

Abstract
The surface force apparatus was used to measure directly the molecular forces between streptavidin and lipid bilayers displaying grafted Mr 2,000 poly(ethylene glycol) (PEG). These measurements provide direct evidence for the formation of relatively strong attractive forces between PEG and protein. At low compressive loads, the forces were repulsive, but they became attractive when the proteins were pressed into the polymer layer at higher loads. The adhesion was sufficiently robust that separation of the streptavidin and PEG uprooted anchored polymer from the supporting membrane. These interactions altered the properties of the grafted chains. After the onset of the attraction, the polymer continued to bind protein for several hours. The changes were not due to protein denaturation. These data demonstrate directly that the biological activity of PEG is not due solely to properties of simple polymers such as the excluded volume. It is also coupled to the competitive interactions between solvent and other materials such as proteins for the chain segments and to the ability of this material to adopt higher order intrachain structures.
Poly(ethylene glycol) (PEG) is used extensively to improve the biocompatibility of foreign materials for both in vivo and ex vivo applications (1–3). Its prevalent use is due largely to its low toxicity and low immunogenicity (1). In addition, due to its protein resistance, it is widely used as a stabilizing surface coating in biological environments (3–7). For example, PEG-functionalization of liposomes increased their blood circulation times by nearly an order of magnitude (4). In the clinic, ethylene oxide surface grafts are used to reduce protein adsorption onto the surfaces of biomedical polymers (1–3, 5–7). This is important for controlling the biological responses to the latter, in part, because protein adsorption is a well established first step in the humoral response against foreign materials (2, 8, 9). Thus, by preventing the unwanted adsorption of bioactive agents onto the surfaces of medical polymers, the surface grafting of hydrophilic polymers is one of the more effective, general strategies used to manipulate the biological activity of medical materials (3, 9–14).
The unusual efficacy of PEG as an apparently biologically passivating surface coating is linked to both the presumed biological inertness of the polymer backbone and to its solvated configuration (1, 3, 5, 7). In most cases, the proposed mechanisms for the protein resistance of PEG borrow from current theories for structureless polymers in isotropic liquids (5, 15–19). For example, PEG’s ability to repel proteins is attributed to its large excluded volume (3, 5, 15–19), its configurational entropy (20, 21), surface coverage by grafted chains (3, 15, 18), and the thickness of grafted layers (1, 3, 9, 17, 18). These latter attributes are all well described theoretically for both terminally anchored and soluble simple chains in simple solvents (22–25). Based on such arguments, it should be a simple matter to design and optimize the performance of PEG-based coatings using state-of-the-art polymer theories.
Yet PEG is not a simple polymer. It forms directional bonds with water (26–28), it adopts higher order intrachain structure (28–31), and it exhibits complex phase behavior (32). None of this behavior is addressed by current theories for simple polymers in simple solvents. The biological inertness of PEG is also unusual in light of its “amphiphilicity.” It is highly soluble both in water and in organic solvents (1, 7, 31), and it forms stable monolayers at the air-water interface (33). By contrast, other closely related polyethers are neither water soluble nor biocompatible (7). Indeed, the polymer’s interactions with water lead to much richer phase and structural behavior than manifested by structureless chains (32). In light of the fact that its properties deviate from the assumptions used to describe simple chains, it is remarkable that measured behavior has been in such good agreement with theoretical predictions (34–36).
The efficacy of PEG coatings and the assumptions regarding its properties have indeed been questioned (37), but the consequences of these unusual characteristics for its biologically relevant properties have not been addressed (19). Apparent discrepancies in the biocompatibility of materials coated with grafted PEG suggest that the properties of such coatings are far from simple (37). Moreover, such complex behavior makes clear that current polymer theories do not adequately describe the variety of phenomenology that determine the interactions of this material with the biological environment. Yet only a few theories considered how the additional degrees of freedom afforded by solvent interactions and the ability to form higher order structures affect PEG behavior in aqueous solutions (28, 38–40).
In this work, we report direct evidence that PEG is not an inert, simple polymer, but that it can bind proteins. The formation of these attractive interactions is linked to rearrangements in the polymer configuration. We measured directly the molecular forces between streptavidin and monolayers of grafted Mr 2,000 methoxy-terminated PEG. The interactions were investigated as a function of polymer grafting density with chain configurations ranging from isolated “mushrooms” to dense polymer brushes. Measurements at both low and high compressive forces and under minimally perturbing conditions determined the repulsive force (interaction energy) required to induce adhesion between protein and PEG at different surface coverages. Results are interpreted in terms of current models for the behavior of PEG in aqueous solutions.
MATERIALS AND METHODS
Chemicals.
Conjugates of distearoylphosphatidyl ethanolamine (DSPE) with Mr 2,000, methoxy-terminated PEG (DSPE-EO45) were purchased from Avanti Polar Lipids. All other phospholipids also were purchased from Avanti. All salts were high purity (>99.5%) and were purchased from Aldrich. Streptavidin was from Calbiochem. Water was purified with a Milli-Q UV filtration system (Millipore). HPLC grade methanol and chloroform were from Mallinckrodt.
Preparation of Supported Lipid Bilayers Displaying Grafted PEG.
Solutions of PEG-lipid (DSPE-EO45) and pure DSPE were prepared in 9:1 chloroform/methanol solutions. Mixtures of 1.5, 4.5, and 9.0 mol % DSPE-EO45 with DSPE were prepared by mixing chloroform solutions of the pure lipid and PEG-lipid in appropriate proportions.
The grafted polymer layers, as shown in Fig. 1, were prepared by the Langmuir–Blodgett deposition of mixed lipid monolayers onto a hydrophobic support as described elsewhere (34). The latter monolayers were composed of DSPE and DSPE-EO45 in the proportions stated above. After spreading the lipids on the water surface, they were compressed to an average area of 43 Å2 per lipid. The monolayer then was deposited at constant pressure onto a hydrophobic, crystalline monolayer of dipalmitoyl phosphatidylethanolamine (DPPE) prepared by its Langmuir–Blodgett deposition from the water-vapor interface onto freshly cleaved mica. The chain grafting density is thereby controlled precisely by adjusting the DSPE-EO45 mole fraction in the bilayer. The transfer ratio—that is, the area transferred relative to the area coated by the film—was greater than 0.95 in all cases.
Figure 1.

Sample configuration used in direct force measurements.
Preparation of Oriented Streptavidin Monolayers.
Force measurements were conducted with oriented monolayers of streptavidin. While this protein will not participate in the in vivo recognition of foreign materials, its surface amino acid composition is not unlike that of other soluble proteins such as those found in blood. Its well known structure and facile formation of oriented protein monolayers (41) also enabled us to relate the measured forces with the protein’s surface composition. These protein monolayers were thus prepared for force measurements by the specific adsorption of soluble streptavidin onto a supported lipid bilayer displaying biotin conjugated to dihexadecyl phosphatidylethanolamine via a six-carbon spacer (biotin-X-DHPE, Molecular Probes). The biotin-lipid was mixed at 5 mol % with ditridecanoyl phosphatidylcholine and spread at the water-vapor interface. The monolayer was compressed to an average lipid area of 65 Å2, and transferred onto a dipalmitoyl phosphatidylethanolamine monolayer on mica. The melting temperature of the neutral lipid ditridecanoyl phosphatidylcholine is 14°C, and the monolayer is in the liquid crystalline state at 25°C. Soluble streptavidin binds to the biotinylated membrane and self-assembles into an oriented monolayer (Fig. 1) (42). The protein coverage on monolayers thus prepared, as determined by radiolabeling methods, was 3,600 ± 100 Å2 per streptavidin. The protein therefore occupies 79% of the bilayer surface.
Force Measurements.
Force measurements were conducted with a Mark II surface force apparatus at 25°C. The chamber of the instrument housing the samples was filled with a solution of 10 mM sodium phosphate buffer (pH 7.0) and 30 mM KNO3. All solutions were saturated with lipid to prevent desorption of the lipid bilayers during the measurements. Before filling the chamber, solutions were filtered through surfactant-free 0.2-μm Durapore membranes (Millipore).
RESULTS
Definition of Zero Separation.
The distances reported refer to the separation between the outer surface of the streptavidin monolayer and that of the opposed, dehydrated bilayer membrane supporting the grafted PEG. The difference in the distance of closest intersurface approach before and after removal of the organic layers from the mica defined their total thickness T. After draining the solution from the chamber of the apparatus, the residual organic material on the mica was burned away by UV irradiation. The compressed polymer thickness was then determined by subtraction of the thicknesses of the two bilayers plus that of the streptavidin from the total thickness T (Fig. 1). Thus, tPEG = T − (tSTA + tDSPE + tDTPC + 2tDPPE). For these calculations, the thickness of the DSPE, streptavidin, and DPPE monolayers were 28 Å (34), 43 ± 2 Å (41), and 27 Å (42), respectively. The 13 Å thickness of the ditridecanoyl phosphatidylcholine layer was determined according to the method of Tanford (43), as described elsewhere (34, 41, 42).
Force Measurements.
The forces between streptavidin and minimally perturbed PEG chains were determined from initial measurements of the forces between them, and successive force measurements in which the polymer was minimally compressed. In the former case, the forces between fresh samples were measured during their first approach. In the second case, the streptavidin was brought into contact with the grafted PEG layer under successively increasing applied loads. To avoid significant polymer perturbations, the materials achieved soft contact at forces below 6–8 mN/m, unless otherwise indicated. At that point, the compression was stopped. The materials then were separated slowly to avoid perturbing the layer by convective fluid flow or mechanical disruption. After the system relaxed for several minutes, the protein was again pushed gently against the polymer layer, but the applied load was increased slightly above that in the previous measurement. This sequence of events was continued, until an attractive force was measured during separation.
Before the onset of attractive interactions, the polymer-protein forces were entirely repulsive. Fig. 2 summarizes the force vs. distance curves between streptavidin and DSPE membranes displaying 1.5 mol % (isolated chains), 4.5 mol % (weakly overlapping chains), and 9.0 mol % PEG-lipid (brushes). The profiles are the superposition of repulsive electrostatic double-layer and steric forces. Under the experimental conditions, both the streptavidin and polymer surfaces were charged. The negative charge on the grafted PEG surface is due to the ionized phosphate group on the anchoring phospholipid (34), and streptavidin is negatively charged at pH 7.0 (41). The steric force was due to the compression of the polymer layer by streptavidin. The range of the latter repulsion was estimated from the onset of the deviation from the exponentially decaying electrostatic double-layer force. Both the range and magnitude of the polymer repulsion increased with the grafting density.
Figure 2.

Force vs. distance profiles were measured during initial approach between streptavidin and DSPE-EO45 monolayers at different polymer grafting densities. Experiments were performed at pH 7.0 and 25°C in 10 mM NaH2PO4 and 30 mM KNO3. The different EO45 grafting densities were 480 (•), 960 (▪), and 2,280 (○) Å2 per site, which corresponded to 9.0 (•), 4.5 (▪), and 1.5 (○) mol % DSPE-EO45 in the DSPE matrix.
The contribution of the electrostatic force to the measured interactions was determined from fits of the data at distances D > 50 Å to the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory of the superposition of electrostatic double-layer and van der Waals forces (44, 45). At those distances, the interactions were uncomplicated by the “non-DLVO” steric polymer forces. A representative fit is shown in Fig. 3 for the interaction between streptavidin and a monolayer of weakly overlapping chains (4.5 mol % DSPE-EO45). The theoretical curves were generated by solving numerically the nonlinear Poisson–Boltzmann equation (41, 44–46). The calculated electrostatic force was superimposed on the van der Waals force, which was calculated using Lifschitz theory, including zero frequency screening by electrolytes (44–46). The electrostatic surface potentials of both the protein and grafted PEG layers were determined from the fits. In these studies, the electrostatic potential of the protein monolayer was constrained by the value determined independently in control measurements between streptavidin and bare DSPE monolayers (see below). Thus, by fixing the electrostatic potential of the protein monolayer at −54 ± 2 mV, and allowing that of the DSPE-EO45 monolayer layer to vary, the electrostatic potential of the latter surface was determined uniquely.
Figure 3.

The force vs. distance profile was measured between a streptavidin and a monolayer of 4.5 mol % DSPE-EO45 under conditions described in the text. The best fits of the data to Derjaguin–Landau–Verwey–Overbeek theory gave −54 ± 2 mV and −41 ± 5 mV for the electrostatic potentials of the streptavidin and PEG-displaying bilayers, respectively. Similar fits were obtained using constant potential (solid line) or constant charge (dashed line) boundary conditions.
The electrostatic surface potentials varied with the PEG grafting density as expected. Similar fits were obtained with either constant potential or constant charge boundary conditions (Fig. 3) (34, 45, 46). The charge density increased nearly linearly with polymer grafting density (Table 1). The greater error in the value for the 1.5 mol % PEG layer is due to the larger relative error in the small measured force used to fit the data. The fitted electrostatic parameters were in remarkably good agreement with the theoretically predicted values. The latter were calculated using the PEG grafting density, the electrolyte concentration, and the Grahame equation (44).
Table 1.
Fitted electrostatic surface parameters and polymer extension as a function of DSPE-EO45 grafting density
| % polymer lipid, mol % | Coverage* Γ, Å2/chain | Potential, mV | Charge density, mC/m2 | Predicted value, mC/m2 | Polymer extension L, Å | Predicted extension L, Å |
|---|---|---|---|---|---|---|
| 1.5 | 3300 | −20 ± 5 | −9 ± 3 | −4 | 35 ± 3 | 35 |
| 4.5 | 960 | −41 ± 5 | −14 ± 1 | −13 | 38 ± 3 | 35 |
| 9.0 | 480 | −67 ± 5 | −28 ± 1 | −26 | 50 ± 5 | 45 |
Based on the excluded area per chain.
The ranges and magnitudes of the steric polymer forces were then determined quantitatively by subtraction of the double-layer force from the overall profile. At 1.5 mol % and 4.5 mol % DSPE-EO45, the onset of the latter repulsion occurred at 35 ± 3 Å and at 38 ± 3 Å, respectively (Table 1). This agrees with the predicted grafted layer thickness, based on polymer scaling theory (24, 25, 47), and with previous measurements (34). In particular, for isolated or weakly interacting chains, the extension of grafted, simple polymers in good solvent should equal the Flory radius RF, which is 35 Å for Mr 2,000 PEG (24, 25, 47). This was the observed thickness for both the 1.5 and 4.5 mol % DSPE-PEG layers (Table 1) (24, 25, 47). The gradient of the force measured with 4.5% layers was steeper than at lower grafting densities, indicative of the stronger repulsion (cf., Fig. 2). At 9.0 mol %, the strongly interacting chains are stretched, and the extension of the brush is determined from (47)
 |
where s is the distance between grafting sites. In the latter case, protein contacted the outer PEG segments farther out at 50 ± 5 Å. This agreed with the predicted chain extension of 45 Å (24, 25, 47).
These findings were exactly as expected for the compression of grafted, unstructured chains by inert, colloidal particles in isotropic solvents (17, 18, 48). While this was indeed the case for weak protein-polymer contact, the behavior differed significantly, if the protein was pushed into or incubated in contact with the PEG layers.
Pushing streptavidin into the grafted chains resulted in two notable changes in the polymer. First, we measured streptavidin-PEG adhesion, as opposed to the purely repulsive forces described in the preceding section. Hysteresis in the measured receding force curves signaled the onset of the attraction between streptavidin and PEG. Before this, the force profiles were completely reversible (Fig. 4A). At the onset of the attraction, the reverse curves became less repulsive, i.e. more attractive (Fig. 4B). Near the adhesive minimum, the protein and polymer separated, and the two surfaces jumped apart. The adhesion was determined from the force required to detach the surfaces.
Figure 4.

Hysteresis measured at the onset of adhesion between streptavidin and PEG. (A) Before the onset of attractive streptavidin-PEG interactions, there was no difference between advancing (○) and receding (•) force profiles. (B) The onset of the attraction, as signaled by hysteresis in the receding force curve (•). The surfaces finally jumped apart at the position indicated by the arrow.
The position of the attractive minimum, the magnitude of the adhesion, and the distance at which the surfaces separated depended on the method of determination. If the materials were initially brought into contact under relatively large loads, then the adhesion was larger and the pull-off distance closer in than when the materials first achieved only soft contact. When streptavidin was pressed into the 1.5 mol % PEG layers at repulsive forces >6 mN/m, the materials separated from 40 ± 10 Å, and the measured adhesion was −1.0 ± 0.3 mN/m (Table 2). Subsequent to that, however, the attractive minima were farther out at 100 ± 10 Å, and the latter adhesion was slightly weaker at −0.5 ± 0.2 mN/m (Table 2). Similarly, streptavidin contact with 4.5 mol % PEG layers at >12 mN/m resulted in pull-off from 28 ± 8 Å, and the adhesion was −2 ± 1 mN/m. By contrast, at the onset of adhesion after a succession of soft contacts, the surfaces separated at 110 ± 10 Å, and the adhesion was −0.7 ± 0.2 mN/m. The minima measured after pull-off from 28 Å were at 110 Å, and the adhesion was similar. Lastly, when protein was brought into contact with 9.0 mol % PEG at repulsive forces >29 mN/m, they separated from 64 ± 6 Å, and the adhesion was −0.5 ± 0.2 mN/m. Both subsequent measurements and the adhesion induced by soft contact were −0.7 ± 0.2 mN/m, and pull-off was at 90 ± 10 Å (Table 2).
Table 2.
Location and depth of adhesive minima as a function of DSPE-EO45 surface coverage
| Density, mol % | Extension L, Å | Pull-off distance Dj, Å | Fadh/R, mN/m |
|---|---|---|---|
| 1.5 | 35 ± 3 | 40 ± 10 | −1.0 ± 0.3 |
| 100 ± 10 | −0.5 ± 0.2 | ||
| 4.5 | 38 ± 3 | 28 ± 8 | −2 ± 1 |
| 110 ± 10 | −0.7 ± 0.2 | ||
| 9.0 | 50 ± 5 | 65 ± 6 | −0.5 ± 0.2 |
| 90 ± 10 | −0.7 ± 0.3 |
This behavior is attributed to the formation of attractive contacts between streptavidin and the ethylene oxide segments and is presumably due to induced or facilitated structural rearrangements in the polymer backbone. A possible mechanism for the shorter range, slightly stronger adhesion is discussed later. However, the broad attractive well near 100 Å is consistent with the sequential rupture of multiple segment-protein interactions. This is as expected for polymer bridging (44), hence, for adhesion between the protein and stretched polymer chains. The pull-off distances were approximately 60% of the fully extended 157 Å chain length, but PEG can reportedly stretch substantially (49).
The pull-off position did not always coincide with the attractive minimum (cf. Fig. 4). The rather sluggish detachment made the precise determination of the pull-off distance difficult, and this caused some uncertainty in its measurement near 100 Å. The variability also can be attributed, in part, to the polydispersity of the polymer preparation. Although low, polydispersity will add to the uncertainty in the determined position at which the polymer and protein finally separate.
The compressive force required to induce adhesion between PEG and streptavidin varied with the polymer density. We estimated the corresponding compressive energy in each case. With 1.5 mol % DSPE-EO45, the attraction set in above repulsive forces of 2 mN/m. Using the Derjaguin approximation (44, 45), which relates the force between curved surfaces to the interaction energy per unit area between them, we estimated an energy of
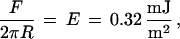 |
or 2 kT per chain. Similarly, streptavidin adhered to the 4.5 mol % and 9 mol % membranes only when the applied load exceeded 12 ± 3 mN/m (4 kT per chain) and 29 ± 5 mN/m (5 kT per chain), respectively. In these calculations, we assumed that all chains were perturbed identically. While this is not the case, it is a reasonable estimate. The protein layer is densely packed, and the polymer effectively “sees” a uniform protein surface. The corresponding energies per protein were 4, 27, and, 65 kT per streptavidin at 1.5, 4.5, and 9.0 mol % PEG-lipid, respectively. Both trends are as expected. The osmotic resistance to protein penetration of the layer increases with the segment density (17, 18, 50, 51). Moreover, at higher grafting densities, proteins perturb more chains (17, 18, 50, 51).
Importantly, the adhesion between protein and PEG was sufficiently robust that, during separation, some DSPE-EO45 pulled from the bilayer rather than detaching at the protein-polymer interface. This was evident from the changes in the force curves measured subsequent to separation. Namely, upon reapproach, the onset of the steric force was farther out, and the maximum compressible polymer layer thickness increased. After the first or second pull-off, the range of the steric force increased by only 10 Å. Indicative of increased polymer pull-out with successive contacts, the range of the steric force continued to increase by up to 35 Å, and the maximum compressible thickness increased by 25 ± 5 Å. Both the magnitude (ca. −0.7 mN/m) and reproducibility of the measured adhesion indicate, however, that the number of polymers extracted was small. Chain perturbations due to polymer stretching during detachment also may have increased the polymer extension, but these measurements cannot distinguish between the two possibilities. However, the relaxation time of such excursions from the equilibrium thickness is on the order of milliseconds (49), and these thickness changes were irreversible.
The second significant change was that the polymer did not rapidly convert back to the repulsive configuration, but continued to readily bind streptavidin. For example, after the onset of the attraction, streptavidin adhered to the weakly overlapping PEG chains (4.5 mol %), even at intersurface repulsive forces below 4 mN/m (<1.5 kT per chain). We observed similar behavior at all polymer densities. Namely, the pull-off was at 100 ± 10 Å, and the adhesion was −0.7 ± 0.3 mN/m. The latter behavior suggests that the outer segments switched from initially repulsive conformations to attractive ones. This might occur if, for example, the backbone structure changed from the more ordered, polar trans-gauche-trans conformation for the COCCOC sequence, to the less polar, mainly gauche configuration (28, 31, 39). With the 9 mol % brushes, the polymer returned to its initial, repulsive state within 3–5 hr, but changes in the 4.5 and 1.5 mol % PEG layers sometimes persisted for more than 5 hr. Thus, interactions with the protein clearly altered the polymer properties.
The forces between streptavidin and bare DSPE monolayers were dominated by the repulsive double-layer force between the negatively charged streptavidin and the neutral lipid at distances D > 10 Å. At smaller separations, the forces were more repulsive, due to the additional contribution by steric solvation. The electrostatic origin of the long-range force was verified by fitting the data to Derjaguin–Landau–Verwey–Overbeek theory (data not shown). The best fit potentials for both the protein and bilayer surfaces were −55 ± 5 mV and 0 ± 2 mV, respectively, in excellent agreement with values reported previously (41, 42). There was no measurable attraction between the streptavidin and bilayer membrane at any separation, and no evidence of protein denaturation at compressive pressures below ca. 50 atm.
A potential source of the measured protein-polymer adhesion, particularly at D < 30 Å, is the double-layer force between the layers. Such attractive electrostatic forces can occur between surfaces with electrostatic potentials of the same sign but different magnitudes (41, 45). To test whether this was responsible for the measured attraction, we calculated the double-layer force between the different samples considered. In all cases, the calculated force was repulsive at distances D > 20 Å (c.f. Fig. 3, solid line). Even at the smallest separation where we measured adhesion (D = 28 ± 8 Å), the double-layer force is repulsive. The measured attraction between streptavidin and the PEG-displaying bilayers is therefore not electrostatic in origin.
To test whether the polymer perturbations were due simply to chain compression, measurements were conducted between 4.5 mol % DSPE-EO45 and bare DSPE monolayers. To reproduce the conditions used to induce protein adhesion, experiments were conducted similar to those performed with streptavidin monolayers. That is, the PEG layers were brought into soft contact with the membrane surface, and then slowly separated. The compressive force was then increased in subsequent measurements up to 20–30 mN/m. Additionally, the PEG layer was pressed against the bilayer membrane for up to 1 hr before separation. The measured forces were repulsive at all separations. In none of these experiments did the polymer thickness change, or the surfaces adhere.
We investigated whether similar attractive interactions could be induced between identical, grafted PEG chains. For example, polymer desolvation and interchain hydrogen-bonding would generate intersurface bridging. We conducted force measurements between identical 4.5 mol % PEG-lipid monolayers under identical conditions and experimental procedures as used in the measurements with streptavidin. The force curves thus determined were identical to those reported previously (34). Additionally, even if the polymers were allowed to incubate in contact under a repulsive force of 15 mN/m for up to 2 hr, there was no hysteresis in the curves. Importantly, none of these treatments resulted in any measured adhesion between the polymer layers.
DISCUSSION
The measured attractive interactions between streptavidin and grafted PEG layers were clearly due to protein-polymer adhesion. First, all measurements were conducted at forces much too low to denature streptavidin. Thus, the measured changes were not due to structural changes in streptavidin. Second, neither the compression of the polymer brush nor the extended incubation with a second polymer layer resulted in similar adhesion. Third, adhesive contacts formed readily with streptavidin, but not with the hydrated bilayer or with PEG chains.
The onset of the adhesion corresponded to fundamental changes in the polymer. The compressive energy required for adhesion was presumably necessary to induce chain rearrangements. These likely involved both changes in the polymer configuration and protein penetration of the polymer core. Regardless of the mechanism, the polymer switched from a protein-resistant to a protein-attractive configuration, and the latter change persisted for several hours. This is inconsistent with single chain behavior. The calculated relaxation times of single Mr 2,000 chains are on the order of milliseconds (49). The perturbations therefore may indicate a cooperative change such as a change of state. Consequently, the persistence of the altered, protein-attractive state is apparently due to an integral change in the PEG.
The adhesion between streptavidin and the polymer layers at 100 Å was relatively independent of the polymer coverage at the grafting densities studied. This is attributed to the fact that the streptavidin density on the monolayer was constant throughout these measurements. At the PEG grafting densities studied, the polymer/protein ratio was always ≥ 1, and each protein could interact with at least one polymer chain. The adhesion thus would be limited by the number of attractive residues on the protein surface and by the protein surface area accessible to the sticky polymer segments.
The average adhesive energy per streptavidin, estimated from the pull-off force at 100 Å and use of the Derjaguin approximation, was ca. 1.6 kT per protein. This exceeds previous estimates of attractive protein-PEG interactions based on indirect neutron scattering measurements of BSA in aqueous polymer solutions (35). The latter estimate of 0.05 kT per segment-protein contact was determined with soluble polymer, and is both model- and protein-dependent. If the polymer indeed envelops the entire protein surface in solution, then interaction energies comparable to those reported here would require 30 segments, or 2/3 of the polymer (n = 45). This is unlikely given the measured pull-off distances. Furthermore, in these studies, the polymer only interacted with a limited region of the protein surface. While we cannot compare directly the individual segment-protein interaction energies, it is obvious from the PEG-lipid pull-out and the average adhesion energies that the attraction is substantial.
The occurrence of PEG-lipid pull-out is not unexpected. The detachment force is not determined by the bond energy, but by its gradient (52, 53). The tensile strengths of short, weak bonds will thus exceed those of stronger, longer bonds such as that between the lipid and the bilayer (52, 53). The rupture of relatively weak, short-range hydrogen bonds between amino acids requires a greater force than uprooting anchoring lipids from the bilayer (M. Tirrell, personal communication). Hydrogen bonding between PEG and streptavidin should give rise to similar behavior.
These findings contrast with theories for structureless, grafted chains that predict that proteins near a polymer brush should encounter a strictly repulsive force (17, 18, 48, 50, 51). However, in several cases PEG indeed appeared to behave as a simple polymer. Forces between grafted DSPE-EO45 layers agreed with predictions based on polymer scaling theory (24, 25, 34, 48). At low polymer-streptavidin interaction forces, as reported here, the forces similarly agreed with expected behavior (17, 18). Single-chain-mean-field (SCMF) calculations of protein interactions with inert, grafted PEG suggest that polymer rearrangements during protein extraction from the brush may generate an apparent attraction (50, 51). This is not inconsistent with our findings (cf. Fig. 2). The SCMF model does not, however, explain the attractive interactions with streptavidin or predict the adhesive minima at 100 Å, far from the core of the grafted layer. We attribute the latter discrepancy to the neglect of the potentiality of attractive segment-solvent or segment-protein interactions.
Our findings show that this polymer is not inert to protein, and that it can adopt different states in water. This deviation from predicted behavior is attributed to ethylene oxide’s ability to bind solvent and other polymers. To better understand the origins of the deviations, we compared our results qualitatively with the few models that consider explicitly hydrogen-bonding interactions between the polymer segments and solvent as well as higher-order intrachain structure (28, 38–40). The current theoretical picture of PEG behavior includes only a limited number of scenarios: namely, segment interactions with water (38), and either the ability of the segments to adopt two different configurations (39) or to form monomer clusters (40) within the chain. These theories entail some ingredients that enabled us to rationalize the observed behavior.
Bekiranov (38) used a modified Flory–Huggins model to account for hydrogen-bonding between solvent and the polymer backbone. They predicted the temperature-dependent phase behavior of PEG solutions. While the model can address protein-polymer bridging, it does not consider higher-order PEG structuring. It is unclear whether it would predict the protein-induced configurational changes. The observed perturbations are also qualitatively consistent with the structural model proposed by Kjellander and Florin (28), and with the theoretical “two-state” (39) and “cluster” (40) models for grafted PEG chains. The first proposes, on the basis of spectroscopic and x-ray data (29, 31, 54), that hydrated PEG is α-helical. Applied pressure could both induce polymer bridging and disrupt the helical structure through the hydrogen bonding or hydrophobic protein contacts. However, these and previous measurements suggest that PEG does not behave as a rigid, helical coil (34–36).
The two-state model predicts the preferential partitioning of segments with nonpolar configurations near the interface and polar segments at the outer surface. The cluster model predicts a similar segment segregation (40). Proteins thus would be repelled from the outer, polar surface, but attracted to the nonpolar, inner segments (39). The measured adhesion was indeed greater when streptavidin was forced into the polymer layer. Protein extraction from the inner core would pull the polymer inside out and further disrupt its structure. Compressive forces could facilitate polymer desolvation and hydrogen bonding to the streptavidin surface. They could as easily force streptavidin into the polymer brush and enable it to bind buried, nonpolar segments. In reality, both mechanisms likely operate.
Clearly, none of the current theories account for all of the biologically relevant properties of PEG (17, 18, 38–40, 48, 50, 51). Nor can its efficacy as a surface coating be understood entirely in terms of properties of structureless polymers such as the excluded volume. Rather its biological activity is due, in part, to the competitive interactions between solvent or other macromolecules and the ethylene oxide segments. To determine not only the mechanism of the induced changes reported in this work, but also the fundamental parameters controlling them, will require further investigations at different temperatures, polymer molecular weights, and solvent conditions. For example, the melting temperature of solvated PEG varies with the molecular weight (27). The polymer solubility is similarly a function of the molecular weight and temperature.
These findings have several important consequences for the use of grafted PEG as passivating coatings. First, the activation energy for protein-polymer adhesion increased with the polymer grafting density. Denser layers thereby increase not only the diffusional barrier but also the energy required to form attractive protein-polymer bonds. Second, while compressive forces were required to induce adhesion, the associated activation barriers for the formation of these adhesive contacts were 2–5 kT per chain and 4–65 kT per streptavidin. The latter value, however, scales with the protein dimensions. In practice, total repulsive potentials of <15 kT per protein can only delay, but not abolish, protein penetration of and adhesion to the brush (45). Thus, repulsive potentials >15 kT are required to stably repel proteins from the underlying surface. Importantly, perturbations that affect the polymer solvation or solubility, such as temperature or molecular weight (33, 38, 40), will similarly alter the repulsive barrier; hence, the protein adsorption kinetics. Indeed, Claesson et al. (55) reported a temperature-dependent attraction between short, ethylene oxide oligomers.
Third, while theories for simple polymers capture some of the phenomenology responsible for its biological activity, they do not predict all of the biologically relevant properties of PEG. Some theories do address the solvation and structural properties of this polymer (38–40), but they are still at an early stage of development. Thus, to quantitatively predict and optimize the performance of PEG-based, protein-resistant coatings, we clearly require more comprehensive theories that consider all of the degrees of freedom peculiar to this material.
Acknowledgments
We thank M. Tirrell and J. Israelachvili for sharing their preliminary data with us and A. Halperin for many provocative discussions. This work was supported by the Whitaker Foundation.
ABBREVIATIONS
- PEG
poly(ethylene glycol)
- DSPE
distearoyl phosphatidylethanolamine
- DSPE-EO45
poly(ethylene glycol) conjugated to distearoyl phosphatidylethanolamine
- DDPE
dipalmitoyl phosphatidylethanolamine
References
- 1.Harris, J. M., ed. Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications (Plenum, New York).
- 2.Barenberg S A. MRS Bull. 1991;16:26–33. [Google Scholar]
- 3.Elbert L, Hubbell J A. Annu Rev Mater Sci. 1996;26:365–394. [Google Scholar]
- 4.Papahadjopoulos D, Allen T, Gabizon A, Mayhew E, Matthay K, Huang S, Lee K, Woodle M, Lasic D, Redemann C, Martin F. Proc Natl Acad Sci USA. 1991;88:11460–11465. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amiji M, Park K. J Biomater Sci Polym Ed. 1993;4:217–234. doi: 10.1163/156856293x00537. [DOI] [PubMed] [Google Scholar]
- 6.Llanos G, Sefton M V. J Biomed Mater Res. 1993;27:1383–1391. doi: 10.1002/jbm.820271105. [DOI] [PubMed] [Google Scholar]
- 7.Lee J H, Lee H B, Andrade J. Progr Polym Sci. 1995;20:1043–1079. [Google Scholar]
- 8.Peppas N, Langer R. Science. 1994;263:1715–1720. doi: 10.1126/science.8134835. [DOI] [PubMed] [Google Scholar]
- 9.Hubbell J A, Langer R. Chem Eng News. 1995;March:13. , 1995, 42–54. [Google Scholar]
- 10.Tseng Y-C, McPherson T, Yuan C S, Park K. Biomaterials. 1995;16:963–972. doi: 10.1016/0142-9612(95)94902-w. [DOI] [PubMed] [Google Scholar]
- 11.Pitt W G, Cooper S L. J Biomed Mater Res. 1988;22:383–404. doi: 10.1002/jbm.820220502. [DOI] [PubMed] [Google Scholar]
- 12.Andrade J, Hlady V, Wei A-P, Ho C-H, Lea A S, Jeon S I, Lin Y S, Stroup E. Clin Mater. 1992;11:1–17. [Google Scholar]
- 13.Gölander C-G, Herron J N, Lim K, Claesson P, Stenius P, Andrade J D. In: Poly(Ethylene Glycol) Chemistry. Harris J M, editor. New York: Plenum; 1992. pp. 221–245. [Google Scholar]
- 14.Lee J H, Kopecek J, Andrade J D. J Biomed Mater Res. 1989;23:351–368. doi: 10.1002/jbm.820230306. [DOI] [PubMed] [Google Scholar]
- 15.Hermans J. J Chem Phys. 1982;77:2193–2203. [Google Scholar]
- 16.McPherson T B, Lee S J, Park K. In: Proteins at Interfaces II: Fundamentals and Applications. Horbett T, Brash J, editors. Washington, DC: Am. Chem. Soc; 1995. pp. 256–268. [Google Scholar]
- 17.Jeon S I, Lee J H, Andrade J D, deGennes P G. J Colloid Interface Sci. 1991;142:149–158. [Google Scholar]
- 18.Jeon S I, Andrade J D. J Colloid Interface Sci. 1991;142:159–166. [Google Scholar]
- 19.Arakawa T, Timasheff S N. Biochemistry. 1985;24:6756–6762. doi: 10.1021/bi00345a005. [DOI] [PubMed] [Google Scholar]
- 20.Reichert W M, Filisko E E, Barenberg S A. In: Biomaterials: Interfacial Phenomena and Applications. Brash J, Horbett T, editors. Washington, DC: Am. Chem. Soc.; 1982. pp. 177–193. [Google Scholar]
- 21.Li J-T, Caldwell K D, Rapoport N. Langmuir. 1994;10:4475–4482. [Google Scholar]
- 22.deGennes P G. Scaling Concepts in Polymer Physics. Ithaca, NY: Cornell Univ. Press; 1979. [Google Scholar]
- 23.Grosberg, A. (1994) Statistical Physics of Macromolecules; trans. Atanov, Y. A. (Am. Inst. Phys. Press, New York).
- 24.Milner S. Science. 1991;251:905–914. doi: 10.1126/science.251.4996.905. [DOI] [PubMed] [Google Scholar]
- 25.Halperin A, Tirrell M, Lodge T P. Adv Polym Sci. 1992;100:31–47. [Google Scholar]
- 26.Bailey F E, Callard R W. J Appl Polym Sci. 1959;1:56–62. [Google Scholar]
- 27.Antonson K P, Hoffman A S. In: Poly(Ethylene Glycol) Chemistry. Harris J M, editor. New York: Plenum; 1992. pp. 15–28. [Google Scholar]
- 28.Kjellander R, Florin E. J Chem Soc Faraday Trans I. 1981;77:2053–2077. [Google Scholar]
- 29.Koenig J L, Angood A C. J Polym Sci. 1970;8:1787–1796. [Google Scholar]
- 30.Miyazawa T, Fukoshima K, Ideguchi Y. J Am Chem Soc. 1962;37:2764–2776. [Google Scholar]
- 31.Liu K-J, Parsons J L. Macromolecules. 1969;2:529–533. [Google Scholar]
- 32.Saeki S, Kuwahara N, Nakata M, Kaneko M. Polymer. 1976;17:685–689. [Google Scholar]
- 33.Cao B H, Kim M W. Faraday Discuss. 1994;98:245–252. [Google Scholar]
- 34.Kuhl T L, Leckband D E, Lasic D D, Israelachvili J N. Biophys J. 1994;66:1479–1488. doi: 10.1016/S0006-3495(94)80938-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abbott N, Blankschtein D, Hatton A T. Macromolecules. 1992;25:3932–3941. [Google Scholar]
- 36.Noppl-Simson D A, Needham D. Biophys J. 1996;70:1391–1401. doi: 10.1016/S0006-3495(96)79697-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Llanos G, Sefton M V. J Biomater Sci Polymer Ed. 1993;4:381–400. doi: 10.1163/156856293x00069. [DOI] [PubMed] [Google Scholar]
- 38.Bekiranov S, Bruinsma R, Pincus P. Europhys Lett. 1993;24:183–188. [Google Scholar]
- 39.Björling M, Linse P, Karlström G. J Phys Chem. 1990;94:471–481. [Google Scholar]
- 40.Wagner M, Brochard-Wyart F, Hervet H, deGennes P-G. Colloid Polym Sci. 1993;271:621–628. [Google Scholar]
- 41.Leckband D, Schmitt F-J, Knoll W, Israelachvili J. Biochemistry. 1994;33:4611–4624. doi: 10.1021/bi00181a023. [DOI] [PubMed] [Google Scholar]
- 42.Marra J, Israelachvili J. Biochemistry. 1985;24:4608–4618. doi: 10.1021/bi00338a020. [DOI] [PubMed] [Google Scholar]
- 43.Tanford C. J Phys Chem. 1972;93:917–922. [Google Scholar]
- 44.Israelachvili J. Intermolecular and Surface Forces. 2nd Ed. New York: Academic; 1991. [Google Scholar]
- 45.Hunter R. Foundations of Colloid Science. New York: Oxford Univ. Press; 1989. [Google Scholar]
- 46.Grabbe A. Langmuir. 1993;9:797–801. [Google Scholar]
- 47.Alexander S. J Phys. 1977;38:983–987. [Google Scholar]
- 48.Subramaniam G, Williams D R M, Pincus P A. Macromolecules. 1996;29:4045–4050. [Google Scholar]
- 49.Wong J Y, Kuhl, Israelachvili J, Zalipsky S, Mullah N. Science. 1997;275:820–823. doi: 10.1126/science.275.5301.820. [DOI] [PubMed] [Google Scholar]
- 50.Szleifer I. Biophys J. 1997;72:595–612. doi: 10.1016/s0006-3495(97)78698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szleifer I, Carignano M A. Adv Chem Phys. 1996;94:165–260. [Google Scholar]
- 52.Bell G. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 53.Leckband D E, Müller W, Schmitt F-J, Ringsdorf H. Biophys J. 1995;69:1162–1169. doi: 10.1016/S0006-3495(95)79990-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi Y, Takadoro H. Macromolecules. 1973;6:672–675. [Google Scholar]
- 55.Claesson P, Kjellander R, Stenius P, Christenson H K. J Chem Soc Faraday Trans 1. 1986;82:2735–2746. [Google Scholar]