 h postinfection. (D) Vero cells were infected (MOI of 1) with Sindbis virus (SV) or vesicular stomatitis virus (VSV), and PABP and GAPDH levels were determined at specific time points up to 12 h postinfection. The average ratios of PABP to GAPDH at each time point (from two independent experiments) are plotted as a function of time. The inset shows the time-dependent expression of SV capsid and VSV G protein at various time points postinfection (in hours).
We next investigated whether PABP upregulation occurred in cells that are infected with other RNA viruses. As mentioned above, it is well documented that PABP is an important target of enterovirus and some retrovirus proteases, but to our knowledge, it is not known how other enveloped RNA viruses affect PABP levels. We first monitored the relative levels of PABP in cells that were infected with the type alphavirus Sindbis virus, which like RV, is a member of the Togaviridae family. From Fig. 3D, it can be seen that the levels of PABP did not increase in response to Sindbis virus infection. In contrast, PABP expression decreased by approximately 15% after 12 h. We also quantitated PABP expression in cells that were infected with the negative-strand RNA virus, vesicular stomatitis virus (Fig. 3D). In this case, the levels of PABP had decreased by approximately 25% at 12 h postinfection. Note that for Sindbis virus and vesicular stomatitis virus infection experiments, the last point in the time course was 12 h as opposed to 48 h for RV. The reason for this is that significant cell death had occurred in the Sindbis virus- and vesicular stomatitis virus-infected samples at 24 h and beyond. Together, our results indicate that upregulation of PABP is unique to RV infection.
We noticed that the sharp increase in PABP levels coincided with expression of the capsid protein (Fig. 3A); therefore, we next investigated whether expression of capsid in the absence of other viral proteins affected PABP localization. Cells were transfected with plasmids encoding capsid alone or the viral glycoproteins E2 and E1 and then processed for indirect immunofluorescence. As previously reported, the RV glycoproteins accumulated in the Golgi complex where they function in virus budding (21); however, the distribution and expression of PABP were unaffected by the presence of RV glycoproteins (Fig. 4). Conversely, the PABP staining pattern was markedly changed in cells expressing capsid protein. In addition to the punctate staining, PABP was distributed in a diffuse cytoplasmic pattern as well as juxtanuclear foci that partially colocalized with capsid (Fig. 4, arrowheads). These data indicate that capsid expression is sufficient to cause redistribution of PABP.
FIG. 4.

Capsid expression is associated with redistribution of PABP. Vero cells were transfected with plasmids encoding capsid or glycoproteins E2 and E1. Forty hours posttransfection, cells were processed for indirect immunofluorescence using rabbit anticapsid (α-capsid) or human anti-RV serum (α-PABP) to detect E1 and E2. PABP was detected using a mouse monoclonal antibody. Nuclei were stained with DAPI. The arrowheads indicate areas with extensive colocalization between PABP and capsid. Note because the PABP signal was so much higher in cells expressing capsid, to avoid saturation in the red channel, it was necessary to use shorter exposure times for the images shown in the top row. As a result, the PABP signal in the untransfected cells appears lower than those in the bottom two rows. Bar, 10 μm.
Other members of the preinitiation complex are not upregulated by RV infection.
It is possible that in addition to PABP, other members of the translation initiation machinery are upregulated in response to viral infection. To investigate this possibility, immunoblot analyses was used to analyze the levels of eIF4G, eIF4E, and PABP-interacting protein 1 (Paip1) in lysates prepared from mock- and RV-infected cell lysates. In contrast to PABP, the levels of these three translation initiation factors were relatively unchanged at times when capsid levels reached their peaks (Fig. 5A). Moreover, in Fig. 5B, it can be seen that the intracellular distribution of eIF4E did not change in response to RV infection. As such, it is likely that the pool of PABP, which is localized at or near the mitochondria, is not available for translation.
FIG. 5.

RV infection does not cause upregulation of other translation initiation factors. (A) A549 cells were infected with RV or mock infected, and cell lysates were prepared at 36 and 48 h postinfection, when capsid and PABP levels are high. The levels of capsid, Paip1, eIF4G, eIF4E, and GAPDH were determined by immunoblot analyses. (B) Forty-eight hours postinfection, cells were processed for indirect immunofluorescence using mouse anticapsid (α-capsid) and goat anti-eIF4E (α-eIF4E) antibodies. Nuclei were stained with DAPI. Bar, 10 μm.
Capsid protein binds to the C terminus of PABP and inhibits translation.
PABP is a modular protein (Fig. 6A) composed of multiple domains. The N-terminal region contains four tandemly arranged RNA recognition motifs that interact with the poly(A) tails of mRNAs, and the C terminus contains a PABC domain [poly(A)-binding protein C-terminal domain] that is required for interaction with the PABP-interacting motif 2 (PAM2) domains of translational regulators, including Paip1 and Paip2 (2, 29). To determine which region of PABP is required for binding to capsid, the N- and C-terminal regions of PABP were expressed as GST fusion proteins in bacteria and then immobilized on glutathione agarose beads (Fig. 6B). The beads were incubated with histidine-tagged capsid protein that was purified from bacteria. The beads were washed, and then bound proteins were eluted and detected by immunoblotting with antibodies to capsid. From the data in Fig. 6C, it can be seen that capsid binds to the C terminus of PABP but not the N terminus nor GST alone. Moreover, these results confirm the results from Fig. 1C indicating that RNA is not required for interaction between capsid and PABP.
FIG. 6.

Capsid binds to the C terminus of PABP. (A) Schematic of human PABP. The N terminus (NT) contains four tandemly RNA recognition motifs (RRMs), and the C terminus (CT) contains a PABC domain that mediates protein-protein interactions with regulatory proteins. (B) GST-tagged N-terminal (NT) and C-terminal (CT) PABP constructs were expressed in bacteria and purified on glutathione agarose. GST fusion proteins (indicated by an asterisk) were separated by SDS-PAGE and then stained with Coomassie blue. The positions of molecular mass markers (in kilodaltons) are indicated to the right of the gel. (C) Histidine-tagged capsid that was purified from bacteria was incubated with glutathione Sepharose prebound with PABP NT, PABP CT, or GST alone. Eluates were subjected to SDS-PAGE and immunoblotting with anticapsid antibodies.
The finding that capsid binds to the PABC-containing region of PABP prompted us to test whether capsid inhibits translation. Purified capsid was added to coupled transcription/translation assays, and the effect of this viral protein on synthesis of radiolabeled luciferase was determined. The addition of capsid to the coupled reactions resulted in a potent dose-dependent inhibition of protein synthesis. When 150 pmol of capsid was added to the reaction mixtures, no 35S-labeled luciferase was detected (Fig. 7A). The negative effect of capsid was not specific to luciferase, as similar results were obtained using 35S-labeled GFP as a reporter (data not shown). Histidine-tagged Sec17 was used as a negative control for these assays. Sec17 (32.8 kDa) is a peripheral membrane protein that is required for membrane transport between the endoplasmic reticulum and Golgi complex (16). It is similar in size to the soluble form of RV capsid protein (30.6 kDa) that was used for these experiments. The addition of up to 150 pmol of Sec17 to the transcription/translation reactions did not have a noticeable effect on synthesis of 35S-labeled luciferase. Because we used a coupled transcription/translation assay, it was not possible to discern whether capsid was inhibiting transcription rather than translation. To differentiate between these possibilities, we first synthesized capped luciferase-specific RNA in the absence of capsid protein. The luciferase RNA was then added to a rabbit reticulocyte lysate system, and synthesis was assayed in the presence or absence of capsid. In the presence of 150 pmol of capsid, translation of 35S-labeled luciferase was almost completely inhibited (Fig. 7B). These results indicate that capsid protein inhibits translation in a dose-dependent fashion. Finally, to determine whether the block in translation was the result of capsid-dependent sequestration of PABP, excess recombinant PABP was added to in vitro translation reaction mixtures containing a fixed inhibitory amount of capsid. In Fig. 7C, it can be seen that the addition of PABP, but not Sec17, partially relieves the capsid-dependent block in translation.
FIG. 7.
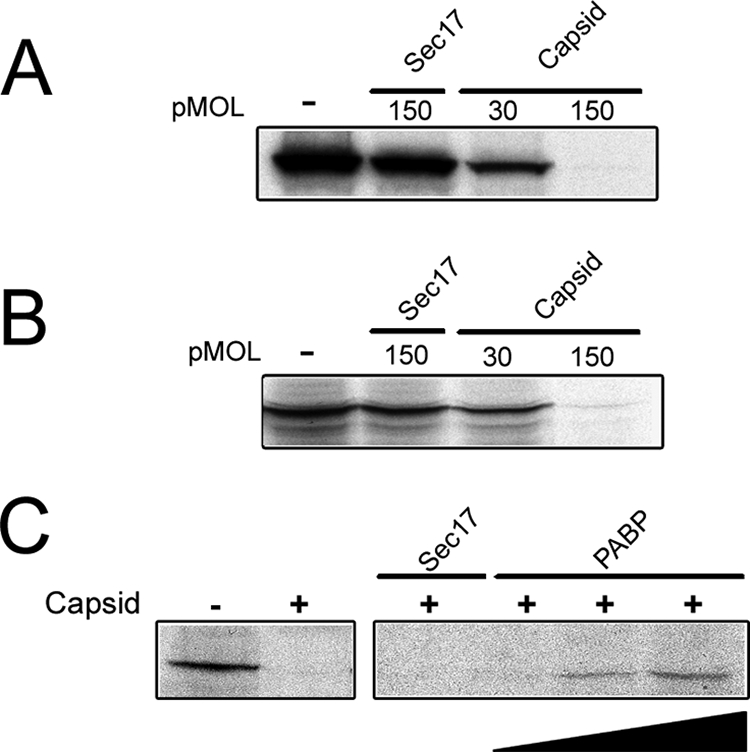
Capsid protein inhibits translation by a PABP-dependent mechanism. (A) 35S-labeled luciferase was synthesized in a coupled transcription/translation reaction in the presence or absence (−) of purified capsid (the amount of purified capsid is shown in picomoles [pMOL]). Sec17, a similarly sized protein was used as a negative control. Radiolabeled luciferase was detected by SDS-PAGE and fluorography. Similar results were obtained using 35S-labeled GFP as a reporter (data not shown). (B) To rule out the possibility that capsid was inhibiting transcription, luciferase RNA was added directly to rabbit reticulocyte lysates, and translation was conducted in the presence or absence of capsid. Capsid, but not Sec17, inhibits translation of luciferase. (C) Increasing amounts (0 to 45 pmol) of recombinant PABP were added to transcription/translation reaction mixtures that contained inhibitory amounts of capsid. Radiolabeled luciferase was detected by SDS-PAGE and fluorography.
DISCUSSION
Similar to other viral capsid proteins, the RV capsid has well-defined roles in nucleocapsid assembly and virus budding. More recent studies indicate that this protein also has functions that are not directly related to virus assembly. For example, data from Tzeng et al. in the Frey laboratory revealed that synthesis of viral RNAs is affected by capsid expression (55-57). Specifically, RV capsid binds to the viral replicase subunit p150 and through an unknown mechanism modulates production of genomic and subgenomic RNAs. In addition to binding other viral proteins, we have reported that capsid binds to two host cell proteins, p32 and Par-4 (6, 7). The significance of Par-4 as a capsid-binding protein has yet to be elucidated, but given the well-known role of Par-4 as a proapoptotic factor (51), it is tempting to speculate that capsid modulates virus-induced apoptosis. p32 is a multifunctional protein, the bulk of which localizes to the mitochondrial matrix and functions in maintaining oxidative phosphorylation (45). Interestingly, p32 was originally identified as a splicing factor-associated protein that is important for regulating RNA splicing (31). Subsequently, it was reported that p32 plays a pivotal role in modulating splicing of certain retroviral RNAs (31, 61). The precise role of p32 in RV biology is not known, but indirect evidence suggests that capsid-p32 interactions are important for subgenomic RNA synthesis (6).
In the present study, we have identified a third host cell-encoded capsid-binding protein, PABP. This protein plays a critical role in regulating initiation of translation and is one of the central targets of viruses that cause shutdown of host cell protein synthesis. It has been well established for years that many RNA and DNA viruses partially or completely inhibit host cell translation as a means to interfere with cellular defense systems (reviewed in reference 50). Most, if not all, of the known ways in which viruses target PABP are through proteolytic cleavage (3, 24, 33). The proteases of picornaviruses, caliciviruses, and certain retroviruses can cleave PABP at multiple sites, thereby reducing or eliminating the translation of cellular mRNAs without significantly affecting production of viral proteins. With respect to alphaviruses, it is known that capsid proteins play a central role shutting down host cell translation (11, 58). Although alphavirus capsids have protease activity (41), there is no evidence to suggest that PABP is a substrate for these proteases. Rather, alphavirus capsids reportedly inhibit protein synthesis by inducing activation of interferon-inducible protein kinase PKR and/or reducing mRNA accumulation (1, 12).
The RV capsid does not possess protease activity (9) nor any other type of catalytic activity, and therefore, its negative effect on translation would seem to be stoichiometric. Indeed, the inhibitory effect of capsid on translation in vitro was largely abrogated by the addition of excess PABP. To understand how this occurs, it is important to consider the nature of the capsid-PABP interaction. In vitro binding assays using purified protein fragments revealed that capsid binds to the C terminus of PABP. This region of PABP contains a PABC domain that interacts with PAM2 motifs (2, 29) which are found in positive (Paip1) and negative (Paip2) regulators of translation (10, 27, 28, 30, 49). When capsid levels are high, it may compete with positive regulators of translation, such as Paip1, for binding to PABP. The Paip1-PABP complex is thought to function in ribosome recruitment and translation initiation through interactions with eIF4A and other translation factors (49).
The effect of RV infection on translation in primary and cultured cell lines has been examined by a number of independent laboratories (reviewed in reference 14). Of relevance to the present study, infection of Vero cells causes cellular translation to decrease by approximately 50% when capsid levels peak between 48 and 72 h postinfection (17, 47). It remains to be determined whether other RV proteins directly affect protein synthesis. A lingering question that stems from our observations is why do levels of PABP increase during the capsid expression phase of RV infection. We were at first surprised to find that PABP mRNA levels were not affected by virus infection, but given that PABP expression is regulated at the translational level by an autoregulatory negative-feedback loop (4, 22, 46, 59), it is possible to explain the effect of capsid on PABP levels through this mechanism. Because of its central role in regulating gene expression, the levels of PABP are controlled in part through interaction of PABP with an adenine-rich sequence (ARS) that is present in the 5′ untranslated region of PABP mRNA. Although PABP is best known for its role as an initiator of translation, when it is idle, PABP binds to the ARS and represses translation of its own mRNA. Mapping studies revealed that the same region of PABP that binds the RV capsid, the C terminus, is required for this autoregulatory function (42). Under this scenario, capsid-mediated sequestration of PABP would result in upregulation of PABP, but not other proteins whose mRNAs lack ARS elements. Given that most cellular mRNAs do not contain ARS elements, it is mechanistically possible for global translation to decrease even when levels of PABP increase.
Finally, it remains to be determined whether the RV-mediated reduction in host cell translation is sufficient to thwart antiviral defenses. However, because RNA replication and virus assembly are confined to specific intracellular sites, it is tempting to speculate that if the capsid-mediated translational inhibition is also localized, the virus may still benefit from this effect. Viral RNA replication sites are associated with endocytic vacuoles that are concentrated together with Golgi membranes and mitochondria in the perinuclear region of the cell (13, 38, 39). The clustering of these organelles is expected to bring the sites of viral RNA synthesis into close proximity with the Golgi complex where nucleocapsid assembly and virus budding occur, an arrangement that may facilitate the interaction of nascent viral genomic RNA with capsid protein. Indeed, because the RV capsid protein is membrane anchored (52), having the sites viral of RNA synthesis (endosomes) located close to where virus assembly occurs (Golgi) would be advantageous for the virus. Late in the virus replication cycle when the levels of viral proteins and newly synthesized genomes are high, there is little need for additional replicase components. As such, the temporal rise in capsid levels may trigger a negative-feedback mechanism to prevent translation of nascent viral genomes. Specifically, binding of capsid to PABP that is localized near the sites of viral RNA synthesis and nucleocapsid formation could prevent recruitment of ribosomes to the nascent 40S RNA. This scenario would favor packaging of the 40S genomic RNA into nucleocapsids. On the basis of the results from this study, it is logical to propose that the pool of capsid that is associated with the surface of perinuclear mitochondria carries out this function.
Acknowledgments
We thank Gary Eitzen, Charles Rice, Nahum Sonenberg, and Jerry Wolinsky for their gifts of reagents.
The work was funded by a grant to T.C.H. from the Canadian Institutes of Health Research. T.C.H. is the recipient of a Medical Scientist Award from the Alberta Heritage Foundation for Medical Research (AHFMR). C.S.I. was supported by graduate studentship awards from AHFMR and the Faculty of Medicine and Dentistry.
Footnotes
Published ahead of print on 27 February 2008.
REFERENCES
- 1.Aguilar, P. V., S. C. Weaver, and C. F. Basler. 2007. Capsid protein of Eastern equine encephalitis virus inhibits host cell gene expression. J. Virol. 813866-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albrecht, M., and T. Lengauer. 2004. Survey on the PABC recognition motif PAM2. Biochem. Biophys. Res. Commun. 316129-138. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez, E., A. Castello, L. Menendez-Arias, and L. Carrasco. 2006. HIV protease cleaves poly(A)-binding protein. Biochem. J. 396219-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bag, J., and J. Wu. 1996. Translational control of poly(A)-binding protein expression. Eur. J. Biochem. 237143-152. [DOI] [PubMed] [Google Scholar]
- 5.Baron, M. D., T. Ebel, and M. Suomalainen. 1992. Intracellular transport of rubella virus structural proteins expressed from cloned cDNA. J. Gen. Virol. 731073-1086. [DOI] [PubMed] [Google Scholar]
- 6.Beatch, M. D., J. C. Everitt, L. J. Law, and T. C. Hobman. 2005. Interactions between rubella virus capsid and host protein p32 are important for virus replication. J. Virol. 7910807-10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beatch, M. D., and T. C. Hobman. 2000. Rubella virus capsid associates with host cell protein p32 and localizes to mitochondria. J. Virol. 745569-5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, M. H., and J. P. Icenogle. 2004. Rubella virus capsid protein modulates viral genome replication and virus infectivity. J. Virol. 784314-4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke, D. M., T. W. Loo, I. Hui, P. Chong, and S. Gillam. 1987. Nucleotide sequence and in vitro expression of rubella virus 24S subgenomic messenger RNA encoding the structural proteins E1, E2 and C. Nucleic Acids Res. 153041-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craig, A. W., A. Haghighat, A. T. Yu, and N. Sonenberg. 1998. Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature 392520-523. [DOI] [PubMed] [Google Scholar]
- 11.Elgizoli, M., Y. Dai, C. Kempf, H. Koblet, and M. R. Michel. 1989. Semliki Forest virus capsid protein acts as a pleiotropic regulator of host cellular protein synthesis. J. Virol. 632921-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Favre, D., E. Studer, and M. R. Michel. 1996. Semliki Forest virus capsid protein inhibits the initiation of translation by upregulating the double-stranded RNA-activated protein kinase (PKR). Biosci. Rep. 16485-511. [DOI] [PubMed] [Google Scholar]
- 13.Fontana, J., W. P. Tzeng, G. Calderita, A. Fraile-Ramos, T. K. Frey, and C. Risco. 2007. Novel replication complex architecture in rubella replicon-transfected cells. Cell. Microbiol. 9875-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey, T. K. 1994. Molecular biology of rubella virus. Adv. Virus Res. 4469-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gingras, A. C., B. Raught, and N. Sonenberg. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68913-963. [DOI] [PubMed] [Google Scholar]
- 16.Griff, I. C., R. Schekman, J. E. Rothman, and C. A. Kaiser. 1992. The yeast SEC17 gene product is functionally equivalent to mammalian alpha-SNAP protein. J. Biol. Chem. 26712106-12115. [PubMed] [Google Scholar]
- 17.Hemphill, M. L., R. Y. Forng, E. S. Abernathy, and T. K. Frey. 1988. Time course of virus-specific macromolecular synthesis during rubella virus infection in Vero cells. Virology 16265-75. [DOI] [PubMed] [Google Scholar]
- 18.Hobman, T., and J. K. Chantler. 2007. Rubella virus, p. 1069-1100. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 19.Hobman, T. C., M. L. Lundstrom, and S. Gillam. 1990. Processing and intracellular transport of rubella virus structural proteins in COS cells. Virology 178122-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hobman, T. C., M. L. Lundstrom, C. A. Mauracher, L. Woodward, S. Gillam, and M. G. Farquhar. 1994. Assembly of rubella virus structural proteins into virus-like particles in transfected cells. Virology 202574-585. [DOI] [PubMed] [Google Scholar]
- 21.Hobman, T. C., L. Woodward, and M. G. Farquhar. 1993. The rubella virus E2 and E1 spike glycoproteins are targeted to the Golgi complex. J. Cell Biol. 121269-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hornstein, E., A. Git, I. Braunstein, D. Avni, and O. Meyuhas. 1999. The expression of poly(A)-binding protein gene is translationally regulated in a growth-dependent fashion through a 5′-terminal oligopyrimidine tract motif. J. Biol. Chem. 2741708-1714. [DOI] [PubMed] [Google Scholar]
- 23.Imataka, H., A. Gradi, and N. Sonenberg. 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 177480-7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joachims, M., P. C. Van Breugel, and R. E. Lloyd. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 73718-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahvejian, A., Y. V. Svitkin, R. Sukarieh, M. N. M'Boutchou, and N. Sonenberg. 2005. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 19104-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerekatte, V., B. D. Keiper, C. Badorff, A. Cai, K. U. Knowlton, and R. E. Rhoads. 1999. Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J. Virol. 73709-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khaleghpour, K., A. Kahvejian, G. De Crescenzo, G. Roy, Y. V. Svitkin, H. Imataka, M. O'Connor-McCourt, and N. Sonenberg. 2001. Dual interactions of the translational repressor Paip2 with poly(A) binding protein. Mol. Cell. Biol. 215200-5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khaleghpour, K., Y. V. Svitkin, A. W. Craig, C. T. DeMaria, R. C. Deo, S. K. Burley, and N. Sonenberg. 2001. Translational repression by a novel partner of human poly(A) binding protein, Paip2. Mol. Cell 7205-216. [DOI] [PubMed] [Google Scholar]
- 29.Kozlov, G., G. De Crescenzo, N. S. Lim, N. Siddiqui, D. Fantus, A. Kahvejian, J. F. Trempe, D. Elias, I. Ekiel, N. Sonenberg, M. O'Connor-McCourt, and K. Gehring. 2004. Structural basis of ligand recognition by PABC, a highly specific peptide-binding domain found in poly(A)-binding protein and a HECT ubiquitin ligase. EMBO J. 23272-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozlov, G., J. F. Trempe, K. Khaleghpour, A. Kahvejian, I. Ekiel, and K. Gehring. 2001. Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc. Natl. Acad. Sci. USA 984409-4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krainer, A. R., A. Mayeda, D. Kozak, and G. Binns. 1991. Functional expression of cloned human splicing factor SF2: homology to RNA-binding proteins, U1 70K, and Drosophila splicing regulators. Cell 66383-394. [DOI] [PubMed] [Google Scholar]
- 32.Kreis, T. E. 1986. Microinjected antibodies against the cytoplasmic domain of vesicular stomatitis virus glycoprotein blocks its transport to the cell surface. EMBO J. 5931-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuyumcu-Martinez, M., G. Belliot, S. V. Sosnovtsev, K. O. Chang, K. Y. Green, and R. E. Lloyd. 2004. Calicivirus 3C-like proteinase inhibits cellular translation by cleavage of poly(A)-binding protein. J. Virol. 788172-8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law, L. J., C. S. Ilkow, W. P. Tzeng, M. Rawluk, D. T. Stuart, T. K. Frey, and T. C. Hobman. 2006. Analyses of phosphorylation events in the rubella virus capsid protein: role in early replication events. J. Virol. 806917-6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Law, L. M., J. C. Everitt, M. D. Beatch, C. F. Holmes, and T. C. Hobman. 2003. Phosphorylation of rubella virus capsid regulates its RNA binding activity and virus replication. J. Virol. 771764-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le, H., R. L. Tanguay, M. L. Balasta, C. C. Wei, K. S. Browning, A. M. Metz, D. J. Goss, and D. R. Gallie. 1997. Translation initiation factors eIF-iso4G and eIF-4B interact with the poly(A)-binding protein and increase its RNA binding activity. J. Biol. Chem. 27216247-16255. [DOI] [PubMed] [Google Scholar]
- 37.Lee, J. Y., J. A. Marshall, and D. S. Bowden. 1999. Localization of rubella virus core particles in Vero cells. Virology 265110-119. [DOI] [PubMed] [Google Scholar]
- 38.Lee, J. Y., J. A. Marshall, and D. S. Bowden. 1992. Replication complexes associated with the morphogenesis of rubella virus. Arch. Virol. 12295-106. [DOI] [PubMed] [Google Scholar]
- 39.Magliano, D., J. Marshall, D. S. Bowden, N. Vardaxis, J. Meanger, and J.-Y. Lee. 1998. Rubella virus replication complexes are virus-modified lysosomes. Virology 24057-63. [DOI] [PubMed] [Google Scholar]
- 40.Marr, L. D., A. Sanchez, and T. K. Frey. 1991. Efficient in vitro translation and processing of the rubella virus structural proteins in the presence of microsomes. Virology 180400-405. [DOI] [PubMed] [Google Scholar]
- 41.Melancon, P., and H. Garoff. 1987. Processing of the Semliki Forest virus structural polyprotein: role of the capsid protease. J. Virol. 611301-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melo, E. O., R. Dhalia, C. Martins de Sa, N. Standart, and O. P. de Melo Neto. 2003. Identification of a C-terminal poly(A)-binding protein (PABP)-PABP interaction domain: role in cooperative binding to poly(A) and efficient cap distal translational repression. J. Biol. Chem. 27846357-46368. [DOI] [PubMed] [Google Scholar]
- 43.Mizushima, S., and S. Nagata. 1990. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 185322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohan, K. V., B. Ghebrehiwet, and C. D. Atreya. 2002. The N-terminal conserved domain of rubella virus capsid interacts with the C-terminal region of cellular p32 and overexpression of p32 enhances the viral infectivity. Virus Res. 85151-161. [DOI] [PubMed] [Google Scholar]
- 45.Muta, T., D. Kang, S. Kitajima, T. Fujiwara, and N. Hamasaki. 1997. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 27224363-24370. [DOI] [PubMed] [Google Scholar]
- 46.Patel, G. P., S. Ma, and J. Bag. 2005. The autoregulatory translational control element of poly(A)-binding protein mRNA forms a heteromeric ribonucleoprotein complex. Nucleic Acids Res. 337074-7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Payment, P., D. Ajdukovic, and V. Pavilanis. 1975. Rubella virus. II. Replication in Vero calls and effects of actinomycin D and cycloheximide. Can. J. Microbiol. 21710-717. (In French.) [PubMed] [Google Scholar]
- 48.Rice, C. M., R. Levis, J. H. Strauss, and H. V. Huang. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 613809-3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy, G., G. De Crescenzo, K. Khaleghpour, A. Kahvejian, M. O'Connor-McCourt, and N. Sonenberg. 2002. Paip1 interacts with poly(A) binding protein through two independent binding motifs. Mol. Cell. Biol. 223769-3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider, R. J., and T. Shenk. 1987. Impact of virus infection on host cell protein synthesis. Annu. Rev. Biochem. 56317-332. [DOI] [PubMed] [Google Scholar]
- 51.Sells, S. F., D. P. Wood, Jr., S. S. Joshi-Barve, S. Muthukumar, R. J. Jacob, S. A. Crist, S. Humphreys, and V. M. Rangnekar. 1994. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ. 5457-466. [PubMed] [Google Scholar]
- 52.Suomalainen, M., H. Garoff, and M. D. Baron. 1990. The E2 signal sequence of rubella virus remains part of the capsid protein and confers membrane association in vitro. J. Virol. 645500-5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suomalainen, M., P. Liljestrom, and H. Garoff. 1992. Spike protein-nucleocapsid interactions drive the budding of alphaviruses. J. Virol. 664737-4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tarun, S. Z., Jr., and A. B. Sachs. 1996. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J. 157168-7177. [PMC free article] [PubMed] [Google Scholar]
- 55.Tzeng, W. P., and T. K. Frey. 2003. Complementation of a deletion in the rubella virus p150 nonstructural protein by the viral capsid protein. J. Virol. 779502-9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tzeng, W. P., and T. K. Frey. 2005. Rubella virus capsid protein modulation of viral genomic and subgenomic RNA synthesis. Virology 337327-334. [DOI] [PubMed] [Google Scholar]
- 57.Tzeng, W. P., J. D. Matthews, and T. K. Frey. 2006. Analysis of rubella virus capsid protein-mediated enhancement of replicon replication and mutant rescue. J. Virol. 803966-3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Steeg, H., M. Kasperaitis, H. O. Voorma, and R. Benne. 1984. Infection of neuroblastoma cells by Semliki Forest virus. The interference of viral capsid protein with the binding of host messenger RNAs into initiation complexes is the cause of the shut-off of host protein synthesis. Eur. J. Biochem. 138473-478. [DOI] [PubMed] [Google Scholar]
- 59.Wu, J., and J. Bag. 1998. Negative control of the poly(A)-binding protein mRNA translation is mediated by the adenine-rich region of its 5′-untranslated region. J. Biol. Chem. 27334535-34542. [DOI] [PubMed] [Google Scholar]
- 60.Yao, J., and S. Gillam. 1999. Mutational analysis, using a full-length rubella virus cDNA clone, of rubella virus E1 transmembrane and cytoplasmic domains required for virus release. J. Virol. 734622-4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng, Y.-H., H.-F. Yu, and B. M. Peterlin. 2003. Human p32 protein relieves a post-transcriptional block to HIV replication in murine cells. Nat. Cell Biol. 5611-618. [DOI] [PubMed] [Google Scholar]