

Abstract
Sepsis induced lethality is characterized by amplified host innate immune response. Nrf2, a bZIP transcription factor regulates a battery of cellular antioxidative genes and maintains cellular redox homeostasis. This study demonstrates that increasing Nrf2 activity by a potent small molecule activator, CDDO-Im (1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole), protects from deregulation of lipopolysaccharide (LPS) induced innate immune response. In response to LPS stimuli, nrf2-deficient (nrf2 −/−) peritoneal neutrophils showed increased NADPH oxidase dependent ROS generation, proinflammatory cytokines (Tnf-α and Il-6) and chemokines (Mip2 and Mcp-1) relative to wild-type (nrf2 +/+) cells. Pretreatment of peritoneal neturophils with CDDO-Im induced antioxidative genes (Ho-1, Gclc, Gclm and Nqo1) and attenuated LPS induced ROS generation as well as expression of proinflammatory cytokines exclusively in nrf2 +/+ neutrophils but not in nrf2 −/− cells. In corroboration with in vitro studies, pretreatment with CDDO-Im induced Nrf2 dependent antioxidative genes, attenuated LPS induced proinflammatory cytokine expression and decreased septic shock induced mortality specifically in the nrf2 +/+ mice. In conclusion, the results suggest that Nrf2 is associated with oxidative regulation of LPS induced innate immune response in neutrophils. Activation of Nrf2 dependent compensatory antioxidative pathways by CDDO-Im protects from LPS induced inflammatory response and mortality due to septic shock.
Keywords: Nrf2, CDDO-im, neutrophils, macrophages, innate immune response, antioxidant, ROS, LPS, Septic shock
INTRODUCTION
Sepsis is a complex syndrome characterized by an amplified innate immune response to infection. In case of bacterial infection, the endotoxin lipopolysaccharide (LPS) triggers a acute and early release of proinflammatory mediators from macrophages and neutrophils through TLR4-NF-κB signaling pathway that mediates host damage [1]. Although the causes of inappropriate immune response of macrophages and neutrophils in septic patients is far from clear, lately the role of redox regulation of LPS-TLR4 signaling in priming innate immune cells, macrophages and neutrophils for increased responsiveness to subsequent stimuli is being well recognized [2-5]
Nuclear factor-erythroid 2-p45-related factor 2 (Nrf2) a basic leucine zipper redox sensitive transcription factor, is a master regulator of cellular antioxidative and other cytoprotective genes [6]. Disruption of Nrf2 causes a decrease in the constitutive expression as well as loss of capacity to adaptively respond to stresses through upregulation of cellular antioxidants. Our and other laboratories have demonstrated enhanced sensitivity of nrf2-deficient mice to various oxidative as well as inflammatory insults [6-9]. More recently, we have demonstrated enhanced sensitivity of nrf2-deficient mice to LPS induced septic shock and cecal ligation and puncture induced sepsis [10]. LPS insult triggered dramatic increases in the infiltration of neutrophils and macrophages into lungs of nrf2 −/− mice when compared to wild-type litter mates. Further, global gene expression analysis by microarray of lungs of nrf2 −/− mice challenged with LPS demonstrated exaggerated expression of inflammatory mediators (such as cytokines, chemokines, adhesion molecules) compared to genotype controls [10]. However, it was far from clear whether the exaggerated lung inflammation observed in the nrf2 −/− mice due to LPS is because of deregulated responses of immune cells and/ or non-immune cells. The present study was designed to determine how Nrf2 influences the response of innate immune cells, particularly neutrophils towards LPS stimulation. A second objective was to determine whether prior activation of Nrf2 protects against an exaggerated LPS induced innate immune response by neutrophils as well as septic shock-induced death. We have shown previously that the synthetic triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole (CDDO-Im) is a extremely potent activator of the Nrf2 signaling pathway resulting in a dramatic increases in expression of number of Nrf2-dependent antioxidative and cytoprotective genes [11, 12]. In summary, the results of this study suggest that Nrf2 is a critical oxidative regulator of LPS induced TLR4 signaling in innate immune cells and that activation of Nrf2 by CDDO-Im protects mice from LPS induced inflammatory response.
MATERIAL AND METHODS
Animals
Nrf2-deficient mice [CD-1 (ICR); nrf2 −/−] were generated as described [10]. Wild-type [CD-1 (ICR); nrf2 +/+] and nrf2 −/− were fed on regular sterile chow diet and water ad libitum and were housed under controlled conditions (25 ± 2°C; 12-h light-dark periods). All experimental protocols conducted on the mice were performed in accordance with NIH guidelines.
Isolation and culture of peritoneal neutrophils
Peritoneal neutrophils were isolated from nrf2 +/+ and nrf2 −/− mice 4 h after intraperitoneal injection of 1 ml of 4% sterile aged thioglycolate broth (Difco Laboratories, Detroit, Mich.). Isolated cells were incubated in a 5% CO2 humidified culture incubator at 37°C for 1 h. Non-adherent cells were > 95% neutrophils as determined by the morphologic appearance of Diff-Quick-stained preparations. Cell were either used immediately or plated into 6-well cell culture plates (Costar) at a density of 4 × 106 cells per well.
LPS treatment of cells
For measurement of ROS, 1 million cells were suspended in PBS supplemented with 0.1% glucose immediately after harvesting, and stimulated with LPS (E. coli, serotype 055.B5; Sigma) (100 ng/ml) just before ROS measurement. For analyzing the expression of cytokines and chemokines, neutrophils were stimulated with LPS (100 ng/ml) for 3 h. For inhibition of NADPH oxidase, cells were pretreated with 10 μM diphenyleneiodonium (DPI) (Sigma) for 20 min followed by LPS stimulation.
CDDO-Im and N-acetyl cysteine treatment of cells
Nrf2 +/+ and nrf2 −/− neutrophils were treated with CDDO-Im at a concentration of 10 nM (diluted from a 5 mM stock dissolved in acetonitrile) and/ or N-acetyl cysteine (NAC) (10mM) for 12 h. For measuring LPS (100 ng/ml) induced ROS generation and inflammatory responses, cells were pretreated with CDDO-Im for 12 h followed by LPS stimulation.
LPS shock and intervention with CDDO-Im
Male mice (8-10 weeks old) of both genotypes were pretreated either with 3 doses of CDDO-Im (3 μmol / kg body weight, dissolved in 10% DMSO, 10% cremophor-EL, PBS) or vehicle 24 h apart. LPS shock was induced by ip injection of LPS (E. coli, serotype 055.B5; Sigma) at dose 50 mg/kg body weight. In addition, another group of nrf2 +/+ was administered with a higher dose of LPS, 60 mg/kg body weight. After LPS injection, the mice were monitored for 5 days and survival was recorded every 12 h.
ROS measurement
ROS levels was assessed using luminol-dependent chemiluminescence as previously described [13].
QPCR
Total RNA was extracted using Trizol (Invitrogen) according to the manufacturer's instructions, and 2 μg total RNA was used for cDNA synthesis. Quantitative PCR analyses were performed by using assay on demand probe sets commercially available from Applied Biosystems. Assays were performed by using the ABI 7000 Taqman system (Applied Biosystems). GAPDH was used for normalization. The cycle threshold (CT) value indicates the number of PCR cycles that are necessary for the detection of a fluorescence signal exceeding a fixed threshold. The fold change (FC) was calculated by using the following formulas: ΔCT = CT (GAPDH) – CT (target gene) and FC = 2 −(ΔCT2 − ΔCT1), in which ΔCT1 represents the highest CT value among all the samples and ΔCT2 represents the value of a particular sample. Results are expressed as mean ± SD, (n=3).
ELISA
Levels of serum TNF-α was measured by enzyme immunoassays by using murine ELISA kits (R&D Systems, Minneapolis, MN).
Statistics
The Students' t-test was used to evaluate the differences between the control and treatment groups within a single genotype as well as between genotypes. Survival studies were analyzed by using the log rank test. Statistical significance was accepted at P < 0.05.
RESULTS
Nrf2-deficient neutrophils show elevated levels of ROS generation in response to LPS stimulus
To detect ROS (superoxide and hydrogen peroxide) in neutrophils in response to LPS, we used the luminol-based chemiluminescence assay. LPS stimulation induced a 4-fold increase in the levels of ROS in nrf2 −/− neutrophils relative to nrf2 +/+ cells (Fig. 1A). NADPH oxidase is a major source of ROS generation in LPS stimulated neutrophils [2, 4]. Pretreatment with DPI (an inhibitor of flavoenzymes including NADPH oxidase) at a concentration of 10 μM for 20 min completely blocked ROS generation in neutrophils of both genotypes suggesting NADPH oxidase as possible major source of ROS. However, mitochondrial ROS cannot be excluded since DPI has also been shown to inhibit mitochondrial ROS [14]. Similar results were obtained in LPS stimulated peritoneal macrophages (Supplemental data Fig 1A).
Fig.1.
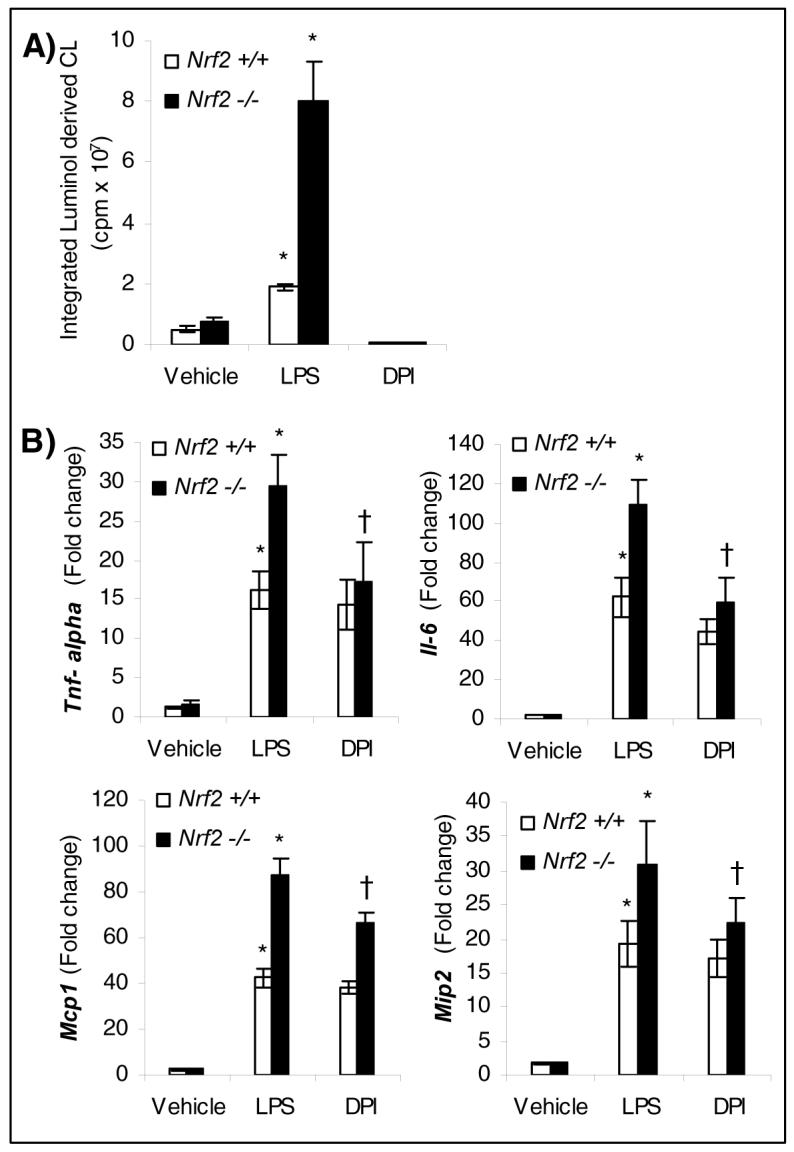
Elevated levels of ROS and proinflammatory cytokines and chemokines in nrf2-deficient neutrophils in response to LPS stimulus. A) ROS levels in nrf2 +/+ and nrf2 −/− neutrophils stimulated either with vehicle or LPS (100 ng/ml) for 1 h. Each bar is the mean ± SD (n=3) of values presenting the integration of the area under curve expressed in counts. The luminol-dependent chemiluminescence of untreated cells is at the level of the instrument background. B) mRNA expression by QPCR of cytokines (Tnf-α and Il-6) and chemokines (Mcp-1 and Mip2) in nrf2 +/+ and nrf2 −/− neutrophils 3 h after LPS stimulation (100 ng/ml). For inhibiting NADPH oxidase, cells were pretreated with DPI (10 μM) for 20 min, before stimulating with LPS. Data represented are mean fold change ± SD, n=3. * Differs from vehicle control of the same genotype; †differs from LPS treated of the same genotype.
Exacerbated expression of proinflammatory cytokines and chemokines by nrf2-deficient neutrophils in response to LPS stimulus
LPS stimulation induced significantly greater expression of cytokines (TNF-α, Il-6) and chemokines (monocyte chemotactic protein-1 (Mcp-1) and macrophage inflammatory protein 2 (Mip2)) in neutrophils of both genotypes (Fig. 1B). However, the expression levels of the measured genes were significantly greater in nrf2 −/− neutrophils when compared to the genotype controls (Fig. 1B). To elucidate whether increased levels of ROS generated by LPS stimulation is the main contributing factor in augmenting LPS response, we pretreated cells with DPI and then measured the expression of cytokines and chemokines. Pretreatment with DPI, significantly reduced the expression of Tnf-α, Il-6, Mcp1 and Mip2 in nrf2 −/− neutrophils to levels comparable to wild-type cells (Fig.1B). DPI provoked a modest decrease in the expression of cytokines and chemokines in wild-type cells. Taken together, these results suggest that exacerbated expression of cytokines and chemokines by nrf2 −/− neutrophils compared to wild-type cells is predominantly mediated through ROS signaling. Similar results were obtained in LPS stimulated peritoneal macrophages (Supplemental data Fig 1B).
Induction of Nrf2-dependent antioxidative genes by CDDO-Im attenuates LPS induced ROS levels as well as expression of proinflammatory cytokines and chemokines in neutrophils
Next, we investigated whether Nrf2-dependent antioxidative genes influence LPS induced ROS levels and expression of cytokines and chemokines. Constitutively, the expression of glutathione biosynthesizing enzymes- glutamate cysteine ligase catalytic subunit (Gclc) and glutamate cysteine ligase modifier unit (Gclm); and NAD(P)H dehydrogenase, quinone 1 (Nqo1) were significantly higher in wild-type neutrophils compared to nrf2 −/− cells (Table 1). Treatment of neutrophils of both genotypes for 12 h with CDDO-Im induced significantly Gclc, Gclm, heme oxygenase −1 (Ho-1) and Nqo1 exclusively in nrf2 +/+ neutrophils as measured by QPCR (Table 1). LPS stimulation of neutrophils for 1 h pretreated with CDDO-Im significantly reduced ROS levels only in wild-type cells whereas ROS concentration in CDDO-pretreated nrf2 −/− cells was comparable to vehicle pretreated cells (Fig 2A). Neutrophils pretreated with CDDO-Im also exhibited selective decreases in LPS induced expression of cytokines (TNF-α, Il-6) and chemokines (Mcp1, Mip2) only in wild-type neutrophils (Fig. 2B).
Table 1.
Expression of Nrf2 dependent antioxidative genes in nrf2 −/− and nrf2 +/+ neutrophils treated with CDDO-Im .
| Gene name | Basal expression | CDDO-Im induction | |
|---|---|---|---|
|
Nrf2 +/+ ÷ Nrf2 −/− |
Nrf2 +/+ (CDDO-Im / Vehicle) |
Nrf2 −/− (CDDO-Im / Vehicle) |
|
| Gclc | 2.0 | 2.1 | 1.2 |
| Gclm | 2.0 | 3.7 | 1.6 |
| Ho-1 | 0.9 | 5.9 | 2.0 |
| Nqo1 | 13.0 | 3.2 | 1.0 |
Peritoneal neutrophils of both genotypes were treated with CDDO-Im (10 nM) for 12 h and gene expression measured by QPCR. Each value is a ratio of mean of 3 biological replicate samples of nrf2 +/+ and nrf2 −/− cells.
Fig 2.

CDDO-Im pretreatment attenuates LPS induced ROS levels as well as expression of cytokines (Tnf-α, Il-6) and chemokines (Mcp1, Mip2) exclusively in nrf2 +/+neutrophils. A) LPS induced ROS levels in nrf2 +/+ and nrf2 −/− peritoneal neutrophils pretreated with CDDO-Im. Peritoneal neutrophils were pretreated with CDDO-Im for 12 h and then stimulated either with vehicle or LPS (100 ng/ml). Each bar is the mean ± SD (n=3) of values presenting the integration of the area under curve for 60 min expressed in counts. B) LPS induced cytokines and chemokines expression in nrf2 +/+ and nrf2 −/− peritoneal neutrophils pretreated with CDDO-Im and or NAC. Peritoneal neutrophils were pretreated either with CDDO-Im or NAC for 12 h then stimulated either with vehicle and or LPS (100 ng/ml) for 3 h. Data presented are mean fold change ± SD (n=3). * Differs from vehicle control of the same genotype; †, differs from LPS treated of the same genotype (P<0.05).
To clarify that lower expression levels of antioxidative genes in nrf2 −/− neutrophils predispose the transcription factor deficient cells to an exaggerated LPS response, we treated nrf2 −/− neutrophils with N-acetyl cysteine (NAC), a well known intracellular antioxidant. Pretreatment of nrf2 −/− neutrophils for 2 h with NAC greatly reduced LPS induced mRNA expression of Tnf-α, Il-6, Mcp1 and Mip2 (Fig. 2B). These results suggest that Nrf2 attenuates LPS induced inflammatory response in neutrophils possibly by regulating ROS signaling. Similar results were obtained in LPS stimulated peritoneal macrophages (Supplemental data Table 1; Fig 2A and B).
Induction of Nrf2- dependent antioxidative genes by CDDO-Im in lungs attenuates expression of proinflammatory cytokines and chemokines and protects from LPS induced septic shock
Pretreatment of mice of both genotypes by CDDO-Im for 3 days greatly induced expression of Gclc, Gclm and Nqo1 in wild-type lungs while no induction was observed in nrf2 −/− mice (Fig. 3A; Supplemental data Fig. 3). Subsequently, we assessed whether induction of Nrf2 dependent antioxidative genes by pretreatment with CDDO-Im altered the exacerbated expression of cytokines and chemokines induced by LPS shock. Nrf2 +/+ and nrf2 −/− mice were pretreated with CDDO-Im intraperitoneally for 3 days and then challenged with a lethal dose of LPS (50 mg/kg bodyweight). At 1 h after LPS treatment, the expression of measured cytokines (TNF-α, Il-6) and chemokines (Mcp-1, Mip2) were drastically higher in nrf2−/− mouse lungs when compared to wild-type mice lungs (Fig 3B). However, CDDO-Im pretreatment significantly attenuated expression of cytokines and chemokines selectively in wild-type lungs while failing to do so in nrf2 −/− lungs (Fig. 3B). In addition, serum levels of TNF-α were significantly reduced exclusively in wild-type mice pretreated with CDDO-Im when compared to vehicle pretreated wild-type mice, suggesting a significant reduction in systemic inflammation (Fig 4A).
Fig. 3.

CDDO-Im pretreatment attenuates LPS shock induced expression of cytokines and chemokines by up-regulating antioxidative genes specifically in the lungs of nrf2 +/+, but not nrf2 −/− mice A) mRNA levels of Gclc, Gclm, and Gpx2 in the lungs of nrf2 +/+ and nrf2 −/− mice after treatment with CDDO-Im. Mice were treated with 3 doses of CDDO-Im (3 μmol/kg bodyweight) 24 h apart by intraperitoneal injection. Lungs were isolated 6 h after the last dose of CDDO-Im. The fold change was calculated using the formula described in methods. Data presented is mean fold change ± SD (n=3). B) LPS induced mRNA expression of cytokines (Tnf-α and Il-6) and chemokines (Mcp1 and Mip2) in the lung of nrf2 +/+ and nrf2 −/− mice pretreated with 3 doses of CDDO-Im. Mice were treated with 3 doses of CDDO-Im (3 μmol/kg bodyweight) 24 h apart by intraperitoneal injection. A lethal dose of LPS (50 mg/kg bodyweight) was administered peritoneally 6 h after the last dose of CDDO-Im. Lungs were isolated for measuring cytokine expression 1 h after LPS treatment. Data represented are mean fold change ± SD (n=3). * Differs from vehicle control of the same genotype; †, differs from LPS treated of the same genotype (P<0.05).
Fig. 4.
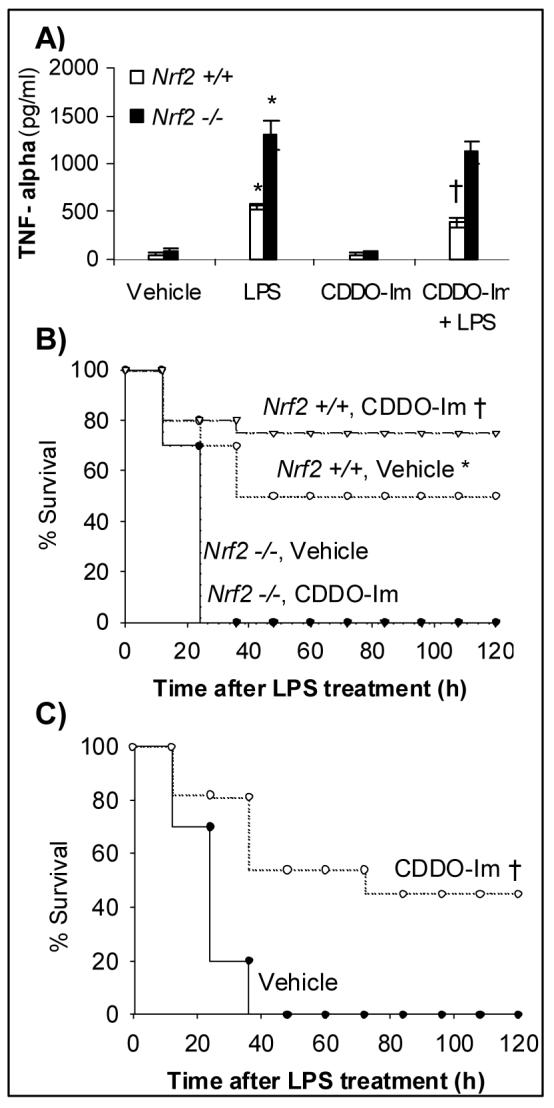
CDDO-Im pretreatment protects nrf2 +/+ mice mortality from LPS induced septic shock mortality. A) Serum TNF-α levels in CDDO-Im pretreated nrf2 +/+ and nrf2 −/− mice after 1 h of LPS challenge. Data represented are mean ± SD, n=5. B) Mortality after LPS administration in nrf2 +/+ and nrf2 −/− mice. Age-matched male nrf2 +/+ (n=10) and nrf2 −/− mice (n=10) were either pretreated with 3 doses of vehicle or CDDO-Im (3 μmol/kg bodyweight) 24 h apart followed by single intraperitoneal injection of LPS at a dose of 50 mg/kg bodyweight. C) Protection of nrf2 +/+ mice pretreated with CDDO-Im after a higher dose of LPS administration. Nrf2 +/+ (n=10) were either pretreated with 3 doses of vehicle or CDDO-Im (3 μmol/kg bodyweight) 24 h apart followed by single intraperitoneal injection of LPS at a dose of 60 mg/kg bodyweight. Mortality was monitored every 12 h for 5 days.*, Nrf2 +/+ mice had improved survival compared to nrf2 −/− mice †, Nrf2 +/+ mice pretreated with CDDO-Im had improved survival compared to vehicle pretreated mice (P<0.05).
Next we explored whether CDDO-Im pretreatment protected mice from mortality from septic shock. Injection of LPS at a dose of 50 mg/ kg bodyweight caused 100 % death in nrf2 −/− mice while 50 % of Nrf2 +/+ mice survived, as previously observed [10] . Pretreatment with CDDO-Im failed to protect nrf2 −/− mice from LPS induced septic shock as all nrf2 −/− mice pretreated with CDDO or the vehicle died by 48h after LPS challenge (Fig 4B), while 80% of the wild-type mice survived (Fig. 4B). To elaborate on the protective effect of CDDO-Im on wild-type mice, we administered a higher dose of LPS (60 mg/kg body weight), which caused 100% mortality in vehicle treated mice. However, pretreatment with CDDO-Im reduced mortality to 50% (Fig. 4C).
DISCUSSION
Major findings of the current study are: i) deficiency of the transcription factor Nrf2 exacerbates the response of innate immune cells, neutrophils and macrophages, to LPS insult; ii) Nrf2-dependent compensatory pathways modulate expression of cytokines and chemokines of neutrophils and macrophages, perhaps by attenuating levels of ROS; iii) prior activation of Nrf2 by CDDO-Im attenuates ROS levels and expression of proinflammatory cytokines and chemokines exclusively in nrf2 +/+ neutrophils and macrophages presumably by up-regulation of antioxidative genes; iv) activation of Nrf2 by CDDO-Im protects only wild-type mice from LPS induced septic shock. Taken together, the results suggest that Nrf2 is associated with oxidative regulation of LPS induced innate immune response in neutrophils and macrophages. Activation of Nrf2 by CDDO-Im protects LPS induced inflammatory response and mortality due to LPS induced septic shock.
Macrophages and neutrophils are the primary innate immune cells that recognize pathogen-associated molecular patterns such as LPS in case of gram negative bacterial infection and trigger innate immune response through TLR signaling [1]. Binding of LPS to TLR4 receptor activates an intracellular tyrosine kinase system that eventually leads to activation of an redox-sensitive transcription factor NF-κB, which in turn induces expression of number of inflammatory mediators including Tnf-α and Il-6 [1]. In addition to theses proinflammatory mediators, macrophages and neutrophils produce ROS. Besides causing tissue damage, ROS can alter LPS–TLR4 signaling at multiple levels and prime the innate immune cell for increase responsiveness [2]. Major source of ROS in innate immune cells in response to LPS is NADPH oxidase activity [4]. Numerous studies have shown ROS such as superoxide and hydrogen peroxide regulate NF-κB activation [5, 15, 16]. More recently, ROS has been demonstrated to mediate trafficking of TLR4 receptor to lipid rafts and downstream signaling adapter molecules (MYD88, TRIF, TRAF6, IRAK) during LPS as well as hydrogen peroxide stimulus [4, 5]. Inhibition of ROS generation or scavenging of ROS greatly suppresses LPS-TLR4 signaling [4, 5, 16]. Deficiency of Nrf2 predisposed neutrophils to greater responsiveness to LPS resulting in enhanced expression of cytokines and chemokines relative to wild-type cells. Greater responsiveness of nrf2 −/− neutrophils to LPS was found to be mainly mediated by increase ROS generation. Inhibition of NADPH oxidase by DPI attenuated ROS generation as well as expression of cytokines and chemokines in nrf2 −/− cells that was comparable to wild-type cells.
Variability or inductive changes in the expression of antioxidative enzymes is another important factor that can influence cellular ROS levels. Nrf2 regulates a battery of antioxidative genes constitutively as well as in response to stress [6]. To investigate whether the Nrf2 dependent antioxidants are the key molecules dampening the LPS response, we activated Nrf2 with CDDO-Im. We have previously demonstrated that CDDO-Im is a highly potent activator of Nrf2 [11, 12]. Deficiency of Nrf2 in neutrophils as well as macrophages (Supplemental data) showed significantly reduced constitutive expression of antioxidative genes as well as loss of inducibility when stimulated with CDDO-Im. CDDO-Im treatment only induced expression of Gclc, Gclm, Ho-1 and Nqo1 in wild-type neutrophils. Further, CDDO-Im pretreatment significantly reduced LPS induced ROS levels in wild-type neutrophils (as well as in nrf2 +/+ macrophages (Supplemental data)), while there were no significant changes in ROS levels of nrf2 −/− cells. In conjunction with reduction of ROS levels in nrf2 +/+ cells, CDDO-Im specifically attenuated expression of cytokines and chemokines in wild-type cells. On contrary, pretreatment of cells with NAC, an intracellular antioxidant dramatically decreased cytokine and chemokine expression in nrf2 −/− neutrophils. Taken together these results suggest that Nrf2 regulates LPS signaling perhaps by decreasing ROS levels through maintenance of cellular antioxidants. However, further studies are required to determine how precisely Nrf2 is modulating the TLR4 signaling in innate immune cells.
Inappropriate innate immune response is a characteristic feature of clinical sepsis that is associated with multiple organ failure and high mortality [1]. To find whether upregulation of Nrf2 dependent antioxidants protects against sepsis, we used a murine model of LPS induced septic shock. Since lung is the most frequent organ to fail during septic shock, we focused on lung as our target organ to study the regulation of the LPS response by activating Nrf2. CDDO-Im treatment significantly upregulated expression of antioxidants (Gclc, Gclm, Gpx2, Ho-1 and Nqo1) only in the lungs of wild-type mice. Pretreatment with CDDO-Im dramatically decreased the LPS induced expression of cytokines and chemokines in the lungs of nrf2 +/+ mice while no significant changes were observed in the lungs of nrf2 −/− mice. Likewise, LPS induced serum TNF-α levels were also significantly decreased only in CDDO-Im pretreated wild-type mice. Furthermore, CDDO-Im pretreatment significantly improved the survival of only nrf2 +/+ mice after lethal doses of LPS while no improvement was observed in the survival of nrf2 −/− mice. However, pretreatment of nrf2 −/− mice with NAC have been shown to provide significant protection from LPS induced septic shock [10]. These genetic and pharmacological approaches establish that Nrf2 protects against LPS induced inflammatory response and death from septic shock. Several published studies have demonstrated substantial modifying effects of individual antioxidative genes such as Ho-1 and glutathione peroxidase in protection from LPS induced inflammatory response [4, 17]. The observed protection of LPS induced inflammatory response in nrf2 +/+ mice likely reflects the impact of a network of Nrf2 dependent cellular antioxidants.
In conclusion, our study demonstrates that Nrf2 is a critical transcription factor in the regulation of LPS-TLR4 signaling, probably by modulating oxidative stress. Attenuation of LPS inflammatory response by CDDO-Im further strengthens the critical role of Nrf2 in regulation of LPS signaling and provides rationale for targeting Nrf2 dependent compensatory pathways in intervening sepsis pathogenesis. A decline in transcriptional activity of Nrf2 has been demonstrated in aged rats [18]. Thus, it will be worth investigating to what extent variation in the constitutive and adaptive capacity of the Nrf2 signaling pathway influences the incidence of sepsis and septic shock induced death in elderly patients. In addition, correction of low Nrf2-mediated transcriptional activity through the use of pharmacological activators of Nrf2 signaling such as CDDO-Im may offer a novel therapeutic strategy for intervening in early stages of sepsis.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grants- HL081205 (SB), P50 CA058184, CA94076 (TWK), NIEHS center grant P30 ES 038819, Young Clinical Scientist award from Flight Attendant Research Institute (SB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 2.DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, Nauseef WM. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. 1998;101:455–463. doi: 10.1172/JCI949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 4.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006;203:2377–2389. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powers KA, Szaszi K, Khadaroo RG, Tawadros PS, Marshall JC, Kapus A, Rotstein OD. Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J Exp Med. 2006;203:1951–1961. doi: 10.1084/jem.20060943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kensler TW, Wakabayashi N, Biswal S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu Rev Pharmacol Toxicol. 2006 doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 7.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohe R, Kensler TW, Yamamoto M, Biswal S. Glutathione Peroxidase 2, the Major Cigarette Smoke-inducible Isoform of GPX in Lungs is Regulated by Nrf2. Am J Respir Cell Mol Biol. 2006 doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005;65:4789–4798. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 12.Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, Baumgartner KJ, Roebuck BD, Liby KT, Yore MM, Honda T, Gribble GW, Sporn MB, Kensler TW. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 2006;66:2488–2494. doi: 10.1158/0008-5472.CAN-05-3823. [DOI] [PubMed] [Google Scholar]
- 13.Traore K, Trush MA, George M, Jr., Spannhake EW, Anderson W, Asseffa A. Signal transduction of phorbol 12-myristate 13-acetate (PMA)-induced growth inhibition of human monocytic leukemia THP-1 cells is reactive oxygen dependent. Leuk Res. 2005;29:863–879. doi: 10.1016/j.leukres.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 15.Mirochnitchenko O, Inouye M. Effect of overexpression of human Cu,Zn superoxide dismutase in transgenic mice on macrophage functions. J Immunol. 1996;156:1578–1586. [PubMed] [Google Scholar]
- 16.Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in toll-like receptor 4-dependent activation of NF-kappaB. J Immunol. 2004;172:2522–2529. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- 17.Mirochnitchenko O, Prokopenko O, Palnitkar U, Kister I, Powell WS, Inouye M. Endotoxemia in transgenic mice overexpressing human glutathione peroxidases. Circ Res. 2000;87:289–295. doi: 10.1161/01.res.87.4.289. [DOI] [PubMed] [Google Scholar]
- 18.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.