

Abstract
Keap1 and Cul3 constitute a unique ubiquitin E3 ligase that degrades Nrf2, a key activator of cytoprotective genes. Upon exposure to oxidants/electrophiles, the enzymatic activity of this ligase complex is inhibited and the complex fails to degrade Nrf2, resulting in the transcriptional activation of Nrf2 target genes. Keap1 possesses several reactive cysteine residues that covalently bond with electrophiles in vitro. To clarify the functional significance of each Keap1 cysteine residue under physiological conditions, we established a transgenic complementation rescue model. The transgenic expression of mutant Keap1(C273A) and/or Keap1(C288A) protein in Keap1 null mice failed to reverse constitutive Nrf2 activation, indicating that cysteine residues at positions 273 and 288 are essential for Keap1 to repress Nrf2 activity in vivo. In contrast, Keap1(C151S) retained repressor activity and mice expressing this molecule were viable. Mouse embryonic fibroblasts from Keap1(C151S) transgenic mice displayed decreased expression of Nrf2 target genes both before and after an electrophilic challenge, suggesting that Cys151 is important in facilitating Nrf2 activation. These results demonstrate critical roles of the cysteine residues in vivo in maintaining Keap1 function, such that Nrf2 is repressed under quiescent conditions and active in response to oxidants/electrophiles.
Transcription factor Nrf2, as a heterodimer with small Maf, coordinately regulates the inducible expression of cytoprotective genes (10) via cis-acting antioxidant-responsive elements (26) or electrophile response elements (6, 14). Since Nrf2 regulates a multitude of detoxification and antioxidant enzymes, mice deficient in Nrf2 are susceptible to a variety of xenobiotic and oxidative insults (reviewed in references 22 and 33). Previously, investigators in our laboratory identified a cytoplasmic regulatory protein that represses Nrf2 activity and subsequently named this protein Keap1 (10). Keap1 in a complex with Cullin 3 (Cul3) forms an E3 ubiquitin ligase that accelerates the proteasomal degradation of Nrf2 (18, 33) under unstressed conditions (10). When the murine Keap1 gene is disrupted, constitutive Nrf2 activation and the induction of Nrf2 target genes are observed, which manifest in vivo as juvenile lethality due to the Nrf2-mediated formation of hyperkeratotic obstructive lesions in the esophagus and forestomach (35). Importantly, the simultaneous deletion of Nrf2 (35) or genes for small Maf proteins (23) and Keap1 can rescue Keap1 null mice from juvenile lethality, demonstrating that the dimeric complex formed between Nrf2 and a small Maf protein is responsible for the lethal hyperkeratotic phenotype. When Keap1 is disrupted in hepatocytes (24), mice are viable and markedly resistant to acetaminophen hepatotoxicity, indicating the critical involvement of Keap1 as a negative regulator of Nrf2 in xenobiotic responses.
Exposure to electrophiles inhibits Keap1-Cul3 E3 ligase activity and stabilizes Nrf2 (16). Keap1 is a thiol-rich protein possessing 25 cysteine residues, some of which are highly reactive (3). Several previous in vitro studies have revealed that electrophiles form direct covalent bonds with thiol groups of the reactive cysteine residues, suggesting that Keap1 may serve as a sensor of electrophiles (3-5, 8, 9). However, the question still remains as to whether cysteine modifications really turn on or turn off E3 ligase activity and, if so, which cysteine residues are responsible for the signaling.
Keap1 harbors two canonical domains: the N-terminal BTB (broad complex-tramtrack-bric a brac) and the C-terminal DC (double glycine repeat, or Kelch, plus C terminus) domains, which are connected by an intervening region (IVR) (11). BTB domains are typically conserved regions that serve in protein-protein interactions. The Keap1 BTB has been deemed necessary for Keap1 homodimerization by researchers in our laboratory. The DC domain is critical for maintaining the Nrf2-Keap1 interface that recruits the N-terminal region of Nrf2 (the Neh2 domain). The IVR, which links the BTB and DC domains, contains several key cysteines that have been proposed to regulate Keap1 activity as a component of the E3 ligase. Thus, each of the three domains is thought to play a unique role in mediating Nrf2 repression and ubiquitination.
Previous studies attempted to evaluate the functional contribution of individual cysteine residues by testing the abilities of specific Keap1 cysteine mutant proteins to inhibit Nrf2 activity in transient transfection assays (19, 36, 38). Many Keap1 cysteine residues were found to react with electrophiles in in vitro binding assays, yet only a few appeared to be required for Keap1 function when evaluated by site-directed mutagenesis in transient transfection assays (19, 36, 38). This discrepancy seems to be inherent in the model system. In vitro binding assays adopt a simplified system that utilizes much higher than physiological concentrations of each substrate, whereas in transfecto experiments typically overexpress the desired protein and rely on cell lines that have already adapted to culture conditions with high oxygen tensions (∼20%), which may easily cause the functional impairment of each Keap1 mutant protein. Thus, a refined evaluation system was an urgent requirement for clarifying Keap1 function in vivo.
We previously utilized a transgenic complementation rescue approach to evaluate the in vivo functions of regulator proteins (29). In the present study, we identified a 5.7-kb genomic region of Keap1 harboring regulatory sequences sufficient for recapitulating wild-type Keap1 expression. Keap1 was expressed in mice under the regulatory influence of this 5.7-kb cassette, and these transgene-positive mice were crossed with Keap1 null mutant mice. The transgenic expression of wild-type Keap1 fully rescued Keap1 null mice from juvenile lethality, indicating that the 5.7-kb cassette recapitulates the endogenous expression of Keap1. Various Keap1 mutant molecules were expressed in a Keap1 null background instead of a wild-type Keap1 background, and mice were screened for an improvement of the Keap1 null phenotype.
While the BTB domain was shown previously to be dispensable for Keap1 function in transfecto (12, 38), other experiments have identified this domain as a component mandatory for the recruitment of the Cul3-ubiquitin complex, either directly or indirectly (1, 7, 15, 39). The results of our study clearly demonstrated that the BTB domain is absolutely required for Keap1 to act as a repressor of Nrf2. Within the BTB domain, a single cysteine, Cys151, has been suggested to be critical for oxidative stress-inducible modifications of Keap1 leading to Nrf2 activation (27, 38, 39). Other studies have suggested that several different cysteines, such as Cys273 and Cys288 in the IVR, may be essential for Nrf2 degradation. The roles of these cysteine residues under physiological conditions are not fully understood, however. In the transgene complementation analysis, Cys273 and Cys288 seemed to be key to the constitutive repression of Nrf2 activity whereas Cys151 was obligatory for the full activation of Nrf2. This study thus revealed the physiological significance of each reactive cysteine residue of Keap1 in vivo.
MATERIALS AND METHODS
Plasmid construction.
The Keap1 gene regulatory domain (KRD)-LacZ transgene, including the 5.7-kbp upstream regulatory region of the Keap1 gene and cDNA encoding the nuclear localization signal and LacZ, was prepared similarly to the targeting vector of the gene (35) and inserted into the XbaI and PstI sites of pBluescript II SK(+) (Stratagene). A PCR-amplified sequence encoding a hemagglutinin (HA) tag (YPYDVPDYAA) was fused to wild-type or mutant Keap1 cDNAs generated by PCR-based mutagenesis at the ends corresponding to the N termini (12, 36). The HA gene-tagged cDNAs were subcloned into the BssHII site of pEF-BOS (21) for the construction of expression plasmids, ligated to the regulatory region of the Keap1 gene, and cloned into the SalI and EcoRI sites of pBluescript II SK(+) to generate a KRD construct encoding wild-type Keap1 under KRD regulation (KRD-Keap1), a KRD construct encoding a product with the mutations C273A and C288A [KRD-Keap1(C273A-C288A)], a KRD construct encoding a product lacking the BTB domain [KRD-Keap1(ΔBTB)], and a KRD construct encoding a product with the mutation C151S [KRD-Keap1(C151S)]. KRD-Keap1 without a HA tag sequence was also generated by ligating cDNAs encoding Keap1 to the regulatory region of the Keap1 gene and cloning the ligated products into the SalI and EcoRI sites of pBluescript II SK(+). Replacing KRD-Keap1 with the BstEII fragment from cDNAs encoding Keap1(C273A) and Keap1(C288A) generated KRD-Keap1(C273A) and KRD-Keap1(C288A), respectively. Cytomegalovirus-based expression vectors for FLAG-tagged wild-type or mutated Keap1 were generated by inserting the HindIII/XbaI fragments of wild-type or mutant Keap1 cDNAs into the HindIII and XbaI sites of p3xFLAG-CMV-10 (Sigma). The sequences of the primers used for PCR are available upon request.
Generation of transgenic mice.
Transgene constructs were injected into fertilized eggs derived from BDF1 parents. Transgenic mice were generated by standard methods. Transgenic mice expressing wild-type or mutant Keap1 proteins were mated into a Keap1 null background to obtain compound mutant mice (Keap1−/−::transgene). All compound mutant mice examined in this study were from a mixed genetic background, with contributions from 129Sv/J, C57BL/6J, and ICR strains. The primer sequences used for genotyping PCR are shown in Table 1. More than three independent animals from each line were used for examining body weights.
TABLE 1.
Primer sequences for PCR genotypinga
| Primer name | Sequence |
|---|---|
| mKE2aF | 5′-AAAATAAAGTGGCAGGGTCTGGTC-3′ |
| LacZ4R | 5′-CCTGTAGCCAGCTTTCATCAAC-3′ |
| Keap1-4exF | 5′-GGAATGAGTGGCGGATGATCAC-3′ |
| Keap1-6exR | 5′-TGCTTCAGCAGGTACAGTTTTG-3′ |
| Keap1-2ndintR | 5′-CAGTTTTCCTCCAGCCTGTC-3′ |
| Keap1-d132 | 5′-CGGGATCCCCATGGAAAGGCTTATT GAGTTC-3′ |
| TV-Neo | 5′-TCAGAGCAGCCGATTGTCTGTTGTG CCCAGTCAT-3′ |
| Keap1-2exF | 5′-CCGGGAGTATATCTACATGCAC-3′ |
| Keap1-3exR | 5′-CAGCGTGAGCTCCTGGAATATC-3′ |
The genotypes of the mice were determined by PCR. Primers for PCR genotyping were as follows: mKE2aF and LacZ4R for the KRD-LacZ transgene; Keap1-4exF and Keap1-6exR for KRD-Keap1 and its mutant transgenes; and Keap1-2ndintR, Keap1-d132, and TV-Neo for wild-type and targeted Keap1 alleles. The presence of the KRD-Keap1(C273A) or KRD-Keap1(C288A) transgene was detected by PCR with primers Keap1-2exF and Keap1-3exR, followed by digestion with SnaBI. PCR products derived from KRD-Keap1(C273A) and KRD-Keap1(C288A) were distinguished by the presence and absence of the SnaBI site, respectively.
Histological analysis.
β-Galactosidase staining was performed as described previously (21). For hematoxylin and eosin staining, stomachs and esophagi of postnatal day 10 (P10) pups were fixed in 3.7% formalin and embedded in paraffin. For immunohistochemical staining, the stomachs of P10 pups were processed as described previously (23). Samples were treated with an anti-Neh2 antibody at a 40,000-fold dilution or an anti-Nrf2 antibody (C-20; Santa Cruz) at a 400-fold dilution and visualized with biotinylated anti-rabbit immunoglobulin G (IgG) or goat IgG and an ABC kit (Vector Laboratories). Methyl green was used for counterstaining nuclei.
Real-time PCR and RNA blot analysis.
Real-time PCR was performed using an ABI 7700 (Applied Bioscience). Total RNAs were extracted from the forestomachs of P10 pups by using ISOGEN (Nippon Gene). From 1 μg of total RNA, cDNAs were synthesized using Superscript II (Life Technologies). Primers for PCR were as follows: Keap1F (5′-GAT CGG CTG CAC TGA ACT G-3′) with Keap1R (5′-GGA CTC GCA GCG TAC GTT-3′) and Keratin6F (5′-TTG GAC CAG TCA ACA TCT CTG TG-3′) with Keratin6R (5′-ACC CCC GGC ACT GCC-3′). Probes were as follows: Keap1Taq (5′-6-carboxyfluorescein-CTG GCC ACG CTC ATC AGC CG-6-carboxytetramethylrhodamine-3′ ) and Keratin6 (5′-6-carboxyfluorescein-CAG TCC ACC GTG TCC AGC GGC TA-6-carboxytetramethylrhodamine-3′). The primers and probe for Nqo1 detection were described previously (14). Primers and a probe for rRNA (Applied Bioscience) were used as an internal control. Ten micrograms of total RNA per lane was applied in RNA blot analyses. The probes for keratin 6, Nqo1, and glutathione S-transferase class pi (GST-P) mRNAs have been described previously (23).
MEFs.
Mouse embryonic fibroblasts (MEFs) were prepared from embryonic day 13.5 (E13.5) embryos, and stable lines were established by standard procedures (31).
Immunoblot and immunoprecipitation analyses.
Whole-cell extracts from mouse forestomach cells or MEF cells were prepared by harvesting the cells in sodium dodecyl sulfate sample buffer or lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40, 0.4% sodium deoxycholate, protease inhibitor cocktail [1× concentration in the manufacturer's instructions; Roche Diagnostic], 10 μM MG132, and 1 μM phenylmethylsulfonyl fluoride). Immunoblot analysis was performed with rat monoclonal antibody against mouse Keap1 (clone 144) (37) and then anti-rat IgG conjugated with horseradish peroxidase (Zymed). Nuclear extracts from MEFs were prepared as described earlier (2), and immunoblot analyses were performed with rat monoclonal antibody against mouse Nrf2 (clone 103) (34). Anti-lamin B (M-20; Santa Cruz) and anti-α-tubulin (Sigma) antibodies were utilized as markers for nuclear and cytoplasmic fractions, respectively.
To examine Keap1 protein dimerization, 293T cells were cotransfected with expression vectors coding for HA-tagged and FLAG-tagged Keap1 proteins. Whole-cell extracts were prepared in lysis buffer as described above and subjected to immunoprecipitation using an anti-FLAG M2-agarose affinity gel (A-2220; Sigma). After being washed in lysis buffer, immune complexes were visualized by immunoblot analysis using anti-HA (Y-11; Santa Cruz) and anti-FLAG (A-8592; Sigma) antibodies.
Serum chemical analysis.
Blood samples were collected from the retro-orbital venous plexuses of 2-week-old mice and analyzed by using FDC7000V (Fujifilm). Data were obtained from three to five independent mice.
RESULTS
A 5.7-kb upstream sequence of Keap1 allows for the transgenic recapitulation of Keap1 expression.
Regulatory domains of the Keap1 gene were examined by a transgene reporter mouse assay, in which Keap1 knock-in mice harbored the β-galactosidase (lacZ) gene in the Keap1 locus (Keap1+/− mice) (Fig. 1A) (35) as a positive control. In Keap1+/− mice, LacZ reporter expression was observed in a wide range of tissues (Table 2). LacZ was expressed in skin, esophagus, and forestomach tissues (Fig. 1C, F, and I, respectively) of Keap1+/− embryos, in which Keap1 expression is particularly important for avoiding juvenile lethality (23). LacZ expression was also observed in alveolar cells, hepatocytes, and myocardial cells of Keap1+/− mice (Fig. 1L, O, and R).
FIG. 1.

The 5.7-kb upstream region of the Keap1 gene (KRD) recapitulates the endogenous expression profile. (A) Schematic structures of the wild-type Keap1 gene, its targeted allele, and the KRD-LacZ transgene. The 5.7-kb genomic fragment of the Keap1 gene contains the upstream region with three alternative noncoding first exons and part of the second exon. Neo, neomycin resistance cassette. (B to S) LacZ expression observed in KRD-LacZ mice (D, G, J, M, P, and S), Keap1+/− mice (C, F, I, L, O, and R), and wild-type mice (B, E, H, K, N, and Q). The LacZ expression profiles observed for KRD-LacZ mice were compared to those for Keap1+/− mice, in which LacZ was expressed from the targeted allele under the regulation of the endogenous Keap1 gene. Skin (B to D), esophagus (E to G), and forestomach (H to J) tissue samples were examined at E18.5. Lung (K to M), liver (N to P), and heart (Q to S) tissue samples were examined at P1. Scale bars correspond to 400 μm (B to G), 200 μm (H to P), and 2 mm (Q to S).
TABLE 2.
Summary of the LacZ staining pattern of five independent lines of KRD-LacZ micea
| Tissue | Staining of samples from KRD-LacZ mice of:
|
Staining of samples from Keap1+/− mice | ||||
|---|---|---|---|---|---|---|
| Line 2 | Line 4 | Line 5 | Line 7 | Line 8 | ||
| Skin | + | + | + | + | + | + |
| Forestomach | + | − | + | + | + | + |
| Esophagus | + | + | + | + | + | + |
| Lung | + | + | + | + | + | + |
| Heart | + | + | + | + | + | + |
| Liver | + | − | − | + | + | + |
A + signifies that the frequency of LacZ-positive cells in the indicated tissue in a field viewed at a magnification of ×200 was greater than 10%, while a − signifies that the frequency was less than 10%.
We prepared transgenic mouse lines with a 5.7-kb upstream sequence of Keap1 (KRD) driving lacZ reporter expression and then examined LacZ staining in the tissues described above (Fig. 1A). Of the nine transgenic founder mice generated with this construct, five mice were used to establish transgenic mouse lines while four mice were analyzed directly at E18.5. In the latter analysis, LacZ expression was observed in epithelial cells of the esophagi and forestomachs of all four embryos and in skin keratinocytes of three of the embryos (Table 3). The profiles of LacZ expression for KRD and Keap1+/− embryos were similar.
TABLE 3.
KRD directs transcriptional activation in keratinocytesa
| Mouse generation(s) | No. of LacZ-positive samples/no. of KRD-LacZ transgene-positive samples of:
|
||
|---|---|---|---|
| Skin | Esophagus tissue | Forestomach tissue | |
| F0 | 3/4 | 4/4 | 4/4 |
| Lines | 3/5 | 3/5 | 3/5 |
F0 indicates the founder generation of transgenic mice, and “lines” indicates the generations of mice after the transgenic lines were established. LacZ staining of tissue samples from the former and the latter was examined at E18.5 and P1, respectively. The staining patterns were compared with those for Keap1+/− mice, which contain a knock-in LacZ gene, and the similarities between the expression profiles were confirmed.
LacZ expression in a wider range of tissues, including forestomach epithelia, skin keratinocytes, alveolar cells, myocardial cells, and hepatocytes, from the KRD transgenic mouse lines at P1 was also examined. The five transgenic lines showed similar expression profiles, albeit with some minor differences. Three of the lines displayed expression patterns similar to that of the Keap1+/− founder mice (Fig. 1D, G, and J and M, P, and S; Table 2). The LacZ expression profiles were consistent among the three KRD transgenic mouse lines and Keap1+/− mice during adulthood (data not shown). These results demonstrate that KRD is sufficient for recapitulating Keap1 expression in vivo.
KRD-mediated Keap1 expression rescues Keap1 null mice from juvenile lethality.
The most stringent verification that KRD is fully functional is to prove that the KRD-Keap1 transgene sufficiently restores Keap1 null mice to a viable state. Therefore, we generated five independent lines of KRD-Keap1 transgenic mice expressing Keap1 under KRD regulation (Fig. 2A). These mice were crossed with Keap1+/− mice to obtain Keap1+/−::KRD-Keap1 transgene (Keap1+/−::TgKeap1) mice, which were further crossed with Keap1+/− mice to generate Keap1−/−::TgKeap1 mice. Five lines of Keap1−/−::TgKeap1 mice were viable and fertile, with body weights indistinguishable from those of wild-type mice, and one line (line 27) had the wild-type phenotype partially restored (Fig. 2B). Transgene-derived Keap1 mRNA expression levels in the forestomachs of these Keap1−/−::TgKeap1 mice was examined by quantitative reverse transcription-PCR and found to be fivefold (line 34), threefold (line 28), and twofold (lines 20 and 38) higher than endogenous Keap1 mRNA expression levels (Fig. 2C). The levels in line 12 were comparable to and those in line 27 were lower than the endogenous levels.
FIG. 2.

Keap1 driven by KRD rescues Keap1-deficient mice from lethality. (A) Schematic structure of the KRD-Keap1 transgene that expresses Keap1 under the regulation of KRD. (B) Growth curves for compound mutant mice expressing Keap1 in a Keap1 null background (Keap1−/−::TgKeap1). More than three animals from each line were examined, and average body weights and standard errors are indicated. (C) Quantitative expression analysis of Keap1 mRNA in the forestomachs of Keap1−/−::TgKeap1 mice by real-time PCR. Values relative to the endogenous Keap1 levels in wild-type mice are presented. Triplicate samples were analyzed, and the means and standard errors are indicated. (D) Immunoblot analysis of Keap1 expression in forestomachs. Whole-cell extracts of the forestomachs of wild-type (lane 1) and Keap1+/− (lane 2) mice and of Keap1−/−::TgKeap1 mice of lines 34, 28, 20, 38, and 27 (lanes 3, 4, 5, 6, and 7, respectively) were analyzed. α-Tubulin was used as a control. (E to M) Hematoxylin and eosin staining of the forestomachs and esophagi of wild-type (E to G), Keap1−/− (H to J), and Keap1−/−::TgKeap1 (K to M) mice at P10. (E, H, and K) Sagittal sections of whole stomachs. (F, I, and L) High-power magnifications of the rectilinear regions converting the limiting ridges in panels E, H, and K, respectively. White and black double-ended arrows indicate the cornified layers and spinous layers of the stomachs, respectively. The scale bar corresponds to 400 μm. (G, J, and M) Transverse sections of esophagi. The white double-ended arrow in panel J indicates the thickened cornified layer. The scale bar corresponds to 100 μm. (N) Serum biochemical data monitoring the nutritional status. Glucose and cholesterol levels and blood urea nitrogen (BUN)/creatinine ratios in wild-type, Keap1+/−, Keap1−/−, and Keap1−/−::TgKeap1 (line 38) mice were measured. Three to five animals from each line were examined, and means and standard errors are indicated.
We also examined the levels of Keap1 protein expression in forestomachs by immunoblotting. However, we found that the expression levels of transgene-derived Keap1 protein were lower than those of endogenous Keap1 protein (Fig. 2D), suggesting that the production of transgene-derived Keap1 was affected at the posttranscriptional stage by some unknown factors. In the low-level expressers (line 27), Keap1 protein was hardly detected, yet Keap1−/−::TgKeap1 line 27 mice had the wild-type phenotype partially restored (Fig. 2B). It should be noted that Keap1−/−::TgKeap1 line 27 mice were viable and fertile, indicating that this low level of Keap1 expression can rescue Keap1 null mice from lethality.
Histological analyses of P10 mice were performed to examine whether or not KRD-driven Keap1 eliminated the upper digestive tract abnormalities found in Keap1 null mice. In agreement with our previous observation (35), Keap1−/− mice suffered from severe hyperkeratinization of the forestomach (Fig. 2H and I), unlike wild-type mice (Fig. 2E and F). Hyperkeratinization and muscle layer atrophy were also seen in the esophagi of Keap1−/− mice (Fig. 2J), unlike those of wild-type mice (Fig. 2G). In contrast, forestomachs (Fig. 2K and L) and esophagi (Fig. 2M) of Keap1−/−::TgKeap1 mice were indistinguishable from those of wild-type mice (Fig. 2E to G). Serum analyses revealed that Keap1 null mice suffered from high cholesterol, low glucose, and a high ratio of blood urea nitrogen levels to creatinine levels, indicative of severe malnutrition (Fig. 2N). In contrast, Keap1−/−::TgKeap1 mice had values within the normal range, indicating full rescue from starvation. Taken together, these results indicate that KRD-driven Keap1 can rescue Keap1 null mice from juvenile lethality.
The BTB domain is indispensable for Keap1 function in vivo.
The BTB domain, of which the function still remains unclear, was examined for its physiological significance by adopting the transgenic complementation rescue approach. We generated a KRD construct with a BTB deletion [KRD-Keap1(ΔBTB)], and the resulting transgenic mice were crossed with Keap1 heterozygous mice and bred back into a Keap1 null background. Four independent lines of Keap1−/−::KRD-Keap1(ΔBTB) mutant (Keap1−/−::TgΔBTB) mice were established (Fig. 3A), with expression levels of the ΔBTB transgene that were 1.5- to 4-fold higher than those of the endogenous Keap1 gene. An immunoblot analysis of whole-cell lysates derived from line 48 with anti-Keap1 antibody showed that the expression of the Keap1(ΔBTB) gene was comparable to that of the wild-type Keap1 transgene (as measured in whole-cell lysates derived from Keap1−/−::TgKeap1 line 38 mice) (Fig. 3B).
FIG. 3.

The BTB domain is indispensable for the in vivo function of Keap1. (A) Quantitative expression analysis of the Keap1(ΔBTB) transgene in the forestomachs of Keap1−/−::TgΔBTB mice by real-time PCR. Values relative to endogenous Keap1 levels in wild-type mice are presented. Triplicate samples were analyzed, and the means and standard errors are indicated. (B) Immunoblot analysis of Keap1 and Keap1(ΔBTB) expression in forestomachs. Whole-cell extracts of the forestomachs of wild-type (lanes 1 and 2) and Keap1 null (lanes 3 and 4) mice and of Keap1−/−::TgKeap1 mice (line 38) (lanes 5 and 6) and Keap1−/−::TgΔBTB mice (line 48) (lanes 7 and 8) were analyzed. α-Tubulin was used as a control. Black and white arrowheads indicate wild-type Keap1 and Keap1(ΔBTB), respectively. (C) Growth curves for Keap1−/−::TgΔBTB mice. (D) RNA blot analysis of total RNAs purified from forestomachs. The abundance of Nqo1 mRNAs was examined. Two animals were examined independently. rRNA was utilized as an internal control.
We found that Keap1−/−::TgΔBTB compound mutant mice died within 3 weeks after birth (Fig. 3C) due to severe hyperkeratosis in the upper digestive tracts (data not shown), a scenario opposite to the transgenic complementation rescue seen with the wild-type Keap1 transgene (Fig. 3C). These results demonstrate that the Keap1(ΔBTB) mutant transgene cannot rescue Keap1−/− mice from lethality. We also discovered that Nrf2 target genes were highly expressed in Keap1−/−::TgΔBTB mice (Fig. 3D and data not shown), indicating that the BTB domain is indeed essential for Keap1 to repress Nrf2 activity.
Two reactive cysteines in the IVR are indispensable for Keap1 activity.
While a number of cysteine residues in Keap1 have been shown previously to react with electrophiles in vitro (Fig. 4A), in transfecto experiments have identified two of these cysteines, Cys273 and Cys288, to be critical for Keap1 to repress Nrf2 (19, 36, 38). To evaluate the in vivo functions of these cysteine residues, we adopted the transgenic complementation rescue approach. If these cysteine residues are truly functional in vivo, cysteine mutant Keap1 molecules will fail to rescue Keap1 null mutant mice from lethality. Three constructs expressing Keap1 mutant proteins with cysteine-to-alanine substitutions (C273A, C288A, and C273A and C288A) (Fig. 4B) under KRD regulation were generated in a manner similar to that for the KRD-Keap1 construct described above.
FIG. 4.

(A) Alignment of Keap1 cysteine residues modified by electrophiles in vitro. Three kinds of electrophiles, dexamethasone 21-mesylate, biotinylated iodoacetamide, and sulforaphane, were applied, and the cysteine residues modified by each reagent are indicated. Physiologically relevant cysteine residues identified in the transgenic complementation rescue analysis in our present study are highlighted in blue or red. #a, #b, #c, #d, and #e indicate references 3, 4, 8, 9, and 5, respectively. (B) Schematic structures of the mutant Keap1 molecules examined in this study. By using these mutant Keap1 molecules, the evaluation of the Keap1 repressor function was executed as an index of the survival of Keap1 null mice carrying a mutant Keap1 transgene expressing a KRD under the regulation of DGR, double glycine repeat.
Five independent lines of KRD transgenic mice harboring alanine substitutions at both positions 273 and 288 of Keap1 [mice expressing the construct KRD-Keap1(C273A-C288A)] were established and showed no apparent abnormalities or growth retardation (Fig. 5F). Mutant Keap1 mRNA expression in line 72 mice was fourfold higher, that in line 26 mice was threefold higher, and that in mice of lines 29, 64, and 23 was up to twofold higher than endogenous levels in wild-type mice (Fig. 5A). When mice were bred to generate Keap1−/− mice expressing the KRD-Keap1(C273A-C288A) construct (Keap1−/−::TgC273A-C288A mice), the compound mutant mice showed severe growth retardation and died approximately 3 weeks after birth (Fig. 5B), a time frame corresponding to that of the deaths of Keap1 null mice. The abnormalities observed in Keap1−/−::TgC273A-C288A mice (Fig. 5C to E) were histologically identical to those in Keap1 null mice.
FIG. 5.

The Keap1(C273A-C288A) mutant form does not rescue Keap1 null mice from lethality. (A) Quantitative expression analysis of the Keap1(C273A-C288A) transgene in the forestomachs of Keap1−/−::TgC273A-C288A mice by real-time PCR. Values relative to the endogenous Keap1 levels in littermate wild-type mice are presented. Triplicate samples were analyzed, and the means and standard errors are indicated. (B) Growth curves for the Keap1−/−::TgC273A-C288A mice. (C to E) Histological examination of hematoxylin- and eosin-stained forestomachs (C and D) and esophagi (E) of line 64 Keap1−/−::TgC273A-C288A mice at P10. (C) Sagittal section of a whole stomach. White and black arrowheads indicate the cardiac part and the pyloric segment of the stomach, respectively. (D) High-power magnification of the limiting ridge region (the region within the square in panel C). White and black double-ended arrows indicate the cornified layer and the spinous layer of the stomach, respectively. (E) Transverse section of the esophagus. A white double-ended arrow indicates the thickened cornified layer. The scale bar corresponds to 400 μm. (F) Growth curves for TgC273A-C288A transgenic mice with a wild-type background.
To ascertain whether or not the double alanine mutation truly affects the ability of Keap1 to repress Nrf2 or not, the forestomachs of Keap1−/−::TgC273A-C288A line 64 mice were immunostained with anti-Nrf2 antibody. Like those in the nuclei from Keap1 null mice (Fig. 6B), intense signals in the nuclei of Keap1−/−::TgC273A-C288A mouse forestomach cells were observed (Fig. 6D), indicating abundant Nrf2 accumulation. In contrast, no Nrf2 signal in wild-type (Fig. 6A) or Keap1−/−::TgKeap1 (line 38) (Fig. 6C) mice was detected.
FIG. 6.

Constitutive nuclear accumulation of Nrf2 caused by the C273A-C288A mutation of Keap1. (A to D) Immunohistochemical staining of forestomach epithelia of wild-type (A), Keap1−/− (B), Keap1−/−::TgKeap1 (C), and Keap1−/−::TgC273A-C288A (D) mice with anti-Nrf2 antibody. White lines indicate the positions of the basement membranes. Brown staining can be seen in the nuclei of cells from Keap1−/− and Keap1−/−::TgC273A-C288A mice (arrows). (E, top panel) Immunoblot analysis showing the expression levels of Keap1 and the Keap1(C273A-C288A) mutant form by using anti-Keap1 antibody. Whole-cell lysates of forestomachs derived from Keap1−/−::TgKeap1 (line 38) and Keap1−/−::TgC273A-C288A (line 64) mice were analyzed. Arrowheads indicate Keap1. (E, bottom panel) α-Tubulin was used as an internal control. (F) RNA blot analysis of total RNAs purified from forestomachs. The abundances of keratin 6, Nqo1, and GST-P mRNAs were examined, and these mRNAs were shown to be highly expressed in Keap1−/− and Keap1−/−::TgC273A-C288A mice. rRNA was utilized as an internal control.
We examined the expression levels of Keap1 and the Keap1 mutant proteins in the forestomachs of the compound mutant mice that were histologically analyzed as described above. The transgene expression levels were lower than the endogenous Keap1 gene expression levels but were comparable between Keap1−/−::TgC273A-C288A line 64 and Keap1−/−::TgKeap1 line 38 mice (Fig. 6E). The former died around the weaning age, with abundant Nrf2 accumulation, while the latter normally survived into adulthood without any detectable Nrf2 accumulation. Therefore, we concluded that Keap1 activity was hampered by the C273A-C288A mutation.
We also examined the expression levels of Nrf2 target genes, such as the keratin 6, Nqo1, and GST-P genes, which were all significantly elevated in Keap1 null forestomachs. The expression levels of these genes were returned to normal in Keap1−/−::TgKeap1 mice, while they were still elevated, albeit slightly repressed, in Keap1−/−::TgC273A-C288A mice (Fig. 6F). Considering the lower expression level of the transgene than of the endogenous gene, the expression of Nrf2 target genes may be further repressed if the amount of C273A-C288A mutant Keap1 is equal to that of the endogenous Keap1. The intact double glycine repeat domain of the C273A-C288A mutant Keap1, which sequesters Nrf2, may contribute to partial remaining repressor activity.
Both Cys273 and Cys288 are required for Keap1 repression of Nrf2 in vivo.
To determine the importance of Cys273 and Cys288 individually in repressing Nrf2, constructs with the single cysteine mutation C273A or C288A were generated and their repressor activities were tested. Two independent lines each of KRD-Keap1(C273A) (line 22 and 32) and KRD-Keap1(C288A) (lines 42 and 45) mice were generated and crossed with Keap1 null mice. The transgene expression levels of the resulting Keap1−/−::TgC273A line 22 mice and Keap1−/−::TgC288A line 42 mice were comparable to those of Keap1−/−::TgKeap1 line 7 mice (Fig. 7D, bottom panel). Neither Keap1−/−::TgC273A nor Keap1−/−::TgC288A mice survived past weaning (Fig. 7A), indicating that a single mutation of either cysteine residue inhibits the ability of Keap1 to repress Nrf2.
FIG. 7.

The simultaneous presence of Cys273 and Cys288 in a single Keap1 molecule is essential for Keap1 repressor activity. (A) Growth curves for Keap1−/−::TgC273A, Keap1−/−::TgC288A, and Keap1−/−::TgC273A::TgC288A mice. (B) PCR products derived from nontransgenic mice (lane 1), KRD-Keap1(C273A) transgenic mice (TgC273A) (lanes 2 and 3), KRD-Keap1(C288A) transgenic mice (TgC288A) (lanes 4 and 5), and KRD-Keap1(C273A)::KRD-Keap1(C288A) double transgenic mice (TgC273A TgC288A) (lanes 6 and 7), either digested with SnaBI (lanes 3, 5, and 7) or undigested (−) (lanes 1, 2, 4, and 6). Arrowheads and the arrow indicate PCR products digested by SnaBI and undigested products, respectively. (C) Hematoxylin and eosin staining of an esophageal section from a Keap1−/−:: TgC273A::TgC288A mouse at P10. The white double-ended arrow indicates an abnormally thickened cornified layer. (D) Quantitative expression analysis of mRNAs encoding Nqo1, keratin 6, and Keap1 and Keap1 mutant proteins from the transgenes KRD-Keap1(C273A) and/or KRD-Keap1(C288A). Real-time PCR was performed using forestomach RNA from each line of mice. Triplicate samples were analyzed, and the means and standard errors are indicated.
In transfection experiments, we previously found that the compound expression of C273A and C288A mutant proteins restores the repressor activity of Keap1, which is lost when the C273A or C288A mutant protein is expressed independently (36). To address this point, we generated Keap1−/−::TgC273A::TgC288A triple compound mutant mice by crossing KRD-Keap1(C273A) mice, KRD-Keap1(C288A) mice, and Keap1+/− mice. The presence and absence of a SnaBI site differentiated between C273A mutant and C288A mutant transgenes, respectively (Fig. 7B). When the transgene products were quantified by real-time reverse transcription-PCR with a common set of primers, the abundance of the triple compound mutant transgene was approximately equal to the summed abundance of the Keap1−/−::TgC273A and Keap1−/−::TgC288A transgenes (Fig. 7D, bottom panel), suggesting that both transgenes were sufficiently expressed. Nonetheless, the Keap1−/−::TgC273A::TgC288A mice did not survive (Fig. 7A) and displayed severe hyperkeratosis in their esophagi (Fig. 7C) and highly elevated expression of Nrf2 target genes (Fig. 7D, top and middle panels), indicating that Cys273 and Cys288 residues are both essential for the repressor activity of Keap1.
Cys151 is dispensable during homeostatic conditions.
Although Cys273 and Cys288 in the IVR domain are critical for repressor activity, Cys151 has been reported previously to be dispensable for repressor activity but important for the activation of Nrf2 in transfection assays (27, 38, 39). To verify the dispensability of Cys151 in vivo, we mutated the reactive cysteine Cys151 into a serine (Fig. 4B) to generate KRD-Keap1(C151S) mice based on previous work (27, 38, 39). Six independent KRD-Keap1(C151S) transgenic mouse lines were obtained, with transgene mRNA expression levels ranging from one- to sixfold higher than the endogenous Keap1 mRNA expression levels in wild-type mice (Fig. 8A). When these transgenic mice were crossed with Keap1 null mutant mice, the resulting compound mutant mice grew normally, with body weight gains comparable to those of littermate wild-type mice (Fig. 8B). Esophagi and forestomachs were histologically indistinguishable from those of wild-type mice (Fig. 8C-E). These results thus demonstrate that Cys151 is not required for Keap1 to repress Nrf2.
FIG. 8.

The Keap1 C151S mutant molecule rescues Keap1 null mice from lethality but compromises Nrf2 activation. (A) Quantitative expression analysis of the Keap1 C151S mutant transgene in the forestomachs of Keap1−/−::TgC151S mice. Values relative to the endogenous Keap1 levels in wild-type mice are presented. Triplicate samples were analyzed, and the means and standard errors are indicated. (B) Growth curves for independent lines of Keap1−/−::TgC151S mice. (C to E) Histological examination of forestomachs and esophagi of Keap1−/−::TgC151S mice. Forestomachs (C and D) and esophagi (E) of line 16 Keap1−/−::TgC151S mice at P10 were stained with hematoxylin and eosin. (C) Sagittal section of a whole stomach. White and black arrowheads indicate the cardiac part and the pyloric segment of the stomach, respectively. (D) High-power magnification of the limiting ridge region (the region within the square in panel C). The scale bar corresponds to 400 μm.
To verify the contributions of the BTB domain and Cys151 residue to Keap1 homodimer formation, we examined the ability of Keap1(ΔBTB) and Keap1(C151S) mutant molecules to form Keap1 homodimers. 293T cells were cotransfected with plasmids expressing HA-tagged and FLAG-tagged Keap1 proteins. Whole-cell extracts were prepared, subjected to immunoprecipitation with anti-FLAG beads, and tested with anti-HA and anti-FLAG antibodies. An association between HA-tagged and FLAG-tagged molecules was observed for Keap1 and Keap1(C151S) (Fig. 9A, lanes 1 and 2) but not for Keap1(ΔBTB) (lane 3), specifying that the BTB domain, but not Cys151, is essential for Keap1 homodimer formation. These results, together with those of the transgenic complementation rescue analyses (see above), argue that BTB-mediated homodimerization of Keap1 is essential for Keap1 to repress Nrf2.
FIG. 9.

Cys151 is important for the full activation of Nrf2 in response to tBHQ. (A) Immunoprecipitation analysis showing the ability of Keap1 and its mutant molecules to dimerize. 293T cells were cotransfected with HA-tagged and FLAG-tagged molecules. Whole-cell extracts were prepared and subjected to immunoprecipitation with anti-FLAG beads (IP:αFLAG) and immunoblotted with anti-HA and anti-FLAG antibodies. An association between HA-tagged and FLAG-tagged molecules can be seen for Keap1 and the Keap1 C151S mutant form but not for Keap1(ΔBTB). (B, top panel) Immunoblot analysis showing the expression levels of Keap1 and the Keap1 C151S mutant form by using anti-Keap1 antibody. Whole-cell lysates of MEFs derived from Keap1−/−::TgKeap1 (line 34) and Keap1−/−::TgC151S (line 16) mice were analyzed. (B, bottom panel) α-Tubulin was used as an internal control. (C) Immunoblot analysis showing Nrf2 accumulation. MEF cells established from each line of mice were treated with the vehicle alone or 25 μM tBHQ for 2 h prior to cell lysis. Nuclear proteins were prepared and examined by immunoblot analysis using anti-Nrf2 antibody. Anti-lamin B antibody was used as an internal control. +, present; −, absent. (D) Quantitative analysis of the inducible expression of Nqo1 mRNA in MEF cells of various genotypes after treatment with tBHQ. MEF cells were treated with the vehicle alone or 25 μM tBHQ for 16 h prior to cell lysis. Real-time PCR was performed using total RNAs. Triplicate samples were analyzed, and the means and standard errors are indicated.
Cys151 is important for activating Nrf2 in response to tBHQ.
Previous in vitro binding assays suggested that Cys151 is a critical reactive cysteine (4, 5). A transfection assay showed that Cys151 is necessary for the activation of Nrf2 in response to chemicals such as the electrophile tert-butylhydroquinone (tBHQ) (38). Transgenic complementation demonstrated that Cys151 is redundant in basal regulation, but it remains plausible that Cys151 may contribute to the sensor function of Keap1. To determine this issue, we first prepared MEFs from wild-type, Keap1−/−, Keap1−/−::TgKeap1 (line 34), and Keap1−/−::TgC151S (line 6 and 16) mice. Levels of wild-type Keap1 and C151S mutant Keap1 in MEF cells from line 34 wild-type Keap1 mice and line 16 KRD-Keap1(C151S) mice, respectively, were comparable (Fig. 9B).
Nrf2 nuclear accumulation in each MEF cell line in the absence or presence of tBHQ was examined. Nrf2 protein was almost undetectable in the nuclei of Keap1−/−::TgC151S, wild-type, and Keap1−/−::TgKeap1 MEF cells under unstressed conditions (Fig. 9C). This observation is consistent with the finding that the expression of the KRD-Keap1(C151S) transgene counteracted Keap1−/− phenotypical death (Fig. 8B). Upon tBHQ treatment, abundant nuclear accumulation of Nrf2 in Keap1−/−::TgKeap1 and wild-type MEF cells was observed, but this response was markedly decreased in Keap1−/−::TgC151S MEF cells (line 16) (Fig. 9C). These results indicate that Cys151 contributes to the induction of nuclear Nrf2 accumulation under electrophilic stress conditions.
Consistent with the decrease in Nrf2 accumulation in Keap1−/−::TgC151S MEF cells, Nqo1 expression in these cells was also significantly reduced compared to that in Keap1−/−::TgKeap1 and wild-type MEF cells (Fig. 9D, black bars). The basal Nqo1 expression level in Keap1−/−::TgC151S MEF cells under unstressed conditions was also decreased (Fig. 9D, gray bars). Despite the marked decrease in both basal and induced Nqo1 expression, Nqo1 induction could still be observed upon treatment with tBHQ (Fig. 9C and D). These results were fully reproducible in other KRD-Keap1(C151S) transgene-rescued mouse lines (e.g., line 6; data not shown). Keap1(C151S) facilitated the degradation of Nrf2 more efficiently than wild-type Keap1. This result indicates that the Cys151 replacement locks Keap1 in a state more active than that of the wild type and supports our contention that Cys151 is one of the sensor residues mediating Nrf2 activation in response to electrophiles and oxidants.
DISCUSSION
The Nrf2-Keap1 regulatory system is a major pathway of defense against oxidative and electrophilic insults, with Keap1 functioning as a sensor for these stress stimuli. In this study, we examined in vivo the functions of the complete BTB domain and individual reactive cysteines within the BTB and IVR domains by using the transgenic complementation rescue method. This new approach revealed that, in contrast to the results obtained in transfecto (12, 38), the BTB domain is indispensable for Keap1 activity in vivo. We also found that Cys151 in the BTB domain is essential for Nrf2 activation in response to electrophiles while cysteine residues Cys273 and Cys288 in the IVR are essential for Keap1 repression of Nrf2 activity under unstressed conditions (as summarized in Fig. 10). These results support our contention that Keap1 utilizes the differential contribution of each reactive cysteine residue to attain its multiple functions as a sensor and a switch.
FIG. 10.
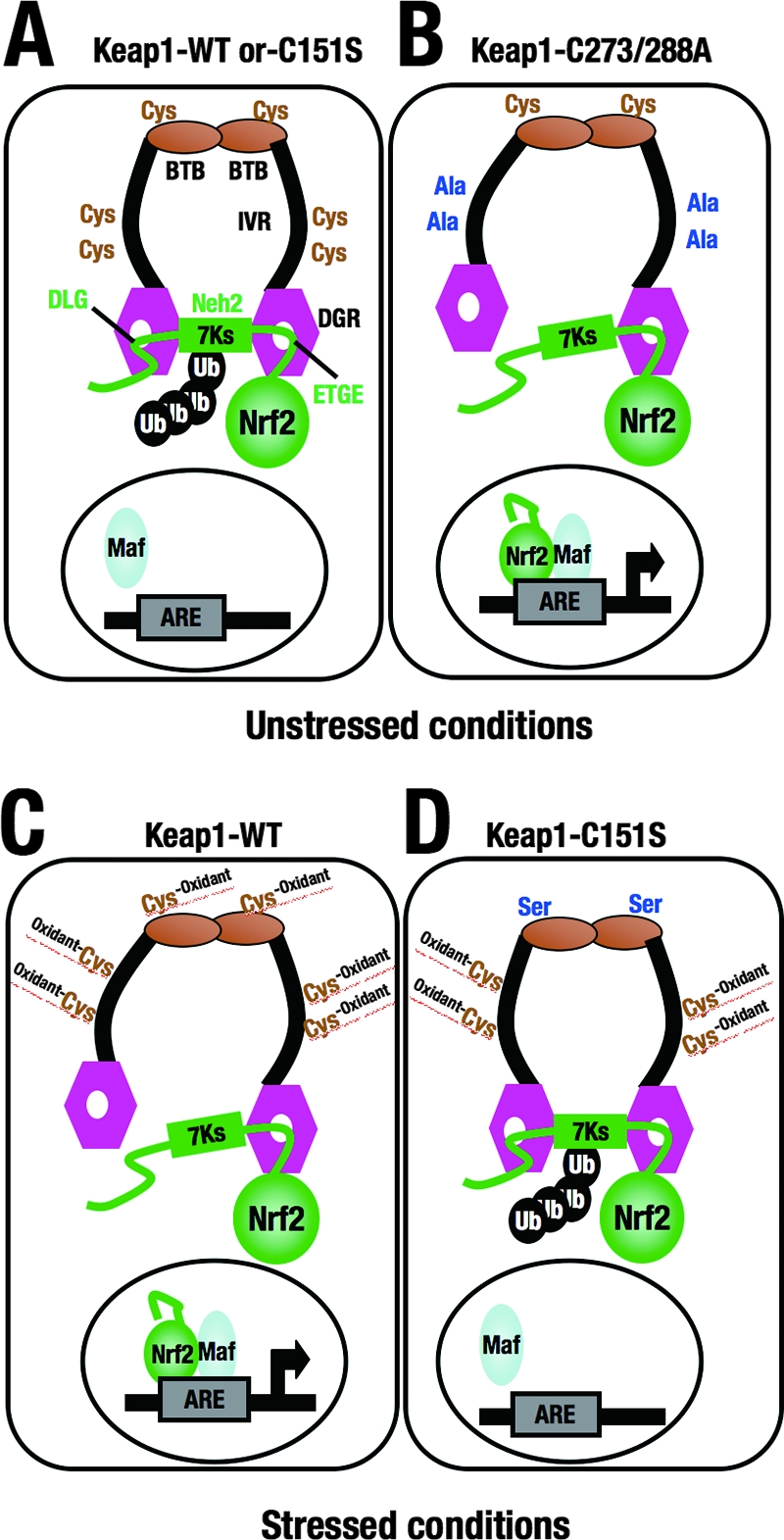
Models based on the results of the transgenic complementation rescue experiments illustrating the distinct role of each cysteine residue in the function of Keap1. Models for unstressed conditions (A and B) and stressed conditions (C and D) are shown. In the absence of stress, wild-type Keap1 (Keap1-WT) and the Keap1 C151S mutant form properly homodimerize and promote the polyubiquitination and proteasomal degradation of Nrf2 (A), whereas the C273A-C288A mutation locks the conformation of Keap1 in the induced state, resulting in the constitutive activation of Nrf2 (B). Under conditions of stress, Keap1 alters its conformation and loses ubiquitin ligase activity (C), while the C151S mutation locks the conformation of Keap1 in the uninduced state, resulting in the constitutive degradation of Nrf2 (D). DGR, double glycine repeat; Ub, ubiquitin; ARE, antioxidant-responsive element; 7Ks, seven lysine residues that are ubiquitinated for degradation.
In Keap1 null mutant mice, severe keratin accumulation obstructs esophageal and forestomach lumens, leading to malnutrition and juvenile lethality (35). When the Keap1 null alleles are complemented with transgene Keap1 driven by the KRD cassette, the level of malnutrition is lessened and the Keap1−/−::TgKeap1 mice start growing normally. This result reflects that the KRD contains regulatory elements sufficient for Keap1 gene expression to rescue Keap1 null mice from juvenile lethality. We surmise that the identification of the KRD is the key achievement enabling us to evaluate the true Keap1 function in vivo.
Our present analysis enabled the assessment of more physiological conditions in terms of expression level and cellular environments than have been evaluated in previous studies. We infer that highly overexpressed Keap1 mutant proteins in transfecto might have overcome their functional impairment per single molecule. Similarly, in in transfecto studies, the cell lines utilized might already have adapted to 20% oxygen, which is higher than the oxygen tension in normal tissues in vivo, such that various cytoprotective systems might have been activated to a certain level. Indeed, we recently found that some immortalized cell lines derived from human carcinoma harbor mutations in the KEAP1 gene that result in the constitutive activation of NRF2 (25, 30). Thus, for authenticity, the real function of Keap1 must be evaluated under physiological conditions, such as those that were exploited in this study.
One striking difference between the results of this in vivo analysis and those from previous in transfecto studies is the functional contribution of Cys273 and Cys288. Previous reporter transfection studies unequivocally demonstrated that the concomitant replacement of Cys273 and Cys288 with alanine in a single Keap1 molecule (a C273A-C288A mutant protein) inhibits the ability of Keap1 to repress Nrf2. When these residues are individually replaced with alanine in separate Keap1 molecules (C273A and C288A mutant proteins) and the mutant proteins are expressed simultaneously in cells, Keap1 activity is not affected (36). This result implied a possible intermolecular interaction between these cysteine residues. In this study, however, the simultaneous expression of C273A and C288A mutant proteins in mice by exploiting the allelic combination of the two mutant transgenes could not rescue Keap1 null mice from juvenile lethality at all. These results indicate that both Cys273 and Cys288 are indispensable for Keap1 repression of Nrf2.
Another significant outcome of this work was the revelation of the indispensable contribution of the BTB domain. Although it was shown previously that the BTB domain mediates Keap1 dimerization (40) and that Keap1 remains as a homodimer in a solution (20, 32), the physiological contribution of the BTB domain to the function of Keap1 was still poorly understood. In previous in transfecto studies, a Keap1(ΔBTB) mutant was found to repress Nrf2 activity, implying a limited or nonexistent contribution from the BTB domain to the role of Keap1. In stark contrast, the present transgenic complementation rescue analysis unequivocally demonstrated the requirement for the BTB domain for the in vivo function of Keap1.
A nuclear magnetic resonance analysis revealed that the Neh2 domain of Nrf2 is an extended rod-like protein and that in the Neh2 domain, the DLG and ETGE motifs flank the central α-helix that contains seven lysine residues, which are targets for ubiquitination by Keap1-Cul3 (32). We found previously that the ETGE motif binds to the Keap1 DC domain (17), and the three-dimensional structure of this complex has been clarified by X-ray crystallography (23). We also recognized previously that the DLG motif binds to the Keap1 DC domain (13, 34). Thus, the ETGE and DLG motifs allow Nrf2 to bind to Keap1 at two sites (32), with a binding stoichiometry between Keap1 and Nrf2 of 2:1. Based on these and other lines of evidence, we proposed a two-site substrate recognition model, in which Nrf2 binds to the Keap1 homodimer through the ETGE and DLG motifs, locking the central α-helix of the Neh2 domain into a position suitable for ubiquitin ligation (20, 32). Our present results demonstrate that the BTB domain is absolutely required for Nrf2 repression, and this finding strongly supports the validity of this model.
It is intriguing that the DLG and ETGE motifs interact with the Keap1 DC domain with disparate binding affinities. The DLG motif has a binding affinity approximately 2 orders of magnitude lower than that of the ETGE motif (32). Keap1 and the Keap1(C273A-C288A) mutant protein bound to Nrf2 with similar binding affinities, but the ability of the mutant protein to mediate Nrf2 degradation was severely impaired (16). Similarly, both Cys273A and Cys288A mutations did not affect the binding of mutant Keap1 to Nrf2 (15). Immunoprecipitation assays usually detect the interaction of the tightly binding ETGE motif but not that of the weakly binding DLG motif. Therefore, we suspected that Cys273A and Cys288A mutant proteins bound to the ETGE motif but not to the DLG motif due to a disrupted conformational relationship between Keap1 and Nrf2, resulting in reduced enzymatic activity of Keap1-Cul3. Given the weak binding affinity between the DLG motif and Keap1, any distortions within the IVR or BTB domain caused by cysteine modifications may prompt the dissociation of the DLG motif from Keap1 while the ETGE motif remains bound. Therefore, we presented a hinge-and-latch mechanism, in that the DLG motif acts as a latch to either enable or disable Nrf2 polyubiquitination depending on the redox state of the cell while the ETGE motif acts as a hinge. In light of the hinge-and-latch mechanism, we envisage that the C151S mutation did not disrupt the conformational relationship between Keap1 and Nrf2 but may have inhibited the dissociation of the DLG motif from Keap1 in response to electrophiles.
An important difference between the Keap1 Cys273-Cys288 and Cys151 mutations is that the former inhibits, while the latter maintains, the ability of Keap1 to repress Nrf2 (Fig. 10). These observations assert that each cysteine residue, individually or in combination, contributes to the overall activity of Keap1 in a distinct manner, such that the role of Keap1 as a redox sensor may be a function of the “cysteine code.” The cysteine residues reported to be attacked by electrophiles in vitro, but not tested in the present study, may also be important participants in the cysteine code operative in vivo. Indeed, quite recently nitric oxide was found to modify cyclic GMP (cGMP) into S-nitorosil-cGMP, which is strongly electrophilic and binds covalently to Keap1 to transduce the stress signal into an increase in cytoprotective enzymes via the Nrf2 pathway (28). It would be intriguing to determine which cysteine in Keap1 is modified by S-nitorosil-cGMP.
In conclusion, Keap1 appears to utilize multiple cysteine residues differentially as acceptors of electrophiles and oxidants to achieve its functional conversion from the active to the inactive state. Considering the distinct patterns of adduct formation depending on the reagents examined, each chemical should attack an inherently determined set of optimal acceptor thiols, i.e., the cysteine code. By establishing the in vivo significance of the three reactive cysteine residues, we have taken the first step in determining the role of these cysteine residues so as to decipher the higher-order conformation of the Nrf2-Keap1-Cul3 complex in both the active and inactive states.
Acknowledgments
We are grateful to N. Wakabayashi and M. I. Kang for critical advice. We also thank N. Kaneko and M. Kimura for technical assistance.
This work was supported by grants from JST-ERATO (M.Y.), the Ministry of Education, Science, Sports and Culture (A.K., H.M., and M.Y.), the Yamanouchi Foundation (H.M.), and the Uehara Memorial Foundation (H.M.).
Footnotes
Published ahead of print on 11 February 2008.
REFERENCES
- 1.Cullinan, S. B., J. D. Gordan, J. Jin, J. W. Harper, and J. A. Diehl. 2004. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 248477-8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dignam, J. D. 1990. Preparation of extracts from higher eukaryotes. Methods Enzymol. 182194-203. [DOI] [PubMed] [Google Scholar]
- 3.Dinkova-Kostova, A. T., W. D. Holtzclaw, R. N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, M. Yamamoto, and P. Talalay. 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 9911908-11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eggler, A. L., G. Liu, J. M. Pezzuto, R. B. V. Breemen, and A. D. Mesecar. 2005. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 10210070-10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eggler, A. L., Y. Luo, R. B. Breeman, and A. D. Mesecar. 2007. Identification of the highly reactive cysteine 151 in the chemopreventive agent-sensor Keap1 protein is method-dependent. Chem. Res. Toxicol. in press. [DOI] [PubMed]
- 6.Frilling, R. S., A. Bensimon, Y. Tichauer, and V. Daniel. 1990. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 876258-6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furukawa, M., and Y. Xiong. 2005. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 25162-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong, F., K. R. Sekhar, M. L. Freeman, and D. C. Liebler. 2005. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 28031768-31775. [DOI] [PubMed] [Google Scholar]
- 9.Hong, F., M. L. Freeman, and D. C. Liebler. 2005. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 181917-1926. [DOI] [PubMed] [Google Scholar]
- 10.Itoh, K., T. Chiba, S. Takahashi, T. Ishii, K. Igarashi, Y. Katoh, T. Oyake, N. Hayashi, K. Satoh, I. Hatayama, M. Yamamoto, and Y. Nabeshima. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236313-322. [DOI] [PubMed] [Google Scholar]
- 11.Itoh, K., N. Wakabayashi, Y. Katoh, T. Ishii, K. Igarashi, J. D. Engel, and M. Yamamoto. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1376-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang, M. I., A. Kobayashi, N. Wakabayashi, S. G. Kim, and M. Yamamoto. 2004. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 1012046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katoh, Y., K. Iida, M. I. Kang, A. Kobayashi, M. Mizukami, K. I. Tong, M. McMahon, J. D. Hayes, K. Itoh, and M. Yamamoto. 2005. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 433342-350. [DOI] [PubMed] [Google Scholar]
- 14.Katsuoka, F., H. Motohashi, T. Ishii, H. Aburatani, J. D. Engel, and M. Yamamoto. 2005. Genetic evidence that small Maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 258044-8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi, A., M. I. Kang, H. Okawa, M. Ohtsuji, Y. Zenke, T. Chiba, K. Igarashi, and M. Yamamoto. 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 247130-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi, A., M. I. Kang, Y. Watai, K. I. Tong, T. Shibata, K. Uchida, and M. Yamamoto. 2006. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 26221-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi, M., K. Itoh, T. Suzuki, H. Osanai, K. Nishikawa, Y. Katoh, Y. Takagi, and M. Yamamoto. 2002. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells 7807-820. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi, M., and M. Yamamoto. 2006. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 46113-140. [DOI] [PubMed] [Google Scholar]
- 19.Levonen, A. L., A. Landar, A. Ramachandran, E. K. Ceaser, D. A. Dickinson, G. Zanoni, J. D. Morrow, and V. M. Darley-Usmar. 2004. Cellular mechanisms of redox cell signaling: role of cysteine modification in controlling antioxidant defense in response to electrophilic lipid oxidation products. Biochem. J. 378373-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMahon, M., N. Thomas, K. Itoh, M. Yamamoto, and J. D. Hayes. 2006. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 28124756-24768. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima, S., and S. Nagata. 1990. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 185322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Motohashi, H., and M. Yamamoto. 2004. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10549-557. [DOI] [PubMed] [Google Scholar]
- 23.Motohashi, H., F. Katsuoka, J. D. Engel, and M. Yamamoto. 2004. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 1016379-6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okawa, H., H. Motohashi, A. Kobayashi, H. Aburatani, T. W. Kensler, and M. Yamamoto. 2005. Hepatic-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 33979-88. [DOI] [PubMed] [Google Scholar]
- 25.Padmanabhan, B., K. I. Tong, T. Ohta, Y. Nakamura, M. Scharlock, M. Ohtsuji, M. I. Kang, A. Kobayashi, S. Yokoyama, and M. Yamamoto. 2006. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 21689-700. [DOI] [PubMed] [Google Scholar]
- 26.Rushmore, T. H., M. R. Morton, and C. B. Pickett. 1991. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. J. Biol. Chem. 26611632-11639.1646813 [Google Scholar]
- 27.Sakurai, T., M. Kanayama, T. Shibata, K. Itoh, A. Kobayashi, M. Yamamoto, and K. Uchida. 2006. Ebselen, a seleno-organic antioxidant, as an electrophile. Chem. Res. Toxicol. 191196-1204. [DOI] [PubMed] [Google Scholar]
- 28.Sawa, T., M. H. Zaki, T. Okamoto, T. Akuta, Y. Tokutomi, S. Kim- Mitsuyama, H. Ihara, A. Kobayashi, M. Yamamoto, S. Fujii, H. Arimoto, and T. Akaike. 2007. Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclic monophosphate. Nat. Chem. Biol. 3727-735. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu, R., S. Takahashi, K. Ohneda, J. D. Engel, and M. Yamamoto. 2001. In vivo requirements for GATA-1 functional domains during primitive and definitive erythropoiesis. EMBO J. 205250-5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh, A., V. Misra, R. K. Thimmulappa, H. Lee, S. Ames, M. O. Hoque, J. G. Herman, S. B. Baylin, D. Sidransky, E. Gabrielson, M. V. Brock, and S. Biswal. 2006. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 3e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tiemann, F., and W. Deppert. 1994. Immortalization of BALB/c mouse embryo fibroblasts alters SV40 large T-antigen interactions with the tumor suppressor p53 and results in a reduced SV40 transformation-efficiency. Oncogene 91907-1915. [PubMed] [Google Scholar]
- 32.Tong, K. I., Y. Katoh, H. Kusunoki, K. Itoh, T. Tanaka, and M. Yamamoto. 2006. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell. Biol. 262887-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong. K. I., A. Kobayashi, F. Katsuoka, and M. Yamamoto. 2006. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol. Chem. 3871311-1320. [DOI] [PubMed] [Google Scholar]
- 34.Tong, K. I., B. Padmanabhan, A. Kobayashi, C. Shang, Y. Hirotsu, S. Yokoyama, and M. Yamamoto. 2007. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 277511-7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wakabayashi, N., K. Itoh, J. Wakabayashi, H. Motohashi, S. Noda, S. Takahashi, S. Imakado, T. Kotsuji, F. Otsuka, D. R. Roop, T. Harada, J. D. Engel, and M. Yamamoto. 2003. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 35238-245. [DOI] [PubMed] [Google Scholar]
- 36.Wakabayashi, N., A. T. Dinkova-Kostova, W. D. Holtzclaw, M. I. Kang, A. Kobayashi, M. Yamamoto, T. W. Kensler, and P. Talalay. 2004. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 1012040-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watai, Y., A. Kobayashi, H. Nagase, M. Mizukami, J. McEvoy, J. D. Singer, K. Itoh, and M. Yamamoto. 2007. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 121163-1178. [DOI] [PubMed] [Google Scholar]
- 38.Zhang, D. D., and M. Hannink. 2003. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 238137-8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang, D. D., S. C. Lo, J. V. Cross, D. J. Templeton, and M. Hannink. 2004. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2410941-10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zipper, L. M., and R. T. Mulcahy. 2002. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 27736544-36552. [DOI] [PubMed] [Google Scholar]