

Abstract
Polycomb group (PcG) transcription regulatory proteins maintain cell identity by sustained repression of numerous genes. The differentiation of embryonic stem (ES) cells induces a genome-wide shift in PcG target gene expression. We investigated the effects of differentiation and protein interactions on CBX family PcG protein localization and dynamics by using fluorescence imaging. In mouse ES cells, different CBX proteins exhibited distinct distributions and mobilities. Most CBX proteins were enriched in foci known as Polycomb bodies. Focus formation did not affect CBX protein mobilities, and the foci dispersed during ES cell differentiation. The mobilities of CBX proteins increased upon the induction of differentiation and decreased as differentiation progressed. The deletion of the chromobox, which mediates interactions with RING1B, prevented the immobilization of CBX proteins. In contrast, the deletion of the chromodomain, which can bind trimethylated lysine 27 of histone H3, had little effect on CBX protein dynamics. The distributions and mobilities of most CBX proteins corresponded to those of CBX-RING1B complexes detected by using bimolecular fluorescence complementation analysis. Epigenetic reprogramming during ES cell differentiation is therefore associated with global changes in the subnuclear distributions and dynamics of CBX protein complexes.
During differentiation, the pluripotency of embryonic stem (ES) cells is restricted by epigenetic changes (7, 11). Polycomb group (PcG) proteins contribute to the stable inheritance of both pluripotent and differentiated cell states (48, 50). These functions implicate PcG proteins in the control of the transition between pluripotency and differentiation. Genome-wide studies of PcG protein binding in mammalian cells have identified hundreds of genes that bind PcG proteins (8, 9, 31). The expression of many of these genes is altered during ES cell differentiation.
Biochemical studies of PcG proteins have identified two Polycomb repressive complexes (PRCs), PRC1 and PRC2 (12, 28, 52). PRC2 has lysine methyltransferase activity for K27 of histone H3; PRC1 contains a subunit (Pc in Drosophila and CBX family proteins in mammals) that can bind trimethyl-K27 H3 in vitro (6, 12). Many genes that are bound by PRC1 are enriched in H3 K27 trimethylation (8, 9). These observations, in combination with epistatic relationships among mutations in Drosophila PcG genes (59), have given rise to the model that histone H3 K27 trimethylation by PRC2 is required for the recruitment of the PRC1 complex to specific genes. These results have also been interpreted to indicate that PRC2 initiates silencing and that PRC1 maintains the silenced state.
Genetic studies of mice indicate that the functions of PcG proteins are at least in part nonoverlapping since the ablation of genes encoding different PcG proteins produces distinct phenotypes (2, 14, 17, 25, 32, 38, 39, 49, 54-57). Null mutations in the EED and Suz12 subunits of PRC2 eliminate histone H3 K27 trimethylation, but do not prevent the recruitment of PRC1 proteins to either the inactive X or to many of their target genes (39, 46). It is therefore unclear whether the recognition of trimethyl-K27 of H3 is necessary or sufficient for stable chromatin association by CBX proteins or whether other interactions, potentially mediated by additional components of the PRC1 complex, are involved.
Many PcG proteins accumulate in subnuclear foci known as Polycomb bodies (5, 10, 18, 21, 23, 44, 58). In Drosophila, several genes that are repressed by PcG proteins are localized to Polycomb bodies, suggesting that gene repression is associated with localization to these foci (22, 33). The number of Polycomb bodies is orders of magnitude smaller than the number of genetic loci targeted by PcG proteins. Therefore, either a large number of genes must be associated with each Polycomb body or some genes repressed by PcG proteins must reside outside Polycomb bodies. The relationship between Polycomb body formation and the stability of PcG protein association with chromatin in mammalian cells remains to be established.
Mammals have several CBX family proteins (CBX2, CBX4, CBX6, CBX7, and CBX8) that contain a highly conserved chromodomain at the amino terminus (3, 5, 21, 40). The isolated chromodomains from all of these proteins, with the exception of CBX6, can bind trimethyl-K27 H3 (6). CBX family proteins also contain a chromobox sequence near the carboxy terminus that can mediate interactions with the Ring1B subunit of the PRC1 complex (5, 43, 47). Different members of the CBX family are thought to have distinct biological functions (18, 21, 26, 51). However, the properties of these proteins have not been compared directly in cells.
The roles of PcG proteins in the stable maintenance of gene silencing over the life span of the organism as well as in the rapid transition between pluripotent and differentiated states suggest that the dynamics of PcG proteins can provide insight into their functions. Subpopulations of mammalian BMI1 and Drosophila Pc and Ph subunits of PRC1 exchange with half times of 20 to 250 s in Polycomb bodies and on salivary gland polytene chromosomes (20, 23). Here we investigate the distributions and dynamics of CBX family proteins during mouse ES cell differentiation and the roles of trimethyl-K27 H3 binding and interactions with other PRC1 proteins in the control of CBX protein distributions and dynamics.
MATERIALS AND METHODS
Plasmid construction.
The Venus fluorescent protein was fused to the N terminus of each CBX protein in the same position relative to the conserved chromodomain to increase the likelihood that any effects of the fusion would be identical for all CBX proteins. For a list of the primers used for amplification of the sequences encoding CBX2, CBX4, CBX6, CBX7, CBX8, RING1B, and H3.1 as well as the enzymes used to digest the amplified fragments, see Table S5 in the supplemental material. The coding sequences of CBX2, CBX4, CBX6, CBX7, CBX8, and RING1B were fused after the coding sequence of the Venus fluorescent protein (36) in plasmid pCDNA3.1(+) (Invitrogen) to produce plasmids pVenusCBX2, pVenusCBX4, pVenusCBX6, pVenusCBX7, pVenusCBX8, and pVenusRING1B. The coding sequence of H3.2 was fused before the coding sequence of Venus in plasmid pGACCS-M (37) to produce pH3.2Venus. The corresponding fusion to Tetrahymena H3 is able to substitute for the endogenous protein (15). For a list of the primers used to produce mutated CBX proteins, see Table S6 in the supplemental material. To produce fusion proteins for bimolecular fluorescence complementation (BiFC) analysis, the sequence encoding residues 1 to 172 of Venus was fused to the coding regions of CBX proteins to produce plasmids pVN173CBX2, pVN173CBX4, pVN173CBX6, pVN173CBX7, and pVN173CBX8. The sequence encoding residues 173 to 238 of yellow fluorescent protein was fused to the coding region of RING1B to produce pYC173RING1B. The sequences encoding all fusion proteins and adjacent control sequences were verified and are available upon request.
Isolation of stable ES cell lines.
The PGK12.1 mouse ES cell line (41) was electroporated with linearized plasmid DNA (pVenusCBX2, pVenusCBX4, pVenusCBX6, pVenusCBX7, pVenusCBX8, or pH3.2Venus). After electroporation, the cells were cultured for 48 h before selection in the presence of 400 μg/ml G418 (Invitrogen). After 6 or 7 days, individual surviving clones were transferred to 96-well plates and screened for fluorescence. The fluorescent cells were expanded and tested for CBX fusion protein expression by Western blot analysis using antibodies against corresponding CBX family proteins (see Fig. S1 and materials and methods in the supplemental material).
ES cell culture and differentiation.
ES cells were cultured and induced to differentiate as described previously (53). To favor either neuronal or adipocyte lineages, embryoid body differentiation protocols were used in combination with selective culture conditions (4, 16).
Immunological methods.
For the antibodies and methods used for chromatin immunoprecipitation, coimmunoprecipitation, immunoblotting, and immunofluorescence analysis, see Tables S6 and S7 and materials and methods in the supplemental material.
Epifluorescence microscopy.
For live-cell imaging, cells were grown in gelatin-coated, cover glass chambers (Nalge Nunc International) and transferred into Dulbecco's modified Eagle's medium lacking phenol red (Invitrogen). To image fixed cells, cells grown on cover glass were fixed in 3.7% formaldehyde for 10 min at room temperature, washed with phosphate-buffered saline, and mounted for microscopy. The epifluorescence images were acquired by using a Nikon TE300 inverted fluorescence microscope equipped with a 60× oil objective (numerical aperture, 1.4) and a Hamamatsu ER II charge-coupled device camera.
FRAP analysis and quantitation.
Fluorescence recovery after photobleaching (FRAP) analysis was performed by using an Olympus FV500 confocal microscope with a 60× water objective (numerical aperture, 1.2) at 37°C. For the experimental conditions and instrument settings, see Table S3 and materials and methods in the supplemental material. Control experiments demonstrated that these conditions resulted in the bleaching of more than 99% of the fluorescence. No significant fluorescence recovery was observed in fixed samples, demonstrating that molecular motion was required for recovery. The images were aligned, and fluorescence intensities were quantified by using ImageJ and TurboReg software. The fluorescence intensity of the bleached area was corrected for background and normalized (see materials and methods in the supplemental material) to obtain the fluorescence recovery (IR) in each image. The best fit of a single-exponential function to IR as a function of time (t) was determined for each individual cell by using equation 1.
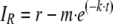 |
(1) |
where m is the major fraction. The fast-recovery fraction (f) was calculated based on equation 2
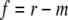 |
(2) |
The nonrecovery fraction (n) was calculated based on equation 3
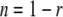 |
(3) |
The time required for recovery of half of the major fraction (t1/2) was calculated based on equation 4
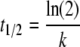 |
(4) |
FLIP analysis and quantitation.
Fluorescence loss in photobleaching (FLIP) analysis was performed under conditions that were the same as those for FRAP analysis. For a description of the instrument settings, see Table S4 and materials and methods in the supplemental material. A region located at a distance from the region of interest was bleached. The bleach cycle was repeated every 30 s to allow redistribution of the mobile fusion proteins between bleach cycles and to minimize photodamage to the cell during the protracted analysis. An image of the cell was acquired before each bleach cycle. The fluorescence intensity in the region of interest was measured, corrected for background, and normalized (see materials and methods in the supplemental material) to obtain the fluorescence remaining (IL) in each image. The best fit of a single-exponential function to IL as a function of the number of bleach cycles (c) was determined for each individual cell by using equation 5
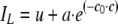 |
(5) |
where u was the nonexchangeable fraction. The exchangeable fraction (b) was calculated based on equation 6
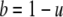 |
(6) |
The number of cycles required to bleach half of the fluorescence in the unbleached region (c1/2) was calculated based on equation 7.
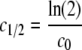 |
(7) |
Transient expression and BiFC analysis.
HEK293T cells were cultured and transfected (see materials and methods in the supplemental material for details). BiFC analysis was performed as described previously (24), and cells with fluorescence intensities comparable to the stable ES cell lines that expressed CBX fusions were imaged 24 h after transfection.
RESULTS
Stable expression of fluorescent CBX fusion proteins in mouse ES cells.
To investigate CBX family protein distributions and dynamics in mouse ES cells, we generated cell lines that stably expressed CBX proteins fused to a fluorescent protein. Two cell lines that expressed each CBX fusion at levels similar to those of endogenous CBX proteins were selected for these studies (see Fig. S1 in the supplemental material). These cell lines had growth rates indistinguishable from that of the parental cell line. No spontaneous differentiation was observed under the conditions used for ES cell culture, and all lines were able to differentiate into several different progenitor cell types under conditions that induced differentiation of the parental cell line (see below). Each line produced the full-length fusion protein. No truncated fragments were detected by Western blot analysis using antibodies directed against either the CBX proteins or the fluorescent protein fusions (Fig. 1; see Fig. S1, S2B, and S2C in the supplemental material). The fluorescence in these cells therefore reflected characteristics of the intact fusion proteins.
FIG. 1.

Analysis of CBX fusion protein binding to PcG target genes and interactions with endogenous Ring1B during ES cell differentiation. (A) Changes in CBX fusion binding to PcG target genes during ES cell differentiation. CBX fusion binding to Hoxa11, Gata6, Olig2, and Egfr promoters was evaluated before (day 0) and 6 days after inducing differentiation (day 6) of the CBX2#16, CBX4#11, CBX6#7, CBX7#9, and CBX8#1 ES cell lines using chromatin immunoprecipitation. The total chromatin (T) and chromatin precipitated by using anti-GFP antibody (α) were analyzed by using primers specific for each of the promoters. MW, molecular weight. The parental PGK12.1 ES cell line was used as a control. All reactions for each promoter were performed in parallel. (B) Interactions between CBX fusions and endogenous Ring1B during ES cell differentiation. Lysates prepared before (day 0) or 6 days after inducing differentiation (day 6) of the CBX2#16, CBX4#11, CBX6#7, CBX7#9, and CBX8#1 ES cell lines were precipitated by using anti-GFP antibody. The precipitates were analyzed by Western blotting using anti-Ring1B antibody (top panel). After detection, the blot was stripped and analyzed by using anti-GFP antibody (second panel) to determine the amounts of CBX fusions precipitated from the extracts. The cell lysates were also analyzed by Western blotting using anti-Ring1B antibody (third panel), antibodies against CBX2, CBX6, CBX7, or CBX 8 (fourth panel), and anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (fifth panel). All precipitations and blots were performed in parallel. A shorter exposure of the anti-Ring1B blot of precipitates from the CBX7#9 and CBX8#1 lines is shown. The CBX4 fusion was not consistently detected by using available anti-CBX4 antibodies. IgH, immunoglobulin H; IP, immunoprecipitate.
We examined whether the CBX fusions bound to known PcG target genes in undifferentiated and differentiating cells using chromatin immunoprecipitation. The promoter regions of the Gata6, Hoxa11, Olig2, and Egfr genes were precipitated by anti-green fluorescent protein (GFP) antibodies from cells that expressed each of the CBX fusions (Fig. 1A). No precipitation above background of the Gata1 or other control promoter regions was detected (see Fig. S2A in the supplemental material). Following the induction of differentiation, binding by the CBX2 and CBX8 fusions decreased at all promoters tested. There was little change in binding by the CBX7 fusion at the Gata6, Hoxa11, or Olig2 promoters, whereas binding at the Egfr promoter decreased. The CBX4 and CBX6 fusions precipitated all promoters tested with lower efficiencies, possibly because of the lower levels of expression of these fusions. Thus, all CBX fusions bound specifically to known PcG target genes and different CBX fusions exhibited distinct changes in binding to different genes during ES cell differentiation.
We examined whether the CBX fusions associated with other PRC1 components during differentiation by immunoprecipitation analysis. Endogenous Ring1B was coprecipitated with each CBX fusion by anti-GFP antibodies (Fig. 1B). The amounts of endogenous Ring1B coprecipitated with the CBX fusions varied in parallel with the amounts of CBX fusions precipitated, with the exception of the CBX6 fusion. The efficiency of Ring1B coprecipitation with the CBX6 fusion was approximately 10-fold lower than for the other CBX fusions. The lower efficiency of Ring1B coprecipitation with the CBX6 fusion was independent of the antibody used for precipitation (see Fig. S2B and S2C in the supplemental material). No precipitation of Ring1B from the parental PGK12.1 cell line was observed. Thus, endogenous Ring1B was present in complexes formed by all CBX fusions both before and after the induction of differentiation. The stably expressed CBX fusions therefore recapitulated many characteristics of endogenous CBX proteins.
Redistribution of CBX fusions during ES cell differentiation.
Different CBX fusions had distinct subnuclear distributions in undifferentiated ES cells (Fig. 2). The CBX2 fusion was enriched in small and large foci resembling Polycomb bodies (Fig. 2Aa). The CBX4 fusion was localized to numerous small foci scattered unevenly in the nucleus (Fig. 2Ab). The CBX6 fusion was dispersed in a granular pattern throughout the nucleus (Fig. 2Ac). The CBX7 and CBX8 fusions were enriched in small foci unevenly distributed in the nucleus (Fig. 2Ad and Ae). The distinct distributions of different CBX fusions in undifferentiated ES cells suggest that different factors determined their localization.
FIG. 2.

Changes in the distributions and mobilities of CBX fusions during ES cell differentiation. (A) Fluorescence images of CBX2#16, CBX4#11, CBX6#7, CBX7#9, and CBX8#1 ES cell nuclei were acquired before differentiation (a to e) and 1 (f to j), 3 (k to o), and 6 (p to t) days after the induction of differentiation. The fluorescence of the CBX fusions is shown in green, and the fluorescence of the Hoechst DNA stain is shown in red. (B) Fluorescence images of CBX2#16 ES cells during FRAP analysis. The images were captured before, and at the indicated times after, bleaching the area outlined in red. (C) Quantitation of fluorescence recovery in the cell lines shown in panel A and ES cells that stably expressed histone H3.2 fused to a fluorescent protein. The curves on the left show the normalized fluorescence intensities of the bleached areas as a function of time after bleaching, together with the best fit of a single-exponential function (see Materials and Methods). Cells were analyzed before differentiation (black) and at 1 (orange), 2 (magenta), and 6 (blue) days after the induction of differentiation. Standard deviations are shown for the data for the fastest and slowest recoveries; others were of similar magnitudes. The graphs on the right show line plots of the rates of fluorescence recovery for the major fraction (t1/2 [right axis]) superimposed on stacked histograms showing the relative sizes of the fast-recovery (green), major (yellow), and nonrecovery (red) fractions (left axis). The derivation of these fractions is graphically depicted by dashed gray lines connecting the recovery curves to the histograms (see Materials and Methods). Statistical significance of the difference between the rates of fluorescence recovery in differentiating ES cells and undifferentiated cells: *, P value was <0.05 by using Student's t test; **, P value was <0.01 by using Student's t test. The data represent mean values and standard deviations (error bars) from six independent differentiation experiments. The images are representative of the majority of cells in each population (see Table S1 in the supplemental material).
The CBX fusions were redistributed following the induction of ES cell differentiation by the withdrawal of leukemia inhibitory factor and the addition of all-trans retinoic acid (Fig. 2Af to At). The foci resembling Polycomb bodies disappeared as each CBX fusion dispersed in a granular pattern. Six days after the induction of differentiation, more than 80% of the cells expressing each CBX fusion were devoid of foci resembling Polycomb bodies (see Table S1 in the supplemental material). The cells continued to proliferate during differentiation, and the fluorescence intensities changed less than twofold, indicating that the differences in distribution were not solely due to changes in cell cycle progression or CBX fusion expression.
The foci formed by each CBX fusion in undifferentiated ES cells did not colocalize with Hoechst-stained chromocenters (Fig. 2Aa ′ to Ae′). The chromocenters underwent characteristic changes in size and number during ES cell differentiation, and 95% of the cells that expressed each CBX fusion exhibited a change in the pattern of Hoechst staining 6 days after the induction of differentiation (Fig. 2Af′ to At′). The CBX2 fusion was relocalized to the chromocenters during the first day after the induction of differentiation and colocalized with Hoechst staining in 50% of the cells 1 day after the induction of differentiation (Fig. 2Af and Af′). Subsequently, the CBX2 fusion dispersed to form a granular distribution that was not preferentially associated with chromocenters (Fig. 2Ak, Ak′, Ap, and Ap′). The CBX6 fusion was excluded from chromocenters during differentiation and was reduced in Hoechst-stained foci in 60% of the cells 6 days after the induction of differentiation (Fig. 2Ar and Ar′). Thus, CBX family proteins exhibited at least two stages of redistribution during ES cell differentiation, and different members of the family exhibited unique changes in localization. There was no consistent relationship between the changes in CBX fusion distributions and the reorganization of heterochromatin during ES cell differentiation. Thus, the changes in CBX fusion distributions did not strictly track the global changes in chromatin organization during ES cell differentiation.
Changes in CBX fusion mobilities during ES cell differentiation.
Studies of protein mobilities can provide information about their interactions with other cellular components. Foci formed by the CBX2 fusion in undifferentiated ES cells appeared nearly stationary for at least 30 min (see movie S1 in the supplemental material). However, the static appearance of these foci does not provide information about the dynamics of the proteins that constitute them. FRAP analysis of cells that expressed individual CBX fusions demonstrated that a major fraction of each CBX fusion was mobile (Fig. 2B). In cells that expressed CBX2, CBX4, CBX7, or CBX8 fusions, half of the fluorescence recovered in 15 to 30 s (Fig. 2C). The rate of fluorescence recovery was twice as fast in cells that expressed the CBX6 fusion. The fluorescence recovery in the time frame examined accounted for a major fraction of the original fluorescence intensity and was well described by a single exchange rate for each CBX fusion. A fraction of the fluorescence recovered before the first image after bleaching was acquired and another fraction did not recover during the experiment. These fast-recovery and nonrecovery fractions were estimated by extrapolation of the fluorescence recovery function for the major fraction to zero and infinite time, respectively (Fig. 2C).
We investigated whether ES cell differentiation affected the mobilities of CBX fusions. Each CBX fusion exhibited an increase in the rate of fluorescence recovery for the major fraction during the first day following the induction of differentiation and a gradual decrease in this rate during subsequent differentiation (Fig. 2C). Six days after the induction of differentiation, the half time for fluorescence recovery by CBX fusions was 40% longer on average than that in undifferentiated cells and twice as long as the value for 1 day after the induction of differentiation (by Student's t test, P values were <0.05 for all CBX fusions). Thus, the dynamics of all CBX fusions were coordinately regulated and exhibited an initial stage of increased mobility, followed by a subsequent decrease in mobility during differentiation.
The sizes of the nonrecovery and fast-recovery fractions changed in concert with the changes in the mobility of the major faction. There was a close correspondence between the size of the nonrecovery fraction and the recovery time of the major fraction at different times after induction of differentiation for each CBX fusion (average R2 of 0.9) (Fig. 2C). Since both the size of the nonrecovery fraction and the mobility of the major fraction of each CBX fusion changed during ES cell differentiation, the changes in CBX fusion dynamics could not be caused by changes in the viscosity of the nucleoplasm or chromatin condensation. There was also no detectable change in the mobility of histone H3.2 fused to a fluorescent protein, indicating that the dynamics of core nucleosomes containing this histone were not altered during ES cell differentiation (Fig. 2C, bottom). The distinctive pattern of changes in CBX fusion mobilities during ES cell differentiation was therefore unlikely to reflect global changes in chromatin dynamics during ES cell differentiation.
Effects of ES cell differentiation into specific progenitor cells on CBX fusion distributions and mobilities.
ES cells can differentiate into all germ cell layers. Under the conditions used in the previous experiments, many different progenitor cell types are produced. Under some culture conditions, differentiation into specific progenitor cell types is favored (4, 16). We examined whether the conditions used to induce ES cell differentiation or the progenitor cell types produced under those conditions affected the distributions or mobilities of CBX fusions. ES cell embryoid bodies were induced to differentiate under conditions that promote differentiation into either neuronal (4) or adipocyte (16) progenitors. Neuronal differentiation was monitored by staining with nestin and neurofilament antibodies, and adipocyte differentiation was monitored by staining lipid droplets using Oil Red O (Fig. 3A). The distributions of all CBX fusions changed during both neuronal and adipocyte differentiation. We focused on an analysis of the distributions and dynamics of CBX2 and CBX6 fusions. The foci resembling Polycomb bodies disappeared from more than 90% of the cells during neuronal differentiation, whereas small foci were detectable in half of the cells during adipocyte differentiation (Fig. 3A).
FIG. 3.

CBX fusion distributions and mobilities in ES cells induced to differentiate into different cell types. (A) Distributions of CBX fusions in ES cells induced to differentiate into neuronal and adipocyte precursors. Epifluorescence images of the CBX2#16 and CBX6#8 cell lines were acquired before differentiation (a and h), 13 days after the induction of neuronal differentiation (b and i), and 23 days after the induction of adipocyte differentiation (c and j). The cells were also stained by using nestin antibody (ES cells [d and k] and neuronal progenitors [f and m]) or Oil Red O (ES cells [e and l] and adipocyte progenitors [g and n]). The fluorescence of the CBX fusion proteins is shown in green, and the signals corresponding to nestin and Oil Red O are shown in red. (B) The fluorescence recovery in cells comparable to those shown in panel A was quantified and plotted as described for Fig. 1. The curves on the left show the fluorescence recovery in undifferentiated ES cells (black) and in adipocyte (red) as well as neuronal (blue) precursors. The data shown represent mean values and standard deviations (error bars). The images are representative of the majority of cells in each population (see Table S1 in the supplemental material). **, statistically significant difference between the rates of fluorescence recovery in differentiating ES cells and undifferentiated cells (P < 0.01 by Student's t test).
We compared the dynamics of CBX fusions in undifferentiated ES cells and in neuronal and adipocyte precursors by using FRAP analysis. The rates of fluorescence recovery in cells that expressed the CBX2 and CBX6 fusions were reduced during both neuronal and adipocyte differentiation (Fig. 3B). In neuronal progenitors, the time required for recovery of half of the fluorescence of the major fraction of CBX2 and CBX6 fusions was more than twice as long as in undifferentiated ES cells. During adipocyte differentiation, a similar decrease in the rate of fluorescence recovery was observed in cells that expressed the CBX6 fusion and about half of this decrease was observed in cells that expressed the CBX2 fusion. The size of the nonrecovery fraction increased in parallel with the recovery time of the major fraction, and the size of the fast-recovery fraction decreased to the point of being nearly eliminated in neuronal progenitors. The changes in CBX fusion distributions and mobilities are therefore a general characteristic of ES cell differentiation into different progenitor cell types.
Comparison of the mobilities of CBX fusions using different photobleaching approaches.
To further investigate the relationship between CBX fusion localization and dynamics, we used FLIP analysis. FLIP is particularly useful for the visualization of proteins that do not exchange with the bleached region. Cells that expressed the CBX2, CBX7, or CBX8 fusions required two to four times as many bleach cycles as did cells that expressed the CBX6 fusion for half-maximal loss of fluorescence, consistent with a higher mobility of the CBX6 fusion (Fig. 4B). The ranking of the CBX fusions by the number of FLIP cycles and the FRAP recovery time required for a half-maximal change in fluorescence was the same. These values are not predicted to be directly proportional because discontinuous bleaching was used during FLIP analysis to minimize photodamage. The nonexchanging fraction was estimated by extrapolation to an infinite number of bleach cycles (Fig. 4B). For each CBX fusion, the nonexchanging fraction in FLIP analysis was equal in size to the nonrecovery fraction in FRAP analysis. The fast-recovery fraction was not distinguished from the major fraction in FLIP analysis because both fractions were redistributed between bleach cycles. The distribution of the fluorescence remaining after 40 FLIP cycles was similar to the distribution of the fluorescence prior to FLIP analysis, indicating that the nonexchanging fraction had a distribution similar to that of the total population of CBX fusions (Fig. 4A).
FIG. 4.

Mobilities of CBX fusions detected using FLIP analysis. (A) Fluorescence images of CBX2#16, CBX6#8, CBX7#9, and CBX8#1 ES cells were acquired before (a, b, c, and d) and after (a′, b′, c′, and d′) 40 cycles of FLIP. The mobility of the CBX4 fusion was not investigated by using FLIP analysis because of the lower fluorescence intensities of cell lines that expressed this fusion. (B) Quantitation of fluorescence loss in the cell lines shown in panel A. The normalized fluorescence intensities of areas outlined in yellow in panel A that did not overlap the bleach area were plotted as a function of the number of FLIP cycles, together with the best fit of a single-exponential function (curves on the top) (see Materials and Methods). Standard deviations are shown for the data for the fastest and slowest recoveries; others were of similar magnitudes. The graph on the bottom shows the mean number of FLIP cycles required to bleach half of the initial fluorescence (c1/2 [right axis]) superimposed on a stacked histogram of the nonexchanging (red) and exchanging (yellow) fractions (left axis). The data represent mean values and standard deviations (see Table S2 in the supplemental material).
Influence of focus formation on the mobilities of CBX fusions.
To probe the relationship between CBX protein localization and dynamics, we compared the mobilities of CBX fusions localized to Polycomb bodies with their mobilities in regions lacking these foci. A quantitative comparison of the rates of fluorescence recovery and loss in foci and outside foci was possible only for cells that expressed the CBX2 fusion because of the small size and low contrast of foci formed by other CBX fusions. There was no significant difference between the rates of fluorescence recovery in foci and those in regions adjacent to foci (Fig. 5A; see Table S1 in the supplemental material). The size of the nonrecovery fraction was marginally larger in foci than in adjacent regions devoid of foci, but this difference was smaller than the variation between different foci. There was also no significant difference either in the number of FLIP cycles required for half-maximal loss of fluorescence or in the size of the nonexchanging fraction in foci and in regions lacking foci (Fig. 5B; see Table S2 in the supplemental material). Thus, there was no significant difference in the dynamics of CBX2 fusions localized to foci and those localized to regions lacking foci.
FIG. 5.

Effects of localization to Polycomb bodies on the mobility of CBX2 fusions. (A) Comparison of CBX2 fusion mobilities in foci (red) and outside foci (green) by using FRAP analysis. The fluorescence recovery of areas within foci and outside foci was quantified and plotted as described for Fig. 1. (B) Comparison of CBX2 fusion mobilities in foci (red) and outside foci (green) by using FLIP analysis. The fluorescence intensities of areas within foci and outside foci in the unbleached region were quantified and plotted as described for Fig. 2. Both the FRAP and FLIP data represent mean values and standard deviations of paired measurements in foci and adjacent regions outside foci in the same cell (see Tables S1 and S2 in the supplemental material).
Roles of conserved CBX protein domains in their distributions and mobilities.
CBX proteins contain an amino-terminal chromodomain that can bind methyl-K27 H3 and a carboxy-terminal chromobox that can interact with RING proteins (5, 6, 12, 43, 47). We investigated the effects of mutations in these conserved regions on the distributions and mobilities of CBX fusions expressed transiently in HEK293T cells. The distributions of wild-type CBX fusions in HEK293T cells were similar to the distributions observed in undifferentiated ES cells (compare Fig. 6Aa to Ae and 2Aa to Ae). The majority of the mutations in either the chromodomain or the chromobox eliminated or reduced focus formation by CBX fusions (Fig. 6Af to Ay). Notable exceptions included chromobox deletions in CBX2 and CBX7.
FIG. 6.

Distributions and mobilities of CBX fusions with mutations in conserved regions. (A) Fluorescence images of live HEK293T cells that transiently expressed the CBX fusions indicated above the images containing a point mutation in the chromodomain (I1716F) or deletions of the chromodomain (ΔChr) or chromobox (ΔBox) separately or in combination (ΔChrΔBox) as indicated to the left of the images. The image in panel o is shown in half-scale relative to the other panels. WT, wild type. (B) The fluorescence recovery in cells similar to those shown in panel A was quantified and plotted as described for Fig. 1. Statistical significance of the differences between the rates of fluorescence recovery observed for the mutated CBX proteins compared to the wild-type proteins: *, P value was <0.05 using Student's t test; **, P value was <0.01 using Student's t test. The data represent mean values and standard deviations (error bars), and the images are representative of the majority of cells in each population (see Table S1 in the supplemental material).
The mobilities of the wild-type CBX fusions in transiently transfected HEK293T cells were similar to those observed in ES cells (compare Fig. 6B and 2C). The deletion of the chromobox eliminated nearly the entire nonrecovery fraction of all CBX fusions. The deletion of the chromodomain had a smaller effect, but caused cytoplasmic localization of the CBX8 fusion. The deletion of conserved regions in CBX fusions therefore had distinct effects on their distributions and mobilities.
Effects of CBX protein interactions with RING1B on their distributions and mobilities.
To investigate whether the dynamics of the CBX fusions reflected the mobilities of complexes containing other PRC1 proteins, we examined the mobilities of CBX-RING1B complexes by combining FRAP with BiFC analysis. BiFC analysis is based on the formation of a fluorescent complex by an interaction between two proteins fused to nonfluorescent fragments of a fluorescent protein (24). Complexes formed by CBX proteins fused to one fragment of a fluorescent protein and RING1B fused to a complementary fragment had distributions very similar to those observed for the corresponding CBX proteins fused to full-length fluorescent proteins (compare Fig. 7A and 6A). The differences in CBX fusion distributions therefore reflected differences in the distributions of CBX complexes containing RING proteins.
FIG. 7.

Distributions and mobilities of CBX-RING1B BiFC complexes. (A) Fluorescence images of live HEK293T cells that transiently coexpressed the CBX proteins (indicated above the images) fused to VN173 and RING1B fused to YC173 (see Materials and Methods). The fluorescence represents CBX-RING1B complexes. (B) The fluorescence recovery in cells comparable to those shown in panel A was quantified and plotted as described for Fig. 1. The data represent mean values and standard deviations (error bars), and the images are representative of a majority of the cells in each population (see Table S1 in the supplemental material).
The mobilities of BiFC complexes formed by CBX2, CBX4, CBX7, and CBX8 with RING1B were similar to those of the corresponding CBX proteins fused to an intact fluorescent protein (compare Fig. 7B and 6B). The mobilities of BiFC complexes formed by CBX6 with RING1B varied over a broad range among different cells in the population. This heterogeneity may be caused by the stabilization of indirect or transient interactions between CBX6 and RING1B by association of the fluorescent protein fragments. These proteins exhibited much weaker interaction during immunoprecipitation (Fig. 1; see Fig. S2B and S2C in the supplemental material). With the exception of CBX6, the formation of BiFC complexes with RING1B did not affect the mobilities of CBX proteins. The similar mobilities of these CBX fusions and CBX-RING1B BiFC complexes suggest that these CBX fusions quantitatively associate with endogenous RING proteins.
DISCUSSION
PcG proteins regulate a large number of genes whose expression is altered during ES cell differentiation (8, 9, 31). The concerted changes in the distributions and mobilities of CBX fusions during ES cell differentiation suggest that the changes in PcG protein regulatory functions are coupled to changes in the subnuclear organization and dynamics of CBX protein complexes.
Coordinate versus independent regulation of different CBX proteins.
The distributions and dynamics of different CBX family proteins were regulated by both common and unique mechanisms. The parallel changes in localization and mobility by the CBX fusions during ES cell differentiation suggest that they were regulated in concert. All of the CBX fusions bound to the Olig2, Gata6, Egfr, and Hoxa11 promoters and associated with Ring1B in ES cells. CBX fusion binding to these genes changed during ES cell differentiation, whereas there was no change in the efficiency of Ring1B coprecipitation with CBX fusions. The size of the PRC1 complex is also not sufficient to account for the increased immobilization of CBX fusions during ES cell differentiation. The changes in CBX fusion dynamics during ES cell differentiation are therefore likely to reflect changes in chromatin binding.
Different CBX fusions had distinct distributions and mobilities in ES cells. The CBX6 fusion in particular had a distribution and mobility dissimilar from those of the other CBX fusions. These differences correlated with weak interactions of the CBX6 fusion with endogenous Ring1B and PcG target genes as well as with the lack of trimethyl-K27 H3 binding by the isolated CBX6 chromodomain in vitro (6). The mutation of homologous regions of different CBX proteins had distinct effects on their distributions and mobilities, suggesting that interactions with different partners affected their properties. Different CBX fusions also exhibited distinct changes in binding to different promoters during ES cell differentiation, indicating that gene-specific binding by different family members was regulated independently. The distinct characteristics of the CBX fusions in living cells therefore correlated with differences in their interactions with Ring1B, with specific promoters, and with histone peptides in biochemical assays.
Polycomb bodies do not affect CBX protein mobilities and disperse during differentiation.
Several independent lines of evidence indicate that the interactions that cause Polycomb body formation do not affect CBX protein mobilities. The mobilities of CBX fusions in Polycomb bodies did not differ from their mobilities outside the foci. Upon ES cell differentiation, Polycomb bodies dispersed, while the mobilities of all CBX fusions decreased, demonstrating that their mobilities were constrained by mechanisms unrelated to Polycomb bodies. Many of the mutations in conserved regions of CBX proteins had distinct effects on focus formation and on the mobilities of CBX fusions. The interactions that affected CBX protein mobilities were therefore distinct from those that mediated Polycomb body association.
With the exception of chromobox mutations in CBX2 and CBX7, mutations in the chromodomains and the chromoboxes of CBX proteins eliminated their association with Polycomb bodies. These results are consistent with previous results indicating that a knockdown of BMI1 expression reduced the number of foci formed by CBX8 in extracted nuclei (18) and that knockdown of KMT6 resulted in the dispersal of foci formed by BMI1 (23). Focus formation by CBX proteins can therefore be affected both by interactions with other PRC1 components as well as by H3 K27 trimethylation.
The dispersal of CBX fusions during ES cell differentiation was unexpected given the enrichment of PcG proteins within foci in many cells (5, 10, 21, 22, 33, 44). The foci may correspond to gene clusters such as the Hox loci that are associated with PcG complexes over extended regions in ES cells, whereas the dispersed fluorescence may correspond to genes with PcG occupancy only at the promoter (8, 31). This hypothesis predicts that the number of PcG complexes associated with such loci decreases during ES cell differentiation. Most previous studies of PcG protein localization in mammalian cells have been performed with transformed cell lines. In primary cells, Polycomb bodies appear much less distinct (58). The accumulation of mammalian PcG proteins in large foci may be restricted to specific cell types and cells engineered to express high concentrations of PcG proteins.
Changes in CBX protein distributions and dynamics during ES cell differentiation.
The CBX fusion proteins exhibited concerted changes in distributions and dynamics during ES cell differentiation. The distributions of the fusions became more disperse, and the mobilities were more constrained as differentiation progressed. The dispersal of foci and the reduction in mobilities were observed whether ES cells were induced to differentiate into neuronal or adipocyte progenitors. The details of the changes in distributions and mobilities of different CBX fusions varied during different differentiation programs, but the overall tendency toward reduced focus formation and diminished mobility was consistently observed, suggesting that these changes were a general characteristic of ES cell differentiation. We hypothesize that the disappearance of foci and the diminished dynamics of CBX fusions during ES cell differentiation were caused by competition for CBX complexes by new PcG target genes in differentiating cells (Fig. 8).
FIG. 8.

Hypothetical model for the mobilities and distributions of CBX protein complexes. Mobile and immobile complexes containing CBX fusions are represented by squares and circles, respectively. Differentiation induces a shift from a clustered to a disperse distribution and increases the proportion of immobile complexes. The thicker arrows indicate a slower rate of PRC1 complex exchange in differentiated cells.
There are concurrent changes in the organization of pericentric heterochromatin and in the dynamics of other chromatin-associated proteins (35). The changes in CBX fusion distributions did not correlate with the changes in heterochromatin organization. Whereas Hoechst-stained heterochromatin condensed to form more discrete foci during differentiation, the foci formed by CBX fusions in pluripotent ES cells dispersed during differentiation.
All CBX fusions exhibited an initial increase in mobility during the first day following the induction of differentiation and a subsequent decrease in mobility to a level lower than that observed in pluripotent ES cells. The initial increase in mobility may reflect the mobilization of chromatin-associated proteins involved in the transition from a pluripotent state to a differentiated state. Conversely, the subsequent decrease in mobility may reflect a greater stability of PRC1 complex association with genes that are repressed in differentiated cells than with genes that are poised for expression in pluripotent cells (Fig. 8). These reciprocal shifts in CBX protein mobilities are unique among chromatin-associated proteins whose mobilities have been studied during ES cell differentiation. We observed no change in the dynamics of stably expressed histone H3.2 fused to the N terminus of Venus. CBX fusions therefore exhibited distinctive changes in mobility during ES cell differentiation that did not correlate with changes in the mobilities of other chromatin components.
Multiple subpopulations of CBX proteins with distinct dynamics.
Three subpopulations of each CBX fusion with distinct dynamics were observed. Several independent lines of evidence validate these subpopulations, including (i) coordinate regulation during differentiation, (ii) corroboration by FRAP and FLIP analysis, (iii) consistent dynamics in stable ES cell lines and transiently transfected cells, (iv) depletion of individual fractions by deletion of the chromobox and by differentiation into neuronal progenitor cells, and (v) equivalent characteristics of most CBX-RING1B BiFC complexes and CBX fusions. We attribute the nonrecovery, major, and fast-recovery fractions to CBX protein complexes that are stably, transiently, and indirectly associated with chromatin, respectively. The low mobility levels of CBX proteins stably associated with histones were corroborated by direct measurement of the mobilities of CBX-H3.1 BiFC complexes by using FRAP analysis (less than 40% recovery in 10 min [data not shown]). Stable versus transient association with chromatin may be due to differences in the chromatin, the composition or conformation of the complexes containing CBX proteins, or interactions with other chromatin-binding proteins (Fig. 8).
The mobilities of CBX proteins observed in mouse ES cells and human 293T cells were of the same order of magnitude as the exchange rates of Pc and Ph proteins in Drosophila wing disks and on salivary gland chromosomes and the mobility of BMI1 in U2OS osteosarcoma-derived cell lines (20, 23). The changes in the mobility of the major fraction of CBX fusions during ES cell differentiation were also comparable in magnitude to the differences in exchange rates of Ph and Pc at different polytene chromosome bands and the differences in BMI1 mobility at different stages of the cell cycle (20, 23). The mobility levels of PRC1 proteins are high considering that one of their major functions is long-term gene silencing. However, their mobility levels are low compared to those of many other chromatin-associated proteins and transcription factors. Different transcription regulatory proteins have dramatically distinct mobilities (13, 19, 34, 42, 60). This variation and the multiple dynamic fractions of CBX complexes may reflect the contrasting requirements of stable gene expression under static conditions and rapid responses to external stimuli. However, the significance of differences in transcription factor dynamics for the regulation of individual genes remains to be established.
Distributions and dynamics of complexes formed by CBX proteins with RING1B.
Most studies of protein dynamics using photobleaching approaches monitor the total population of fluorescent fusion proteins and therefore provide information about the aggregate mobilities of different types of complexes containing the protein. Analysis of the dynamics of BiFC complexes enables the determination of the mobilities of complexes formed by specific combinations of interaction partners (24, 29, 45). The association of the fluorescent protein fragments is likely to stabilize the complex (24, 27). Therefore, the mobility of the BiFC complex does not account for possible effects of complex dissociation on mobility. The similar distributions and dynamics of most CBX-RING1B BiFC complexes and CBX fusions expressed alone indicate that these CBX fusions were quantitatively associated with endogenous RING proteins and that their dissociation did not contribute to the mobilities of CBX fusions. These results also indicate that the multiple dynamic subpopulations of CBX fusions did not result from differential association with RING proteins. Thus, the association with RING proteins was necessary, but not sufficient, for the immobilization of CBX fusions. The efficiency of Ring1B coprecipitation with CBX fusions was also unaffected by ES cell differentiation, suggesting that the changes in CBX fusion dynamics during ES cell differentiation did not involve changes in their interactions with Ring1B.
CBX protein immobilization requires interactions with Ring1B, but not direct binding to trimethyl-K27 H3.
The deletion of the chromobox, which can mediate interactions with Ring1B, eliminated the immobile subpopulation of each CBX fusion. In contrast, deletion of the chromodomain, which can directly bind trimethyl-K27 H3, had only a small effect on the mobility of each CBX fusion and did not eliminate the immobile subpopulations. These results indicate that stable binding by CBX proteins does not require direct recognition of trimethyl-K27 of histone H3, but does require interactions with Ring1B that mediate PRC1 complex assembly. This interpretation is consistent with previous observations that many functions of PRC1 complexes are maintained in EED1 and Suz12 null mutants that have no detectable trimethylation of H3 K27 (39, 46).
The association of PRC1 complexes with many genes is correlated with an enrichment of trimethyl-K27 H3 (8, 9). However, the causal basis of this correlation has not been unambiguously established in mammalian cells. PRC1 binding to some genes is reduced in ES cells derived from EED1 as well as Suz12 null mutants (8, 39). The overexpression of KDM6B H3 trimethyl-K27 demethylase in U2OS cells reduces binding by several PRC1 proteins to chromatin (1). Conversely, the knockdown of KDM6A H3 trimethyl-K27 demethylase in HEK293 cells increases PRC1 occupancy at some genes and reduces their expression (30). One interpretation of the apparent contradiction between stable chromatin binding by CBX mutants lacking the chromodomain and reduced PRC1 occupancy at some genes in Eed and Suz12 null mutants as well as in cells overexpressing H3-K27 demethylases is that other subunits of the PRC1 complex recognize trimethyl-K27 H3. Additionally, H3 K27 trimethylation could be required for the stable recruitment of PRC1 complexes only at some promoters or in specific cell types. Further studies are required to determine the mechanisms of CBX protein recruitment independent of chromodomain recognition of trimethyl-K27H3.
Supplementary Material
Acknowledgments
We thank J'aime Manion, Zunair Mahmood, and Shoumei Bai for developing reagents for these studies. We thank Neil Brockdorff and Atsushi Miyawaki for generously sharing materials and members of the Kerppola laboratory for constructive criticisms. Fluorescence photobleaching was performed at the University of Michigan Microscopy and Image Analysis Core supported by the Michigan Diabetes Research and Training Center.
Footnotes
Published ahead of print on 3 March 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Agger, K., P. A. Cloos, J. Christensen, D. Pasini, S. Rose, J. Rappsilber, I. Issaeva, E. Canaani, A. E. Salcini, and K. Helin. 2007. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449731-734. [DOI] [PubMed] [Google Scholar]
- 2.Akasaka, T., M. Kanno, R. Balling, M. A. Mieza, M. Taniguchi, and H. Koseki. 1996. A role for mel-18, a Polycomb group-related vertebrate gene, during the anteroposterior specification of the axial skeleton. Development 1221513-1522. [DOI] [PubMed] [Google Scholar]
- 3.Alkema, M. J., J. Jacobs, J. W. Voncken, N. A. Jenkins, N. G. Copeland, D. P. Satijn, A. P. Otte, A. Berns, and M. van Lohuizen. 1997. MPc2, a new murine homolog of the Drosophila Polycomb protein is a member of the mouse Polycomb transcriptional repressor complex. J. Mol. Biol. 273993-1003. [DOI] [PubMed] [Google Scholar]
- 4.Bain, G., D. Kitchens, M. Yao, J. E. Huettner, and D. I. Gottlieb. 1995. Embryonic stem cells express neuronal properties in vitro. Dev. Biol. 168342-357. [DOI] [PubMed] [Google Scholar]
- 5.Bárdos, J. I., A. J. Saurin, C. Tissot, E. Duprez, and P. S. Freemont. 2000. HPC3 is a new human Polycomb orthologue that interacts and associates with RING1 and Bmi1 and has transcriptional repression properties. J. Biol. Chem. 27528785-28792. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein, E., E. M. Duncan, O. Masui, J. Gil, E. Heard, and C. D. Allis. 2006. Mouse Polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 262560-2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyer, L. A., D. Mathur, and R. Jaenisch. 2006. Molecular control of pluripotency. Curr. Opin. Genet. Dev. 16455-462. [DOI] [PubMed] [Google Scholar]
- 8.Boyer, L. A., K. Plath, J. Zeitlinger, T. Brambrink, L. A. Medeiros, T. I. Lee, S. S. Levine, M. Wernig, A. Tajonar, M. K. Ray, G. W. Bell, A. P. Otte, M. Vidal, D. K. Gifford, R. A. Young, and R. Jaenisch. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441349-353. [DOI] [PubMed] [Google Scholar]
- 9.Bracken, A. P., N. Dietrich, D. Pasini, K. H. Hansen, and K. Helin. 2006. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 201123-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchenau, P., J. Hodgson, H. Strutt, and D. J. Arndt-Jovin. 1998. The distribution of Polycomb-group proteins during cell division and development in Drosophila embryos: impact on models for silencing. J. Cell Biol. 141469-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buszczak, M., and A. C. Spradling. 2006. Searching chromatin for stem cell identity. Cell 125233-236. [DOI] [PubMed] [Google Scholar]
- 12.Cao, R., L. Wang, H. Wang, L. Xia, H. Erdjument-Bromage, P. Tempst, R. S. Jones, and Y. Zhang. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2981039-1043. [DOI] [PubMed] [Google Scholar]
- 13.Cheutin, T., A. J. McNairn, T. Jenuwein, D. M. Gilbert, P. B. Singh, and T. Misteli. 2003. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299721-725. [DOI] [PubMed] [Google Scholar]
- 14.Coré, N., S. Bel, S. J. Gaunt, M. Aurrand-Lions, J. Pearce, A. Fisher, and M. Djabali. 1997. Altered cellular proliferation and mesoderm patterning in Polycomb-M33-deficient mice. Development 124721-729. [DOI] [PubMed] [Google Scholar]
- 15.Cui, B., Y. Liu, and M. A. Gorovsky. 2006. Deposition and function of histone H3 variants in Tetrahymena thermophila. Mol. Cell. Biol. 267719-7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dani, C., A. G. Smith, S. Dessolin, P. Leroy, L. Staccini, P. Villageois, C. Darimont, and G. Ailhaud. 1997. Differentiation of embryonic stem cells into adipocytes in vitro. J. Cell Sci. 1101279-1285. [DOI] [PubMed] [Google Scholar]
- 17.del Mar Lorente, M., C. Marcos-Gutierrez, C. Perez, J. Schoorlemmer, A. Ramirez, T. Magin, and M. Vidal. 2000. Loss- and gain-of-function mutations show a Polycomb group function for Ring1A in mice. Development 1275093-5100. [DOI] [PubMed] [Google Scholar]
- 18.Dietrich, N., A. P. Bracken, E. Trinh, C. K. Schjerling, H. Koseki, J. Rappsilber, K. Helin, and K. H. Hansen. 2007. Bypass of senescence by the Polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J. 261637-1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Festenstein, R., S. N. Pagakis, K. Hiragami, D. Lyon, A. Verreault, B. Sekkali, and D. Kioussis. 2003. Modulation of heterochromatin protein 1 dynamics in primary mammalian cells. Science 299719-721. [DOI] [PubMed] [Google Scholar]
- 20.Ficz, G., R. Heintzmann, and D. J. Arndt-Jovin. 2005. Polycomb group protein complexes exchange rapidly in living Drosophila. Development 1323963-3976. [DOI] [PubMed] [Google Scholar]
- 21.Gil, J., D. Bernard, D. Martinez, and D. Beach. 2004. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 667-72. [DOI] [PubMed] [Google Scholar]
- 22.Grimaud, C., F. Bantignies, M. Pal-Bhadra, P. Ghana, U. Bhadra, and G. Cavalli. 2006. RNAi components are required for nuclear clustering of Polycomb group response elements. Cell 124957-971. [DOI] [PubMed] [Google Scholar]
- 23.Hernández-Muñoz, I., P. Taghavi, C. Kuijl, J. Neefjes, and M. van Lohuizen. 2005. Association of BMI1 with Polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol. Cell. Biol. 2511047-11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu, C. D., Y. Chinenov, and T. K. Kerppola. 2002. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9789-798. [DOI] [PubMed] [Google Scholar]
- 25.Isono, K., Y. Fujimura, J. Shinga, M. Yamaki, J. O-Wang, Y. Takihara, Y. Murahashi, Y. Takada, Y. Mizutani-Koseki, and H. Koseki. 2005. Mammalian polyhomeotic homologues Phc2 and Phc1 act in synergy to mediate Polycomb repression of Hox genes. Mol. Cell. Biol. 256694-6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kagey, M. H., T. A. Melhuish, and D. Wotton. 2003. The Polycomb protein Pc2 is a SUMO E3. Cell 113127-137. [DOI] [PubMed] [Google Scholar]
- 27.Kerppola, T. K. 2006. Visualization of molecular interactions by fluorescence complementation. Nat. Rev. Mol. Cell Biol. 7449-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzmichev, A., K. Nishioka, H. Erdjument-Bromage, P. Tempst, and D. Reinberg. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Genes Dev. 162893-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laricchia-Robbio, L., T. Tamura, T. Karpova, B. L. Sprague, J. G. McNally, and K. Ozato. 2005. Partner-regulated interaction of IFN regulatory factor 8 with chromatin visualized in live macrophages. Proc. Natl. Acad. Sci. USA 10214368-14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, M. G., R. Villa, P. Trojer, J. Norman, K. P. Yan, D. Reinberg, L. Di Croce, and R. Shiekhattar. 2007. Demethylation of H3K27 regulates Polycomb recruitment and H2A ubiquitination. Science 318447-450. [DOI] [PubMed] [Google Scholar]
- 31.Lee, T. I., R. G. Jenner, L. A. Boyer, M. G. Guenther, S. S. Levine, R. M. Kumar, B. Chevalier, S. E. Johnstone, M. F. Cole, K. Isono, H. Koseki, T. Fuchikami, K. Abe, H. L. Murray, J. P. Zucker, B. Yuan, G. W. Bell, E. Herbolsheimer, N. M. Hannett, K. Sun, D. T. Odom, A. P. Otte, T. L. Volkert, D. P. Bartel, D. A. Melton, D. K. Gifford, R. Jaenisch, and R. A. Young. 2006. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125301-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorente, M., C. Perez, C. Sanchez, M. Donohoe, Y. Shi, and M. Vidal. 2006. Homeotic transformations of the axial skeleton of YY1 mutant mice and genetic interaction with the Polycomb group gene Ring1/Ring1A. Mech. Dev. 123312-320. [DOI] [PubMed] [Google Scholar]
- 33.Martinez, A. M., S. Colomb, J. Dejardin, F. Bantignies, and G. Cavalli. 2006. Polycomb group-dependent cyclin A repression in Drosophila. Genes Dev. 20501-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNally, J. G., W. G. Muller, D. Walker, R. Wolford, and G. L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 2871262-1265. [DOI] [PubMed] [Google Scholar]
- 35.Meshorer, E., D. Yellajoshula, E. George, P. J. Scambler, D. T. Brown, and T. Misteli. 2006. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell 10105-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagai, T., K. Ibata, E. S. Park, M. Kubota, K. Mikoshiba, and A. Miyawaki. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2087-90. [DOI] [PubMed] [Google Scholar]
- 37.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108193-199. [DOI] [PubMed] [Google Scholar]
- 38.O'Carroll, D., S. Erhardt, M. Pagani, S. C. Barton, M. A. Surani, and T. Jenuwein. 2001. The Polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 214330-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasini, D., A. P. Bracken, J. B. Hansen, M. Capillo, and K. Helin. 2007. The Polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 273769-3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pearce, J. J., P. B. Singh, and S. J. Gaunt. 1992. The mouse has a Polycomb-like chromobox gene. Development 114921-929. [DOI] [PubMed] [Google Scholar]
- 41.Penny, G. D., G. F. Kay, S. A. Sheardown, S. Rastan, and N. Brockdorff. 1996. Requirement for Xist in X chromosome inactivation. Nature 379131-137. [DOI] [PubMed] [Google Scholar]
- 42.Reid, G., M. R. Hubner, R. Metivier, H. Brand, S. Denger, D. Manu, J. Beaudouin, J. Ellenberg, and F. Gannon. 2003. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 11695-707. [DOI] [PubMed] [Google Scholar]
- 43.Satijn, D. P., and A. P. Otte. 1999. RING1 interacts with multiple Polycomb-group proteins and displays tumorigenic activity. Mol. Cell. Biol. 1957-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saurin, A. J., C. Shiels, J. Williamson, D. P. Satijn, A. P. Otte, D. Sheer, and P. S. Freemont. 1998. The human Polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J. Cell Biol. 142887-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt, U., K. Richter, A. B. Berger, and P. Lichter. 2006. In vivo BiFC analysis of Y14 and NXF1 mRNA export complexes: preferential localization within and around SC35 domains. J. Cell Biol. 172373-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schoeftner, S., A. K. Sengupta, S. Kubicek, K. Mechtler, L. Spahn, H. Koseki, T. Jenuwein, and A. Wutz. 2006. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 253110-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoorlemmer, J., C. Marcos-Gutierrez, F. Were, R. Martinez, E. Garcia, D. P. Satijn, A. P. Otte, and M. Vidal. 1997. Ring1A is a transcriptional repressor that interacts with the Polycomb-M33 protein and is expressed at rhombomere boundaries in the mouse hindbrain. EMBO J. 165930-5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuettengruber, B., D. Chourrout, M. Vervoort, B. Leblanc, and G. Cavalli. 2007. Genome regulation by Polycomb and Trithorax proteins. Cell 128735-745. [DOI] [PubMed] [Google Scholar]
- 49.Schumacher, A., C. Faust, and T. Magnuson. 1996. Positional cloning of a global regulator of anterior-posterior patterning in mice. Nature 383250. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz, Y. B., and V. Pirrotta. 2007. Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 89-22. [DOI] [PubMed] [Google Scholar]
- 51.Scott, C. L., J. Gil, E. Hernando, J. Teruya-Feldstein, M. Narita, D. Martinez, T. Visakorpi, D. Mu, C. Cordon-Cardo, G. Peters, D. Beach, and S. W. Lowe. 2007. Role of the chromobox protein CBX7 in lymphomagenesis. Proc. Natl. Acad. Sci. USA 1045389-5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao, Z., F. Raible, R. Mollaaghababa, J. R. Guyon, C. T. Wu, W. Bender, and R. E. Kingston. 1999. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 9837-46. [DOI] [PubMed] [Google Scholar]
- 53.Smith, A. G. 1991. Culture and differentiation of embryonic stem cells. J. Tissue Cult. Methods 1389-94. [Google Scholar]
- 54.Suzuki, M., Y. Mizutani-Koseki, Y. Fujimura, H. Miyagishima, T. Kaneko, Y. Takada, T. Akasaka, H. Tanzawa, Y. Takihara, M. Nakano, H. Masumoto, M. Vidal, K. Isono, and H. Koseki. 2002. Involvement of the Polycomb-group gene Ring1B in the specification of the anterior-posterior axis in mice. Development 1294171-4183. [DOI] [PubMed] [Google Scholar]
- 55.Takihara, Y., D. Tomotsune, M. Shirai, Y. Katoh-Fukui, K. Nishii, M. A. Motaleb, M. Nomura, R. Tsuchiya, Y. Fujita, Y. Shibata, T. Higashinakagawa, and K. Shimada. 1997. Targeted disruption of the mouse homologue of the Drosophila polyhomeotic gene leads to altered anteroposterior patterning and neural crest defects. Development 1243673-3682. [DOI] [PubMed] [Google Scholar]
- 56.van der Lugt, N. M., J. Domen, K. Linders, M. van Roon, E. Robanus-Maandag, H. te Riele, M. van der Valk, J. Deschamps, M. Sofroniew, M. van Lohuizen, et al. 1994. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 8757-769. [DOI] [PubMed] [Google Scholar]
- 57.Voncken, J. W., B. A. Roelen, M. Roefs, S. de Vries, E. Verhoeven, S. Marino, J. Deschamps, and M. van Lohuizen. 2003. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc. Natl. Acad. Sci. USA 1002468-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Voncken, J. W., D. Schweizer, L. Aagaard, L. Sattler, M. F. Jantsch, and M. van Lohuizen. 1999. Chromatin-association of the Polycomb group protein BMI1 is cell cycle-regulated and correlates with its phosphorylation status. J. Cell Sci. 1124627-4639. [DOI] [PubMed] [Google Scholar]
- 59.Wang, L., J. L. Brown, R. Cao, Y. Zhang, J. A. Kassis, and R. S. Jones. 2004. Hierarchical recruitment of Polycomb group silencing complexes. Mol. Cell 14637-646. [DOI] [PubMed] [Google Scholar]
- 60.Yao, J., K. M. Munson, W. W. Webb, and J. T. Lis. 2006. Dynamics of heat shock factor association with native gene loci in living cells. Nature 4421050-1053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.