

Abstract
Although the net benefits of tamoxifen in adjuvant breast cancer therapy have been proven, the recurrence of the cancer in an aggressive and hormone independent form has been highly problematic. We previously demonstrated the important role mitochondrial DNA (mtDNA) plays in hormone-independence in prostate cancer. Here, the role of mtDNA in breast cancer progression was investigated. We established hydroxytamoxifen (4-OHT) resistant HTRMCF by growing MCF-7, a human breast adenocarcinoma cells, in the presence of 4-OHT. HTRMCF was cross-resistant to 4-OHT and ICI182,780 concurrent with the depletion of mtDNA. To further investigate the role of mtDNA depletion, MCF-7 was depleted of mtDNA by treatment with ethidium bromide. MCFρ0 was resistant to both 4-OHT and ICI182,780. Furthermore, cybrid (MCFcyb) prepared by fusion MCFρ0 with platelet to transfer mtDNA showed susceptibility to anti-estrogen. Surprisingly, after withdrawal of 4-OHT for 8 weeks, HTRMCF and their clones became susceptible to both drugs concurrent with a recovery of mtDNA. Herein, our results substantiated the first evidence that the depletion of mtDNA induced by hormone therapy triggers a shift to acquired resistance to hormone therapy in breast cancer. In addition, we showed that mtDNA depletion can be reversed, rendering the cancer cells susceptible to anti-estrogen. The hormone independent phenotype can be reversed should be a step toward more effective treatments for estrogen-responsive breast cancer.
Keywords: Mitochondrial DNA, Breast cancer, 4-Hydroxytamoxifen, hormone therapy resistance, MCF-7
Introduction
The American Cancer Society estimates that over 210,000 women in the US were newly diagnosed with breast cancer in 2005. Approximately 60–80% of those cancers were positive for estrogen receptors (ER) and/or progesterone receptors (PgR) and therefore were candidates for hormone therapy.
For over three decades, tamoxifen (4-OHT), a selective estrogen receptor modulator (SERM) with non-steroidal structure, has been widely used as an adjuvant therapy to inhibit estrogen from fueling breast cancer growth. In addition, tamoxifen has been introduced as a prophylactic drug for the prevention of breast cancer in high-risk women 1–3. Although the net benefits of tamoxifen have been proven 4, 5, the risk of tamoxifen-resistant recurrent cancer and an increased risk of endometrial cancer 6–9 have been problematic. Approximately 40% of patients with estrogen-responsive breast cancer die due to the recurrence of breast cancer after hormone therapy 10.
Mitochondrial DNA (mtDNA) encodes 13 oxidative phosphorylation (OXPHOS) proteins. Alterations of mtDNA have recently been recognized as playing a possible roles in the pathogenesis of so-called common diseases such as neuronal disorder, heart failure, diabetes, and cancer, as well as in aging 11. Mutations of mtDNA in various types of cancer, including breast cancer, colon carcinoma, prostate cancer, and pancreatic cancer have been reported 12–14. The growth advantage of cancer cells with specific mutations of mtDNA was also demonstrated 15, 16. In an earlier study, we showed that tumor necrosis factor and serum starvation could not induce apoptosis in respiration-deficient cells, whereas they induced apoptosis in parental cells and cells reconstituted with normal mtDNA 17. Amuthan et al. 18 demonstrated that mtDNA-depleted murine skeletal myoblasts C2C12 showed invasive phenotypes. It is thus likely that mutation and depletion of mtDNA affects initiation, progression and metastasis of cancer cells by preventing apoptosis and generating cancer-related signaling.
Most breast cancer cells exhibit point mutation, deletion mutation 19, and large-deletion mutation in mtDNA 20. Bhat measured the mtDNA content in estrogen-induced and estrogen-dependent hamster kidney tumors and showed the importance of mtDNA depletion in human breast cancer cells 21.
In a previous study, we demonstrated that C4-2, an androgen-independent prostate cancer cell line derived from LNCaP, lost mtDNA, and LNρ0, a mtDNA-depleted prostate cell line derived from LNCaP, had a phenotype of androgen independence 22. These results substantiated that hormone dependence/independence in prostate cancer is regulated by mtDNA.
In this study, we established HTRMCF, human breast cancer cells that were resistant to 4-hydroxytamoxifen (4-OHT), by culturing MCF-7, a human breast adenocarcinoma cell line. We found that HTRMCF also showed cross-resistance to ICI182,780 which is the first of a new type of ER antagonists with no known agonist effects 23, 24. We observed that the amount of mtDNA in HTRMCF was reduced significantly. In addition, we directly reduced mtDNA amount without using anti-estrogen. The established mtDNA-depleted MCF-7 (MCFρ0) by treating MCF-7 with ethidium bromide (EtBr) was resistant to both 4-OHT and ICI182,780. In addition, MCFρ0 fused with platelets (MCFcyb) for replete of mtDNA showed susceptibility to anti-estrogens. Surprisingly, after HTRMCF was cultured in the absence of 4-OHT for 8 weeks, it became susceptible to 4-OHT and ICI182,780 concurrent with a recovery in the level of mtDNA recovered. Thus, in this study, we substantiated that the depletion of mtDNA caused by hormone therapy or other independent means triggers resistance to hormone therapy. We also established that mtDNA depletion due to hormone therapy, depletion of mtDNA is reversible and mtDNA restores estrogen dependence and sensitivity to hormone therapy.
Materials and Methods
Materials
4-hydroxytamoxifen (4-OHT) and bovine insulin were purchased from Sigma-Aldrich (St. Louis, MO). Fetal calf serum (FCS) was obtained from Hyclone (Logan, UT). ICI182,780 was purchased from Tocris Cookson (Bristol, UK).
Cell culture
The parent cell line MCF-7 was obtained from ATCC (Manassas, VA). The cells were cultured at 37°C in a humidified atmosphere of 5% CO2 with Dulbecco’s modified Eagle medium (DMEM) containing 10% FCS supplemented with 0.01 mg/ml bovine insulin.
Establishment of the HTRMCF and HTR(−)
For the first 10 days, MCF-7 were continuously treated with 10−9 M 4-OHT. Subsequently, the cells were treated with 10−8 M 4-OHT for the next 8 weeks, 10−7 M for another 6 weeks, and 10−6 for 6 weeks, and 4-OHT-resistant HTRMCF was established. HTRMCF cells were cultured continuously in the presence of 10−6 M 4-OHT. The medium containing each concentration of 4-OHT was freshly prepared and changed every 3 to 4 days. The cells were passaged when they reached about 75% confluence. HTRMCF was cloned by limiting dilution. Among nine clones established, two of them were randomly selected and further analyzed.
HTR(−) cells were established by culturing HTRMCF then removing 4-OHT from the culture for 8 weeks.
Establishment of mtDNA-deficient MCFρ0 from MCF-7
MtDNA-depleted cells were established by culturing MCF-7 in DMEM supplemented with 10% FCS, 50μg/ml uridine, and 100μg/ml sodium pyruvate in the presence of 200ng/ml ethidium bromide for 8 weeks to select mtDNA-deficient cells as described previously 25. After 8 weeks, the amount of mtDNA was analyzed by PCR, which confirmed mtDNA-deficient status. The cells were maintained in DMEM supplemented with 10% FCS, 50μg/ml uridine, and 100μg/ml sodium pyruvate.
Fusion of MCFρ0 with platelets
Heparinized whole blood was centrifuged for 15 min at 150 g at 4°C, and platelet-rich plasma was recovered. PBS was added and the mixture was centrifuged for 15 min at 150 g at 4°C. The platelet fraction, which contained no other cell types, was centrifuged for 30 min at 2000 g at 4°C. Platelets (1 × 107) and MCFρ0 (5 × 105) cells were mixed in PBS and centrifuged at 160 g for 10 min. The supernatant was aspirated and a polyethylene glycol solution (45% of polyethylene glycol 2000 in Hanks balanced salt solution) was added. After 1 min incubation at 37°C, 9 ml of RPMI 1640 medium was added slowly. The cells were then centrifuged for 5 min at 150 g, and suspended in DMEM supplemented with 10% FCS. In these conditions only cybrid cells could grow.
Detection ofcell viability by crystal violet
The cells were cultured at a concentration of 2.5 × 103 cells/well in DMEM containing 5% FCS in 96-well plates with or without 4-OHT (1nM, 10nM, 100nM and 1000nM) or ICI182,780 (1nM, 10nM, 100nM and 1000nM) for 5 days. Crystal violet assay was previously described 22.
Southern blotting
Genomic DNA from each cell line was isolated using a Qiagen Blood and Cell Culture DNA Midi kit (Qiagen Science, Germantown, MD) according to the manufacturer’s instructions. Two micrograms of each genomic DNA sample were digested with Bam HI (New England BioLabs, Ipswich, MA). Southern blotting analysis was performed as previously described 22. The amount of intact mtDNA in each cell line was measured by ImageJ 1.36b software (NIH).
Detection of the mitochondrial membrane potential (ΔΨ m) by flow cytometry
The cells were sub-cultured overnight in 6 well plates at a concentration of 5 × 105 cells/ml. Two μM of JC-1 (Invitrogen) were added to the medium and incubated for 20min. After cells were harvested, the fluorescence intensities were measured by using FACScan (Becton Dickinson, Franklin Lakes, NJ).
Amplification of whole mtDNA at the single cell level
The cell suspensions were diluted at a concentration of 1cell/μl, and 1 μl of cell suspension was plated into a 384-well plate by Hamilton syringe. Then, 1μl of 20mM EDTA pH 8.0, 1% SDS, and 4 mg/ml proteinase K were added to the well containing a single cell observed by microscope, followed by incubation for 30min at 37°C. The samples were then diluted to 10μl. One μl of the DNA solution was mixed with 1 μl of 10x TaKaRa LA buffer and 8μl of H2O incubated for 1 min at 95°C for the inactivation of proteinase K. The PCR were performed according to the method developed by Khrapko et al. 26.
Western blotting
Twenty-five micrograms of total cell lysate were applied and transferred onto PVDF membrane (Immobilon P, Millipore, Billerica, MA). The primary antibodies used were rabbit anti β-actin, and mouse anti ERα (Cell Signaling, Danvers, MA). Subsequently the corresponding horseradish peroxidase labeled secondary antibody (Cell Signaling) was applied. The signals were developed by ECL plus (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) using X-ray films.
RT-PCR
Total RNA was isolated by AGPC method. One microgram of RNA was reverse transcribed by Superscipt III (Invitrogen) primed with oligo (dT)18–20 (Invitrogen). ERα cDNA was amplified using the primers 5′-AACCAGGGAAAATGTGTAGAGGG-3′ and 5′-CAAGGAATGCGATGAAGTAGAGC-3′ covering from 1237–1737. Ex Taq (Takara Mirus) was used for the amplification at 56 °C for the annealing with 32 cycles. GAPDH cDNA covering whole coding sequence was amplified as a control.
Results
Establishment of 4-OHT–resistant MCF-7
We established 4-OHT-resistant MCF-7, designated as HTRMCF (see Materials and Methods). HTRMCF cells were resistant to more than 1,000 times greater concentration of 4-OHT (10−6 M) than MCF-7 (10−9 M) (Figure 1A, left panel). We also tested whether HTRMCF is cross-resistant to ICI182,780, a pure anti-estrogen (Figure 1A, right panel). HTRMCF cells were resistant to 100 to 1,000 times greater concentration of ICI182,780 than MCF-7. Therefore, we concluded that HTRMCF has a phenotype of cross-resistance to both 4-OHT and ICI182,780.
Figure 1.
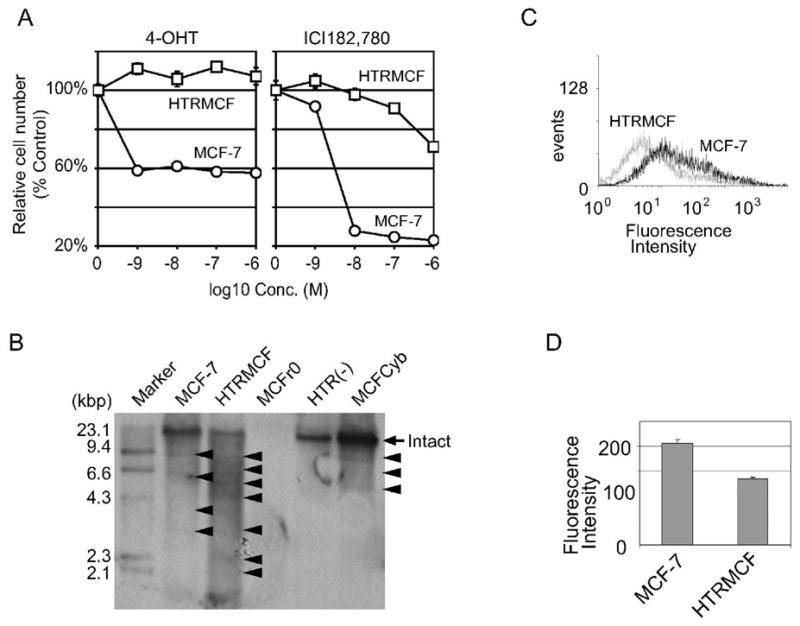
HTRMCF showed anti estrogen resistance with decreasing mtDNA amounts. A, The cell number of MCF-7 and HTRMCF cultured under 4-OHT or ICI182,780. MCF-7 and HTRMCF cells were cultured under the indicated concentrations of 4-OHT(left panel) or ICI182,780 (right panel). Cell proliferation was assayed by crystal violet as described in Materials and Methods. The relative cell numbers were plotted. The numbers of the cells cultured without reagents were calculated as 100%. The error bars indicate +/− SEM of 6 replicates. B, MtDNA analysis by Southern blotting in cell lines. Two micrograms of total DNA from each cell were applied. The signal at 16kbp indicates normal mtDNA (arrow) and other signals below 16kbp indicate large deletion mutant mtDNA (arrowheads). C, Mitochondrial membrane potential on MCF-7 and HTRMCF cells. The cells were stained with JC-1 and analyzed by flow cytometry. Representative FACS analysis of JC-1-stained cells. X-axis represents red fluorescence intensity. Solid line represents MCF-7 and dotted line represents HTRMCF. D, Quantitation of the fluorescence intensity of cells stained with JC-1. Data are expressed as the mean of three independent experiments; bars, SD. *, P<0.01 compared with untreated cells, calculated by Student’s t test.
MtDNA depletion in 4-OHT-resistant HTRMCF
We analyzed mtDNA in MCF-7 and HTRMCF by Southern blotting analysis showed that the band intensity of the intact mtDNA was reduced in HTRMCF compared to MCF-7 (Figure 1B, arrow), but HTRMCF exhibited significantly more deletion mutations, indicated as fragments with M.W. less than 16kbp (Figure 1B, arrow heads). The signal intensity of intact mtDNA in HTRMCF was a half of that in MCF-7 (Table 1).
Table 1.
mtDNA amount
| Cell line | MCF-7 | HTRMCF | MCFρ0 | MCFCyb | HTR(−) |
|---|---|---|---|---|---|
| Fold changes | 1.0 | 0.5 | 0 | 0.8 | 1.9 |
The amount of intact mtDNA in each cell line (Figure 1B) was measured by ImageJ 1.36b software (NIH). The intensity of mtDNA signal in MCF-7 was calculated as 1.0.
Reduction of membrane potential in HTRMCF
A decrease in mtDNA will lead to less transcription of mtDNAs encoding proteins involved in oxidative phosphorylation, resulting in decrease in mitochondrial membrane potential. Therefore, to investigate whether mitochondrial function in mtDNA less cells is downregulated, we measured mitochondrial membrane potential by flow cytometer using a JC-1 fluorescent probe. JC-1 is a cationic carbocyanine dye that accumulates in mitochondria. When the dye exists as a monomer at low membrane potential, it yields green fluorescence, while at higher membrane potential, the dye forms J-aggregates that exhibit a red fluorescence. Higher red fluorescence intensity represents the higher mitochondrial membrane potential. Representative histograms of red fluorescence intensity by JC-1 in MCF-7 and HTRMCF are shown in Figure 1C. The mean intensity of triplicate samples was statistically lower in HTRMCF than MCF-7 (p<0.01, Figure 1D), clearly indicating loss of membrane potential due to mtDNA reduction.
Mitochondrial DNA depletion caused hormone-independent phenotype
In order to show the relationship between 4-OHT resistance and mtDNA, we established mtDNA-depleted MCF-7 by culturing in the presence of EtBr. MtDNA in EtBr-resistant MCF-7 was under detectable range by PCR (data not shown) and was designated as MCFρ0. No mtDNA was also detected by Southern blotting (Figure 1B). We analyzed the sensitivity of MCFρ0 to 4-OHT (Figure 2A, left panel) and ICI182,780 (Figure 2A, right panel). Estimated from the dose-response of 4-OHT and ICI182,780 to each cell lines, MCFρ0 cells were resistant to more than 1,000 greater concentration of 4-OHT and ICI182,780 than MCF-7 and more resistant than HTRMCF. These findings indicated that hormone independence could be achieved by a means of quantity depleting mtDNA that is not related to hormone antagonism.
Figure 2.
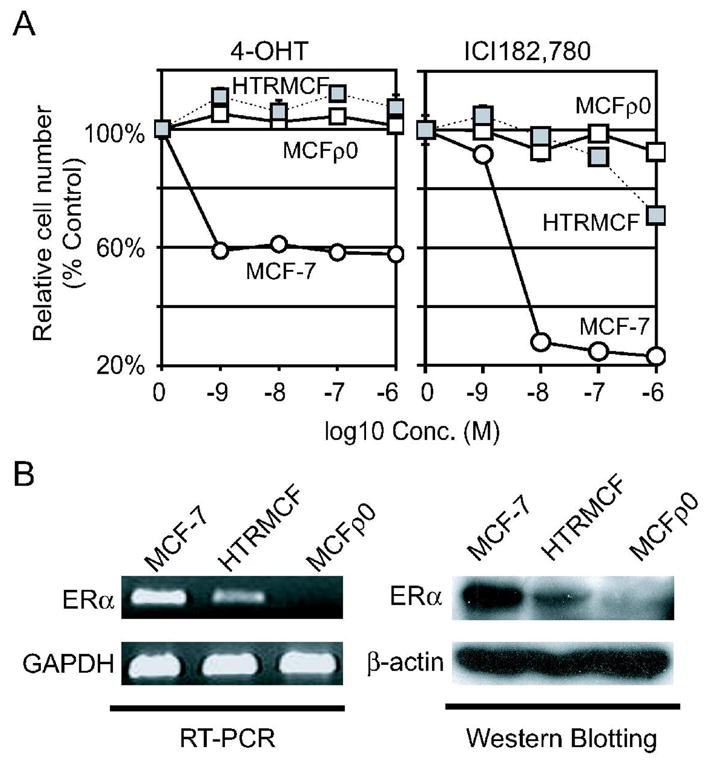
A, , The cell number of MCFρ0 cultured under 4-OHT or ICI182,780. MCFρ0 cells cultured under the indicated concentrations of 4-OHT or ICI182,780 were plotted in left and right panel respectively. The relative cell numbers of MCF-7 and HTRMCF were also plotted for comparison. See figure 1 legend for more details. B, The expression of ERα. The expression of ERα in MCF-7, HTRMCF and MCFρ0 was analyzed by RT-PCR (left panel) and Western blotting (right panel).
Reduction of ERα in HTRMCF and MCFρ0
One of the possible causes for the resistance to anti-estrogen is the reduction of estrogen receptor. The expression level of ERαin mRNA and protein level were reduced in HTRMCF and completely lost in MCFρ0 cells (Figure 2B).
Repletion of mtDNA into MCFρ0 cells restored the susceptibility to anti –estrogen
In order to show the causal relationship of mtDNA to hormone resistance, we fused MCFρ0 cells with platelets to obtain cybrid (MCFcyb). The amount of mtDNA was 1.9 folds greater than MCF-7 by Southern blotting (Figure 1B and Table 1). MCFcyb showed higher susceptible to both 4-OHT and ICI182,780 (Figure 3).
Figure 3.
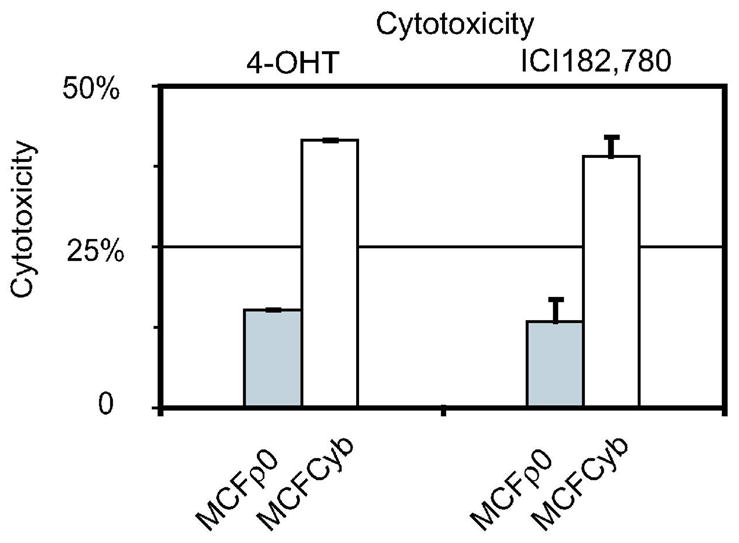
Cytotoxicity of 4-OHT (10−6 M) or ICI182,780 (10−6 M) in MCFcyb. The cytotoxicity of either 4-OHT (10−6 M) or ICI182,780 (10−6 M) of MCFcyb (white) and MCFρ0 (gray), were measured by crystal violet for 5 days. The numbers of the cells cultured without reagents were calculated as 0.
Withdrawal of 4-OHT from culture led to recovery of mtDNA copies and hormone-dependent phenotype in HTRMCF
EtBr treatment had been shown to reduce mtDNA copies, and withdrawal of EtBr can reverse the reduction in mtDNA copies in established mtDNA-less cells 18. We tested whether withdrawal of 4-OHT from the culture medium resulted in a recovery in number of mtDNA copies in HTRMCF. HTRMCF cells were cultured for 8 weeks without 4-OHT. Those cells were designated as HTR(−). To analyze the recovery of mtDNA, Southern blotting was performed. The mtDNA copies were apparently recovered during the withdrawal of 4-OHT from the culture medium (Figure 1B and Table 1). We then investigated the phenotype of hormone-dependence. HTR(−) recovered sensitivity to 4-OHT and ICI182,780, comparable to the sensitivity of MCF-7 (Figures 4A). In order to eliminate the possibility that HTRMCF with higher mtDNA contents and higher susceptible to 4-OHT already existed in HTRMCF, had growth advantage and was selected as HTR(−), we used clones of HTRMCF and analyzed their mtDNA contents and susceptibility to 4-OHT after withdrawal of 4-OHT. After 4 weeks of withdrawal of 4-OHT from the culture, the HTRMCF clones had more mtDNA contents and were more susceptible to 4-OHT (Figure 4B and C). Thus, we not only denied the clonal selection, but also proved that mtDNA content is highly related to 4-OHT resistance.
Figure 4.
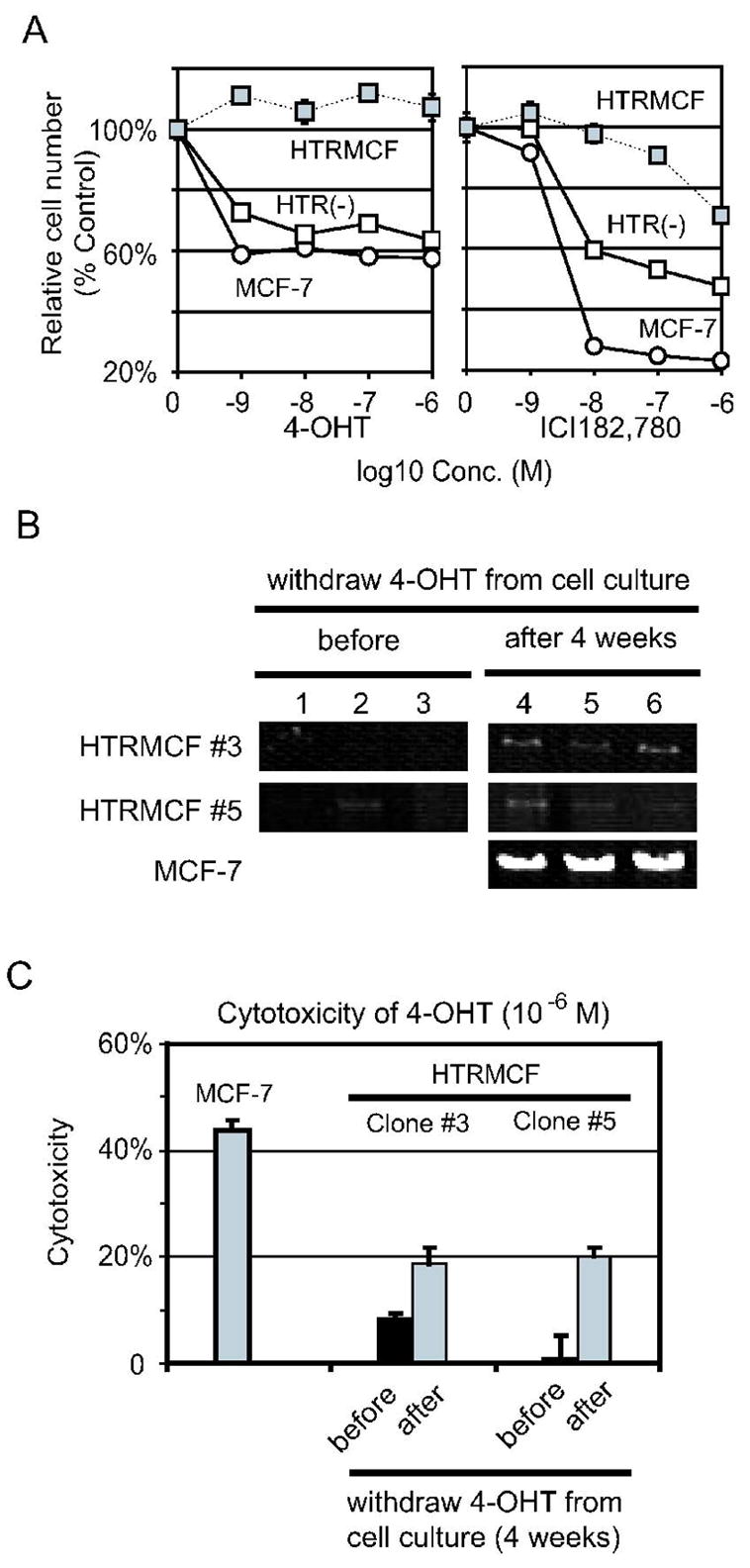
A, The cell number of HTR(−) cultured under 4-OHT or ICI182,780. HTR(−) cells cultured under the indicated concentrations of 4-OHT or ICI182,780 were plotted in left and right panel respectively. The relative cell numbers of MCF-7 and HTRMCF were also plotted for comparison. See legend of figure 1 for more details. B, Single-cell PCR analysis in MCF-7, and HTRMCF clones after withdrawal of 4-OHT. DNA was collected from single cell of each clone, cultured continuously with 4-OHT (left panel) or 4 weeks after withdrawal 4-OHT (right panel), and mtDNA was amplified by PCR as mentioned above. MCF-7 was used as a control. C, Cytotoxicity of 4-OHT (10−6 M) after withdrawal of 4-OHT in HTRMCF clones. The cytotoxicity of 4-OHT (10−6 M) of HTRMCF clones, cultured continuously with 4-OHT (black) or 4 weeks after withdraw 4-OHT (gray), was measured by crystal violet. The numbers of the cells cultured without reagents were calculated as 0. MCF-7 was used as a control.
Discussion
Our study provides the first in vitro model that the depletion of mtDNA induced by hormone-therapy plays a significant role in the recurrence of the breast cancer due to the acquisition of resistance to hormone therapy. In addition, we demonstrated that reversing mtDNA depletion by means of both fusion with platelet and releasing from anti-estrogen pressure results in the restoration of hormone dependence. In our study of androgen independent prostate cancer we established that hormone dependence in prostate cancer is regulated by mtDNA 22. In our current study, we observed a reduction in mtDNA in hormone therapy-resistant breast cancer cells (HTRMCF) compared to MCF-7 parental cells. In addition, mtDNA-depleted MCF-7 cells (MCFρ0) showed 4-OHT resistance MCFρ0 cells fused with platelet to compensate depleted mtDNA (MCFcyb) showed susceptibility to anti-estogens. These results substantiated that mtDNA depletion triggers a shift to estrogen independence, resulting in acquired resistance to tamoxifen treatment. However, we also showed that mtDNA depletion can be reversed, leading to the cancer cells’ return to estrogen dependence and sensitivity to hormone tamoxifen therapy.
The mechanism of mtDNA decrease by 4-OHT is recently proposed by Larosche et.al27. They showed 4-OHT was preferentially accumulated in mitochondria and inhibited mtDNA synthesis and topoisomerase. It may be possible that 4-OHT treatment also triggers mtDNA deletions by inhibiting topoisomerase which causes insufficient relaxation to complete mtDNA replication. Chen et al. reported that ER localized in mitochondria28, bound to mtDNA and enhanced the mtDNA transcription29. Considering mtDNA replication is coupled with transcription, it is possible that down-regulation of ERα in HTRMCF reduces the number of mtDNA copies. However, since MCFρ0 lost ERα expression, it is likely that mtDNA biogenesis is located upstream of ERαexpression. Seidel-Rogol and Shadel reported that mtDNA amount recovered after removal of EtBr from the culture being used to treat mtDNA-less cells, although the mtDNA level in the cells without mtDNA could not recover 30. From our results using clones, it is clear that 4-OHT inhibited mtDNA replication and reduced mtDNA copies in HTRMCF. Therefore, the withdrawal of 4-OHT have released HTRMCF from the selective pressure of the inhibition of mtDNA replication, allowing mtDNA to recover and causing the cells to return to the hormone-sensitive phenotype. Brunner et al. reported that 4-OHT–resistant cells are still sensitive to the induction of cell death induced by ICI182,780 31. However, the sensitivity Brunner observed might be explained by the fact that ICI182,780 has a much stronger effect than 4-OHT 32. Another factor that might generate the variation in experimental results is that in our study HTRMCF was continuously cultured in the presence of 4-OHT because we found that resistance to 4-OHT gradually declined when we removed 4-OHT from the medium. In contrast, Brunner et al. stated that their cells did not lose resistance to 4-OHT after removing 4-OHT from the medium. Contradictory results regarding resistance of MCF-7 to ICI182,780 have also been reported as follows. Brunner et al. reported that ICI182,780-resistant MCF-7 cells were resistant to 4-OHT 33. This phenotype is very similar to HTRMCF. In contrast, Lykkesfeldt et al. reported that ICI182,780-resistant cells were as sensitive to 4-OHT as parental MCF-7 34. Lykkesfeldt cultured MCF-7 in 1% FCS, whereas our cells were cultured in 10% FCS, and we observed that reducing FCS in the culture medium greatly reduced sensitivity to 4-OHT. Although, we do not know the exact mechanism for the reduced sensitivity, this variance in FCS concentration may explain the discrepancy between the results of these two studies.
In an earlier study, we showed that tumor necrosis factor and serum starvation could not induce apoptosis in respiration-deficient cells, whereas they induced apoptosis in parental cells and cells reconstituted with normal mtDNA 17. Amuthan et al. 18 demonstrated that mtDNA-depleted murine skeletal myoblasts C2C12 showed invasive phenotypes. It is thus likely that mutation and depletion of mtDNA affects initiation, progression and metastasis of cancer cells by preventing apoptosis and generating cancer-related signaling. We found that ERα expression was reduced in HTRMCF and completely lost in MCFρ0 cells, indicating that induction of the estrogen-independent phenotype is correlated with the reduction of both normal mtDNA and ERα. The apoptotic cell death after 4-OHT treatment was not detected by conventional apoptosis assays (DNA fragmentation and Annexin-V staining, data not shown), suggesting that the observed resistance is likely due to insensitivity to cell growth inhibition by 4-OHT. Indeed the growth retardation was observed by counting the cells after 4-OHT treatment (data not shown). Thus, one of the causes for the resistance to anti-estrogen is the reduction of estrogen receptor. Due to highly susceptibility of ρ0 cells to transfection reagents, we could not compensate ERα expression to ρ0 cells for the confirmation that reduction of ERα expression by mtDNA depletion led 4-OHT resistance. It is also possible that the depletion of ERα from breast cancer cells results from the continuous stimulation with other growth signaling such as EGFR signaling. MtDNA depletion may cause the up-regulation of growth factor receptor signaling, leading to a bypass of ERα signaling and down-regulation of ERα. Additionally, we did not detect overexpression of Her2/neu in HTRMCF and MCFρ0 by western blotting (Data not shown) and could eliminate the possibility that overexpression of Her2/neu plays a role in 4-OHT resistance in HTRMCF.
Although the mechanisms underlying mtDNA reduction need to be further investigated, it is possible that mtDNA status would be useful data in the prognosis for acquiring resistance to hormone therapy. This evidence of relationship between mtDNA and breast cancers acquired resistance to hormone therapy and the discovery that the hormone independent phenotype can be reversed should be a step toward more effective treatments for estrogen-responsive breast cancer.
Acknowledgments
This work was supported by Taiho Pharmaceutical Co. Ltd, State of Arkansas Tobacco Settlement, and NIH grant CA100846 (M. Higuchi). We would like to express our gratitude to Dr. Thomas Kelly for his advice during preparation of the manuscript.
Abbreviations
- 4-OHT
4-hydroxytamoxifen
- ICI182
780 fulvestrant
- DMEM
Dulbecco’s modified Eagle medium
- ER
estrogen receptor
- EtBr
ethidium bromide
- FCS
fetal calf serum
- mtDNA
mitochondrial DNA
- OXPHOS
oxidative phosphorylation
- PCR
polymerase chain reaction
- PgR
progesterone receptors
- SERM
selective estrogen receptor modulator
Footnotes
Category
Cancer Cell Biology
Our results substantiated the first evidence that the depletion of mtDNA induced by hormone therapy triggers a shift to acquired resistance to hormone therapy in breast cancer. The hormone independent phenotype can be reversed should be a step toward more effective treatments for estrogen-responsive breast cancer.
References
- 1.Fisher B. Highlights from recent National Surgical Adjuvant Breast and Bowel Project studies in the treatment and prevention of breast cancer. CA Cancer J Clin. 1999;49:159–77. doi: 10.3322/canjclin.49.3.159. [DOI] [PubMed] [Google Scholar]
- 2.Fisher B, Costantino JP, Wickerham DL, Cecchini RS, Cronin WM, Robidoux A, Bevers TB, Kavanah MT, Atkins JN, Margolese RG, Runowicz CD, James JM, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst. 2005;97:1652–62. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 3.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–88. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 4.Group EBCTC. Effects of adjuvant tamoxifen and of cytotoxic therapy on mortality in early breast cancer. An overview of 61 randomized trials among 28,896 women. Early Breast Cancer Trialists’ Collaborative Group. N Engl J Med. 1988;319:1681–92. doi: 10.1056/NEJM198812293192601. [DOI] [PubMed] [Google Scholar]
- 5.Group EBCTC. Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet. 1998;351:1451–67. [PubMed] [Google Scholar]
- 6.Assikis VJ, Neven P, Jordan VC, Vergote I. A realistic clinical perspective of tamoxifen and endometrial carcinogenesis. Eur J Cancer. 1996;32A:1464–76. doi: 10.1016/0959-8049(96)00184-0. [DOI] [PubMed] [Google Scholar]
- 7.Dallenbach-Hellweg G, Schmidt D, Hellberg P, Bourne T, Kreuzwieser E, Doren M, Rydh W, Rudenstam G, Granberg S. The endometrium in breast cancer patients on tamoxifen. Arch Gynecol Obstet. 2000;263:170–7. doi: 10.1007/s004040050276. [DOI] [PubMed] [Google Scholar]
- 8.Rutqvist LE, Johansson H, Signomklao T, Johansson U, Fornander T, Wilking N. Adjuvant tamoxifen therapy for early stage breast cancer and second primary malignancies. Stockholm Breast Cancer Study Group. J Natl Cancer Inst. 1995;87:645–51. doi: 10.1093/jnci/87.9.645. [DOI] [PubMed] [Google Scholar]
- 9.Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM. Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14. J Natl Cancer Inst. 1994;86:527–37. doi: 10.1093/jnci/86.7.527. [DOI] [PubMed] [Google Scholar]
- 10.Faltus T, Yuan J, Zimmer B, Kramer A, Loibl S, Kaufmann M, Strebhardt K. Silencing of the HER2/neu gene by siRNA inhibits proliferation and induces apoptosis in HER2/neu-overexpressing breast cancer cells. Neoplasia. 2004;6:786–95. doi: 10.1593/neo.04313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Booker LM, Habermacher GM, Jessie BC, Sun QC, Baumann AK, Amin M, Lim SD, Fernandez-Golarz C, Lyles RH, Brown MD, Marshall FF, Petros JA. North American white mitochondrial haplogroups in prostate and renal cancer. J Urol. 2006;175:468–72. doi: 10.1016/S0022-5347(05)00163-1. discussion 72–3. [DOI] [PubMed] [Google Scholar]
- 14.Lievre A, Chapusot C, Bouvier AM, Zinzindohoue F, Piard F, Roignot P, Arnould L, Beaune P, Faivre J, Laurent-Puig P. Clinical value of mitochondrial mutations in colorectal cancer. Journal of Clinical Oncology. 2005;23:3517–25. doi: 10.1200/JCO.2005.07.044. [DOI] [PubMed] [Google Scholar]
- 15.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, Marshall FF, Wallace DC. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719–24. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–63. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi M, Aggarwal BB, Yeh ET. Activation of CPP32-like protease in tumor necrosis factor-induced apoptosis is dependent on mitochondrial function. J Clin Invest. 1997;99:1751–8. doi: 10.1172/JCI119339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. Embo J. 2001;20:1910–20. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianchi MS, Bianchi NO, Bailliet G. Mitochondrial DNA mutations in normal and tumor tissues from breast cancer patients. Cytogenet Cell Genet. 1995;71:99–103. doi: 10.1159/000134072. [DOI] [PubMed] [Google Scholar]
- 20.Penta JS, Johnson FM, Wachsman JT, Copeland WC. Mitochondrial DNA in human malignancy. Mutat Res. 2001;488:119–33. doi: 10.1016/s1383-5742(01)00053-9. [DOI] [PubMed] [Google Scholar]
- 21.Bhat HK. Depletion of mitochondrial DNA and enzyme in estrogen-induced hamster kidney tumors: a rodent model of hormonal carcinogenesis. J Biochem Mol Toxicol. 2002;16:1–9. doi: 10.1002/jbt.10017. [DOI] [PubMed] [Google Scholar]
- 22.Higuchi M, Kudo T, Suzuki S, Evans TT, Sasaki R, Wada Y, Shirakawa T, Sawyer JR, Gotoh A. Mitochondrial DNA determines androgen dependence in prostate cancer cell lines. Oncogene. 2006;25:1437–45. doi: 10.1038/sj.onc.1209190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51:3867–73. [PubMed] [Google Scholar]
- 24.Addo S, Yates RA, Laight A. A phase I trial to assess the pharmacology of the new oestrogen receptor antagonist fulvestrant on the endometrium in healthy postmenopausal volunteers. Br J Cancer. 2002;87:1354–9. doi: 10.1038/sj.bjc.6600644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–3. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 26.Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, Perls TT, Upton M, Vijg J, Wei JY. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–41. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larosche I, Letteron P, Fromenty B, Vadrot N, Abbey-Toby A, Feldmann G, Pessayre D, Mansouri A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J Pharmacol Exp Ther. 2007;321:526–35. doi: 10.1124/jpet.106.114546. [DOI] [PubMed] [Google Scholar]
- 28.Chen JQ, Delannoy M, Cooke C, Yager JD. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab. 2004;286:E1011–22. doi: 10.1152/ajpendo.00508.2003. [DOI] [PubMed] [Google Scholar]
- 29.Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem. 2004;93:358–73. doi: 10.1002/jcb.20178. [DOI] [PubMed] [Google Scholar]
- 30.Seidel-Rogol BL, Shadel GS. Modulation of mitochondrial transcription in response to mtDNA depletion and repletion in HeLa cells. Nucleic Acids Res. 2002;30:1929–34. doi: 10.1093/nar/30.9.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brunner N, Frandsen TL, Holst-Hansen C, Bei M, Thompson EW, Wakeling AE, Lippman ME, Clarke R. MCF7/LCC2: a 4-hydroxytamoxifen resistant human breast cancer variant that retains sensitivity to the steroidal antiestrogen ICI 182,780. Cancer Res. 1993;53:3229–32. [PubMed] [Google Scholar]
- 32.Diel P, Smolnikar K, Michna H. The pure antiestrogen ICI 182780 is more effective in the induction of apoptosis and down regulation of BCL-2 than tamoxifen in MCF-7 cells. Breast Cancer Research & Treatment. 1999;58:87–97. doi: 10.1023/a:1006338123126. [DOI] [PubMed] [Google Scholar]
- 33.Brunner N, Boysen B, Jirus S, Skaar TC, Holst-Hansen C, Lippman J, Frandsen T, Spang-Thomsen M, Fuqua SA, Clarke R. MCF7/LCC9: an antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997;57:3486–93. [PubMed] [Google Scholar]
- 34.Lykkesfeldt AE, Larsen SS, Briand P. Human breast cancer cell lines resistant to pure anti-estrogens are sensitive to tamoxifen treatment. Int J Cancer. 1995;61:529–34. doi: 10.1002/ijc.2910610417. [DOI] [PubMed] [Google Scholar]