

Summary
SRC-3/AIB1 is a steroid receptor coactivator with potent growth promoting activity and its overexpression is sufficient to induce tumorigenesis. Previous studies indicate that the cellular level of SRC-3 is tightly regulated by both ubiquitin-dependent and ubiquitin-independent proteasomal degradation pathways. Atypical protein kinase C (aPKC) is frequently overexpressed in cancers. In the present study, we show that aPKC phosphorylates and specifically stabilizes SRC-3 in a selective ER-dependent manner. We further demonstrate that an acidic residue rich region in SRC-3 is an important determinant for aPKC mediated phosphorylation and stabilization. The mechanism of the aPKC mediated stabilization appears due to a decreased interaction between SRC-3 and the C8 subunit of the 20S core proteasome, thus preventing SRC-3 degradation. Our results demonstrate a new and potent signaling mechanism for regulating SRC-3 levels in cells by coordinate enzymatic inhibition of both ubiquitin-dependent and ubiquitin-independent proteolytic pathways.
Introduction
Steroid receptor coactivator-3 (SRC-3/AIB1/ACTR/pCIP/RAC3) is a member of the p160 coactivator family and plays an important role in cell growth, reproduction, metabolism, and cytokine signaling (Wang et al., 2000; Xu et al., 2000; Zhou et al., 2003). SRC-3 was originally found to be overexpressed and amplified in a number of cancers, particularly in breast cancer (Anzick et al., 1997). Accumulating evidence substantiates an important role of SRC-3 in cell growth and oncogenesis (Yan et al., 2006; Zhou et al., 2003). Transgenic mice which overexpress SRC-3 were found to have an extremely high tumor incidence (Torres-Arzayus et al., 2004). In contrast, SRC-3 knockout mice have a significantly lower incidence of ras-induced mammary gland tumorigenesis (Kuang et al., 2004). Thus, it appears that the level of SRC-3 protein is critical for tumorigenesis and progression.
Protein kinases are often overexpressed in cancers and certain kinases serve as prognostic factors (Kumar and Wang, 2002; Mackay and Twelves, 2003; Scheid and Woodgett, 2001). Select extracellular signals stimulate SRC-3 coactivator activity by activating downstream protein kinases, which eventually lead to SRC-3 phosphorylation and can contribute to the oncogenic potential of SRC-3 (Wu et al., 2004). Distinct patterns of phosphorylation induced by different signals encode the specificity of SRC-3 for formation of different transcription factor complexes (Wu et al., 2004). In agreement with tumorigenesis studies in mice, the cellular levels of SRC-3 appear to be tightly regulated. Both ubiquitin-dependent and ubiquitin-independent mechanisms have been reported to regulate SRC-3 stability (Li et al., 2006; Lonard et al., 2000). Recently, it was shown that phosphorylation of SRC-3 by p38 MAP kinase and GSK3 is associated with increased degradation (Gianni et al., 2006; Wu et al., 2007). However, kinases that stabilize SRC-3 in cancer cells have not hitherto been reported.
Atypical protein kinase C (aPKC) belongs to the PKC superfamily. There are twelve PKC isoforms which can be divided into three classes. Unlike conventional (α, β and γ) and novel PKCs (δ, ε, η and θ), atypical PKCs (ζ and λ/ι) are insensitive to phorbol ester, DAG or calcium stimulation (Mackay and Twelves, 2003). They are important for a variety of physiological processes including immune responses, glucose homeostasis and establishment of cell polarity (Hirai and Chida, 2003; Suzuki et al., 2003). aPKC members can be as homologous as 72% amino acid identity overall (Mackay and Twelves, 2003) and aPKCs are reported to be overexpressed in ovarian, prostate, lung, liver, and bladder carcinomas (Regala et al., 2005; Tsai et al., 2000), indicating they are important players in cancer cell proliferation and migration (Donson et al., 2000; Eder et al., 2005).
Here we demonstrate that aPKC increases the cellular SRC-3 protein level in a selective ER-dependent manner. It induces the phosphorylation of C-terminal residues of SRC-3 and transduces it into a form that only weakly interacts with the C8 subunit of the core 20S proteasome (also named PSMA3), thereby making it more resistant to proteasomal degradation and leading to cellular stabilization of the oactivator. Using this mechanism, aPKC can play an important role inestrogen-dependent growth and tumorigenesis.
Results
Atypical PKC stabilizes cellular SRC-3 protein levels
To determine whether protein kinases can effect the level of cellular SRC-3 protein, we cotransfected flag-SRC-3 with HA tagged PKCζ, PKA catalytic subunit, and IKKα or IKKβ into HeLa cells; flag-estrogen receptor (flag-ER) was co-transfected as a control. As shown in Fig. 1A, PKCζ dramatically increased the flag-SRC-3 protein level while other kinases had minor effects. The effect of PKCζ on SRC-3 protein level also was observed when a different antibody, anti-SRC-3 antibody, was used (Fig. S1). The effect of PKCζ on the SRC-3 protein level requires its kinase activity since the kinase dead mutant of PKCζ did not alter SRC-3 levels (Fig. 1B). Similar results also were obtained with aPKC member, PKCι. RT-PCR results (Fig. 1C) showed that PKCζ did not increase the level of flag-SRC-3 transcripts, suggesting that the increased SRC-3 protein levels likely were due to a change in SRC-3 protein stability and not to increased mRNA transcription. We further performed a pulse-chase experiment to monitor the turnover of flag-SRC-3 protein. As shown in Fig. 1D, the half-life of flag-SRC-3 is significantly increased in the presence of PKCζ. To confirm that PKCζ affects SRC-3 stability, we analyzed the effect of PKCζ on SRC-3 in the presence of MG132, a proteasome inhibitor. As shown in Fig. 1E, MG132 significantly increased the flag-SRC-3 protein level and addition of PKCζ had no further stabilizing effect on SRC-3. Together these results suggest that aPKC stabilizes SRC-3 protein by preventing its proteasome mediated degradation.
Fig. 1.
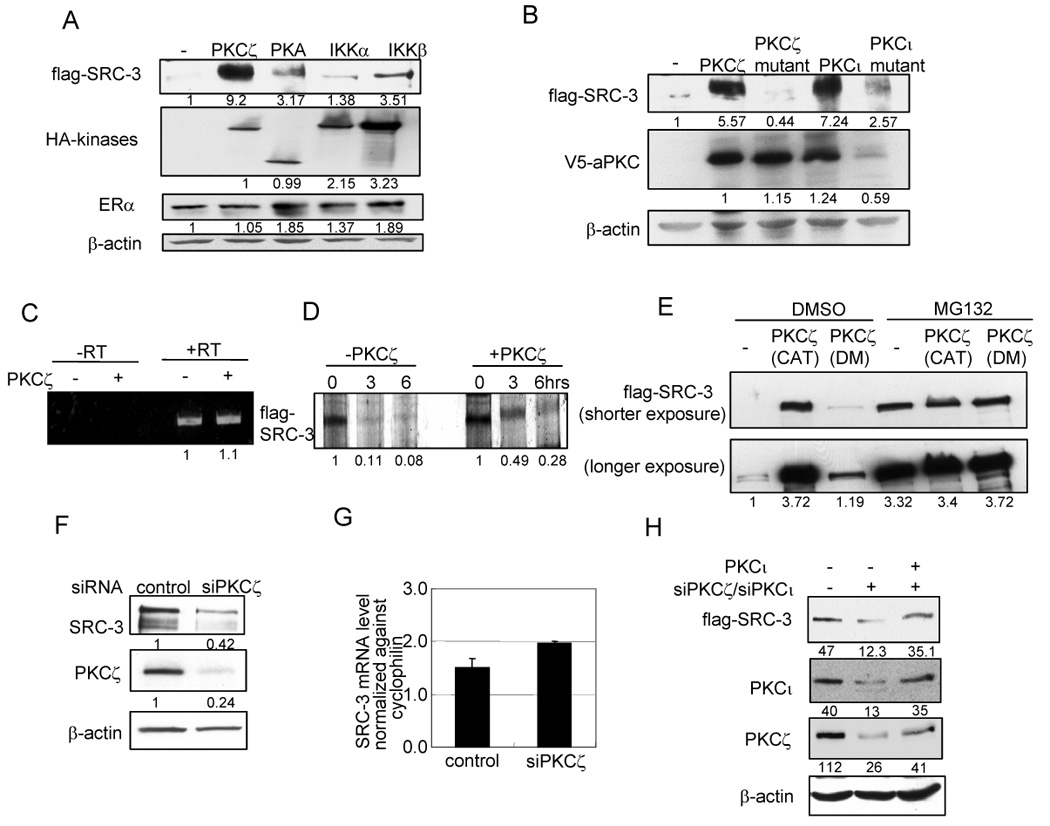
aPKC stabilizes SRC-3 protein. (A) Flag-SRC-3 and ERα expression vectors were co-transfected with HA tagged kinase expression vectors into HeLa cells. (B) Western blot analysis of flag-SRC-3 protein level when cells were co-transfected with constitutively active mutant of PKCζ (A119E) or PKCι (A120D), or their kinase dead mutants (K281W and K274W, respectively). (C) RT-PCR was performed using flag-specific primer and SRC-3-specific primer to detect the mRNA level of transfected flag-SRC-3. A RT-PCR experiment without the addition of reverse transcriptase (RT) was included as a control. (D) Pulse-chase experiment. HeLa cells were transfected with flag-SRC-3 and ERα, with or without co-transfection of PKCζ. Shown is autoradiograph of flag-SRC-3 immunoprecipitated from cell lysates. (E) PKCζ stabilizes SRC-3 protein in proteasome-mediated degradation pathway. Cells were treated with proteasome inhibitor MG132 or vehicle DMSO for 16 hrs. (F) Endogenous SRC-3 and PKCζ protein levels in MCF-7 cells treated with siPKCζ. (G) Real time qRT-PCR was carried out to detect the mRNA level of endogenous SRC-3. (H) Overexpression of PKCι rescued the siRNA knock-down effect on SRC-3 protein level. Details were described in Supplemental Procedures.
We next examined whether PKCζ affects endogenous SRC-3 protein levels. Toward this end, ER positive MCF-7 breast cancer cells were treated with a pool of siRNAs against PKCζ. As shown in Fig. 1F, the PKCζ protein level was significantly reduced by the siRNA treatment. Knock-down of PKCζ also decreased the level of SRC-3 protein. To minimize the possibility of siRNA off-target effects, we also used different individual siRNAs to knock down PKCζ similar results were obtained (data not shown). Real time qRT-PCR showed that endogenous SRC-3 mRNA levels were not decreased by siRNAs against PKCζ (Fig. 1G). Since both PKCζ and PKCι affect SRC-3 stability, we examined whether overexpression of PKCι could rescue the knockdown effect. As shown in Fig. 1H, knockdown of both PKCζ and PKCι decreased flag-SRC-3 levels in Flp-In 293 cells which contain stably expressed tetracycline inducible flag-SRC-3 (Yi et al., 2005). Overexpression of PKCι complemented the knockdown effect on the SRC-3 level. We also treated MCF-7 cells with different kinase inhibitors (Fig. S2A), and confirmed that inhibition of aPKC leads to a significantly reduced amount of SRC-3. In contrast, conventional or novel PKCs function in an opposite way in regulating SRC-3 level (Fig. S2 A, B and C).
The aPKC stabilization effect is selective for SRC-3
SRC-3 has significant sequence homology and distinct as well as redundant functions relative to its companion family members SRC-1 and SRC-2. To examine whether aPKC also stabilizes other SRCs, we co-transfected flag-SRC-1 and flag-SRC-2 with flag-ERα in the absence and presence of PKCζ (Fig. 2A). Interestingly, the levels of SRC-1 and SRC-2 were not altered by PKCζ, indicating that the PKCζ effect appears to be selective for SRC-3.
Fig. 2.
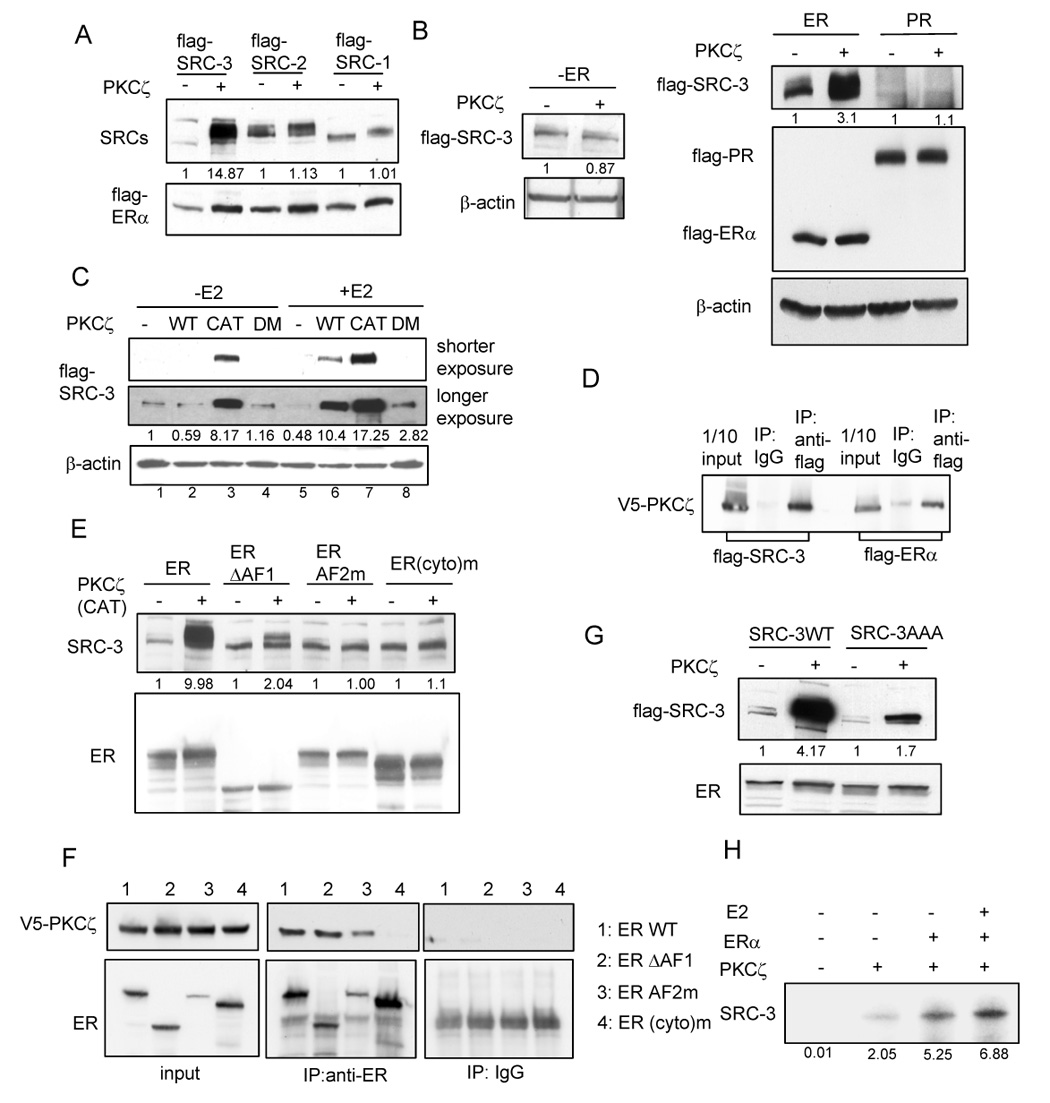
Estrogen and estrogen receptor are required for the aPKC stabilization effect (A) Effects of active PKCζ on SRCs in the presence of ERα. (B) Effects of active PKCζ on flag-SRC-3 protein level in the absence or presence of flag-ERα, or flag-PRB. (C) E2 increases PKCζ mediated stabilization effect. WT: wild type PKCζ, CAT: constitutively active mutant, DM: kinase dead mutant. (D) PKCζ interacts with both SRC-3 and ERα. Cells were transfected with V5- PKCζ and flag-SRC-3 or flag-ERα. Cell lysates were immunoprecipitated by anti-flag antibody or normal IgG and Western blot analysis was performed using anti-V5 antiobody. (E) Effects of different ERα mutants on the PKCζ mediated stabilization effect. ERΔAF1: deletion of 1–178 amino acids; ERAF2m: K362D/V376D/L539A; ER(cyto)m: deletion of 250–303 amino acids. (F) Coimmunoprecipitation experiment detecting the interaction between PKC and different ERα mutants. (G) PKCζ has significantly reduced stabilization effect on SRC-3 AAA mutant. (H) Autoradiograph of in vitro PKCζ kinase assay detecting phosphorylation of purified recombinant SRC-3 by incorporation of γ-32P ATP in the absence and presence of purified ERα and estrogen.
Estrogen and estrogen receptor are required for the aPKC stabilization effect
The experiments performed to this point were carried out in the presence of ERα, either with transfected cells or in cells containing endogenous ERα. We next performed the experiments in the absence of transfected ERα. Surprisingly, we found that active PKCζ did not enhance the level of SRC-3 in the absence of ERα (Fig. 2B). We then tested whether another steroid receptor progesterone receptor (PR) could support this stabilization effect. As shown in Fig. 2B, no stabilization was observed. We also did not observe this effect in the presence of another ER subtype (ERβ) (Fig. S3). Our results suggest that the stabilization effect is ERα-selective.
Since ERα is necessary for the stabilization effect, we tested whether estrogen (E2) enhances the response (Fig. 2C). In the absence of E2, despite the presence of ERα, wild type PKCζ(lane 2) and its kinase dead mutant (lane 4) did not stabilize SRC-3, while the constitutively active mutant increased SRC-3 protein level (lane 3). In the presence of E2, wild type PKCζ increased the SRC-3 protein level (lane 6) while the effect of its constitutively active mutant was further augmented (lane 7 vs. lane 3). Thus, estrogen is not only important for the activation of PKCζ (Fig. 2C, lane 2 vs. lane 3), which is consistent with a previous report (Castoria et al., 2004), but it also plays an important role in the stabilization process mediated by aPKC (lane 3 vs. lane 7).
Next we examined the mechanism by which ERα enhances PKCζ mediated SRC-3 stability. We first investigated whether ERα can bring SRC-3 and PKCζ together. It was reported that PKCζ interacts with ERα (Castoria et al., 2004). Since ERα is required for SRC-3 stabilization, we examined the interaction between PKCζ and SRC-3 or ERα in co-immunoprecipitation experiments. As shown in Fig. 2D, ERα indeed interacts with PKCζ. SRC-3 also interacts with PKCζ without co-transfection of ERα, suggesting that the interaction between SRC-3 and PKCζ does not require ERα. Thus, ERα does not simply support the aPKC stabilization effect by mediating the interaction between PKCζ and SRC-3.
We then tested the effects of different ERα mutants on SRC-3 stabilization. ERα wild type, a mutant with its AF1 domain deleted (ERΔAF1), an AF2-defective mutant (ERAF2m), and a cytoplasm-only ER mutant (ER(cyto)m) each were co-transfected with flag-SRC-3. As shown in Fig. 2E, ERΔAF1 had a significantly reduced capability to stabilize SRC-3 while ERAF2m and ER(cyto)m had no effect on SRC-3 levels. To determine whether the inability of ERAF2m and ER(cyto)m to stabilize SRC-3 is due to an inability to effect an interaction with PKCζ or SRC-3, we performed co-immunoprecipitation experiments (Fig. 2F). V5- PKCζ and different ERα mutants were co-transfected into HeLa cells. The cell lysates were immunoprecipitated with an anti-ERα antibody and immunoblotted with anti-V5 antibody. Our results show that ERAF2m was still able to interact with PKCζ while the ER(cyto)m failed to interact. Since ERAF2m is not capable of an E2-dependent interaction with SRC-3 (Zheng et al., 2005), the combined results suggest that this interaction between ERα and SRC-3 is important for the PKCζ stabilization effect, and that, the stabilization likely occurs in the nucleus. To further confirm the importance of the ER-SRC-3 interaction, we examined the effect of PKCζ on an SRC-3 AAA mutant which contains mutations in three LXXLL motifs (L to A mutations) that are important for its interaction with nuclear receptors. In this case, stabilization of SRC-3 is greatly reduced (Fig. 2G). The AAA mutant still is functional in activating transcription factors other than nuclear receptors (Fig. S4).
Since SRC-3 was shown to be a phosphoprotein, we examined whether PKCζ can directly phosphorylate SRC-3 and whether ER plays a role in this phosphorylation. To test this, we performed an in vitro cell free PKCζ kinase assay using purified SRC-3 as a substrate. As shown in Fig. 2H, PKCζ is capable of phosphorylating SRC-3. The presence of ER and E2 further enhanced its phosphorylation. Since SRC-3 can interact with PKCζ and they both can interact with ER, our results suggest that the formation of this ternary complex may place SRC-3 in a more optimal conformation to facilitate its phosphorylation by PKCζ.
PKCζ enhances SRC-3 coactivator activity
To answer the question as to whether the increase in SRC-3 protein is functionally significant, the transcription of an endogenous ER target gene, pS2, was measured when cells were co-transfected with SRC-3 and PKCζ (Fig. 3A, left panel). Addition of PKCζ alone only moderately increased ER-activated pS2 transcription. When SRC-3 was co-transfected, the wild type PKCζ and the constitutively active mutant, but not the kinase dead mutant, significantly enhanced pS2 transcription in the presence of E2. As shown in Fig. 3A right panel, wild type PKCζ and its mutants are expressed in cells at similar levels. These results suggest that the SRC-3 protein stabilized by PKCζ is transcriptionally active.
Fig. 3.
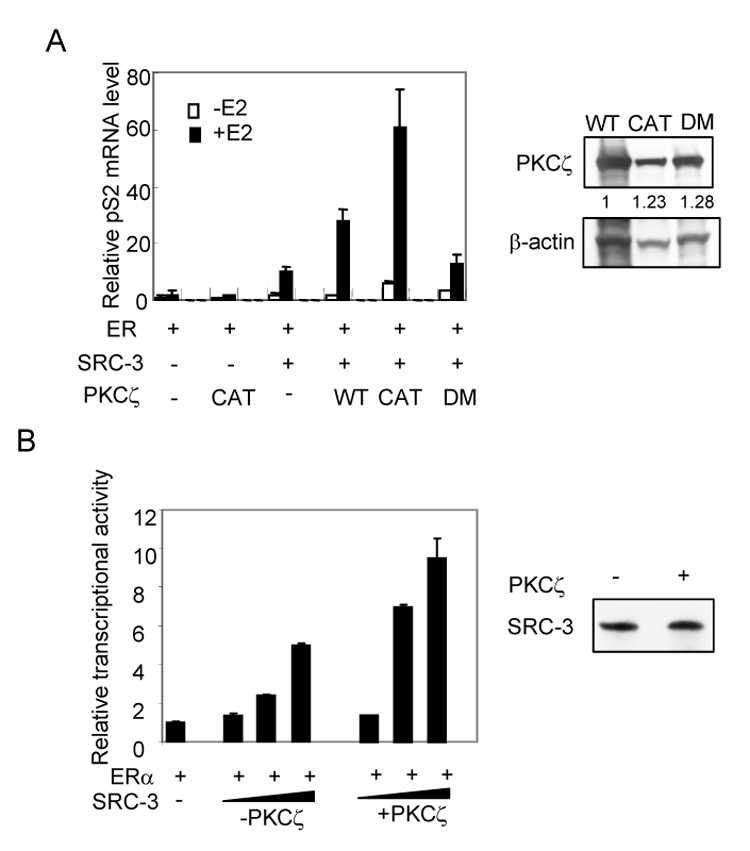
PKCζ enhances SRC-3 coactivator activity. (A) PKCζ enhances SRC-3 activated ER targeted gene transcription. HeLa cells were transfected with ERα, SRC-3, PKCζ or its mutants in the absence or presence of E2. The transcription of pS2 gene was measured by real time qRT-PCR (left panel). The expression levels of PKCζ and its mutants were shown in the right panel. (B) aPKC stabilized SRC-3 is transcriptionally active in cell-free transcription assay. Plasmid ERE-E4 was assembled into chromatin and transcribed as described in Materials and Methods. ER and E2 were added in the reaction. Triangles represent increasing amounts of purified flag-SRC-3 (1.5, 9 and 37.5 ng SRC-3 protein, respectively) from HeLa cells without (−PKCζ) or with (+PKCζ) cotransfection of active PKCζ. The relative level of E4 transcript was measured using real time qPCR (left panel). Right panel: comparable amounts of flag-SRC-3 (−PKCζ or +PKCζ, 9 ng protein) were used in the assay by Western blot analysis. Error bars indicate the standard error of the means from three independent experiments.
To further confirm this result, we performed in vitro cell free chromatin transcription assays using purified flag-SRC-3 from cell lysates prepared with or without the co-transfection of PKCζ but in the presence of ERα. Comparable amounts of SRC-3 proteins were used in the assays (Fig. 3B right panel). As shown in Fig. 3B left panel, the level of in vitro transcription increased with increasing amounts of SRC-3. SRC-3 protein purified from cells in the presence of PKCζ co-transfection was more active than in the case of no PKCζ co-transfection. In each case, an eventual squelching effect was observed when too much SRC-3 protein was added (data not shown). These results substantiate that aPKC is able to stabilize functionally active SRC-3.
An important C-terminal region in SRC-3 is required for the aPKC stabilization effect
It was recently shown by our lab that GSK3 phosphorylates Ser505 of SRC-3 and promotes its degradation (Wu et al., 2007). To explore the mechanism of aPKC stabilization of SRC-3, we first determined whether this GSK3 pathway is involved. We tested the effect of aPKC on an SRC-3 A1-6 mutant, which contains Ser/Thr to Ala mutations on six identified phosphorylation sites including Ser505 (Wu et al., 2004). As shown in Fig. 4A, aPKC still is able to stabilize the A1-6 mutant, suggesting that aPKC utilizes a different pathway from GSK3 in regulating SRC-3 turnover.
Fig. 4.
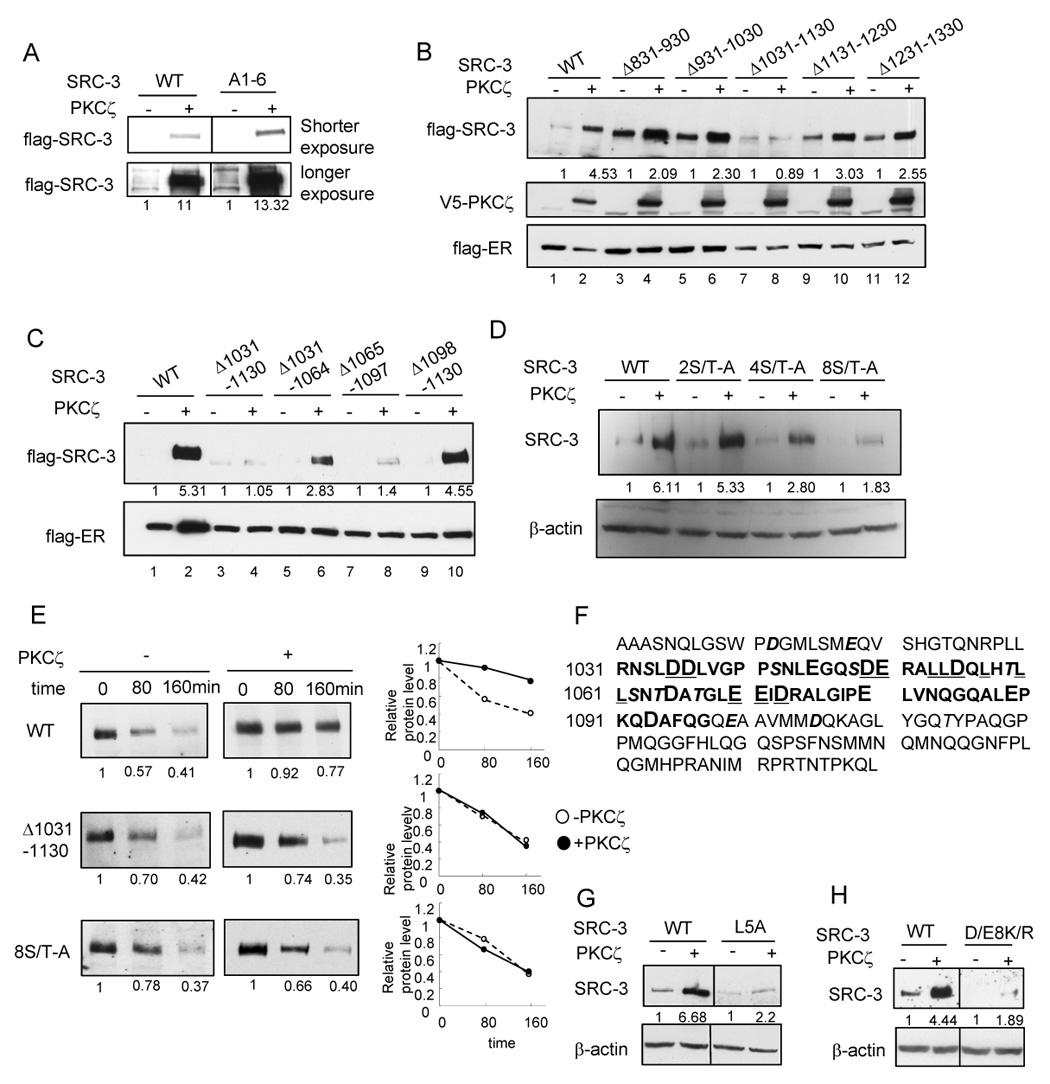
Identification of an important region in SRC-3 for aPKC stabilization effect (A) Effects of aPKC on SRC-3 A1-6 mutant (B) Effects of aPKC on SRC-3 deletion mutants (C) Effects of aPKC on SRC-3 mutants with deletion within the region of 1031–1130 (D) Effects of aPKC on SRC-3 mutants with Ser/Thr-Ala mutations within the region of 1031–1130. Details of the mutants were described in Supplemental Procedures. (E) Protein stability of SRC-3 wild type and the mutants as assayed by treating cells with 200 µg/ml cycloheximide at indicated time period. Shown in the left panel is the Western blot analysis of flag-SRC-3 protein. The right panel shows the quantification of protein intensity. (F) Amino acid sequences of SRC-3 1031–1097 (bolded) and the surrounding sequences. Acidic residues are in larger fonts. Ser/Thr residues are in Italic and shadowed. (G) L5A mutation (shown underlined in panel A) significantly reduced aPKC stabilization effect. (H) D/E8K/R mutation (shown underlined in panel A) significantly reduced aPKC stabilization effect.
We then attempted to determine the region in SRC-3 required for its stabilization by PKCζ. We constructed a series of deletion mutants of SRC-3 and tested the effect of aPKC on these mutants. The deletion of C-terminal 840–1417 amino acids significantly increased the SRC-3 protein level even in the absence of aPKC. No further increase on this mutant protein level was observed when aPKC is present (data not shown). Our result suggested that this region is likely involved in both SRC-3 protein degradation and aPKC stabilization. Consistent with this interpretation, the C-terminal region was reported to be involved in REGγ or E6AP mediated SRC-3 degradation (Li et al., 2006; Mani et al., 2006). To further define this important region, we made a series of deletion mutants at 100 amino acid intervals in the C-terminal region (Fig. 4B). PKCζ did not have any effect on the mutant Δ1031–1130 (lane 7 and 8) while other deletion mutants were stabilized by PKCζ. We further divided this 100-amino acid region into three subregions and determined the effect of deletion of individual subregions on aPKC-dependent stabilization (Fig. 4C). Deletion of 1098–1130 did not affect aPKC mediated stabilization. Deletion of 1031–1064 or 1065–1097 reduced but did not totally abolish this effect. These results suggest that the region involved in aPKC mediated SRC-3 stabilization encompasses aa. 1031–1097.
Since aPKC kinase activity is critical for stabilizing SRC-3 (Fig. 1B), we then asked whether the eight Ser/Thr residues in the 1031–1130 region could be the potential phosphorylation targets for aPKC mediated SRC-3 stabilization. As shown in Fig. 4D, the aPKC stabilization effect gradually decreased with an increasing number of Ser/Thr residues mutated to Ala in this region. The results provide evidence that these Ser/Thr residues are likely involved in aPKC mediated stabilization of SRC-3.
To confirm that deletion of 1031–1130 or 8S/T-A mutation indeed alters SRC-3 stability, we monitored the half-life of SRC-3 wild type or these mutants in the absence or presence of PKCζ after cycloheximide treatment of transfected cells (Fig. 4E). Wild type SRC-3 protein was more stable in the presence of PKCζ while no significant alteration in protein stability was observed for the Δ1031–1130 deletion or the 8S/T-A mutant in the absence or presence of PKCζ.
Acidic residues in region 1031–1097 are important for SRC-3 stabilization
To investigate the means by which the 1031–1097 region contributes to the stabilization effect of PKCζ on SRC-3, we examined the amino acid sequence of this region. The sequence of 1031–1097 (bolded) and surrounding sequences are shown in Fig. 4F. Interestingly, this region is enriched in acidic residues (Asp and Glu, Fig. 4F) and hydrophobic residues (Leu and Ile). It was reported that this region can form an amphipathic α-helical structure (Demarest et al., 2002). By using Geno3D three dimensional structure modeling based on this structure, we observed that the Leu-rich motif in this region is aligned at one side of the α-helices and probably becomes an interface for inter- or intra-molecular interactions due to its hydrophobicity. The majority of acidic residues are located at the opposite side of the α-helices. Mutation of either five Leu to Ala (L5A) (Fig. 4G) or eight acidic residues to basic residues (D/E8K/R) (Fig. 4H) significantly decreased the stabilization effect of PKCζ. Mutation of acidic residues to basic residues does not alter the overall amphipathic α-helical structure in 3D structural remodeling (data not shown). These observations suggest that the α-helix structure formed by region 1031–1097 and the acidic rich area are important for aPKC mediated SRC-3 stabilization.
aPKC stabilizes SRC-3 by preventing an interaction with the 20S proteasome C8 subunit
Since the acidic area in SRC-3 appears to be important for aPKC mediated SRC-3 stabilization, it is possible that aPKC mediated SRC-3 phosphorylation induces a conformational change in SRC-3 that promotes formation of an acidic surface on the SRC-3 protein that either prevents the interaction of SRC-3 with the core proteasome or induces the binding of a blocking protein.
The 20S proteasome core possesses a C8 subunit that has a unique acidic C-terminus not present in any of the other proteasomal α subunits. It was reported (Gao et al., 2000) that a naturally present basic peptide PR39 is able to interact with C8 subunit, presumably through an interaction with the acidic C-terminus. p21 also was found to be directly degraded by the 20S core proteasome through its interaction with the acidic C-terminal region of C8 (Touitou et al., 2001). Interestingly, we noticed that the region in p21 that was found to be important for this interaction is enriched with basic residues. Based on these reports, we reasoned that C8 may be a candidate proteasomal component that interacts with a positive charged region of SRC-3 and that this interaction is inhibited by an aPKC induced exposure of acidic surface in SRC-3.
To test this hypothesis, we examined whether SRC-3 is able to interact with C8 using a GST pull-down assay. As shown in Fig. 5A, SRC-3 interacted with GST-C8 (lane 3) while no interaction was observed between SRC-3 and GST alone (lane 1). We tested the interaction of SRC-3 with another 20S α subunit, PSMA7 (lane 2), which is involved in regulating the transcription factor HIF-1α (Cho et al., 2001). SRC-3 did not interact with GST-PSMA7, suggesting that the interaction between SRC-3 and C8 is selective. We also tested the contribution of the C8 C-terminal acidic region in this interaction. Deletion of its C-terminus (227–255 (Fig. 5A, lane 4) or 211–255 (lane 5)) totally abolished this interaction, indicating the acidic C-terminus of C8 is required for interaction with SRC-3. We observed that GST-PSMA7 but not GST was able to interact with HIF-1α, suggesting that the PSMA7 is a functional protein (Fig. 5B).
Fig. 5.
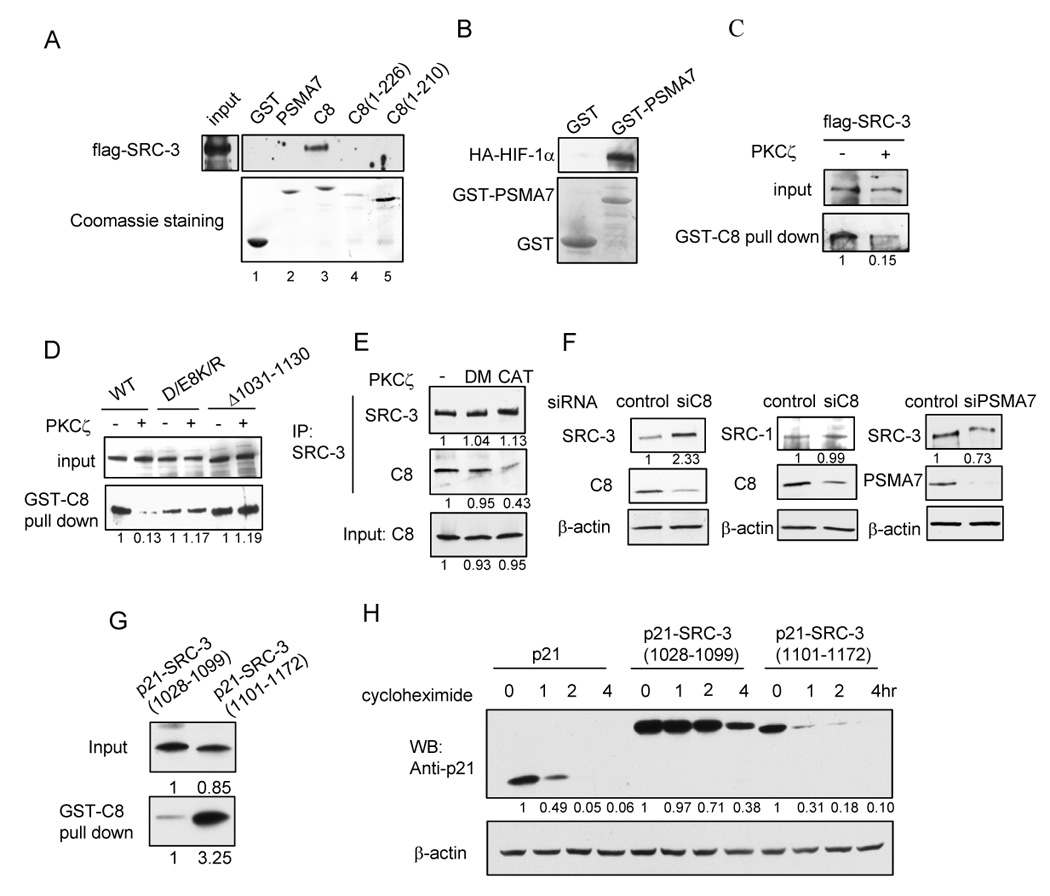
aPKC stabilizes SRC-3 by preventing interaction with the 20S proteasome C8. (A) SRC-3 interacts with GST-C8. 293T cells were transfected with flag-SRC-3 and ERα. Cell lysates were incubated with GST fusion proteins and the bound flag-SRC-3 was detected by Western blot. Upper panel: Western blot analysis of flag-SRC-3 pulled-down by GST fusion proteins. Lower panel: Coomassie staining of GST and GST fusion proteins. (B) GST-PSMA7 is a functional protein. In vitro transcribed and translated HA-HIF-1α was incubated with GST or GST-PSMA7 and the bound HA-HIF-1α was detected by anti-HA antibody. (C) PKCζ decreases the interaction between SRC-3 and GST-C8. (D) D/E8K/R mutation or deletion of 1031–1130 in SRC-3 abolishes the effect of PKCζ on the interaction between SRC-3 and C8. (E) PKCζ decreases the interaction between endogenous SRC-3 and C8 in 293T cells. (F) Knocking-down of C8 protein in MCF-7 cells increases the SRC-3 protein level. (G) p21-SRC-3(1028–1099) has weaker interaction with C8. GST-C8 pull-down experiment was carried out using in vitro synthesized p21-SRC-3(1028–1099) and p21-SRC-3(1101–1172) fusion proteins. The bound proteins were detected using anti-p21 antibody. (H) SRC-3 1028–1099 region increased the stability of p21. HeLa cells were transfected with p21, p21-SRC-3(1028–1099) and p21-SRC-3(1101–1172), respectively, and cells were treated with 200µg/ml cycloheximide at an indicated time period one day after transfection. Shown are Western blot analyses using anti-p21 antibody.
We then asked whether aPKC has any effect on the interaction between SRC-3 and C8. 293T cell lysates transfected with flag-SRC-3 and ER with or without co-transfection of aPKC were used in a GST-C8 pull-down experiment. To ensure that equal amounts of flag-SRC-3 were used as inputs, we utilized two different methods. First, the amounts of cell lysates used were adjusted to the same amount of SRC-3 in the absence and presence of PKCζ (Fig. 5C). Second, cells were treated with 10µM MG132 for 16 hrs before harvesting to ensure that the SRC-3 level was the same when using equal amounts of cell lysates (Fig. 5D). In both cases, we found that flag-SRC-3 interacted more weakly with C8 in lysates co-transfected with aPKC compared to that without aPKC co-transfection (Fig. 5C and 5D). Deletion of amino acids 1031–1130 or mutation of acidic residues to basic residues in this region abolished this aPKC effect (Fig. 5D). To confirm that aPKC also affects the interaction between endogenous SRC-3 and C8 in cells, we performed coimmunoprecipitation assays. Cells were treated with MG132 for 16 hrs to ensure the same level of SRC-3 and C8 in the absence and presence of PKCζ. Consistent with in vitro results, we found that endogenous SRC-3 interacts more weakly with endogenous C8 in 293T cells when active aPKC is co-transfected, while the kinase dead mutant does not have any effect (Fig. 5E). Similar results also were obtained in HeLa cells (Fig. S5). Our results strongly implicate C8 in the aPKC-regulated degradation of SRC-3.
To further confirm this result, we knocked-down the endogenous C8 protein level in MCF-7 cells using a C8-specific siRNA and monitored endogenous SRC-3 protein levels. As shown in Fig. 5F (left panel), the SRC-3 level was increased by the knock-down of C8 while the level of SRC-1, which was not altered by aPKC (Fig. 2A), did not change (Fig. 5F middle panel). As a control, knock-down of PSMA7 did not increase the level of the SRC-3 protein (Fig. 5F right panel).
To further test whether the acidic residue rich region of SRC-3 is sufficient to interfere in C8 mediated protein degradation, we chose p21 as a C8 targeted protein and tested the interaction between C8 and p21 when fused to the SRC-3 acidic region (1028–1099) and also the stability of the fusion protein. Another region in SRC-3 (1101–1172) was fused to p21 as a control. As shown in Fig. 5G, p21-SRC-3(1028–1099) interacts with C8 much more weakly than p21-SRC-3(1101–1172). The stability of p21-SRC-3(1028–1099) but not the p21-SRC-3(1101–1172) also was significantly increased compared to p21 (Fig. 5H). These results suggest that the acidic residue rich region in SRC-3 is important for protein stability, probably through preventing efficient interaction with 20S C8 subunit. Taken together, our results imply that the 20S C8 subunit mediates SRC-3 degradation and that aPKC is able to inhibit the interaction between SRC-3 and C8, thereby stabilizing the SRC-3 protein.
PKCζ protects SRC-3 from proteasome mediated degradation in vitro
To ‘directly’ demonstrate that aPKC indeed converts SRC-3 into a “degradation-resistant” form in the presence of ERα, we carried out in vitro proteasome degradation assays. Flag-SRC-3 was purified from HeLa cell lysates, which were co-transfected with flag-SRC-3 and either PKCζ, ERα, or both PKCζ and ERα, and then subjected to 26S proteasome degradation in vitro. As shown in Fig. 6A, flag-SRC-3 is efficiently degraded by the 26S proteasome when either PKCζ or ERα alone was transfected. When both PKCζ and ERα were present, SRC-3 became more resistant to degradation. This protective effect required PKCζ kinase activity since in the presence of the kinase dead mutant of PKCζ, the stabilization was not observed (data not shown).
Fig. 6.
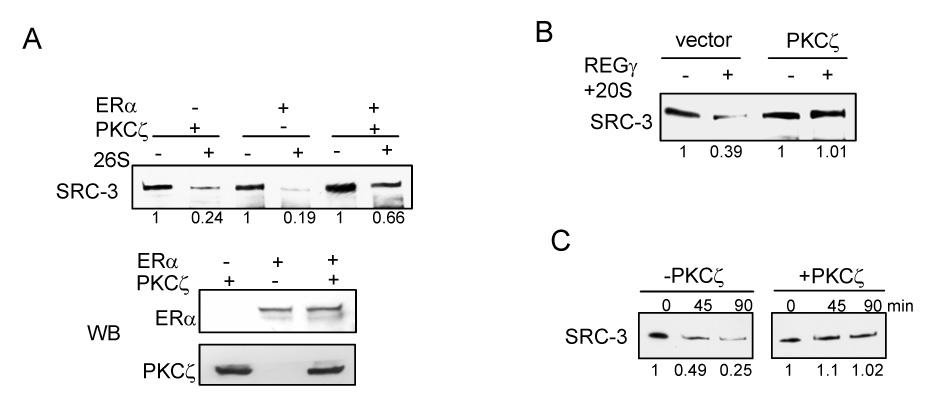
PKCζ protects SRC-3 from proteasome mediated degradation. (A) in vitro 26S proteasome degradation assay. Detailes were described in Experimental Procedures. (B) PKCζ protects SRC-3 from REGγ-mediated proteasome degradation. (C) PKCζ protects SRC-3 from REGγ-mediated proteasome degradation in an in vitro purified system. Purified baculovirus expressed recombinant SRC-3 was phosphorylated by PKCζ in vitro in the presence of ERα and E2. Purified REGγ and 20S proteasome were then added into the reaction and were incubated for an indicated time period.
It was shown recently that SRC-3 also can be degraded through a REGγ-mediated ubiquitin-independent proteasome pathway (Li et al., 2006). Since C8 is a component of the 20S core proteasome, we reasoned that aPKC also should protect SRC-3 from REGγ-mediated degradation if it affects the interaction between C8 and SRC-3. We tested whether the PKCζ modified form of SRC-3 is also resistant to REGγ-mediated degradation (Fig. 6B). Similar to the results shown in Fig. 6A, PKCζ also protected SRC-3 from degradation in this case.
We show in Fig. 2H that PKCζ can directly phosphorylate SRC-3. To test whether this protective effect we observed is due to the direct phosphorylation of SRC-3 by PKCζ, purified baculovirus expressed SRC-3 was phosphorylated by PKCζ in vitro and the protein was then subjected to in vitro proteasome degradation in the presence of purified REGγ, purifed ERα and E2. As shown in Fig. 6C, PKCζ protected SRC-3 from degradation in this purified system. Taken together, our results suggest that PKCζ phosphorylates SRC-3 in a C-terminal degron region and promotes a change of SRC-3 conformation into a form that is more resistant to all proteasome degradation pathways.
Effects of aPKC on ER mediated gene expression and estrogen-dependent cell growth
SRC-3 has been shown to promote cell growth and an increase in cell size (Zhou et al., 2003). To examine whether aPKC could affect the ability of SRC-3 to stimulate ER-dependent cell growth, we generated a pool of stable PKCζ knocked-down MCF-7 cells. As shown in Fig. 7A, PKCζ is reduced in this cell line. The SRC-3 protein level also is decreased. In the PKCζ knockdown cells, SRC-2 level does not change while SRC-1 level is slightly increased, which probably is in part due to a compensation effect (Fig. S6A and B). The MCF-7 cell line is ER-positive and its growth requires estrogen and ER. Two important ER-regulated genes that promote MCF-7 cell proliferation are c-myc and cyclin D1. We showed in Fig. 3A that overexpression of PKCζ significantly increased SRC-3 activated transcription of the ER target gene pS2. Conversely, disruption of PKCζ expression by shRNA reduced E2-dependent transcription of c-myc and cyclin D1, as well as pS2 (Fig. 7B).
Fig. 7.
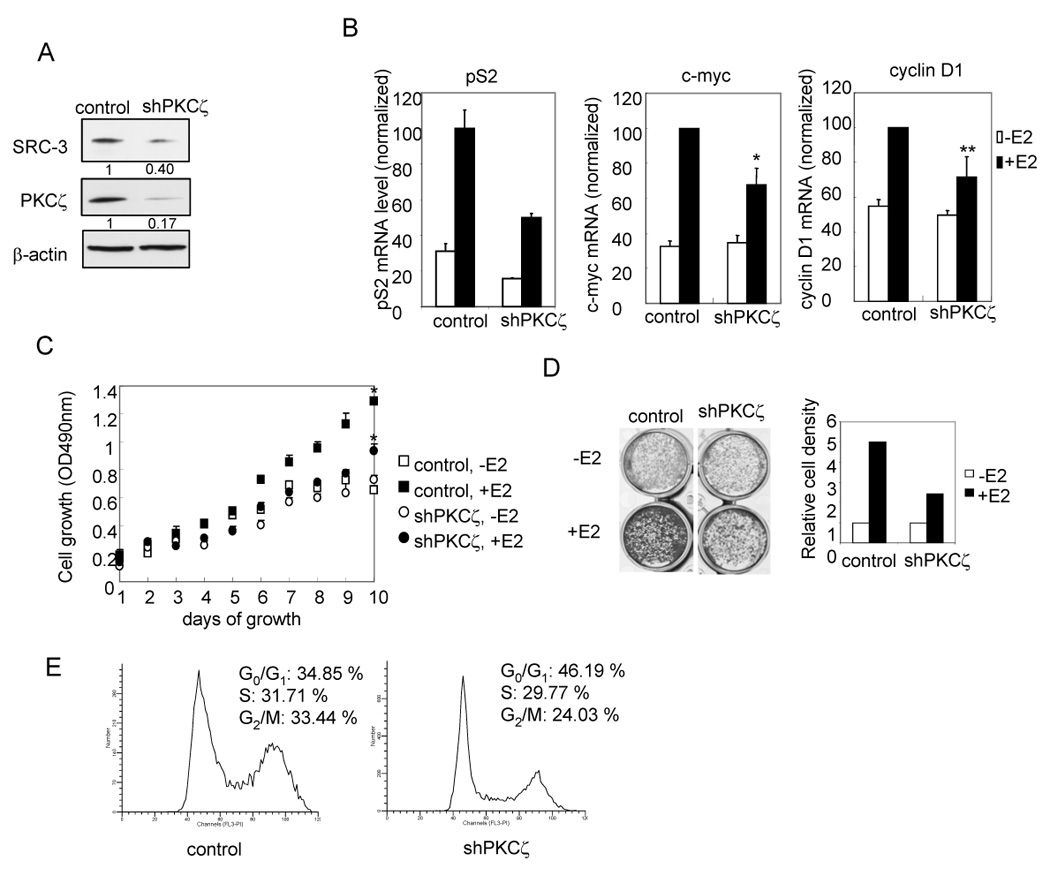
Effects of aPKC on ER mediated gene expression and estrogen-dependent cell growth (A) SRC-3 protein level was decreased in MCF-7 (shPKCζ) stable cell line. (B) Knocking-down of PKCζ reduces estrogen-dependent ER target gene transcription. Cells were treated with 10−8 M E2 for 2 hours (c-myc, cycline D1) or overnight (pS2) before being harvested. The transcription were quantified by real time qRT-PCR and normalized against a cyclophilin mRNA level. * indicates p< 0.01. ** indicates p< 0.05. Error bars indicate the standard error of the means from three independent experiments. (C) MTT assay for detecting estrogen-dependent growth of MCF-7. * indicates p< 0.01 (control vs. shPKCζ cells in the presence of E2). Error bars indicate the standard error of the means from triplicate experiments. (D) Estrogen-dependent cell growth by crystal violet staining. Cells were stained with crystal violet after 9 days of growth in medium with or without 10−8 M E2 (Left panel). The Right panel shows the relative cell densities estimated by Scion Image. Shown is the representative result from three independent experiments._(E) Flow cytometry analysis on cell cycle distribution of MCF-7 cells after two days of growth in the presence of 10−7 M E2.
We then compared estrogen-dependent growth of cells bearing stably integrated shPKCζ and control cells using an MTT proliferation assay. Consistent with lower transcription levels of cmyc and cyclin D1, cells with PKCζ knocked-down (shPKCζ) grow slower (p<0.01) in the presence of E2 (Fig. 7C). Similarly, crystal violet staining also showed that MCF-7 (shPKCζ) cells have a lesser number of cells in the presence of E2 after 9 days of growth in culture compared to control cells when starting from the same number of plated cells (Fig. 7D). We also tested the cell cycle distribution of shPKCζ cells compared to the control cells in the presence of E2. As shown in Fig. 7E, shPKCζ cells have a higher percentage of cells in G0/G1 phase, indicating a slower rate of cell growth. The results suggest that aPKC can affect estrogen-mediated cell growth by modulating SRC-3 protein levels. We also found a correlation between endogenous SRC-3 and PKCζ protein levels in several different cancer cell lines (Fig. S7), indicating that the regulation of aPKC on SRC-3 stability is likely to be physiologically relevant.
Discussion
Protein phosphorylation is an important reversible modification for modulating protein stability. Here we demonstrate that the level of SRC-3 protein can be regulated by aPKC in an ERα and estrogen-dependent manner. The exposure of an acidic surface on SRC-3 induced by aPKC mediated phosphorylation reduces its interaction with the acidic C-terminus of 20S proteasome core α C8 subunit, thereby stabilizing SRC-3 protein. Our data describe a new regulatory mechanism for SRC-3 protein turnover, which may play an important role in regulating SRC-3 levels in normal and oncogenic cell growth.
It is interesting that aPKC-mediated SRC-3 stabilization requires the presence of ER while another steroid receptor PR is not able to support this stabilization, suggesting that this is a receptor-selective effect. SRC-3 is not only a coactivator for nuclear receptors but for certain other transcription factors as well (Korzus et al., 1998; Louie et al., 2004; Wu et al., 2004). Cells require a mechanism to distinguish their needs for SRC-3 in different signaling pathways that utilize different transcription factors. The requirement for association with ERα for this C-terminal phosphorylation and stabilization of SRC-3 defines an intriguing selectivity for aPKC in ER mediated pathway function.
The C8 subunit interacts with a number of proteins and mediates their degradation (Sdek et al., 2005; Shu et al., 2003; Touitou et al., 2001). The acidic C-terminus of C8 appears to be important for the degradation of p21 (Touitou et al., 2001) and for the inhibitory effect of a highly basic peptide PR39 on IκBα degradation (Gao et al., 2000). C8 binds to the C-terminus of p21 and deletion of the C-terminus increases p21 half life (Touitou et al., 2001). Interestingly, the C-terminal region of p21 is highly enriched in basic residues. Fusion of this region of p21 to p27, which does not bind to C8 by itself, then allows binding of p27 to C8. MDM2 was found to promote the interaction of Rb (Sdek et al., 2005) and p21 (Zhang et al., 2004) with C8 through its acidic domain. It was suggested that MDM2 may function either by facilitating the delivery of these proteins to the proteasome or by stabilizing their interaction with C8. This collection of reports led us to propose that C8 mediates the degradation of select proteins through its negative charged C-terminus by electrostatic interactions.
A number of studies have indicated that electrostatic attraction and repulsion are mechanisms to regulate protein-protein interactions and that such reactions can be modulated by phosphorylation (Schlarb-Ridley et al., 2002; Singh and Murray, 2003; Ward et al., 2004). Our data indicate that the acidic C-terminus of C8 is required for interaction with SRC-3 and imply that C8 interacts with the positively-charged region in SRC-3. The findings that aPKC blocks the interaction of SRC-3 with C8 and that mutation of amino acid residues in the 1031–1130 region, or its deletion, abolished this aPKC effect, suggest that aPKC induces a conformational exposure of this negative charged region to the surface of SRC-3 which then weakens the interaction between SRC-3 and C8 through electrostatic repulsion. This region was shown previously (Demarest et al., 2002) to be a disordered structure in solution that is capable of forming α-helices when bound to another coregulator. When aPKC mediated SRC-3 phosphorylation induces a conformational change of SRC-3, ERα, in addition to promoting aPKC mediated SRC-3 phosphorylation, appears to facilitate the correct structure so that acidic residues in this region are presented to C8. Experiments showing that SRC-3 is more resistant to proteasome degradation in vitro when aPKC is present and active (Fig. 6) directly support the hypothesis that SRC-3 is induced to form a functionally different structure in the absence and presence of active aPKC.
SRC-3 can be degraded through both 26S proteasome and REGγ-mediated proteasome pathways. A question arises as to why cells need yet another layer of control in regulating SRC-3 protein levels through interaction with the 20S core. It is important to note that cellular levels of SRC-3 represent the “potential” for its functional activity. As a primary growth promoting protein and oncogene, SRC-3 levels are tightly controlled in cells and phosphorylation appears to play a central role in this process. Different signals induce differential SRC-3 phosphorylations in different pathways so as to trigger selective degradation by either the 26S- or the REGγ-mediated proteasome. Several different kinases appear to affect SRC-3 degradation including p38 MAPK (Gianni et al., 2006), conventional and novel PKC (Fig. S2), and GSK3 (Wu et al., 2007). We also found that peptidyl prolyl isomerase 1 catalyzes a conformational change of phosphorylated SRC-3 to promote its degradation (Yi et al., 2005). In the face of these multiple molecular players in degradation, when cells need to increase acutely the level of SRC-3 protein to accomplish active growth, it would be most efficient if both 26S proteasome and REGγ-mediated degradation were shut down simultaneously. Since the 20S is the final core component for both proteasomeal pathways, controlling its interaction with SRC-3 by regulating SRC-3 phosphorylation status via aPKC would efficiently prevent degradation and allow accumulation of SRC-3 in cells.
aPKC is overexpressed in a number of cancers. We propose that in normal non-malignant cells, SRC-3 is phosphorylated by a variety of kinases. Different phosphorylation patterns of SRC-3 induce different changes in conformation and function. Specific kinases may promote (or prevent) SRC-3 degradation. SRC-3 protein levels are therefore maintained in a normal range by a balanced equilibrium of different kinases. However, when aPKC is overexpressed or is highly active in cancer cells, SRC-3 does not interact efficiently with the 20S proteasome and this “degradation-resistant” form of SRC-3 accumulates in cells. The consequence is increased SRC-3 function and powerful enhancement of ER-dependent target gene transcription, thereby, promoting estradiol-dependent cell growth in cancer cells such as breast.
Experimental Procedures
In vitro proteasome degradation
HeLa cells were transfected with expression vectors containing flag-SRC-3 and PKCζ, or ER, or both. Flag-SRC-3 was purified from cell lysates by immunoprecipitation with 20 µl anti-flag M2-conjuagated agarose (Sigma), washed 3 times with wash buffer (20 mM Hepes, 150 mM KCl, 1 mM DTT, 0.1% NP-40, 8% glycerol and 0.5 mM PMSF), and eluted 4 times with 150 ng/µl 3XFlag peptide (Sigma) solution (50 mM Tris-HCl, 150 mM NaCl, pH 7.4). The purified flag-SRC-3 was then added into 20 µl solutions containing 4 mM ATP, 4 mM creatine phosphate, 0.02 µg/µl creatine phosphate kinase, 0.2 µg/µl ubiquitin, 3 µl rabbit reticulocyte lysate (Promega) and 0.6 µl partially purified 26S proteasome (Boston Biochem) and incubated for 0 and 40 min at 30°C. The reaction was stopped by adding 50 µl dH2O and 5X sample buffer. For REGγ mediated proteasome degradation, 0.125 µg of 20S proteasome (Affinity Bioreagents) and 0.5 µg of REGγ heptamers were pre-incubated for 15 min in 20 mM HEPES, pH 7.5, 100 mM KCl, 0.5 mM EDTA, and 10% glycerol reaction buffer at 4°C. Purified flag-SRC-3 was then added into the mixture for 30 min and the reaction was stopped by adding 5X sample buffer. For REGγ mediated proteasome degradation of purifed recombinant SRC-3 from baculovirus, SRC-3 was incubated with purified ERα, 1 µM E2, 75 mM MgCl2, 0.5 mM ATP in 8 mM Hepes, 0.2 mM EDTA containing solution for 15 min at 4°C. 100 ng PKCζ protein or BSA was then added into the solution and the mixture was incubated at 30°C for 30 min. REGγ and 20S core proteasome were then added into the mixture as described above to start the degradation.
In vitro transcription
The procedure for in vitro transcription was essential the same as described previously (Feng et al., 2006) except that flag-SRC-3 proteins were purified from HeLa cell lysates with the co-transfection of ERα and in the absence or presence of PKCζ using the procedure described above.
Cell growth assay
For MTT assay, 6000 of MCF-7 cells with stable integration of control vector or shPKCζ were seeded in 96-well plates. Cells were grown in phenol red free DMEM medium supplemented with 10% charcoal-stripped FBS in the absence or presence of 10−8 M estradiol. Cell growth was assayed at different days by measuring OD490nm using Promega CellTiter 96 Aqueous One Solution. For crystal violet staining, 24000 of the same MCF-7 cells were seeded in 12-well plates and were grown in the absence or presence 10−8 M estradiol. After 9 days of growth, cells were stained with crystal violet (Sigma) for 10 min at room temperature. For cell cycle analysis, cells were grown in the presence of 10−7 M estradiol for two days. Cells were stained with 10 µg/ml propidium iodide, and then subjected to flow cytometry. The results were analyzed by ModFit LT software.
Repetitions of experiments
Experiments presented in Fig. 1–Fig. 7 were repeated 3–5 times.
Supplementary Material
Acknowledgments
We thank Dr. Gordon B. Mills for kindly providing PKCζ and PKCι expression vectors. This work is supported by NIH grants and NICHD, HD08818 (to B.W.O.), and a postdoctoral fellowship from the Department of Defense Breast Cancer Research Program (W81XWH-04-1-0552) (to P.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional Procedures are described in Supplemental Procedures.
References
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- Castoria G, Migliaccio A, Di Domenico M, Lombardi M, de Falco A, Varricchio L, Bilancio A, Barone MV, Auricchio F. Role of atypical protein kinase C in estradiol-triggered G1/S progression of MCF-7 cells. Mol Cell Biol. 2004;24:7643–7653. doi: 10.1128/MCB.24.17.7643-7653.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Choi YJ, Kim JM, Jeong ST, Kim JH, Kim SH, Ryu SE. Binding and regulation of HIF-1alpha by a subunit of the proteasome complex, PSMA7. FEBS Lett. 2001;498:62–66. doi: 10.1016/s0014-5793(01)02499-1. [DOI] [PubMed] [Google Scholar]
- Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–553. doi: 10.1038/415549a. [DOI] [PubMed] [Google Scholar]
- Donson AM, Banerjee A, Gamboni-Robertson F, Fleitz JM, Foreman NK. Protein kinase C zeta isoform is critical for proliferation in human glioblastoma cell lines. J Neurooncol. 2000;47:109–115. doi: 10.1023/a:1006406208376. [DOI] [PubMed] [Google Scholar]
- Eder AM, Sui X, Rosen DG, Nolden LK, Cheng KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, et al. Atypical PKCiota contributes to poor prognosis through loss of apical-basal polarity and cyclin E overexpression in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:12519–12524. doi: 10.1073/pnas.0505641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Yi P, Wong J, O'Malley BW. Signaling within a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol Cell Biol. 2006;26:7846–7857. doi: 10.1128/MCB.00568-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Lecker S, Post MJ, Hietaranta AJ, Li J, Volk R, Li M, Sato K, Saluja AK, Steer ML, et al. Inhibition of ubiquitin-proteasome pathway-mediated I kappa B alpha degradation by a naturally occurring antibacterial peptide. J Clin Invest. 2000;106:439–448. doi: 10.1172/JCI9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni M, Parrella E, Raska I, Jr., Gaillard E, Nigro EA, Gaudon C, Garattini E, Rochette-Egly C. P38MAPK-dependent phosphorylation and degradation of SRC-3/AIB1 and RARalpha-mediated transcription. Embo J. 2006;25:739–751. doi: 10.1038/sj.emboj.7600981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai T, Chida K. Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. J Biochem (Tokyo) 2003;133:1–7. doi: 10.1093/jb/mvg017. [DOI] [PubMed] [Google Scholar]
- Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McInerney EM, Mullen TM, Glass CK, Rosenfeld MG. Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science. 1998;279:703–707. doi: 10.1126/science.279.5351.703. [DOI] [PubMed] [Google Scholar]
- Kuang SQ, Liao L, Zhang H, Lee AV, O'Malley BW, Xu J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875–1885. doi: 10.1158/0008-5472.can-03-3745. [DOI] [PubMed] [Google Scholar]
- Kumar R, Wang RA. Protein kinases in mammary gland development and cancer. Microsc Res Tech. 2002;59:49–57. doi: 10.1002/jemt.10176. [DOI] [PubMed] [Google Scholar]
- Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai MJ, O'Malley BW. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell. 2006;124:381–392. doi: 10.1016/j.cell.2005.11.037. [DOI] [PubMed] [Google Scholar]
- Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–948. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- Louie MC, Zou JX, Rabinovich A, Chen HW. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol. 2004;24:5157–5171. doi: 10.1128/MCB.24.12.5157-5171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay HJ, Twelves CJ. Protein kinase C: a target for anticancer drugs? Endocr Relat Cancer. 2003;10:389–396. doi: 10.1677/erc.0.0100389. [DOI] [PubMed] [Google Scholar]
- Mani A, Oh AS, Bowden ET, Lahusen T, Lorick KL, Weissman AM, Schlegel R, Wellstein A, Riegel AT. E6AP Mediates Regulated Proteasomal Degradation of the Nuclear Receptor Coactivator Amplified in Breast Cancer 1 in Immortalized Cells. Cancer Res. 2006;66:8680–8686. doi: 10.1158/0008-5472.CAN-06-0557. [DOI] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005;280:31109–31115. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. Phosphatidylinositol 3' kinase signaling in mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2001;6:83–99. doi: 10.1023/a:1009520616247. [DOI] [PubMed] [Google Scholar]
- Schlarb-Ridley BG, Bendall DS, Howe CJ. Role of electrostatics in the interaction between cytochrome f and plastocyanin of the cyanobacterium Phormidium laminosum. Biochemistry. 2002;41:3279–3285. doi: 10.1021/bi0116588. [DOI] [PubMed] [Google Scholar]
- Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, Allday MJ, Xiao ZX. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Shu F, Guo S, Dang Y, Qi M, Zhou G, Guo Z, Zhang Y, Wu C, Zhao S, Yu L. Human aurora-B binds to a proteasome alpha-subunit HC8 and undergoes degradation in a proteasome-dependent manner. Mol Cell Biochem. 2003;254:157–162. doi: 10.1023/a:1027317014159. [DOI] [PubMed] [Google Scholar]
- Singh SM, Murray D. Molecular modeling of the membrane targeting of phospholipase C pleckstrin homology domains. Protein Sci. 2003;12:1934–1953. doi: 10.1110/ps.0358803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Akimoto K, Ohno S. Protein kinase C lambda/iota (PKClambda/iota): a PKC isotype essential for the development of multicellular organisms. J Biochem (Tokyo) 2003;133:9–16. doi: 10.1093/jb/mvg018. [DOI] [PubMed] [Google Scholar]
- Torres-Arzayus MI, De Mora JF, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263–274. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Touitou R, Richardson J, Bose S, Nakanishi M, Rivett J, Allday MJ. A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. Embo J. 2001;20:2367–2375. doi: 10.1093/emboj/20.10.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Hsieh YS, Kuo SJ, Chen ST, Yu SY, Huang CY, Chang AC, Wang YW, Tsai MT, Liu JY. Alteration in the expression of protein kinase C isoforms in human hepatocellular carcinoma. Cancer Lett. 2000;161:171–175. doi: 10.1016/s0304-3835(00)00597-8. [DOI] [PubMed] [Google Scholar]
- Wang Z, Rose DW, Hermanson O, Liu F, Herman T, Wu W, Szeto D, Gleiberman A, Krones A, Pratt K, et al. Regulation of somatic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci U S A. 2000;97:13549–13554. doi: 10.1073/pnas.260463097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DG, Brewer SM, Calvert MJ, Gallon CE, Gao Y, Trayer IP. Characterization of the interaction between the N-terminal extension of human cardiac troponin I and troponin C. Biochemistry. 2004;43:4020–4027. doi: 10.1021/bi036128l. [DOI] [PubMed] [Google Scholar]
- Wu RC, Feng Q, Lonard DM, O'Malley BW. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129:1125–1140. doi: 10.1016/j.cell.2007.04.039. [DOI] [PubMed] [Google Scholar]
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O'Malley BW. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O'Malley BW. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A. 2000;97:6379–6384. doi: 10.1073/pnas.120166297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Yu CT, Ozen M, Ittmann M, Tsai SY, Tsai MJ. Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/AKT signaling pathway. Cancer Res. 2006;66:11039–11046. doi: 10.1158/0008-5472.CAN-06-2442. [DOI] [PubMed] [Google Scholar]
- Yi P, Wu RC, Sandquist J, Wong J, Tsai SY, Tsai MJ, Means AR, O'Malley BW. Peptidyl-prolyl isomerase 1 (Pin1) serves as a coactivator of steroid receptor by regulating the activity of phosphorylated steroid receptor coactivator 3 (SRC-3/AIB1) Mol Cell Biol. 2005;25:9687–9699. doi: 10.1128/MCB.25.21.9687-9699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wang H, Li M, Agrawal S, Chen X, Zhang R. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J Biol Chem. 2004;279:16000–16006. doi: 10.1074/jbc.M312264200. [DOI] [PubMed] [Google Scholar]
- Zheng FF, Wu RC, Smith CL, O'Malley BW. Rapid estrogen-induced phosphorylation of the SRC-3 coactivator occurs in an extranuclear complex containing estrogen receptor. Mol Cell Biol. 2005;25:8273–8284. doi: 10.1128/MCB.25.18.8273-8284.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Hashimoto Y, Kwak I, Tsai SY, Tsai MJ. Role of the steroid receptor coactivator SRC-3 in cell growth. Mol Cell Biol. 2003;23:7742–7755. doi: 10.1128/MCB.23.21.7742-7755.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.