

Abstract
The mRNA stabilizing factor HuR is involved in the posttranscriptional regulation of many genes, including that coding for cyclooxygenase 2 (COX-2). Employing RNA interference technology and actinomycin D experiments, we demonstrate that in human mesangial cells (hMC) the amplification of cytokine-induced COX-2 by angiotensin II (AngII) occurs via a HuR-mediated increase of mRNA stability. Using COX-2 promoter constructs with different portions of the 3′ untranslated region of COX-2, we found that the increase in COX-2 mRNA stability is attributable to a distal class III type of AU-rich element (ARE). Likewise, the RNA immunoprecipitation assay showed AngII-induced binding of HuR to this ARE. Using the RNA pulldown assay, we demonstrate that the AngII-caused HuR assembly with COX-2 mRNA is found in free and cytoskeleton-bound polysomes indicative of an active RNP complex. Mechanistically, the increased HuR binding to COX-2-ARE by AngII is accompanied by increased nucleocytoplasmic HuR shuttling and depends on protein kinase Cδ (PKCδ), which physically interacts with nuclear HuR, thereby promoting its phosphorylation. Mapping of phosphorylation sites identified serines 221 and 318 as critical target sites for PKCδ-triggered HuR phosphorylation and AngII-induced HuR export to the cytoplasm. Posttranslational modification of HuR by PKCδ represents an important novel mode of HuR activation implied in renal COX-2 regulation.
Cyclooxygenases, by catalyzing prostaglandin biosynthesis from arachidonic acid, play an important functional role in a wide spectrum of physiological and pathophysiological cell responses. Up to now, at least two different cyclooxygenase isoforms have been identified, including cyclooxygenase 1 (COX-1), which in most tissues is constitutivly expressed, and COX-2, the abundance of which is strongly induced by a large variety of extracellular stimuli (4; for review, see references 35, 49, and 55). Whereas the constitutive COX-1 expression is mechanistically related to several GC-rich promoter elements present in most housekeeping enzymes, the COX-2 gene resembles a typical immediate-early gene with various prominent transcription factor binding sites in the 5′-flanking region, including a TATA box and cis regulatory elements for transcription factor nuclear factor κB (24), activated protein 1, cyclic AMP-responsive binding protein (27), nuclear factor of interleukin 6 (IL-6) (50), and ETS-1 (17). The control of COX-2 expression is complex and in addition to transcriptional events, underlies a posttranscriptional regulation which is structurally related to a 3′ untranslated region (3′ UTR) encompassing more than 2.0 kb with multiple copies of AU-rich elements (AREs) (8, 9, 48). These cis regulatory elements by recruiting the ARE-containing mRNAs to exosomal mRNA degradation have been implied as major determinants of mammalian mRNA degradation (5, 45, 47). In this regard, the 3′ UTR of COX-2 is a specific target of different RNA-binding proteins, among which the ELAV (embryonic lethal abnormal vision) protein HuR (HuA) is one of the best-characterized ARE binding proteins. Functionally, members of the ELAV protein family have been described to stabilize many inherently unstable mRNAs (14, 61). Unlike its neuron-specific ELAV relatives (HuB, HuC, and HuD), HuR itself is ubiquitously expressed and therefore implicated in a large variety of physiological and pathophysiological processes, including cell growth, differentiation, and inflammation (for review, see references 12, 18, and 34).
Functionally, overexpression of HuR and its subsequent binding to ARE-containing mRNAs leads to a marked increase in the half-life of many short-lived mRNAs, possibly by antagonizing the recruitment of competitive mRNA-destabilizing factors (7, 39, 60). Most intriguingly, increased cytosolic HuR levels as detected in several human cancers coincide with high COX-2 expression. Due to this correlation, the cellular HuR content serves as an eligible indicator for a poor survival prognosis of cancer patients (9, 13, 36).
Since HuR is mainly located within the nucleus, its export to the cytoplasm is an important prerequisite for its protective effects on cognate target mRNAs from rapid RNA decay (14). The structure of HuR revealed a basic hinge region which bears a novel nuclear shuttling sequence denoted as “HNS” (HuR nucleocytoplasmic shuttling sequence) which contains a nuclear export signal as well as a typical nuclear localization sequence, which are both implicated in the nucleocytoplasmic HuR shuttling (14, 26). Physiologically, the regulation of HuR shuttling is a target of different signaling pathways, including the mitogen-activated protein kinases (MAPKs) (33, 53, 57, 58), the AMP-activated kinase (AMPK) (54), and protein kinase Cα (PKCα) (10, 38). A critical contribution of these signaling devices in the stabilization of several labile mRNAs has been experimentally confirmed by various studies (for review, see reference 12). However, the underlying mechanisms of how these pathways discharge into HuR shuttling are still poorly understood.
Previously, we have reported on a PKCα-dependent serine-phosphorylation of HuR and its critical role in the ATP-induced HuR export to the cytoplasm (10). Functionally, the PKC-dependent increase in HuR shuttling is critically involved in the amplification of cytokine-triggered COX-2 expression by ATP but is not required for induction of COX-2 by cytokines (10). Consistent with these findings, PKC stimulation of neuroblastoma cells was shown to induce a nucleocytoplasmic shuttling of the neuronal ELAV proteins HuB, HuC, and HuD, but, in contrast to our study, this modulation occurs via phosphorylation at threonine residues (38). Therefore, a posttranslational modification of HuR by PKC seems to represent an important mode of stimulus-dependent HuR shuttling. In silico analysis of the human HuR protein sequence revealed several target motifs for PKC-dependent phosphorylation and a multitude of putative modification sites for glycosylation and myristoylation. PKC is a family of serine/threonine protein kinases consisting of at least 10 isoforms which comprise the conventional α, β, and γ PKCs; the novel δ, ɛ, η, and θ PKCs; and the atypical ζ, λ, and ι PKCs (3).
In the kidney, PKC participates physiological key functions, most importantly, the control of renal hemodynamics and tubular transport mechanisms (3, 20, 21, 56). Renal mesangial cells (MC) express four PKC isoforms (42) of these and respond to a variety of vasoactive agents, including the strong vasoconstricting peptide angiotensin II (AngII) (3). In addition to the PKC-dependent activation of cPLA2 and subsequent liberation of arachidonic acid (41), the induction of COX-2 complements the capacity of MC to produce vasodilatatory prostaglandins, which are thought as major constituents of a renal feedback mechanism toward AngII (22, 40).
In this study, we report that AngII via involvement of PKCδ can activate a nucleocytoplasmic HuR shuttling in human MC (hMC) and, coincidently, causes an increase in COX-2 mRNA stability with a subsequent rise in prostaglandin E2 (PGE2) formation. Furthermore, our studies suggest that serine phosphorylation of HuR at different target sites can mediate a PKC isotype-specific activation of HuR by different extracellular stimuli.
MATERIALS AND METHODS
Reagents.
Human recombinant IL-1β was from Cell Concept (Umkirch, Germany), and human recombinant TNF-α was from Knoll AG (Ludwigshafen, Germany). AngII, Ponceau red, and an antibody specifically raised against Flag tag were purchased from Sigma-Aldrich (Deisenhofen, Germany). Actinomycin D (Act D; from Streptomyces species) was purchased from Alexis Biochemicals (Laeufelfingen, Switzerland). U0126, SB203580, PD98059, and Gö6976 were obtained from Calbiochem (Schwalbach, Germany). CGP41251 was a kind gift from Novartis Pharma (Basel, Switzerland). Ribonucleotides and modifying enzymes were purchased from Life Technologies (Karlsruhe, Germany). Antibodies raised against β-actin, COX-2, HuR, and histone deacetylase 1 (HDAC-1) and anti-rabbit and anti-mouse horseradish peroxidase-linked immunoglobulin G (IgG) were purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Antibodies for PKCα and PKCδ were obtained from New England Biolabs (Frankfurt am Main, Germany). Fluorescently labeled antibodies were from Molecular Probes (Karlsruhe, Germany). Human recombinant PKCδ was obtained from Biomol GmbH (Hamburg, Germany). All cell culture media and supplements were purchased from Life Technologies (Karlsruhe, Germany).
Cell culture.
Human primary MC were isolated from collagenase IV-treated human glomeruli and cultivated as described previously (43). For experiments, serum-free preincubations were performed in Dulbecco′s modified Eagle′s medium supplemented with bovine serum albumin. For experiments, hMC between passages 4 and 10 were used.
RNA interference.
Gene silencing was performed using small interfering RNAs (siRNAs) for human PKCδ (sc-36253), PKCα (sc-36243), and HuR (sc-35619) from Santa Cruz Biotechnology. Transfection of subconfluent hMC was performed with the Oligofectamine reagent (Invitrogen, Karlsruhe, Germany) according to the manufacturer′s instructions.
Western blot analysis.
Preparation of cytoplasmic and nuclear lysates from cells and subsequent Western blot analysis were performed using standard procedures as described previously (11). After overnight blocking in 5% milk powder in Tris-buffered saline containing 0.05% Tween, Western blots were incubated with the primary antibody overnight at 4°C. Following incubation with a horseradish peroxidase-conjugated secondary antibody, proteins were detected using the enhanced chemiluminescence system (Amersham Bioscience, Freiburg, Germany).
Expression and purification of recombinant protein.
The plasmid pQE-30-His-HuR had been generated by subcloning the plasmid pGEX-HuR encompassing human HuR into the prokaryotic expression plasmid pQE30 (Qiagen, Hilden, Germany) by using internal primers as described previously (10). pGEX-HuR was generated as described previously by Ma et al. (30). The plasmids pQE-His-HuRΔ1, -Δ2, -Δ3, and -Δ4—each containing a single serine-to-alanine substitution at different positions—were generated by changing (underlined) a single base pair (either by changing TCG to GCG, or TCC to GCC) using the following (sense) primers: pQE-His-HuRΔ1 (5′-TTTGACAAACGGGCGGAGGCAGAAGAGGC-3′, corresponding to a region from nucleotides 459 to 488 of the human HuR cDNA; GenBank, accession no. NM_001419), pQE-His-HuRΔ2 (5′-GAGATTCAGGTTCGCCCCCATGGGCGTCG-3′, corresponding to nucleotides 648 to 676), pQE-His-HuRΔ3 (5′-AAATCTTACAGGTTGCCTTCAAAACCAAC-3′, corresponding to a region from nucleotides 938 to 966), and pQE-His-HuRΔ4 (5′-AAAACCAACAAGGCCCACAAATAACTCGC-3′, corresponding to nucleotides 957 to 986). For generation of pQE-30-HuRΔ1/Δ2 bearing two serine-to-alanine substitutions, pQE-His-HuRΔ1 was used as a template and primers were used for generation of pQE-His-HuRΔ2. In a similar way to pQE-30-HuRΔ2/Δ3, bearing two serine-to-alanine substitutions, pQE-His-HuRΔ2 was used as a template with the primers used to generate pQE-His-HuRΔ3. All mutants were generated with the Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA).
In vitro phosphorylation of HuR.
Phosphorylation of HuR by human recombinant PKCs was tested by an in vitro kinase assay using HuR as a substrate and was performed as described previously (10) by using a modified protocol from Geiges et al. (16). Briefly, 5 μg of recombinant PKC was incubated in PKC assay buffer containing 20 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 10 μM Na2ATP, 25 μg/ml phosphatidylserine, 2.5 μg/ml diolein, 1 μl of [γ-32P]dATP (3,000 Ci/mmol), 2 μg of recombinant HuR, and 100 μM CaCl2 in the presence or absence of 100 μM EGTA. Reaction mixtures were incubated at 30°C for 20 min and directly subjected to polyacrylamide gel electrophoresis (PAGE). After fixing, the gels were vacuum dried and radioactive signals were visualized with a PhosphorImager.
Generation of reporter plasmids and transient transfection.
The COX-2 reporter gene pGL3-COX-2P was a generous gift from S. Prescott (Salt Lake City, UT) and cloned as described previously (32). The plasmids pGL3-COX-2P-UTR1 and pGL3P-UTR1 were generated by cloning a 101-bp fragment from the pcDNAI/Amp-COX-2 full-length clone (25) into either the pGL3P or the pGL3-COX2P plasmid, respectively. The 3′ UTR sequence was generated by PCR using the XbaI-flanked (underlined) forward primer 5′-TATTCTAGAAAGTCTAATGATCATATT-3′ and FseI-flanked (underlined) reverse primer 5′-GGCCGGCCGGATCCAGAAGATGTTAAGTAACA-3′, corresponding to a region from nucleotides 1950 to 2051 of the human COX-2 mRNA (GenBank accession no. NM_000963). The plasmids pGL3-COX-2P-UTR2 and pGL3P-UTR2 were generated by cloning a 162-bp fragment from the COX-2 full-length clone into either the pGL3P or the pGL3-COX2P plasmid. The 3′ UTR sequence was generated by PCR using the XbaI-flanked (underlined) forward primer 5′-TATTCTAGATCTAGATGACCTCATAAAATACCT-3′ and FseI-flanked (underlined) reverse primer 5′-GGCCGGCCGGATCCTGTGTCTCTTAGCAAAAT-3′, corresponding to a region from nucleotides 3074 to 3236 of the human COX-2 mRNA. The PCR products were digested with FseI and XbaI and cloned into the XbaI/FseI-cut pGL3P or pGL3-COX-2P plasmid, thereby allowing a forced insertion of the 3′ UTR part of human COX-2 mRNA at the 3′ end of luc+ coding region (CR).
Introduction of a point mutation into the ARE site (TTTCTTTT to CGCCCGCT) to generate pGL3P-UTR2mut was performed using the sense primer 5′- GCATGCTGTTCCTCGCCCGCTCTTCTTTTAGCCATTTTG-3′ (corresponding to a region from nucleotides 3185 to 3223). The mutant was generated by use of the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). For transient transfection, hMC were grown on six-well plates and transfected with 400 ng of plasmid DNA plus 100 ng Renilla luciferase DNA per well by using the Effectene transfection reagent (Qiagen, Hilden, Germany). Luciferase activities were measured with the dual-reporter gene system (Promega, Mannheim, Germany) using an automated chemiluminescence detector (Berthold, Bad Wildbad, Germany).
Construction of Flag-tagged HuR.
Flag-tagged recombinant HuR proteins were generated by PCR using pQE-His-HuR and pQE-30-HuRΔ2/Δ3 as templates. For introduction of the N-terminal Kozak sequence, the following primer pair was used: 5′-GAATTCCCACCATGTCTAATGGTTATGAA-3′ and 5′-CCCGGGCCCTTTTTGTGGGACTTGTTG-3′. PCR products were first cloned into the vector pCR 2.1 TOPO (Invitrogen). Afterwards, the positive ligates were successively digested with EcoRI and ApaI and inserts subsequently subcloned into the pCMV-Flag-N3 expression vector. Transient overexpression in hMC of different pCMV-Flag-N3 expression vectors was performed with the Lipofectamine reagent (Invitrogen).
IP.
For immunoprecipitation (IP), nuclear extracts (400 μg) were incubated overnight at 4°C with either 2 μg of a monoclonal anti-HuR antibody or with the same amount of mouse IgG (both diluted in lysis buffer containing 5% fetal calf serum). Subsequently protein G-Sepharose CL-4B beads (Amersham-Biosciences, Freiburg, Germany) were added and incubated for another 2 h. After centrifugation for 5 min at 3,000 × g, the precipitated complexes were successively washed three times with low-salt buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.2% Triton X-100, 2 mM EDTA, 2 mM EGTA, 0.1% sodium dodecyl sulfate [SDS]) and once with high-salt buffer (50 mM Tris-HCl [pH 7.5], 500 mM NaCl, 0.2% Triton X-100, 2 mM EDTA, 2 mM EGTA, 0.1% SDS). After these washing steps, beads were subjected to SDS-PAGE and coimmunoprecipitated proteins were detected by Western blot analysis.
IP-qRT-PCR analysis (pull-down RT assay).
HuR-bound mRNA was detected by using a pull-down reverse transcription (RT) assay as described previously (10). Briefly, cells were treated with lysis buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 0.1% NP-40, 50 mM NaF, 10 mM Na3VO4, 10 mM sodium pyrophosphate, 50 mM disodium glycerol phosphate, 100 U/ml RNasin) and cell lysates were immunoprecipitated with 2 μg of an anti-HuR antibody or, alternatively, the same amount of mouse IgG in an overnight incubation at 4°C. Subsequently, protein G-Sepharose CL-4B beads were added and the mixture was incubated for further 2 h. After a short centrifugation (3,000 × g) the beads were successively washed with low- and high-salt buffer before total RNA was extracted by the Tri reagent (Sigma-Aldrich). The HuR-bound RNA was reverse transcribed using SuperScript reverse transcriptase (Invitrogen, Karlsruhe, Germany) and subjected to quantitative RT-PCR (qRT-PCR). Normalization of input RNA was confirmed by RT reaction of total cellular RNA isolated from an equal amount of cell extract as was used for IP and subsequent assessment of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels.
Real-time RT-PCR.
Two-step real-time PCR was performed using a Taqman (ABI 7000) from Perkin-Elmer. The mRNA levels for COX-2 and GAPDH were determined by using a protocol according to the “hot start” real-time PCR procedure with “Quanti-Tec” Sybr green (Qiagen, Hilden, Germany). Equal amounts of RNA were reverse transcribed with reverse transcriptase (Invitrogen, Karlsruhe, Germany) by using random hexamer primers. The following oligonucleotides were used for PCR: COX-2 forward (5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′) and reverse (5′-AGATCATCTCTGCCTGAGTATCTT-3′), GAPDH forward (5′-CACCATCTTCCAGGAGCGAG-3′) and reverse (5′-GCAGGAGGCATTGCTGAT-3′), and luciferase forward (5′-GCCCGCGAACGACATTTA-3′) and reverse (5′-TTTGCAACCCCTTTTTGGAA-3′). Calculation of relative COX-2 and luciferase mRNA levels was done by using the following cycle threshold (CT) method: 2[−ΔΔCT] (29). According to this method, the CT values of COX-2 and luciferase mRNA levels were normalized to the CT values of GAPDH mRNA within the same sample.
Detection of mRNA by standard PCR.
RT-PCR was performed as described above. The following primer sets were used: acetylcholine receptor α (AchRα), 5′-TGGGCTCCGAACATGAGACC-3′ and 5′-TGGGGCGTGGCAGATCTACCA-3′; β-actin, 5′-TTGCCGACAGGATGCAGAAGGA-3′ and 5′-AGGTGGACAGCGAGGCCAGGAT-3′; CR of COX-2, 5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′ and 5′-AGATCATCTCTGCCTGAGTATCTT-3′; UTR1, 5′-AAGTCTAATGATCATATTTA-3′ and 5′-GGATCCAGAAGATGTTAAGTAACA-3′; and UTR2, 5′-TCACACATTAATTTTATC-3′ and 5′-GGATCCTGTGTCTCTTAGCAAAAT. The different PCR products (AchRα, 206 bp; β-actin, 128 bp; CR, 304 bp; UTR1, 102 bp; and UTR2, 104 bp) were separated on a 1.5% agarose gel containing 0.5 μg/ml ethidium bromide. The identity of amplicons was confirmed by DNA sequencing using the AbiPrism 310 genetic analyzer from Applied Biosystems.
RNP-IP assay.
IP of protein-RNA complexes was performed using a protocol from Niranjanakumari et al. (37) with slight modification as described previously (64). Briefly, hMC (5 × 107) were harvested by centrifugation at 100 × g for 5 min at 4°C and resuspended in cold phosphate-buffered saline, formaldehyde was added to a final concentration of 0.5% (vol/vol), and the mixture was incubated at room temperature for 10 min. Cross-linking was quenched by the addition of glycine (pH 7.0) at a final concentration of 0.25 M, and the mixture was incubated for a further 5 min. After centrifugation, cells were washed and resuspended in 1 ml of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.5], 1% NP-40, 0.05% SDS, 1 mM EDTA, 150 mM NaCl) supplemented with protease inhibitors. Cell lysis was accomplished by three rounds of sonication for 20 s each. The supernatant was diluted with RIPA buffer (1,000 μl) containing RNasin and protease inhibitors, mixed with a monoclonal anti-HuR antibody (0.2 mg/ml) or with the same amount of mouse IgG (0.2 mg/ml), and incubated overnight before protein G-Sepharose beads were added. After shaking on a rotator for 2 h at 4°C, Sepharose beads were washed several times with different buffers according to the protocol by Niranjanakumari et al. (37). The beads containing the immunoprecipitated samples are collected and resuspended in 100 μl of a mixture of 50 mM Tris-HCl [pH 7.0], 5 mM EDTA, 10 mM dithiothreitol, and 1% SDS and were further incubated at 70°C for 45 min to reverse cross-linking. RNA was extracted from the immunoprecipitates using the TRIzol reagent (Sigma) followed by a DNase I treatment. Recovered nucleic acids were precipitated with ethanol and resuspended in diethyl pyrocarbonate-treated water. The RNA was used as a template with which to synthesize cDNA using random hexamer primers and Superscript reverse transcriptase (Invitrogen) according to the manufacturer's protocol.
Polysome fractionation.
The isolation of different polysomal fractions was performed by a protocol described by Hovland et al. (19). Briefly, cells were lysed on ice in lysis buffer I containing 10 mM Tris-HCl (pH 7.6), 0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 0.5 mM CaCl2, and 0.05% NP-40. Cell lysates were collected by centrifugation at 1,000 × g for 5 min, and supernatants were directly used as free polysomal fractions. The pellet which contained cell nuclei, polymerized cytoskeletal material, and insolube membranes was additionally washed in lysis buffer I, and the cytoskeleton-bound polysomes were extracted by addition of lysis buffer II (10 mM Tris-HCl [pH 7.6], 0.25 M sucrose, 130 mM KCl, 5 mM MgCl2, 0.5 mM CaCl2, 0.05% NP-40) for 10 min. Lysates were centrifuged (2,000 × g for 5 min), and from the supernatants cytoskeleton-bound polysomes were obtained. The pellets were washed with lysis buffer II, and membrane-bound polysomes were released by resuspension of cell pellets in buffer II additionally containing 0.5% deoxycholate. After incubation for 10 min on ice, the supernatants containing the membrane-bound polysomes were collected by a final centrifugation step at 3,000 × g. The activity of lactate dehydrogenase (LDH) in different polysomal fractions was quantified by the CytoTox96 nonradioactive cytotoxicity assay (Promega) and normalized to protein concentrations of corresponding samples.
Determination of PGE2 levels in conditioned media.
Levels of PGE2 in cell culture supernatants were determined by the Correlate-enzyme immunoassay PGE2 enzyme-linked immunosorbent assay kit (Assay Designs, Ann Arbor, MI). For immunodetection, 100 μl of conditioned medium was directly transferred into the microtest strip wells of the enzyme-linked immunosorbent assay plate, and further procedures were performed according to the manufacturer′s instructions. A405 was measured in a microtest plate spectrophotometer, and PGE2 contents were determined by a calibration curve using PGE2 as a standard.
Indirect immunofluorescence microscopy.
For indirect immunofluorescence analysis, cells were grown on coverslips and subsequently fixed for 20 min at room temperature with 4% paraformaldehyde in phosphate-buffered saline and monitoring of HuR shuttling was performed as described previously (10). Stained cells were finally monitored using an inverse immunofluorescence microscope, BZ-7000 (Biozero, Keyence, Neu-Isenburg, Germany), equipped with a Zeiss Apo 20×/0.75 numerical-aperture objective, and images were analyzed by using the BZ-H1TL software from Biozero.
Statistical analysis.
Results are expressed as means ± standard deviations (SD). The data are presented as induction (n-fold) compared to untreated control or compared to stimulated values. Statistical analysis was performed using Student′s t test and analysis of variance. P values of <0.05, <0.01, and <0.005 were considered significant.
RESULTS
AngII potentiates cytokine-induced mRNA expression of COX-2 in hMC.
We have previously shown that the extracellular nucleotide ATP, by activation of PKCα, amplifies the cytokine-induced COX-2 mRNA level in hMC, concomitant with an amplification in the cytokine-evoked induction in PGE2 synthesis (10). Here, we evaluated possible modulatory effects of AngII, which similar to extracellular nucleotides can activate PKC-dependent signaling pathways in MC (for review, see references 3 and 42). Since hMC show only a low constitutive COX-2 expression, cells were treated with TNF-α and IL-1β (both at 2 nM) to ensure transcriptional induction of COX-2 and costimulated with or without AngII (100 nM), or with AngII alone, for different time points before the steady-state mRNA level of COX-2 was monitored by qRT-PCR (Fig. 1A). AngII causes a strong and significant increase in cytokine-evoked COX-2 mRNA level, which is most obvious after 8 to 16 h of stimulation, whereas AngII alone had no significant effect on the basal COX-2 mRNA level (Fig. 1A). Furthermore, the stimulatory effect on COX-2 was completely blocked by valsartan, indicating the involvement of the AngII type 1 receptor (data not shown). Functionally, the increase in COX-2 mRNA by AngII is accompanied by an amplification of the cytokine-induced PGE2 release and, again, AngII had no effect on the constitutive PGE2 synthesis when given alone (see also Fig. 6B).
FIG. 1.

Time-dependent amplification of cytokine-induced COX-2 mRNA by AngII. (A) Confluent hMC were serum starved before being stimulated either with a cytokine mixture (IL-1β and TNF-α; both 2 nM) in the absence or presence of AngII (100 nM) or with AngII (100 nM) alone for the indicated time points. The relative levels of COX-2 mRNA compared with GAPDH mRNA were measured by qRT-PCR. Data represent means ± SD (n = 3) and are representative of three independent experiments. * and **, P ≤ 0.05 and P ≤ 0.01, respectively, compared with control. ## and ###, P ≤ 0.01 and P ≤ 0.005, respectively, compared with cytokine-induced conditions. (B) AngII causes an increase in cytokine-induced COX-2 mRNA stability. Serum-starved hMC were stimulated for 16 h with the cytokine mixture and then washed twice before Act D (5 μg/ml) was added. After a short preincubation of 30 min, cells were additionally treated for the indicated time points with either vehicle or with AngII (100 nM) before cells were harvested and extracted for total cellular RNA. Steady-state mRNA levels of COX-2 were quantified by qRT-PCR using GAPDH as a normalization control. The table shows the percentage of remaining COX-2 mRNA compared to the level of COX-2 transcripts measured at time point 0 h (100%). The upper panel shows an agarose gel from a semiquantitative PCR and is representative of three independent experiments giving similar results.
FIG. 6.

Silencing of HuR inhibits AngII-induced amplification of cytokine-stimulated COX-2 levels and PGE2 synthesis. (A) MC were left untransfected (control) or were transfected with either duplex siRNA of human HuR (siRNA-HuR) or, alternatively, with control duplex siRNA (siRNA-control) as described in Materials and Methods. After transfection, cells were serum starved for 16 h before being treated for a further 24 h with vehicle or with a cytokine mixture (cyt-mix) containing IL-1β and TNF-α (both at 2 nM) in the presence (+ AngII; 100 nM) or absence of AngII or with AngII alone, as indicated. The efficiency of silencing of HuR protein levels was monitored by assessment of the total HuR level by Western blot analysis using an anti-HuR-specific antibody and is depicted as the average amount of remaining HuR contents. To correct for variations in protein loading, the blot was stripped and probed with an anti-β-actin antibody Changes in the COX-2 mRNA levels were determined by qRT-PCR by normalizing COX-2 mRNA to GAPDH mRNA. The results are means ± SD (n = 3) and are presented as induction versus nonstimulated controls (**, P ≤ 0.01) or versus cytokine-stimulated values (##, P ≤ 0.01). n.s., not significant. (B) PGE2 levels in cell supernatants derived from hMC treated as indicated in panel A and described in detail in the legend to panel A. Data represent means ± SD (n = 3).
AngII inhibits the decay of cytokine-induced COX-2 mRNA.
The expression of COX-2 in many cases occurs through a transcriptional activation induced by proinflammatory cytokines and tumor promoters (28, 59). Yet, growing experimental evidence has demonstrated that COX-2 is additionally regulated on a posttranscriptional level (8, 10, 44, 48). To test whether the AngII effects would rely on any posttranscriptional mechanisms, we performed Act D experiments. MC were stimulated for 16 h with IL-1β plus TNF-α (both at 2 nM) to allow a profound induction of COX-2 mRNA expression before transcription was blocked by Act D (5 μg/ml). Subsequently, cells were either directly homogenized (0 h), left untreated (vehicle), or additionally treated with AngII (100 nM) before total RNA was extracted after 2, 4, and 8 h, respectively, and subjected to qRT-PCR. AngII caused a substantial raise in COX-2 mRNA stability, increasing the half-life from almost 3 h to more than 8 h when equilibrated with GAPDH mRNA levels (Fig. 1B).
The 3′ UTR of the COX-2 gene confers promoter activation by AngII.
Previous studies have identified multiple copies of evolutionarily highly conserved AU-rich motifs within the 3′ UTR of the COX-2 mRNA as primary regulatory elements of posttranscriptional COX-2 expression (Fig. 2A) (6, 8). Furthermore, major binding sites for the mRNA-stabilizing factor HuR, regulating COX-2 mRNA, could be deciphered by the use of RNase T1 selection assay (46). Namely, a high binding affinity of recombinant HuR to a proximal region between +1949 and +2051 encompassing three copies of the typical pentameric AUUUA motif (“UTR-site1” construct) and which is also known to confer translational silencing (8) and to a second distal site lying between +3075 and +3236 and containing a nonpentameric destabilizing motif (“UTR-site2”) has previously been demonstrated by electrophoretic mobility shift assay (46). Therefore, we tested the impact of these high-affinity HuR binding sites on AngII-dependent COX-2 mRNA stabilization. To this end, different fusion constructs containing one of these UTRs downstream of the luciferase coding sequence which was either driven by a pGL-3 control promoter (pGL3P-UTR1 and pGL3P-UTR2) or, alternatively, by a 3.9-kb fragment of the human COX-2 promoter region (25) (pGL3-COX-2P-UTR1 and pGL3-COX-2P-UTR2) were transiently transfected in hMC. Subsequent stimulation with AngII (100 nM) for 16 h caused a significant increase in luciferase activity independent of which promoter construct was used (Fig. 2B). However, the strongest induction by AngII was measured with the UTR-site2 construct of COX-2 (pGL3P-UTR2) (Fig. 2B). The induction of luciferase activity by AngII was accompanied by an increased half-life of luciferase mRNA as assessed by Act D experiments (Fig. 2C). These data indicate that the increase in luciferase activity by AngII is mainly due to an increase in mRNA stability. In contrast, AngII had no significant effect on luciferase activity when testing the same COX-2 promoter construct but without corresponding UTRs irrespective of which time was used for stimulation (Fig. 2E). This clearly demonstrates that the cis-regulatory elements conferring any AngII response on COX-2 are mainly attributable to the 3′ UTR of the COX-2 mRNA.
FIG. 2.

Posttranscriptional regulation of COX-2 by AngII is attributed to different AREs. Various parts of the 1,455-nucleotide COX-2 3′ UTR as depicted in panel A (black discs) were fused to the reporter gene luciferase in the absence or presence of COX-2 promoter fragments to generate reporter gene constructs containing the luciferase cDNA fused to the different COX-2 AREs (UTR1/UTR2). Discs represent AREs. (B) Firefly luciferase reporter constructs (0.4 μg/well) were transiently transfected in hMC together with a vector expressing Renilla luciferase (0.1 μg/well) to control for transfection efficiency. Cells were either left unstimulated (black bars) or stimulated with AngII (100 nM) (gray bars) for 16 h prior to measurement of luciferase activities. (C) AngII causes an increase in cytokine-induced luciferase mRNA stability. Firefly luciferase reporter constructs were transfected as described for panel B. Serum-starved transfected hMC were stimulated for 16 h with cytokine mixture and then washed twice before Act D (5 μg/ml) was added. After a short preincubation of 30 min, cells were additionally treated for the indicated time points with either vehicle or with AngII (100 nM) before cells were harvested and extracted for total cellular RNA. Steady-state mRNA levels of firefly luciferase were quantified by qRT-PCR using GAPDH as a normalization control. The table depicts the percentage of remaining luciferase mRNA compared to the level of luciferase transcripts measured at time point 0 h (100%). (D) Promoter activities of reporter constructs containing either a nonmutated UTR2 (pGL3P-UTR2) or a construct of the COX-2 3′ UTR bearing multiple point mutations (underlined nucleotides) in the ARE of region 2 (UTR2) were tested by “dual-luciferase assay.” (E) Modulation of a 3,900-bp fragment of the 5′-flanking region of the human COX-2 gene by AngII. The pGL3-COX-2P plasmid was transfected, and cells were subsequently treated for the indicated time periods with vehicle (black bars) or with 100 nM AngII (gray bars). The values for beetle luciferase were related to the values of Renilla luciferase and are depicted as relative luciferase activities. Data represent means ± SD (n = 3). *, **, and ***, P ≤ 0.05, P ≤ 0.01, and P ≤ 0.005, respectively, compared with control. ns, not significant.
The critical role of UTR-site2 in AngII-induced luciferase activity was furthermore confirmed by site-directed mutagenesis. Mutation of this specific HuR-binding site (pGL3P-UTR2mut) resulted in the total loss of AngII-induced promoter activity, thus corroborating that the UTR-site2 construct of COX-2 mRNA is critically involved in AngII-mediated regulation of COX-2 mRNA turnover (Fig. 2D).
AngII augments HuR binding to site 2 within the 3′ UTR of COX-2.
To prove the in vivo relevance of HuR binding to UTR-site2 of COX-2 mRNA, we performed a RNP-IP assay which combines reversible cross-linking with IP as described previously (37, 64). To this end, hMC were stimulated for 2 h with vehicle or AngII (100 nM) and were subsequently incubated with the chemical cross-linker formaldeyde followed by a sonication step. The anti-HuR antibody-caused precipitation of fragmented RNA from cross-linked cell lysates with a subsequent PCR from reverse-transcribed mRNA revealed a clear binding of HuR to a region encompassing either site 1 (UTR1) or site 2 (UTR2) of the 3′ UTR of the COX-2 mRNA (Fig. 3). Most importantly, the HuR-RNA interaction was not detectable in lysates from untreated cells, thus indicating that binding of HuR to the 3′ UTR depends on a prior stimulation with AngII. In contrast, amplification of a COX-2-specific band was impossible independent of which source of cell lysates was used as template when a primer pair encompassing the CR of COX-2 was utilized for PCR (Fig. 3). These data clearly indicate that HuR specifically binds to the 3′ UTR but not to the CR of COX-2 mRNA (Fig. 3). The inability of control IgG to yield an appropriate PCR product furthermore verified the specificity of RNA binding (Fig. 3). Additionally, the differences in the amplified cDNA were not due to unequal amounts of RNA and protein used for IP, as indicated by an RT-PCR and Western blot analysis of equivalent input levels (Fig. 3).
FIG. 3.
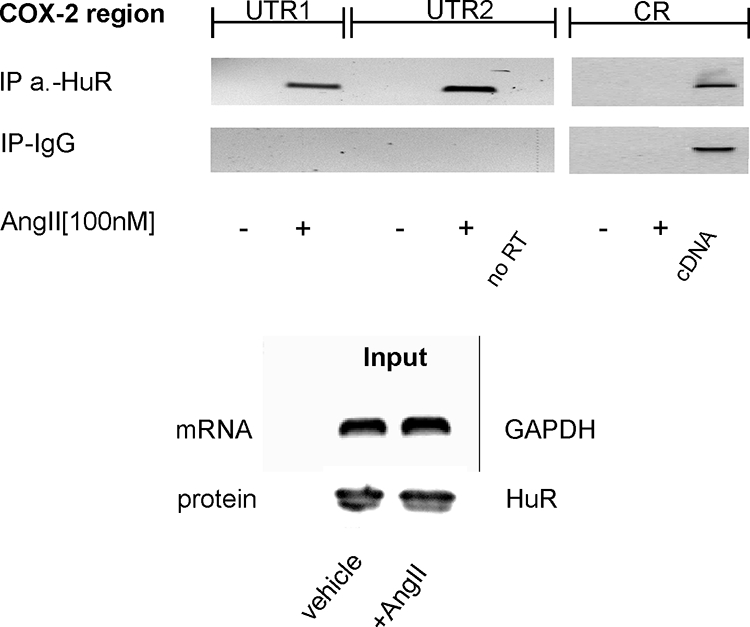
AngII induces the RNA binding of HuR to the UTR of COX-2. hMC were stimulated with vehicle (−) or with 100 nM AngII (+) for 2 h before cells were cross-linked and lysed for IP. HuR-bound mRNA was precipitated by addition of a HuR-specific antibody or, alternatively, the same amount of IgG as a negative control. After elution from Sepharose beads, the isolated RNA samples were analyzed by RT-PCR either by using primer pairs, complementary to sequences encompassing different COX-2 3′ UTR parts (UTR1 and UTR2), or by use of a primer pair complementary to a sequence from the CR as described in Materials and Methods. Input GAPDH levels isolated before the IP were used as a control for equal amounts of input RNA (input) and were assessed by RT-PCR. Furthermore, input HuR levels isolated prior to the IP were used as input protein (input) control. A PCR without RT product was used as a negative control (no RT), whereas COX-2 full-length plasmid used as a template was used as a positive control (cDNA).
AngII increases the constitutive binding of the mRNA-stabilizing factor HuR to cytoplasmic COX-2 mRNA in mesangial cells.
Previous evidence has demonstrated a critical role of the mRNA-stabilizing factor HuR in the posttranscriptional regulation of COX-2 in different cells, including hMC (8, 10, 46). Employing a pull-down assay, we tested whether HuR binding to COX-2 mRNA is modulated upon stimulation of hMC with AngII. To this end, crude cytoplasmic extracts from hMC were first subjected to IP with an anti-HuR antibody before HuR-bound mRNA was amplified by qRT-PCR. Interestingly, the small amount of cytosolic COX-2 mRNA precipitated by the anti-HuR antibody under unstimulated conditions was markedly increased when cells had been treated with AngII (14.4 ± 1.8-fold; mean ± SD; n = 6) (Fig. 4A and B), although equal amounts of mRNA (GAPDH and COX-2) and protein (HuR) were used for IP (Fig. 4A, input IP). In contrast, no COX-2 transcripts were detectable when a non-HuR-related control antibody (p65-NF-κB) or serum IgG were used in the pull-down assay (Fig. 4A). Similarly, no cDNA was amplified when instead of COX-2-specific primers, primers complementary to GAPDH cDNA were applied to the PCR, thus indicating that COX-2 mRNA is a specific target of cytoplasmic HuR and AngII stimulation—a prerequisite for their mutual interaction.
FIG. 4.

AngII increases the intracellular binding of HuR to COX-2 mRNA. hMC were treated for 2 h with vehicle or AngII (100 nM) as indicated before being immunoprecipitated with 2 μg of an anti-HuR antibody (A.b.). RNA bound by HuR was harvested and subjected to qRT-PCR (upper panel) using COX-2-specific primers. Normalization of similar amounts of input RNA added to the IP reaction mixtures was done by assessment of GAPDH and COX-2 levels from input RNA by RT-PCR (A, right panel). Input HuR levels isolated before the IPs were used as a control for equal amounts of input protein (input IP). The upper left panel shows a DNA gel from a representative RT-PCR (B). The lower panel shows a triplicate experiment representative of three independent experiments giving similar results. Results are expressed as means ± SD (n = 3) and are presented as induction compared with vehicle (***, P ≤ 0.005).
Polysomal association of HuR-bound COX-2 transcripts is restricted to the cytoskeleton and cytoplasmic fractions.
To elucidate whether the increase in HuR binding causes COX-2 mRNA to interact with the translation apparatus, we analyzed the presence of HuR-bound COX-2 transcripts within different polysomal fractions, including free polysomes, cytoskeleton-bound polysomes, and membrane-bound polysomes, making use of their different salt and detergent susceptibilities as described in Materials and Methods (19).
Immunoblot analysis of the related fractions revealed that under unstimulated conditions in none of the fractions was HuR detectable (Fig. 5A). In contrast, HuR was abundant in cytoskeletal and free polysomal fractions obtained from AngII-treated cells. In neither case, the membrane-associated ribosomes contained any HuR (Fig. 5A). As expected, β-actin, a marker of cytoplasmic fraction was exclusively detectable in the free and cytoskeleton-bound polysomal fractions and the levels of β-actin were not affected upon AngII treatment (Fig. 5A). In contrast, expression of AchRα, a marker of membrane-bound proteins (19) was specifically found in the membrane-bound polysomal fraction (Fig. 5A). Complementarily, the specificity of different polysomal fractions was proven by measuring LDH, which displayed a maximal activity in the free-polysomal fractions (table in Fig. 5A). A pull-down assay of the same samples revealed that AngII-induced HuR coprecipitated with COX-2 mRNA, indicating that the HuR-bound COX-2 mRNA is specifically transported to free and cytoskeleton-bound polysomes but not to polysomes associated with membranes (Fig. 5B).
FIG. 5.

HuR binds to COX-2 mRNA in response to AngII and is localized with free and cytoskeleton-bound polysomes. hMC were subjected to sequential detergent and salt extraction to release free polysomes (FP), cytoskeleton-bound polysomes (CBP), and membrane-bound (MBP) polysomes as indicated in Materials and Methods. (A) Cells were serum starved for 16 h and were treated either with vehicle (−) or with AngII (100 nM) (+) for 2 h before being isolated and extracted for specific polysomal fractions. Seventy micrograms of the corresponding polysomal fraction was subjected to SDS-PAGE and immunoblotted with an anti-HuR-specific antibody (upper panel). Equal amounts of loaded proteins were ascertained by Ponceau S (PS) staining (second panel). Different markers of the three different pools of polysomes were used to prove the accuracy of the subcellular fractionation procedure. Representative RT-PCR products corresponding to β-actin and AchRα mRNAs were used as specific markers of cytoplasmic (actin) and membrane-bound proteins (AchRα). Aliquots from the same fractions were additionally assayed for LDH content. Data are expressed as a percentage of the activity determined in total protein extracts. (B) RNA extracted from different polysomal fractions was analyzed with COX-2-specific primers by RT-PCR. Normalization of similar amounts of input RNA was done by assessment of GAPDH levels (relative COX-2 mRNA/input GAPDH). Results are means ± SD (n = 3) and are presented as induction compared with nonstimulated control compared with vehicle (***, P ≤ 0.005). ns, not significant.
HuR is indispensable for amplification COX-2 mRNA by AngII but not involved in cytokine-triggered COX-2 expression.
To investigate the functional impact of HuR on the Ang-II-induced amplification of cytokine-induced COX-2 expression (Fig. 1A), we transiently attenuated HuR expression by a corresponding siRNA treatment. For this purpose, hMC were transfected with either an HuR-specific siRNA (siRNA-HuR) or, alternatively, with a non-gene-specific control siRNA (siRNA-control) before cells were stimulated for 24 h with vehicle or with a cytokine mixture (IL-1β plus TNF-α, each at 2 nM) without or with AngII (100 nM) (Fig. 6A, lower panel). Most interestingly, attenuation of HuR expression results in a total loss of the AngII-mediated amplification of cytokine-evoked COX-2 expression without affecting either basal or cytokine-induced COX-2 mRNA contents (Fig. 6A, upper panel). In contrast, hMC transfected with the control siRNA showed an unchanged response, thus indicating that the inhibition of AngII-mediated amplification is due to silencing of HuR expression (Fig. 6A). Interestingly, although AngII causes an increase in HuR binding to COX-2 mRNA (Fig. 4), it did not suffice to increase the basal COX-2 mRNA level on its own. This contrasts with the results obtained from the pull-down assay (see Fig. 4) but may simply indicate that the HuR-triggered increase in the basal COX-2 mRNA levels are below the threshold level detectable by PCR amplification in the experimental setting applied.
Consistent with the changes in the COX-2 mRNA levels, we observed a lack in the AngII-dependent amplification of cytokine-induced PGE2 release after HuR attenuation (Fig. 6B). These results further support the notion that AngII by a HuR-dependent mechanism specifically affects the expression and function of cytokine-induced COX-2 while having no effect on basal COX-2 expression.
AngII-induced recruitment of HuR is mediated via the AT1 receptor and depends on PKCδ.
Various studies have shown that the stimulus-induced stabilization of many ARE-containing mRNAs by HuR is attributable to an increased shuttling of HuR from the nucleus to the cytoplasm, which in turn is controlled by different signaling pathways, including the p38 MAPK (33, 57), the AMP kinase (54), the phosphatidylinositol 3 kinase (PI3K) (33), and finally by PKCα (10).
To test the involvement of either of these pathways in the AngII-induced HuR activation, we analyzed whether the treatment with AngII causes any changes in the subcellular localization of endogenous HuR. To verify the grade of purity of cytoplasmic fractions, we tested for occurrence of HDAC which is used as a marker protein of the nuclear compartment (Fig. 7A, HDACc). We observed that untreated hMC contained low HuR protein levels in the cytoplasm but stimulation with AngII (100 nM) caused a marked increase in cytosolic HuR with a maximal effect seen after 2 h of stimulation (Fig. 7A). Furthermore, we tested the modulatory effects of various pharmacological inhibitors: SB203580 (10 μM), a specific inhibitor of the p38 pathway; PD98059 (30 μM), an inhibitor of the p42/p44 MAPK pathway; U0126 (20 μM), an inhibitor of the MAPK kinase (MEK); rottlerin (10 μM), an inhibitor of the Ca2+-independent PKC isoenzymes; and finally, CGP41251 (0.1 μM), a PKC inhibitor with a high selectivity toward the Ca2+-dependent isoenzymes. Whereas SB203580, PD98059, U0126, and CGP41251 had almost no inhibitory effect on the AngII-induced cytoplasmic HuR accumulation, rottlerin almost completely blocked the AngII-induced HuR export to the cytoplasm (Fig. 7A, HuRc). This indicates that AngII acts specifically via the novel, Ca2+-independent PKC isoenzyme PKCδ or PKCθ. Importantly, the changes in cytoplasmic HuR levels did not result from changes in HuR expression levels, as demonstrated by the unchanged amount of total cellular HuR (Fig. 7A, HuRt). To further substantiate this observation, we tested additional PKC inhibitors, including staurosporine (100 nM), a broad-spectrum PKC inhibitor; and Gö6976 (20 nM), which, similar to CGP41251, displays a high selectively toward the classical, Ca2+-dependent PKC isoenzymes. Staurosporine, similar to rottlerin, substantially reduced the AngII-induced HuR shuttling, whereas the inhibition of Ca2+-dependent PKCs by Gö6976 did not affect the AngII-induced cytoplasmic HuR accumulation (Fig. 7B). Moreover, the PKC dependency of HuR shuttling by AngII was also monitored by indirect immunofluorescence. AngII induces a clear redistribution of HuR from the nuclear compartment to the cytoplasm, as shown in Fig. 7C. The nucleocytoplasmic shuttling of HuR is strongly reduced when cells were pretreated with either rottlerin or valsartan (100 nM), a specific inhibitor of the AT1 receptor, indicating that AngII triggers Ca2+-independent PKCs and in turn HuR shuttling via the AT1 receptor subtype (Fig. 7C).
FIG. 7.

PKC inhibition prevents the cytoplasmic accumulation of HuR by AngII. (A) Western blot analysis showing stimulus-dependent changes in the cytoplasmic (HuRc; upper panel) and total HuR levels (HuRt; lower panel) in hMC. Cells were serum starved for 16 h before being treated for an additional 2 h with vehicle (−) or AngII (100 nM) in the absence (vehicle) or presence of either SB203580 (10 μM), PD98059 (30 μM), rottlerin (10 μM), U0126 (20 μM), or CGP 41251 (100 nM), as indicated. All inhibitors were preincubated for 30 min before the addition of AngII. Protein lysates (50 μg) from cytoplasmic (HuRc and β-actinc; upper panels) fractions or whole-cell extracts (HuRt and β-actint; lower panels) were subjected to SDS-PAGE and immunoblotted with an anti-HuR-specific antibody. The validity of the subcellular fractionation was verified by assessment of HDAC contents; cytoplasmic fraction HDAC (HDACc) was used as a nuclear marker protein. To correct for variations in the protein loading, the blots were stripped and reincubated with an anti-β-actin antibody. (B) Effects of different PKC inhibitors on AngII-induced HuR accumulation in cytoplasmic fractions. hMC were serum starved for 16 h and subsequently either untreated (−) or treated for an additional 2 h with AngII (100 nM) in the absence (vehicle) or presence of either staurosporine (100 nM), rottlerin (10 μM), Gö6976 (20 nM), or CGP 41251 (100 nM) as indicated. The Western blots shown are representative of two independent experiments giving similar results. (C) AngII induces the nuclear export of HuR in hMC. Indirect immunofluorescence was applied to visualize changes in the subcellular localization of HuR by AngII (note the different magnifications in the upper and lower panels). Quiescent hMC were stimulated for 2 h with either vehicle or with AngII (100 nM) in the absence or presence of rottlerin (10 μM) or valsartan (100 nM), as indicated, before cells were fixed and stained with an anti-HuR antibody and anti-mouse Alexa 488 antibody. Data are representative of two independent experiments giving similar results. (D) Confluent hMC were serum starved before being stimulated with either cytokine mixture (IL-1β and TNF-α; both 2 nM) for 16 h and subsequently either left untreated (−) or treated for an additional 2 h with AngII (100 nM) in the absence or presence (+) of either staurosporine (100 nM), rottlerin (10 μM), Gö6976 (20 nM), or CGP 41251 (100 nM) as indicated. The relative levels of COX-2 mRNA compared with GAPDH mRNA were measured by qRT-PCR. Data represent means ± SD (n = 3) and are representative of two independent experiments. *, P ≤ 0.05 compared with control. # and ##, P ≤ 0.05 and P ≤ 0.01, respectively, compared with cytokine-induced conditions. §§, P ≤ 0.01 compared with cytokine- and AngII-induced conditions.
As expected by the changes in cytoplasmic HuR levels, staurosporine and rottlerin caused a complete inhibition of the AngII-triggered amplification of cytokine-induced COX-2 mRNA without affecting the cytokine-induced COX-2 mRNA levels (Fig. 7D).
To further substantiate the results obtained with pharmacological inhibitors, we performed RNA attenuation approaches with silencing RNAs. Whereas silencing of PKCα had no effect on the AngII-induced cytoplasmic HuR accumulation (Fig. 8, left panel), silencing of PKCδ caused a total inhibition of AngII-evoked HuR shuttling (Fig. 8, right panel). In both cases, silencing of either PKC had no effect on the total HuR content (data not shown). We observed that the basal cytoplasmic HuR content in transfected MC is somewhat higher than that measured in untransfected cells, suggesting that the transfection procedure by itself may have a stimulatory effect on HuR shuttling (Fig. 8). Collectively, these data demonstrate that AngII by specifically activating PKCδ induces a nucleocytoplasmic HuR shuttling which in turn increases the binding to the 3′ UTR of COX-2 mRNA.
FIG. 8.

Silencing of PKCδ but not of PKCα impairs the AngII-induced increase in cytosolic HuR levels. MC were transfected with either siRNA duplexes of human PKCα (left panel) or, alternatively, human PKCδ (right panel) or an unrelated control siRNA as described in Materials and Methods. After transfection, hMC were serum starved for 16 h before being stimulated with AngII (100 nM) for 2 h and then harvested for protein extraction. Protein lysates (50 μg) from cytoplasmic fractions were subjected to SDS-PAGE and successively immunoblotted with anti-HuR, anti-PKCα (left panel), or, alternatively, anti-PKCδ-specific antisera (right panel). To correct for variations in the protein loading, blots were subsequently stripped and reprobed with an anti-β-actin antibody. The Western blots shown are representative of three independent experiments giving similar results.
AngII induces colocalization of PKCδ and nuclear HuR.
In order to determine whether the critical role of PKCδ in AngII-induced HuR shuttling relies on a physical interaction of both proteins, we tested for a possible colocalization by immunofluorescence microscopy. A physical interaction between PKCα and members of the ELAV protein family has been demonstrated in neurons (38) and hMC (10).
Under basal conditions (vehicle), PKCδ staining was detected mostly in the cytoplasm and only in some cells did we notice a perinuclear distribution (Fig. 9A). Stimulation with AngII (100 nM) induced a rapid and transient translocation of PKCδ to the nucleus, with maximal staining observed already after 15 min (Fig. 9A). At that time point, PKCδ colocalized with HuR, as shown by the strong yellow fluorescence in Fig. 9A (merged images), thus indicating that treatment with AngII specifically induces assembly of both proteins within the nuclear compartment and, moreover, that this process precedes the nuclear export of HuR to the cytosol. In line with these results, Western blot analysis revealed that AngII induces a rapid translocation of PKCδ from the cytoplasm into the nuclear compartment with a maximal accumulation in the nucleus observed after 20 min (Fig. 9B).
FIG. 9.

AngII promotes translocation of PKCδ to the nucleus and induces a physical interaction between PKCδ and nuclear HuR. (A) Indirect immunofluorescence images show PKCδ (green) and HuR (red) proteins in unstimulated cells and cells stimulated for 15 min with AngII (100 nM) as indicated. The merged images show a colocalization of PKCδ and HuR after AngII exposure (yellow signal). Data are representative of two independent experiments giving similar results. (B) Time course of AngII-induced PKCδ entry to the nucleus. Quiescent hMC were treated with either vehicle (−) or with AngII (100 nM) for the indicated time points and then lysed. Fifty micrograms of cytosolic (PKCδc) and nuclear (PKCδn) extracts was subjected to SDS-PAGE and successively immunoblotted with a phosphorylation-independent PKCδ antibody. To ascertain equal protein contents within the fractions, the blots were stripped and reprobed with an HDAC1-specific or anti-β-actin antibody. The Western blots shown are representative of three independent experiments giving similar results. (C) IP of nuclear PKCδ (PKCδ) and pSer-PKC substrate (pSer) levels in hMC stimulated either without (−) or with (+) 100 nM AngII for 15 min. For IP, a total protein amount (500 μg) of nuclear extracts from differentially treated cells was incubated overnight with 2 μg of either anti-HuR antibody (a.-HuR) or a nonspecific IgG isotype antibody (IgG). Equal amounts of immunoprecipitated HuR were ascertained by stripping the blot and reincubating it with the anti-HuR antibody used for IP. The data shown are representative of three independent experiments giving similar results.
A physical interaction of HuR with PKCδ was proven by coIP studies demonstrating that AngII treatment causes a marked increase in PKCδ coimmunoprecipitated by a monoclonal HuR-specific antibody (Fig. 9C). It is worth noting that the nuclear content of HuR remained unchanged (Fig. 9C). Control experiments demonstrated that PKCδ was undetectable when instead of anti-HuR antibody a similar amount of mouse IgG was used for IP (Fig. 9C). Additionally, no PKCδ was pulled down by the HuR-specific antiserum when instead of nuclear extracts, a similar amount of protein from the corresponding cytoplasmic fraction was subjected to IP (data not shown), thus demonstrating that an AngII-induced interaction of PKCδ with HuR is restricted to the nuclear compartment.
HuR phosphorylation by recombinant PKCδ.
In silico evaluation of the amino acid sequence of HuR predicted four motifs (XX-R/K-X-S-hydrophobic residue-R/K) which allude to putative PKC phosphorylation sites, and we previously had demonstrated that two of these sites are phosphorylated by PKCα (10). To test whether HuR in addition to PKCα is also a direct target of PKCδ, we performed a cell-free PKC assay using bacterially expressed human HuR and human recombinant PKCδ. As shown in Fig. 10A, HuR is clearly phosphorylated by PKCδ and phosphorlyation is accompanied by a strong autophosphorylation. Next we evaluated whether the PKCδ phosphorylation sites differ from those mapped for PKCα. To this end, we generated mutated full-length HuR expression plasmids bearing single mutations of the putative PKC phosphorylation sites in which the amino acid serine was substituted for by alanine (Fig. 10B, left panel). Determination of critical phosphorylation sites revealed that mutation in serine 158 (HuRΔ1), serine 221 (HuRΔ2), or serine 318 (HuRΔ3) caused a clear reduction in phosphorylation and double mutation of serine 221 and 318 sites resulted in an almost complete loss of PKCδ-dependent phosphorylation (Fig. 10B, right panel). In contrast, mutation in serine 324 (HuRΔ4) had only minor effects on HuR phosphorylation by PKCδ (Fig. 10B, right panel).
FIG. 10.

Determination of phosphorylation sites of HuR targeted by PKCδ by an in vitro PKC assay. (A) For the in vitro PKC assay, 2 μg of bacterially expressed and histidine-tagged HuR was incubated with 5 μg of recombinant PKCδ in a cell-free phosphorylation assay with [γ-32P]ATP in the presence (+) or absence (−) of 100 μM EGTA. Loading of equal protein amounts was ascertained by Coomassie blue staining (data not shown). Radioactive bands detected by autoradiography of a representative gel migrate at 97 kDa and represent autophosphorylated PKCδ. (B) Schematic representation of the HuR protein and positions of putative conserved PKC phosphorylation sites (numbers indicate positions of amino acids). For generation of HuR single point mutations (HuRΔ1, HuRΔ2, HuRΔ3, and HuRΔ4) and the double mutations HuRΔ1/2 and HuRΔ2/3, serines at the indicated positions (black circles) were substituted for with alanine (left panel). In vitro PKC phosphorylation in the absence (vehicle) or presence of either wild-type HuR or the indicated point-mutated HuR proteins and recombinant PKCδ was assessed in the absence of EGTA. The addition of equal amounts of recombinant HuR was ascertained by Coomassie blue staining (input HuR) (bottom panel). The data shown are representative of three independent experiments giving similar results. (C) Flag-HuR was detected by immunofluorescence 24 h after transfection of hMC with pCMV-Flag plasmids containing either full-length wild-type HuR (HuR) or full-length HuR bearing a double mutation in serines 221 and 318 (HuRΔ2/3). Images show Flag-HuR (red) and DAPI (4′,6′-diamidino-2-phenylindole) (blue) in hMC either left untreated (vehicle) or stimulated for 2 h with 100 nM AngII (+AngII). Data are representative of two independent experiments giving similar results.
Mutations in serines 221 and 318 impair the AngII-induced nuclear export of HuR.
The above results strongly suggest that phosphorylation sites at serines 221 and 318 play essential roles in the AngII-induced nucleocytoplasmic HuR shuttling. Therefore, we next tested whether a double mutation of both phosphorylation sites would alter the AngII-triggered nuclear export of ectopic Flag-tagged HuR protein. The subcellular localization of Flag-HuR was determined by indirect immunofluorescence utilizing an anti-Flag-specific antibody. In contrast to endogenous HuR, under unstimulated conditions, the ectopically expressed Flag-HuR constructs (pCMV-Flag-wtHuR and pCMV-Flag-HuRΔ2/3) occurred in the nucleus and in the cytoplasm, independent of which Flag-HuR construct was transfected (Fig. 10C). This strongly suggests that the intracellular distribution of Flag-HuR fusion proteins in hMC differs from the endogenous HuR. Importantly, AngII stimulation in cells transiently transfected with Flag-tagged wild-type HuR (pCMV-Flag-wtHuR) caused a clear reduction in the nuclear Flag staining and was paralleled by an overall accumulation of the Flag-fusion protein to the perinuclear region (Fig. 10C, panel 2). In contrast to the wild-type HuR, the cellular localization of the mutated Flag-HuR fusion protein (pCMV-Flag-HuRΔ2/3) was not affected by AngII (Fig. 10C, panel 4), thus demonstrating that PKC phosphorylation sites at serines 221 and 318 are essentially involved in the AngII-induced HuR shuttling.
AngII induces a PKC-specific serine phosphorylation of nuclear HuR.
Finally, we tested whether the PKC-dependent HuR phosphorylation observed by the in vitro kinase assay is also found in living cells. To this end, we applied an antibody specifically recognizing phosphorylated serines within PKC-specific consensus motifs (p-Ser-PKC-substrate antibody) and investigated nuclear extracts from either untreated or AngII-treated hMC. Samples were subjected to IP by an anti-HuR antibody and subsequently probed with the p-Ser-PKC-substrate antibody. As shown in Fig. 9C, PKCδ coimmunoprecipitates with HuR, and HuR in turn is rapidly phosphorylated at PKC-specific serines when cells are exposed to AngII. In contrast, PKCδ was not coimmunoprecipitated and no HuR-associated phosphoserine band was found in the extracts from vehicle-treated cells, although an equal amount of nuclear HuR was used for IP (Fig. 9C). Similarly, no immunoprecipitated p-Ser-PKC substrate could be detected when mouse IgG was used instead of anti-HuR antibody for the IP reaction (Fig. 9C).
Collectively, our data provide experimental evidence that AngII, by activating a rapid and transient translocation of PKCδ to the nucleus, does induce serine phosphorylation of HuR and thereby promotes the recruitment of HuR and its mRNA target (in our case the COX-2 mRNA) to the cytoplasm. Concomitantly, the ARE-containing HuR target COX-2 is bound to and accompanied by the free cytoplasmic and cytoskeleton-bound polysomes, which functionally results in an increase in COX-2-derived PGE2 formation (Fig. 6B).
DISCUSSION
The human COX-2 gene, in addition to transcriptional regulation, underlies a tight control by posttranscriptional events which, in most cases, are attributable to the 3′ UTR containing multiple copies of typical AREs functionally involved in the regulation of mRNA decay (8, 44, 48, 51). More specifically, among these primary destabilizing elements a conserved 116-nucleotide region is bound with high affinity by the ubiquitously mRNA stabilizing factor HuR. By employing an siRNA approach, we demonstrate that the AngII-mediated amplification of cytokine-induced COX-2 expression is mainly due to an HuR-dependent increase in COX-2 mRNA stability. Given the fact that AngII did not cause an equivalent increase in basal COX-2 mRNA level, we suggest that AngII on its own has no effect on the COX-2 transcription rate. Obviously, in human MC none of the signaling pathways activated by AngII suffices to trigger the transcription of COX-2, but additional signaling events induced by the proinflammatory cytokines IL-1β and TNF-α are required. Vice versa, silencing of HuR in MC did not cause an attenuation in cytokine-induced COX-2 expression levels, thus indicating that in this cell type a posttranscriptional regulation by HuR does not contribute to the cytokine-triggered regulation of COX-2. This again is in clear contrast to a previous study with rat MC in which IL-1β induced both transcriptional and also posttranscriptional, HuR-triggered regulation of COX-2 (6). Previously, we have reported on a similar phenomenon found with the extracellular nucleotide ATP, which, similar to AngII, affects a variety of pathophysiological key functions in the kidney, including cell proliferation, inflammation, and apoptosis (10). Strikingly, both mediators likewise can activate the export of HuR from the nucleus to the cytoplasm and thereby increase COX-2 expression levels via an increase in mRNA stability by activating different PKC isozymes.
Although various studies demonstrated the critical involvement of different signaling pathways in the stimulus-specific HuR shuttling, little is known about a direct posttranslational modification of HuR and its role in HuR export to the cytoplasm.
In this study, we provide experimental evidence that HuR is a critical target of PKCδ and is essentially involved in the AngII-stimulated nucleocytoplasmic HuR shuttling.
Similar to the classical PKCs, also for the Ca2+-independent PKCδ, a stimulus-induced translocation to membranes is considered as a hallmark of activation and is physiologically initiated by the generation of diacylglycerol (for review, see reference 52). In addition, the localization to a certain subcellular compartment, which most probably is mediated by binding to specific anchor proteins, critically determines the specificity and function of diverse PKC isoforms (23). On the molecular level, the structural requirements for the nuclear import of PKCδ are attributed to a bipartite nuclear localization sequence within the C terminus of the protein (62). Here we demonstrate that AngII induces a rapid and transient sequestration of PKCδ from the cytoplasm to the nucleus, which is followed by its physical interaction with nuclear HuR, as revealed by coIP and colocalization of both proteins (Fig. 9). A transient translocation of PKCδ to the nucleus in response to AngII has also been demonstrated in a previous study in rat liver WB cells (31). Besides the so far known nuclear targets of PKCδ, which comprise the nuclear DNA-dependent protein kinase, lamin, c-Abl, and p73b, a homolog of the apoptosis regulator p53 (for a review, see reference 52), we have identified the ubiquitous ELAV protein HuR as a further binding partner of PKCδ. Here we demonstrate that PKCδ is indispensable for the AngII-induced nucleocytoplasmic shuttling of HuR, since attenuation of PKCδ either by RNA attenuation or by use of pharmacological inhibitors caused a complete inhibition of this process (Fig. 8). These data suggest that AngII initiates two sequential processes within hMC: a rapid trafficking of PKCδ to the nucleus, which is followed by an export of nuclear HuR to the cytoplasm, especially to either free cytoplasmic or microtuble-associated polysomes (Fig. 11).
FIG. 11.

Schematic overview of AngII-induced COX-2 mRNA stabilization by PKCδ-dependent HuR activation. AngII binds to the AT1 receptor and activates PKCδ by generating diacylglycerol in the plasma and nuclear membrane. Upon activation, PKCδ translocates to the nucleus (step 1), where it physically interacts with the mRNA-stabilizing ELAV protein HuR. The association of both proteins is followed by a PKCδ-dependent phosphorylation of HuR at serine 221 and 318 (step 2). HuR phosphorylation promotes mRNA binding and nucleocytoplasmic shuttling of HuR (as indicated in step 3). The increase in HuR binding to COX-2-specific AREs stabilizes COX-2 mRNA, thereby leading to an increase in HuR-associated RNA translation at the polysomes (step 4). Functionally, the HuR-dependent increase in COX-2 mRNA stability by AngII is linked to a substantial increase in COX-2-protein and subsequent PGE2 synthesis (step 5).
In a cell-free in vitro kinase assay, we were able to demonstrate that HuR is not only a binding partner but even is a direct substrate of PKCδ (Fig. 10Β). Concomitantly, antibodies against HuR immunoprecipitated increased amounts of PKC associated with phosphoserine HuR after stimulation of cells with AngII (Fig. 9C), thus demonstrating that a phosphorylation of HuR by PKC occurs both in vitro and in intact cells. Mechanistically, the nucleocytoplasmic shuttling of HuR relies on a sequence in the hinge region between the RNA recognition motifs (RRMs) 2 and 3, referred to as HNS (14). Interestingly, serine 221 (HuRΔ2), which besides PKCδ is also critically involved in the PKCα-dependent HuR phosphorylation is located within this HNS (10). Therefore, it is tempting to speculate that a common mechanism of phosphorylation by different PKC isozymes within this hinge domain accelerates a conformational change, which in turn allows for a physical interaction between HuR and other binding partners that promote the export of HuR and its target mRNA from the nucleus to the cytoplasm. With regard to the functional consequences of HuR modification by PKCδ, this is the first study demonstrating that serine residues at positions 221 and 318 are critically involved in the stimulus-dependent HuR shuttling (Fig. 10C). Moreover, we cannot exclude that the phosphorylation by PKCδ, in addition to facilitating HuR export to the cytoplasm, may also improve the mRNA binding affinity of HuR to its target mRNAs within the nucleus. Such a mechanism was suggested by a previous study which demonstrated that a phosphorylation of HuR by the cell cycle checkpoint kinase 2 (Chk2) at phosphorylation sites lying between the two proximal RMMs (RRM1 and RRM2) modulates the affinity of HuR to its target ARE (1). However, in this report the phosphorylation of HuR by Chk2 was accompanied by an overall reduction in the RNA binding affinity of HuR (1). In contrast, our data show that phosphorylation of HuR by PKCδ has the opposite effect (increase in ARE binding), indicating that a ligand- and/or kinase-specific action accounts for COX-2 mRNA stabilization by AngII. Irrespective of this observation, the AngII-induced appearance of HuR within different polysomal fractions (Fig. 5A) was linked to COX-2 mRNA, thus indicating that the increased COX-2 mRNA expression rate functionally elicits an increased recuitment of active messenger RNPs (mRNPs) to polysomal compartments and is functionally linked to an overall increase in COX-2 translation. Furthermore, the detection of HuR-containing COX-2 mRNA in the cytoskeletal ribosomal fraction implies a functional role of HuR in the directed transport of soluble RNPs to the polysomes, as has been predicted for different carcinoma cell lines (2). Moreover, these observations imply that HuR binding to specific cytoskeletal elements is necessary for a HuR-promoted mRNA export to the translation machinery. Future experimental work is needed to decipher the functional role of cytoskeletal elements in HuR shuttling.
Functionally, the HuR-mediated increase in COX-2 mRNA stability leads to a substantial increase in PGE2 synthesis. Prostaglandins generated by the glomerular COX-2 enzyme play an important role in counterbalancing the vasoconstrictory effects of AngII and are, furthermore, implicated in tubuloglomerular feedback mechanism (22). Importantly, in addition to regulation of intrarenal vascular tone and water hemostasis, PGE2 elicits several growth-promoting and proinflammatory effects initiated by AngII (15, 22, 63). The AT1 receptor, which is the primary receptor for AngII in MC, can activate multiple signaling pathways including Ras, the MAPKs, the PI3K, the Akt/PKB kinase, and PKC, respectively (3). All of these kinases have been implicated in cellular key functions and, presumably, in a cell- and tissue-specific manner may affect the activity of HuR and, thereby, modulate the expression levels of genes critically involved in renal inflammation and fibrosis. Our study definitely warrants further investigations to identify additional genes whose mRNA stability is regulated by HuR in response to AngII or other key players of renal inflammation.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft grants EB 257/2-1, EB 257/2-2, PF 361/2-2, FOG 784, and EXC 147/1.
We thank Stephen M. Prescott (Department of Internal Medicine, University of Utah, Salt Lake City) for kindly providing us the human COX-2 promoter constructs.
Footnotes
Published ahead of print on 19 February 2008.
REFERENCES
- 1.Abdelmohsen, K., R. Pullmann, Jr., A. Lal, H. H. Kim, S. Galban, X. Yang, J. D. Blethrow, M. Walker, J. Shubert, D. A. Gillespie, H. Furneaux, and M. Gorospe. 2007. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 25543-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antic, D., and J. D. Keene. 1998. Messenger ribonucleoprotein complexes containing human ELAV proteins: interactions with cytoskeleton and translational apparatus. J. Cell Sci. 111183-197. [DOI] [PubMed] [Google Scholar]
- 3.Aschrafi, A., S. Shabahang, J. Pfeilschifter, and A. Huwiler. 2003. Regulatory functions of protein kinase C isoenzymes in the kidney. Curr. Topics Biochem. Res. 527-41. [Google Scholar]
- 4.Chandrasekharan, N. V., and D. L. Simmons. 2004. The cyclooxygenases. Genome Biol. 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, C. Y., and A. B. Shyu. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20465-470. [DOI] [PubMed] [Google Scholar]
- 6.Cok, S. J., S. J. Acton, and A. R. Morrison. 2003. The proximal region of the 3′-untranslated region of cyclooxygenase-2 is recognized by a multimeric protein complex containing HuR, TIA-1, TIAR, and the heterogeneous nuclear ribonucleoprotein U. J. Biol. Chem. 27836157-36162. [DOI] [PubMed] [Google Scholar]
- 7.Dean, J. L. E., R. Wait, K. R. Mahtani, G. Sully, A. R. Clark, and J. Saklatvala. 2001. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol. Cell. Biol. 21721-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon, D. A., C. D. Kaplan, T. M. McIntyre, G. A. Zimmerman, and S. M. Prescott. 2000. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J. Biol. Chem. 27511750-11757. [DOI] [PubMed] [Google Scholar]
- 9.Dixon, D. A., N. D. Tolley, P. H. King, L. B. Nabors, T. M. McIntyre, G. A. Zimmerman, and S. M. Prescott. 2001. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Investig. 1081657-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doller, A., A. Huwiler, R. Muller, H. H. Radeke, J. Pfeilschifter, and W. Eberhardt. 2007. Protein kinase Cα-dependent phosphorylation of the mRNA-stabilizing factor HuR: implications for posttranscriptional regulation of cyclooxygenase-2. Mol. Biol. Cell 182137-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eberhardt, W., M. Schulze, C. Engels, E. Klasmeier, and J. Pfeilschifter. 2002. Glucocorticoid-mediated suppression of cytokine-induced matrix metalloproteinase-9 expression in rat mesangial cells: involvement of nuclear factor-κB and Ets transcription factors. Mol. Endocrinol. 161752-1766. [DOI] [PubMed] [Google Scholar]
- 12.Eberhardt, W., A. Doller, A. E.-S. Akool, and J. Pfeilschifter. 2007. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 11456-73. [DOI] [PubMed] [Google Scholar]
- 13.Erkinheimo, T. L., H. Lassus, A. Sivula, S. Sengupta, H. Furneaux, T. Hla, C. Haglund, R. Butzow, and A. Ristimaki. 2003. Cytoplasmic HuR expression correlates with poor outcome and with cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer Res. 637591-7594. [PubMed] [Google Scholar]
- 14.Fan, X. C., and J. A. Steitz. 1998. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 173448-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faour, W. H., Y. He, Q. W. He, M. de Ladurantaye, M. Quintero, A. Mancini, and J. A. Di Battista. 2001. Prostaglandin E2 regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J. Biol. Chem. 27631720-31731. [DOI] [PubMed] [Google Scholar]
- 16.Geiges, D., T. Meyer, B. Marte, M. Vanek, G. Weissgerber, S. Stabel, J. Pfeilschifter, D. Fabbro, and A. Huwiler. 1997. Activation of protein kinase C subtypes α, γ, δ, ɛ, ζ, and η by tumor-promoting and nontumor-promoting agents. Biochem. Pharmacol. 53865-875. [DOI] [PubMed] [Google Scholar]
- 17.Grall, F. T., W. C. Prall, W. Wei, X. Gu, J. Y. Cho, B. K. Choy, L. F. Zerbini, M. S. Inan, S. R. Goldring, E. M. Gravallese, M. B. Goldring, P. Oettgen, and T. A. Libermann. 2005. The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes. FEBS J. 2721676-1687. [DOI] [PubMed] [Google Scholar]
- 18.Guhaniyogi, J., and G. Brewer. 2001. Regulation of mRNA stability in mammalian cells. Gene 26511-23. [DOI] [PubMed] [Google Scholar]
- 19.Hovland, R., G. Campbell, I. Pryme, and J. Hesketh. 1995. The mRNAs for cyclin A, c-myc and ribosomal proteins L4 and S6 are associated with cytoskeletal-bound polysomes in HepG2 cells. Biochem. J. 310193-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Idris, I., S. Gray, and R. Donnelly. 2001. Protein kinase C activation: isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia 44659-673. [DOI] [PubMed] [Google Scholar]
- 21.Inoguchi, T., T. Sonta, H. Tsubouchi, T. Etoh, M. Kakimoto, N. Sonoda, N. Sato, N. Sekiguchi, K. Kobayashi, H. Sumimoto, H. Utsumi, and H. Nawata. 2003. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol. 14227-232. [DOI] [PubMed] [Google Scholar]
- 22.Jaimes, E. A., R. X. Tian, D. Pearse, and L. Raij. 2005. Up-regulation of glomerular COX-2 by angiotensin II: role of reactive oxygen species. Kidney Int. 682143-2153. [DOI] [PubMed] [Google Scholar]
- 23.Jaken, S., and P. J. Parker.2000. Protein kinase C binding partners. Bioessays 22245-254. [DOI] [PubMed] [Google Scholar]
- 24.Jobin, C., O. Morteau, D. S. Han, and R. Balfour Sartor. 1998. Specific NF-κB blockade selectively inhibits tumour necrosis factor-alpha-induced COX-2 but not constitutive COX-1 gene expression in HT-29 cells. Immunology 95537-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones, D. A., D. P. Carlton, T. M. McIntyre, G. A. Zimmerman, and S. M. Prescott. 1993. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. J. Biol. Chem. 2689049-9054. [PubMed] [Google Scholar]
- 26.Keene, J. D. 1999. Why is Hu where? Shuttling of early-response-gene messenger RNA subsets. Proc. Natl. Acad. Sci. USA 965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirtikara, K., R. Raghow, S. J. Laulederkind, S. Goorha, T. Kanekura, and L. R. Ballou. 2000. Transcriptional regulation of cyclooxygenase-2 in the human microvascular endothelial cell line, HMEC-1: control by the combinatorial actions of AP2, NF-IL-6 and CRE elements. Mol. Cell. Biochem. 20341-51. [DOI] [PubMed] [Google Scholar]
- 28.Kujubu, D. A., B. S. Fletcher, B. C. Varnum, R. W. Lim, and H. R. Herschman. 1991. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 26612866-12872. [PubMed] [Google Scholar]
- 29.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 30.Ma, W. J., S. Cheng, C. Campbell, A. Wright, and H. Furneaux. 1996. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 2718144-8151. [DOI] [PubMed] [Google Scholar]
- 31.Maloney, J. A., O. Tsygankova, A. Szot, L. Yang, Q. Li, and J. R. Williamson. 1998. Differential translocation of protein kinase C isozymes by phorbol esters, EGF, and ANGII in rat liver WB cells. Am. J. Physiol. 274C974-C982. [DOI] [PubMed] [Google Scholar]
- 32.Meade, E. A., T. M. McIntyre, G. A. Zimmerman, and S. M. Prescott. 1999. Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J. Biol. Chem. 2748328-8334. [DOI] [PubMed] [Google Scholar]
- 33.Ming, X.-F., G. Stoecklin, M. Lu, R. Looser, and C. Moroni. 2001. Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol. Cell. Biol. 215778-5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Misquitta, C. M., V. R. Iyer, E. S. Werstiuk, and A. K. Grover. 2001. The role of 3′-untranslated region (3′-UTR) mediated mRNA stability in cardiovascular pathophysiology. Mol. Cell. Biochem. 22453-67. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell, J. A., and T. D. Warner. 2006. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat. Rev. Drug Discov. 575-86. [DOI] [PubMed] [Google Scholar]
- 36.Mrena, J., J. P. Wiksten, A. Thiel, A. Kokkola, L. Pohjola, J. Lundin, S. Nordling, A. Ristimaki, and C. Haglund. 2005. Cyclooxygenase-2 is an independent prognostic factor in gastric cancer and its expression is regulated by the messenger RNA stability factor HuR. Clin. Cancer Res. 117362-7368. [DOI] [PubMed] [Google Scholar]
- 37.Niranjanakumari, S., E. Lasda, R. Brazas, and M. A. Garcia-Blanco. 2002. Reversible cross-linking combined with IP to study RNA-protein interactions in vivo. Methods 26182-190. [DOI] [PubMed] [Google Scholar]
- 38.Pascale, A., M. Amadio, G. Scapagnini, C. Lanni, M. Racchi, A. Provenzani, S. Govoni, D. L. Alkon, and A. Quattrone. 2005. Neuronal ELAV proteins enhance mRNA stability by a PKCα-dependent pathway. Proc. Natl. Acad. Sci. USA 10212065-12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng, S. S., C. Y. Chen, N. Xu, and A. B. Shyu. 1998. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 173461-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfeilschifter, J., and H. Muhl. 1990. Interleukin 1 and tumor necrosis factor potentiate angiotensin II- and calcium ionophore-stimulated prostaglandin E2 synthesis in rat renal mesangial cells. Biochem. Biophys. Res. Commun. 169585-595. [DOI] [PubMed] [Google Scholar]
- 41.Pfeilschifter, J., C. Schalkwijk, V. A. Briner, and H. van den Bosch. 1993. Cytokine-stimulated secretion of group II phospholipase A2 by rat mesangial cells. Its contribution to arachidonic acid release and prostaglandin synthesis by cultured rat glomerular cells. J. Clin. Investig. 922516-2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfeilschifter, J., and A. Huwiler. 1996. Regulatory functions of protein kinase C isoenzymes in purinoceptor signalling in mesangial cells. J. Auton. Pharmacol. 16315-318. [DOI] [PubMed] [Google Scholar]
- 43.Radeke, H. H., B. Meier, N. Topley, J. Floge, G. G. Habermehl, and K. Resch. 1990. Interleukin 1-β and tumor necrosis factor-α induce oxygen radical production in mesangial cells. Kidney Int. 37767-775. [DOI] [PubMed] [Google Scholar]
- 44.Ristimaki, A., K. Narko, and T. Hla. 1996. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem. J. 318325-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ross, J. 1995. mRNA stability in mammalian cells. Microbiol. Rev. 59423-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sengupta, S., B. C. Jang, M. T. Wu, J. H. Paik, H. Furneaux, and T. Hla. 2003. The RNA-binding protein HuR regulates the expression of cyclooxygenase-2. J. Biol. Chem. 27825227-25233. [DOI] [PubMed] [Google Scholar]
- 47.Shaw, G., and R. Kamen. 1986. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46659-667. [DOI] [PubMed] [Google Scholar]
- 48.Sheng, H., J. Shao, D. A. Dixon, C. S. Williams, S. M. Prescott, R. N. DuBois, and R. D. Beauchamp. 2000. Transforming growth factor-beta1 enhances Ha-ras-induced expression of cyclooxygenase-2 in intestinal epithelial cells via stabilization of mRNA. J. Biol. Chem. 2756628-6635. [DOI] [PubMed] [Google Scholar]
- 49.Smith, W. L., D. L. DeWitt, and R. M. Garavito. 2000. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69145-182. [DOI] [PubMed] [Google Scholar]
- 50.Sorli, C. H., H. J. Zhang, M. B. Armstrong, R. V. Rajotte, J. Maclouf, and R. P. Robertson. 1998. Basal expression of cyclooxygenase-2 and nuclear factor-interleukin 6 are dominant and coordinately regulated by interleukin 1 in the pancreatic islet. Proc. Natl. Acad. Sci. USA 951788-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Srivastava, S. K., T. Tetsuka, D. Daphna-Iken, and A. R. Morrison. 1994. IL-1β stabilizes COX II mRNA in renal mesangial cells: role of 3′-untranslated region. Am. J. Physiol. 267F504-F508. [DOI] [PubMed] [Google Scholar]
- 52.Steinberg, S. F. 2004. Distinctive activation mechanisms and functions for protein kinase Cδ. Biochem. J. 384449-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subbaramaiah, K., T. P. Marmo, D. A. Dixon, and A. J. Dannenberg. 2003. Regulation of cyclooxgenase-2 mRNA stability by taxanes: evidence for involvement of p38, MAPKAPK-2, and HuR. J. Biol. Chem. 27837637-37647. [DOI] [PubMed] [Google Scholar]
- 54.Wang, W., J. Fan, J. X. Yang, S. Fürer-Galban, I. Lopez de Silanes, C. von Kobbe, J. Guo, S. N. Georas, F. Foufelle, D. G. Hardie, D. Carling, and M. Gorospe. 2002. AMP-activated kinase regulates cytoplasmic HuR. Mol. Cell. Biol. 223425-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warner, T. D., and J. A. Mitchell. 2004. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J. 18790-804. [DOI] [PubMed] [Google Scholar]
- 56.Whiteside, C. I., and J. A. Dlugosz. 2002. Mesangial cell protein kinase C isozyme activation in the diabetic milieu. Am. J. Physiol. Renal Physiol. 282F975-F980. [DOI] [PubMed] [Google Scholar]
- 57.Winzen, R., M. Kracht, B. Ritter, A. Wilhelm, C. Y. Chen, A. B. Shyu, M. Muller, M. Gaestel, K. Resch, and H. Holtmann. 1999. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 184969-4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Winzen, R., G. Gowrishankar, F. Bollig, N. Redich, K. Resch, and H. Holtmann. 2004. Distinct domains of AU-rich elements exert different functions in mRNA destabilization and stabilization by p38 mitogen-activated protein kinase or HuR. Mol. Cell. Biol. 244835-4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie, W. L., J. G. Chipman, D. L. Robertson, R. L. Erikson, and D. L. Simmons. 1991. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. USA 882692-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu, N., C.-Y. A. Chen, and A.-B. Shyu. 2001. Versatile role for hnRNP D isoforms in the differential regulation of cytoplasmic mRNA turnover. Mol. Cell. Biol. 216960-6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao, K. M., M. L. Samson, R. Reeves, and K. White. 1993. Gene elav of Drosophila melanogaster: a prototype for neuronal-specific RNA binding protein gene family that is conserved in flies and humans. J. Neurobiol. 24723-739. [DOI] [PubMed] [Google Scholar]
- 62.Yoshida, K., Y. Miki, and D. Kufe. 2002. Activation of SAPK/JNK signaling by protein kinase Cδ in response to DNA damage. J. Biol. Chem. 27748372-48378. [DOI] [PubMed] [Google Scholar]
- 63.Yu, C., R. Gong, A. Rifai, E. M. Tolbert, and L. D. Dworkin. 2007. Long-term, high-dosage candesartan suppresses inflammation and injury in chronic kidney disease: nonhemodynamic renal protection. J. Am. Soc. Nephrol. 18750-759. [DOI] [PubMed] [Google Scholar]
- 64.Zalfa, F., B. Eleuteri, K. S. Dickson, V. Mercaldo, S. De Rubeis, A. di Penta, E. Tabolacci, P. Chiurazzi, G. Neri, S. G. Grant, and C. Bagni. 2007. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci. 10578-587. [DOI] [PMC free article] [PubMed] [Google Scholar]