

Abstract
Microfabrication technology has been adapted to produce micron-scale needles as a safer and painless alternative to hypodermic needle injection, especially for protein biotherapeutics and vaccines. This study presents a design that encapsulates molecules within microneedles that dissolve within the skin for bolus or sustained delivery and leave behind no biohazardous sharp medical waste. A fabrication process was developed based on casting a viscous aqueous solution during centrifugation to fill a micro-fabricated mold with biocompatible carboxymethylcellulose or amylopectin formulations. This process encapsulated sulforhodamine B, bovine serum albumin, and lysozyme; lysozyme was shown to retain full enzymatic activity after encapsulation and to remain 96% active after storage for two months at room temperature. Microneedles were also shown to be strong enough to insert into cadaver skin and then to dissolve within minutes. Bolus delivery was achieved by encapsulating molecules just within microneedle shafts. For the first time, sustained delivery over hours to days was achieved by encapsulating molecules within the microneedle backing, which served as a controlled release reservoir that delivered molecules by a combination of swelling the backing with interstitial fluid drawn out of the skin and molecule diffusion into the skin via channels formed by dissolved microneedles. We conclude that dissolving microneedles can be designed to gently encapsulate molecules, insert into skin, and enable bolus or sustained release delivery.
Keywords: Microfabrication, Microneedle, Minimally invasive, Polysaccharide, Protein delivery, Transdermal drug delivery
1. Introduction
To address limitations of oral delivery and hypodermic injection [1–3], arrays of micron-scale needles have been developed to painlessly pierce skin’s outer barrier of stratum corneum with the goal to deliver drugs with the efficacy of a needle and the convenience of a transdermal patch. This approach has demonstrated increased transdermal delivery of small-molecule drugs, proteins, DNA, and vaccines [4, 5]. One approach involves pretreatment of skin with microneedles, followed by application of a transdermal patch for extended drug delivery through the permeabilized skin [6]. Another approach involves coating or encapsulating drug onto or within microneedles. Upon dissolution of the coating or the needle itself, the drug cargo is released within the skin as a bolus or possible controlled-release delivery [7–10].
Microfabrication tools have been leveraged to make microneedles using methods suitable for low-cost, high-volume manufacturing, which is critical to impacting medicine as a disposable device. Microneedles suitable for piercing skin to increase skin permeability or for carrying drug into the skin as a coating have been fabricated from silicon and metal [4, 5]. Microneedles that encapsulate drug and subsequently dissolve or degrade in the skin have been fabricated from polymers, such as slow-degrading polylactic-co-glycolic acid [9] and rapidly-dissolving sugar [8].
To address growing needs of protein delivery, we propose that a microneedle device should (1) encapsulate drug within a biocompatible and mechanically robust material using processes that do not damage protein integrity, (2) enable controlled delivery as a bolus or sustained release, and (3) utilize a device suitable for self-administration without medical training that leaves behind no sharp, biohazardous waste. This study presents microneedles designed to have these attributes using polysaccharide biomaterials; a gentle microneedle molding technique; and study of mechanical, stability, and delivery properties using model proteins and cadaver skin.
2. Materials and Methods
2.1 Fabrication of microneedles
2.1.1 Molding
Micromolds were fabricated using photolithography and molding processes described previously [11]. In brief, a female microneedle master-mold was structured in SU-8 photoresist (SU-8 2025, Microchem, Newton, MA) by UV exposure to create conical (circular cross section) or pyramidal (square cross section) microneedles tapering from a base measuring 300 μm to a tip measuring 25 μm in width over a microneedle length of 600–800 μm. A male microneedle master-structure made of polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, MI) was created using this mold. The male PDMS master-structure was sputter-coated (601 Sputtering System, CVC Products, Rochester, NY) with 100 nm of gold to prevent adhesion with a second PDMS layer cured onto the male master-structure to create a female PDMS replicate-mold. Excess PDMS on the female replicate-mold was trimmed so that the mold fit within the 27-mm inner diameter of a 50 ml conical tube (Corning Inc., Corning, NY). This metal-coated male master-structure was repeatedly used to make replicate-molds that were repeatedly used to make microneedle devices.
2.1.2 Preparation of microneedle matrix
To serve as microneedle matrix materials, ultra-low viscosity carboxymethylcellulose (CMC, Cat No. 360384, Aldrich, Milwaukee, WI), amylopectin (Cat No. 10120, Fluka, Steinheim, Germany) and bovine serum albumin (BSA, Sigma, St. Louis, MO) were dissolved in deionized water. Water was then evaporated off until the concentration of solute (e.g., CMC) was approximately 27 wt%, which resulted in a viscous hydrogel. CMC was concentrated by heating at 60–70° C at ambient pressure or vacuuming at -50 kPa at room temperature. Amylopectin and BSA were concentrated only by the heating method at 60–70° C or 37° C, respectively. Solute concentration was determined by measuring solution mass before and after evaporation. Viscosity of concentrated hydrogels was measured using a Couette viscometer (Physica MCR300, Anton Paar Physica, Ostfildem, Germany).
In some cases, a model drug was added by hand mixing to solubilize or suspend the compound in the concentrated hydrogel. Three model drugs were added at final concentrations of 0.15–30 wt% sulforhodamine B (Molecular Probes, Eugene, OR), 20 wt% BSA (Sigma), or 5 wt% lysozyme (Sigma). The term “model drug” is used to indicate that these compounds have physicochemical and transport properties representative of certain classes of drugs, but not to suggest that these compounds have pharmacological activity representative of drugs.
2.1.3 Casting
To mold microneedles from concentrated hydrogels, 100–300 mg of hydrogel was placed on a female PDMS mold in a conical centrifuge tube (Corning) and centrifuged in a 45° angled rotor (GS-15R, Beckman, Fullerton, CA) at 3000×g and 37° C for up to 2 h to fill the microneedle mold cavities and dry the hydrogel.
To prepare microneedles with model drug encapsulated only within the microneedles and not in the backing layer, 8–10 mg of hydrogel mixed with model drug was filled just into the microneedle cavities in the mold and then dried under centrifugation for up to 30 min. Residual hydrogel on the surface of the mold was removed with dry tissue paper (Kimwipes, Kimberly-Clark, Roswell, GA) and 100–200 mg pure hydrogel without drug was then applied and dried onto the mold to form the backing layer. To prepare microneedles with model drug encapsulated only in the backing layer and not within the microneedles, the same two-step process was followed, except pure hydrogel was filled into the microneedle mold cavities and a hydrogel mixed with model drug was used to form the backing layer.
2.2 Microneedle mechanics
2.2.1 Microneedle failure
Mechanical failure tests were performed with a displacement-force test station (Model 921A, Tricor Systems Inc., Elgin, IL, USA), as described previously [12]. A 3×3 array containing 9 microneedles was attached to the mount of a moving sensor and an axial force was applied to move the mount at a speed of 1.1 mm/s. The mount pressed the microneedles against a flat, rigid surface of stainless steel oriented perpendicularly to the axis of mount movement. The test station recorded the force required to move the mount as a function of distance.
2.2.2 Microneedle skin insertion
To determine if microneedles insert into skin, CMC pyramidal microneedles (600 μm height, 300 μm base width, and 600 μm center-to-center spacing) in a 10×10 array were inserted with IACUC approval into full-thickness cadaver pig skin without subcutaneous fat that was shaved (series 8900, WHAL, Sterling, IL) and affixed under mild tension to a wooden plate using 1 cm long screws. Microneedles were inserted by pressing against the microneedle backing layer with a thumb using a force of approximately 1.5 N and then removed immediately after the insertion. The site of microneedle insertion on the skin surface was exposed for 10 min to a red tissue-marking dye (Shandon, Pittsburgh, PA, USA) that selectively stains sites of stratum corneum perforation. After wiping residual dye from the skin surface with dry tissue paper, skin was viewed by brightfield microscopy (SZX12, Olympus).
Skin samples were prepared for histology by freezing in histology mounting compound (Tissue-Tek, Sakura Finetek, Torrance, CA) and slicing into 20-μm thick sections (Cryo-star HM 560MV, Microm, Waldorf, Germany) and then viewed by brightfield microscopy (E600, Nikon, Tokyo, Japan).
2.2.3 Simulation of microneedle critical buckling load
Critical buckling load, Pcr, of microneedles was simulated during axial loading using analytical methods of Smith [13]. For the fixed-free case, where the microneedle base was fixed in position and the microneedle tip could move freely, the square-based pyramidal and circle-based conical geometries were modeled using the equations for Pcr5 of case 3 and Pcr7 of case 5 in Appendix A of Smith [13], respectively:
| (1) |
| (2) |
Here, E is Young’s modulus; L is microneedle length; H1and H2 are microneedle widths at the base and tip of pyramidal microneedles, respectively; and R1 and R2 are radii at the base and tip of conical microneedles, respectively. Young’s modulus of CMC microneedles was determined to be 1 GPa by direct measurement (MicroTester, Instron 5548, Norwood, MA) using bulk CMC prepared using the same casting process used to make microneedles. Young’s modulus of PLA microneedles was previously determined to be 5 GPa [14]. Tip width and diameter of pyramidal and conical microneedles, respectively, were estimated both to be 25 μm based on microscopic examination.
2.3 Drug release from microneedles
2.3.1 Imaging bolus release from dissolving microneedles
CMC pyramidal microneedles (600 μm height, 300 μm base width, and 600 μm center-to-center spacing) in a 6×6 array were inserted by hand into pig cadaver skin. Just the microneedles, and not the backing layer, contained sulforhodamine B at 0.15 wt% on a dry basis, such that each microneedle contained 0.04 μg of sulforhodamine and the 36-needle array contained 1.44 μg of sulforhodamine. After 5 min, the microneedles were removed from skin and the skin sample was examined histologically. In a separate set of experiments, the shape of microneedles was also observed after 10 s, 1 min, 15 min, and 60 min insertion into the skin by light microscopy (SZX12, Olympus).
2.3.2 Imaging and quantification of sustained release from microneedle patches
To image long-term release from dissolving microneedles into skin, sulforhodamine B was encapsulated within the needles and the backing layer at 0.15 wt% in a 6×6 array of CMC pyramidal microneedles (600 μm height, 300 μm base width, and 600 μm center-to-center spacing). The microneedle device contained 15 μg of sulforhodamine. The microneedles were inserted into pig cadaver skin by hand, covered with dermal tape (Blenderm, 3M Health Care, St. Paul, MN), and kept at room temperature for up to 12 h. Next, the microneedle device was removed and skin was examined histologically.
To quantify sulforhodamine release, a 7×7 array of CMC or amylopectin pyramidal microneedles (600 μm height, 300 μm base width, and 600 μm center-to-center spacing) was prepared with a backing layer of approximately 300 μm thickness. Sulforhodamine B was encapsulated within the needles and the backing layer at 10 wt%, which corresponded to 1 mg of sulforhodamine in the microneedle device weighing 10 mg. Alternatively, sulforhodamine was encapsulated only within the backing layer at 10 wt% and 30 wt%, which corresponded to almost 1 mg and 3 mg of model drug per device, respectively. Microneedles were inserted by hand into heat-stripped human cadaver epidermis (Emory University Body Donor Program, Atlanta, GA) with IRB approval [15]. Microneedles were secured to skin with dermal tape and the microneedle-skin assembly was placed in a Franz diffusion chamber (Permegear, Hellertown, PA) at 32° C. Phosphate-buffered saline (PBS) in the receptor compartment of the Franz chamber contained 0.01 M sodium azide as an anti-bacterial agent and was sampled periodically for up to 7 days to determine sulforhodamine flux by spectrofluorimetry (QM-1, Photon Technology International, South Brunswick, NJ).
2.4 Protein stability and activity
2.4.1 Circular dichroism
The secondary structure of lysozyme was examined by spectropolarimetry (JASCO, J-810, Tokyo, Japan) after encapsulation and release from dissolving microneedles. CMC pyramidal microneedle devices weighing 5 mg that encapsulated lysozyme at a mass fraction of 5 wt% were completely dissolved in 50 ml PBS at room temperature for 10 min and filtered by centrifugal filtration (Centricon YM-50, Millipore, Bedford, MA, USA) at 1000×g and room temperature for 10 min to isolate lysozyme (14.3 kDa) from the dissolved CMC matrix material (90 kDa average molecular mass). After determining lysozyme concentration by Lowry protein assay [16], PBS was added to dilute to lysozyme to 20 μg/ml. CD spectra were taken for (1) untreated lysozyme, (2) lysozyme encapsulated in microneedles that were dissolved 1 h after fabrication, (3) lysozyme encapsulated in microneedles that were dissolved after 60 days of storage at ambient conditions (23±2° C and 38±5% relative humidity), and (4) lysozyme thermally treated at 80° C for 30 min to cause irreversible denaturation [17].
2.4.2 Lysozyme activity
Enzymatic activity of lysozyme encapsulated within CMC microneedle devices was tested with EnzCheck lysozyme assay kit (Molecular Probes). Microneedle devices weighing 5 mg that contained lysozyme encapsulated at a concentration of 5 wt% were completely dissolved in PBS at room temperature for 10 min. PBS was added to dilute each sample to 0.05 μg/ml lysozyme and 0.95 μg/ml CMC. Lysozyme activity was assayed using 1 ml solution samples for: (1) untreated lysozyme, (2) untreated lysozyme (0.05 μg) and CMC hydrogel (0.95 μg) mixed, and dissolved together, (3) lysozyme encapsulated in microneedles that were dissolved 1 h after fabrication, and (4) lysozyme encapsulated in microneedles that were dissolved after 60 days of storage at ambient conditions.
3. Results
3.1 Fabrication of dissolving microneedles
We identified four materials-related criteria to make microneedles for self-administration of biotherapeutics from a minimally invasive patch: (1) gentle fabrication to avoid damaging sensitive biomolecules, (2) sufficient mechanical strength for insertion into skin, (3) controlled release for bolus and sustained drug delivery, and (4) rapid dissolution of microneedles made of safe materials. Guided by these criteria, we selected two polysaccharides – i.e., carboxymethylcellulose and amylopectin – because they are biocompatible materials with a history of use in FDA-approval parenteral formulations [18, 19], are expected to be mechanically strong due to their relatively high Young’s modulus [20, 21], and are highly water soluble for rapid dissolution in the skin [22].
Building off our previous microneedle fabrication methods [11, 14], we fabricated dissolving microneedles using a micromolding approach that faithfully reproduces microneedle structures in an economical manner suitable for scale up to mass production. Female master-molds were first prepared out of SU-8 photoresist by lithography and used to created PDMS male master-structures, shown in Fig. 1a. These master-structures were then molded to make PDMS female molds. PDMS was chosen as the material for master-structures and molds because of its ability to conformally coat microstructures and fill micromolds; its poor adhesion and flexibility to facilitate separation of microstructures from micromolds; and its low cost.
Figure 1.

Dissolving microneedles for transdermal drug delivery. (a) Microneedle master-structure (600 μm in height and 300 μm wide at base) used to mold dissolving microneedles made of (b) CMC, (c) amylopectin and (d) BSA. The master-structure was imaged by scanning electron microscopy and the molded microneedles were imaged by brightfield microscopy.
These micromolds were used to prepare dissolving microneedles by solvent casting with aqueous solutions of CMC and amylopectin. However, simply filling molds with CMC solution and then drying produced weak needles, probably due to structural voids left within the microneedle matrix after water evaporation (data not shown). To avoid this problem, we developed a modified casting method in which the CMC solution was first concentrated by evaporation under vacuum (i.e., -50 kPa) or heating (i.e., 60–70° C) to produce a highly viscous solution that minimized water content, but was still fluid enough to fill the mold. We found that an aqueous CMC solution with a viscosity of 4.5x105 cP (measured with a Couette viscometer at 1/s shear rate at 23° C) met these criteria. In case of amylopectin, the initial solvent removal was carried out at elevated temperature (i.e., 60–70° C) rather than just under vacuum, because amylopectin has poor water solubility at room temperature.
Concentrated CMC and amylopectin solutions were then cast into micromolds and dried completely during centrifugation at 37° C. The elevated temperature increased the speed of evaporation and the centrifugation continuously compressed the mold contents, which minimized void formation during drying. This modified casting method was effective to reproduce polysaccharide microneedles having the same dimensions as their master-structures, as shown in Figs. 1b and 1c for CMC and amylopectin microneedles, respectively. A similar approach was used to make microneedles out of BSA in Fig. 1d, which is a model for making needles out of pure drug, rather than encapsulating drug within a polysaccharide matrix, as discussed below.
As an alternative approach, we tried using high viscosity CMC (1.5 – 3 x 103 cP for a 1% aqueous solution at 25° C) as the matrix material, but found that it required much more water to be solubilized compared to the ultra-low viscosity CMC used above. As a result, high viscosity CMC took longer to dry and produced deformed microneedles that shrank substantially during drying and were mechanically weak (data not shown).
As discussed below, different drug delivery scenarios were addressed by selectively encapsulating model compounds within microneedles, within the microneedle device backing layer, or within both. To encapsulate within the CMC or amylopectin matrix, we simply mixed the model drug into the polysaccharide solution before casting into the molds. To selectively encapsulate within the microneedles and not in the backing layer, a smaller volume of drug-polysaccharide solution was cast into the holes of the micromold to form microneedles. After wiping off excess solution from the micromold surface, polysaccharide solution without model drug was cast onto the micromold and dried.
To selectively encapsulate within the backing layer and not in the microneedles, a similar two-step process was carried out, in which the model drug was only added to the polysaccharide solution applied to the micromold during the second step. Drying of the complete, integrated system or just the backing layer during the second step required 1–2 h, whereas drying of just the microneedles during the first step took approximately 30 min. These process times varied depending on materials and processing conditions.
3.2 Mechanical properties of dissolving microneedles
The design of dissolving microneedles is governed by a number of interdependent materials and fabrication constraints, one of which is the need for microneedles to have sufficient strength to insert into skin without mechanical failure. We therefore measured and simulated microneedle mechanical properties as a function of microneedle material composition and geometry, and then imaged insertion of optimized microneedles into skin.
3.2.1 Measurement of microneedle failure force
We first measured the mechanical behavior of CMC microneedles with a conical shape. As shown in Fig. 2a by the black circle data points, the force-displacement curve (which is analogous to a stress-strain curve) exhibited an initial increase in force with displacement, followed by a discontinuity at a force of approximately 0.1 N/needle. This is interpreted as the point of microneedle failure, which is consistent with previous studies [14]. Moreover, microscopic examination of the microneedles showed little deformation before this failure point and showed microneedles bent up to 90º starting approximately half way up the shaft after this failure point, which is consistent with failure by buckling (data not shown).
Figure 2.
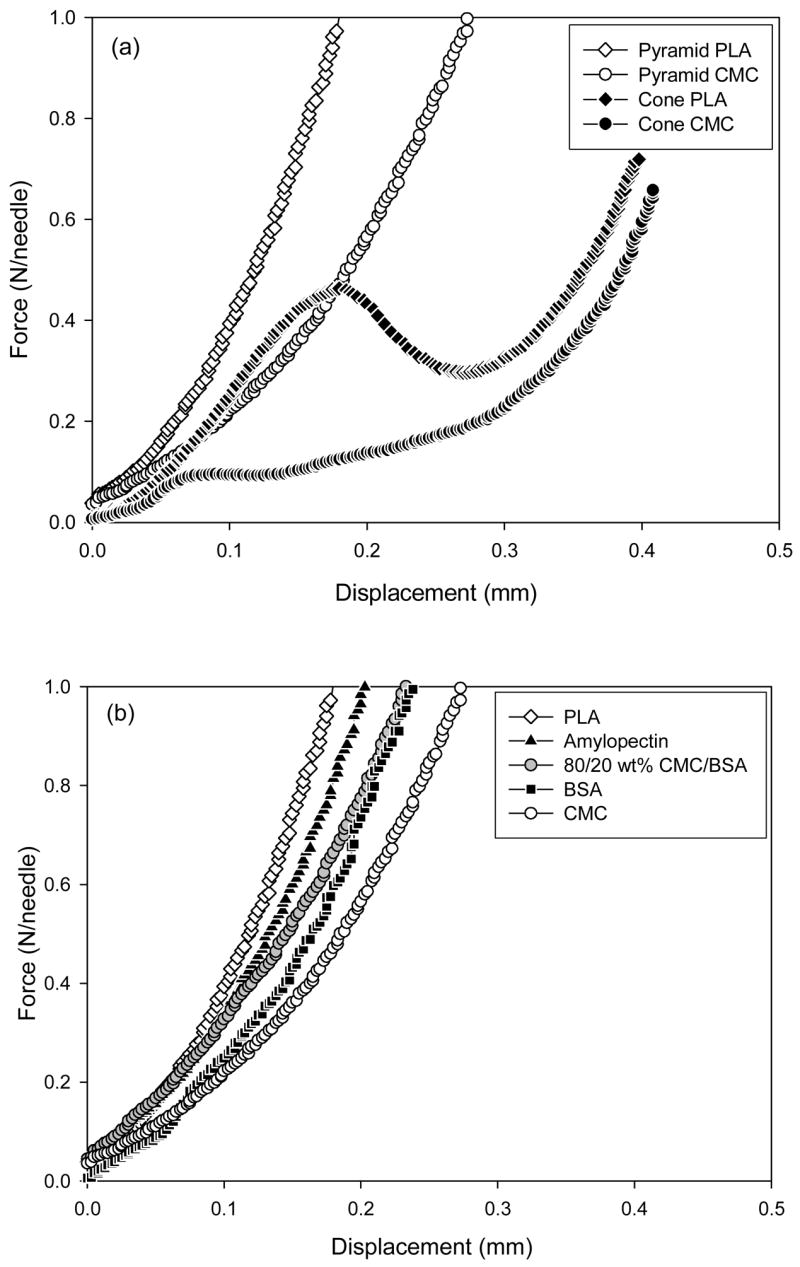
Mechanical behavior of dissolving microneedles. Force measured as a function of microneedle displacement while pressing against a rigid surface for (a) CMC and PLA microneedles having conical and pyramidal geometries and (b) pyramidal microneedles made of PLA, amylopectin, BSA, CMC, and a mixture of 80/20 wt% CMC/BSA. Conical microneedles measured 800 μm in height and 200 μm in base diameter. Pyramidal microneedles measured 600 μm in height and 300 μm in base width. The graphs contain data representative of 5 replicate measurements each.
For comparison, we generated a similar curve for PLA microneedles having the same geometry, which demonstrated a five-fold greater failure force of 0.5 N/needle (black diamonds in Fig. 2a). Previous work showed that conical PLA microneedles similar to those used in this study have a failure force more than 3 times greater that the force needed for insertion into the skin, which indicates that these conical PLA microneedles are suitable for skin insertion without breaking [14]. Given that the conical CMC microneedles are 5 times weaker than their PLA counterparts, this analysis suggests that the conical CMC microneedles are too weak to insert into the skin.
Because microneedle geometry affects mechanical strength, we next examined pyramidal microneedles made of CMC and PLA. In contrast to conical microneedles, pyramidal microneedles did not show a distinct transition point indicating failure over the range of conditions tested. Microscopic examination of pyramidal microneedles showed a progressive deformation of the microneedles, starting near the tip and moving downward with increasing force, but never showed a catastrophic buckling event at a single point of failure (data not shown). This progressive deformation is consistent with the continuous force-displacement curve shown in Fig. 2. The reason for the different behaviors of conical and pyramidal microneedles may have to do with the larger aspect ratio and the smaller cross-sectional area of conical microneedles, as discussed below.
To further study the effect of microneedle composition on mechanical strength, the mechanical behavior of pyramidal microneedles having the same geometry was measured for microneedles made of CMC, PLA, amylopectin, a 20/80 wt% mixture of BSA and CMC, and 100% BSA. As shown in Fig. 2b, these five pyramidal microneedles all showed similar mechanical behavior, although the choice of material influenced microneedle strength (i.e., amount of deformation). The materials can be ranked from strongest to weakest as: PLA, amylopectin, CMC/BSA, BSA, and CMC. Amylopectin microneedles were stronger than CMC microneedles, which can be explained by the higher Young’s modulus of amylopectin (4.5 GPa [21]) compared to CMC (1 GPa, See section 2.2.3). CMC and CMC/BSA microneedles were designed to simulate a CMC microneedle encapsulating a model protein and a microneedle made completely of a model protein, respectively. These two microneedles designs had similar mechanical strength, both of which were greater than for pure CMC microneedles. In this case, encapsulation of protein increased microneedle mechanical strength, but this is unlikely to be true in all cases.
3.2.2 Simulation of microneedle failure force
To better interpret these experimental results, we simulated mechanical behavior of microneedles to predict critical buckling load. As shown in Table 1, CMC microneedles with a conical geometry (800 μm length and 200 μm base diameter) have a predicted failure force of 0.10 N and PLA microneedles with the same geometry have a predicted failure force of 0.51 N, which is in excellent agreement with experimental measurements (Fig. 2a). The pyramidal microneedles (600 μm length, 300 μm base width) made of CMC and PLA have predicted failure forces of 1.8 N and 8.9 N, respectively (Table 1). The 18-fold increase in critical buckling load for these pyramidal microneedles compared to conical microneedles is also consistent with experimental measurements. However, this model accounts only for buckling and does not account for the progressive deformation observed experimentally at smaller forces.
Table 1.
Simulated critical buckling load, Pcr, of CMC and PLA microneedles as a function of microneedle shape, length, and base width/diameter.
| Conical (800 μm length)
|
Pyramidal (600 μm length)
|
Conical (600 μm length)
|
||||||
|---|---|---|---|---|---|---|---|---|
| Diameter* (μm) | Pcr (N)
|
Width* (μm) | Pcr (N)
|
Diameter* (μm) | Pcr (N)
|
|||
| CMC | PLA | CMC | PLA | CMC | PLA | |||
| 50 | 0.0004 | 0.0020 | 50 | 0.0020 | 0.010 | 50 | 0.0007 | 0.0035 |
| 75 | 0.0019 | 0.0096 | 75 | 0.0087 | 0.044 | 75 | 0.0034 | 0.017 |
| 100 | 0.0061 | 0.031 | 100 | 0.026 | 0.13 | 100 | 0.011 | 0.055 |
| 200 | 0.10 | 0.51 | 200 | 0.36 | 1.8 | 200 | 0.18 | 0.91 |
| 300 | 0.5 | 2.6 | 300 | 1.8 | 8.9 | 300 | 0.94 | 4.7 |
| 400 | 1.7 | 8.4 | 400 | 5.5 | 28 | 400 | 3.0 | 15 |
| 600 | 8.6 | 43 | 500 | 13 | 67 | 500 | 7.3 | 37 |
| 800 | 27 | 137 | 600 | 27 | 137 | 600 | 15 | 76 |
Diameters and widths are reported at the base of the microneedles, which taper to a tip of approximately 25 μm diameter.
The above comparison involved longer and thinner conical microneedles versus shorter and wider pyramidal microneedles. To make a comparison that isolates the effect just of microneedle shape, failure force for microneedles of 600 μm length and 300 μm base width/diameter was predicted to be 0.93 N and 4.7 N for conical microneedle made of CMC and PLA, respectively, which is almost two-fold smaller than the corresponding predictions for pyramidal microneedles (Table 1). We therefore conclude that pyramidal microneedles are stronger, probably due to their larger cross-sectional area at the same base width/diameter.
Examination of Table 1 for each microneedle design as a function of base width/diameter also shows that increasing base dimensions (i.e., decreasing aspect ratio) increases needle strength. Thus, using pyramidal microneedles with a small aspect ratio can provide added mechanical strength for mechanically weak biomaterials like CMC. However, microneedles with an aspect ratio that is too small will also have poor insertion due to fabrication difficulties to make a sharp tip and insertion difficulties to force the rapidly widening needle shaft into the small hole made in the skin by the needle tip.
3.2.3 Microneedle insertion into skin
Guided by the expectation that pyramidal CMC microneedles with an aspect ratio of two should be strong, we inserted arrays of these needles into pig cadaver skin and found that 100-needle arrays of microneedles were inserted reliably into the skin using the gentle force of a thumb. The backside of a representative microneedle array made of transparent CMC is shown in Fig. 3a with its microneedles embedded in the skin. After removing the microneedles from the skin after just 3 s, the tips had already begun to dissolve (Fig. 3b), indicating onset of rapid dissolution in the skin. We next treated the skin with a dye that selectively marks sites of skin penetration and found that typically all microneedles in the array inserted into the skin (Fig. 3c).
Figure 3.
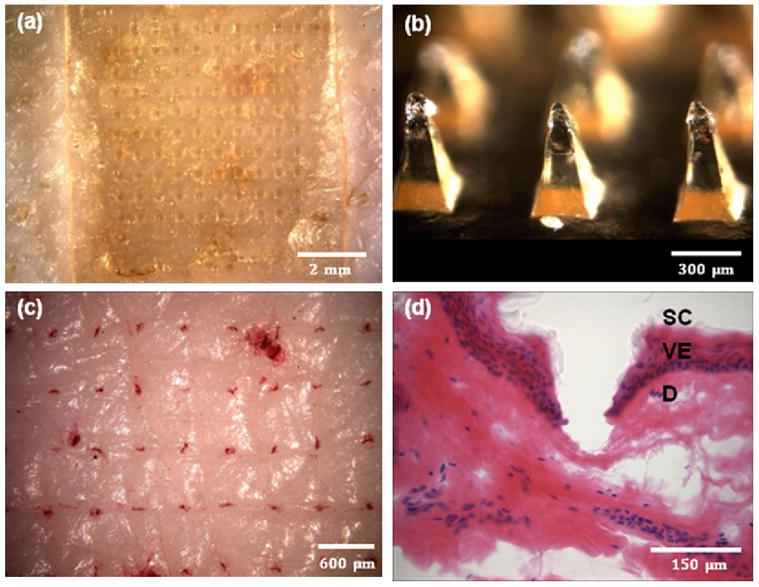
Imaging microneedle insertion into pig cadaver skin. (a) View of the back side of a CMC microneedle patch applied onto the surface of the skin. (b) CMC pyramidal microneedles after insertion into the skin for 3 s. (c) Skin stained with tissue marking dye to identify the sites of needle penetration after insertion of CMC pyramidal microneedles. (d) Cross-sectional image of H&E-stained skin at a site of microneedle penetration (SC: stratum corneum, VE: viable epidermis, and D: dermis). All images viewed by brightfield microscopy.
Histological examination of skin pierced with microneedles showed penetration depths of approximately 150 – 200 μm, which corresponded to insertion across the stratum corneum and viable epidermis and into the superficial dermis (Fig. 3d). Microneedles used in this experiment measured 600 μm in length, which means that one-fourth to one-third of the microneedle shaft penetrated into skin. This can be explained by deformation of skin’s surface that is known to occur during microneedle insertion due to skin’s viscoelasticity [23]. The relatively wide base (i.e., 300 μm) and small aspect ratio (i.e., 2) of the pyramidal microneedles contributed to this incomplete insertion. Further optimization of microneedle geometry, such as aspect ratio, tip sharpness, and spacing between microneedles, and microneedle material may increase depth of insertion. However, as discussed below, partial microneedle insertion may be adequate for drug delivery strategies presented in this study.
3.3 Release of model drugs from dissolving microneedle patches
By loading model drug into dissolving microneedles in different ways, we were able to design systems that achieved either bolus or extended release from a microneedle patch. To achieve bolus release, model drug was selectively incorporated into the microneedles themselves and not into the backing layer. In this way, we hypothesize that microneedles can be inserted into skin and release encapsulated drug during their rapid dissolution. The rate of release in this scenario is controlled largely by microneedle dissolution rate. A limitation is that the total dose administered is small, because microneedles each contain about 25–60 μg of matrix material and typically just a fraction of the microneedle matrix can made of drug in order to maintain microneedle mechanical strength. Thus, bolus delivery from a microneedle patch containing a few hundred microneedles is likely to be limited to less than 1 mg of drug.
To administer larger drug doses as an extended release over at least hours, we incorporated model drug into both the microneedles and backing layer or, alternatively, just the backing layer. This permits much larger doses to be administered, because the backing layer can be large (e.g., 10 – 100 mg) and can be loaded with larger fractions of drug, because backing layer mechanical properties have fewer constraints. In this scenario, we hypothesize that drug can diffuse over time from the drug reservoir in the backing layer and into skin through transdermal pathways created by dissolving microneedles. In this way, the backing layer acts as a drug source similar to a conventional matrix-design transdermal patch.
3.3.1 Bolus Release
To test our hypothesis regarding bolus release, we selectively encapsulated a model drug, sulforhodamine B, in pyramidal CMC microneedles. As shown in Fig. 4a, red-colored sulforhodamine was encapsulated within each microneedle, but the bottom portion of each microneedle and the backing layer did not contain sulforhodamine. After inserting sulforhodamine-loaded microneedles into pig cadaver skin and then removing them after 5 min, inspection of the skin surface showed an array of red spots corresponding to the sites of each microneedle insertion (Fig. 4b). These spots could not be wiped off by cleaning the skin surface and are therefore interpreted as sulforhodamine deposited within skin after microneedle dissolution.
Figure 4.

Dissolving microneedles for bolus delivery into skin. (a) CMC pyramidal microneedles encapsulating sulforhodamine B within the microneedle shafts, but not in the backing layer. (b) Skin surface showing sulforhodamine delivered into the skin by insertion of the microneedles shown in part (a) for 5 min imaged by brightfield microscopy. (c) Cross-sectional histological image of skin at the penetration site of two adjacent microneedles shown in part (a) inserted for 5 min and imaged by brightfield (c1) and fluorescence (c2) microscopy. (d) Cross-sectional histological image of skin pierced by an array of sulforhodamine-containing microneedles for 1 h and imaged by an overlay of brightfield and fluorescence microscopy. Pig cadaver skin was used.
This interpretation is confirmed by histological sections, which show deposition of sulforhodamine within skin at sites of microneedle penetration (Fig. 4c). Microneedle insertion depth was approximately 150–200 μm, which is in agreement with Fig. 3d. The width of each hole was approximately 100 μm (Fig. 4b), which is similar to microneedle width at a distance of 150 to 200 μm up the shaft from the tip. To supplement this information, Fig. 4d shows a lower-magnification histological section of skin 1 h after insertion of bolus-delivery microneedles. In this case sulforhodamine is not located just at sites of microneedle insertion, but has diffused more extensively within the skin.
To generate a better understanding of the kinetics of bolus release from dissolving microneedles, we imaged the microneedle dissolution process after insertion into skin for different times. The tips of microneedles dissolved within 10 s (Fig. 5b), half of the microneedle height disappeared within 1 min (Fig. 5c), and two-thirds disappeared within 15 min (Fig. 5d). After 1 h, microneedles were fully dissolved (Fig. 5e). This kinetics could be altered by changing microneedle geometry and matrix material. For example, we observed that similar microneedles made of amylopectin dissolved more slowly and ones made of polyvinylpyrolide dissolved more quickly based on their different levels of water solubility (data not shown). It is also worth noting that even though microneedles did not penetrate to their full length into the skin, they were nonetheless able to fully dissolve, probably due to transport of interstitial fluid from the skin up the needle shaft, as discussed below.
Figure 5.

Dissolution kinetics of microneedles after insertion in skin. (a) CMC pyramidal microneedles imaged by brightfield microscopy before insertion and (b) 10 sec, (c) 1 min, (d) 15 min, and (e) 1 h after insertion into pig cadaver skin.
3.3.2 Sustained Release
To test our hypothesis regarding sustained release, we encapsulated sulforhodamine in the backing layer and shafts of pyramidal CMC microneedles (Fig. 6a). The microneedle device contained 1 mg of sulforhodamine at a concentration of 10 wt% (on a dry basis). These microneedles designed for sustained release could be inserted into skin (Fig. 6b) and histological examination showed release of sulforhodamine throughout the skin (Fig. 6c).
Figure 6.
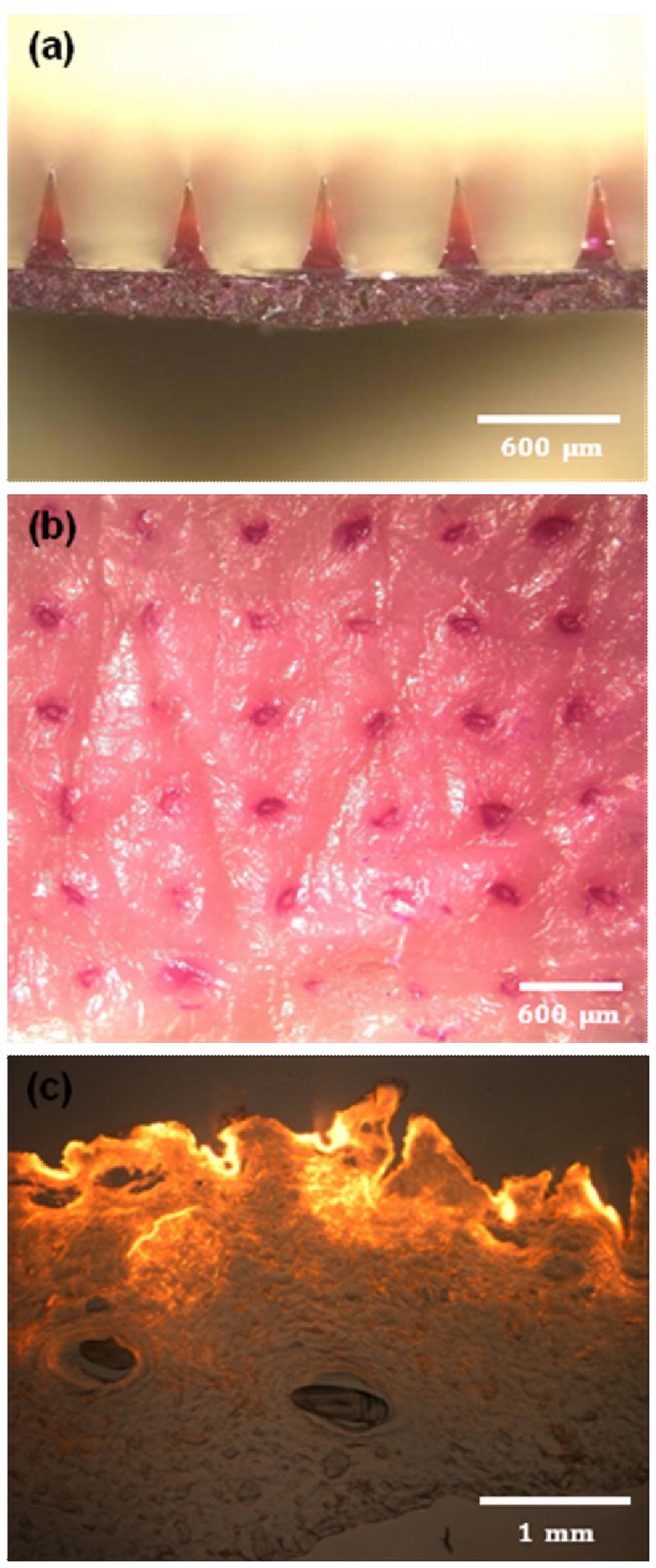
Dissolving microneedles for sustained release. (a) CMC pyramidal microneedles encapsulating sulforhadamine only in the backing layer. (b) Skin surface showing sulforhodamine delivered into the skin by insertion of the microneedles shown in part (a) for 12 h imaged by brightfield microscopy. (c) Cross-sectional histological image of skin pierced by the microneedles shown in part (a) for 12 h and imaged by an overlay of brightfield and fluorescence microscopy. Pig cadaver skin was used.
To quantify sustained release properties in greater detail, we inserted microneedle patches into human cadaver skin and measured transdermal flux. Sulforhodamine release from CMC microneedle patches exhibited an initial lag time of a few hours, followed by steady release for approximately one day (Fig. 7a). Similar behavior was seen for microneedle patches made of amylopectin, but with slower kinetics. In this case, lag time was longer and release took place over a few days (Fig. 7a).
Figure 7.
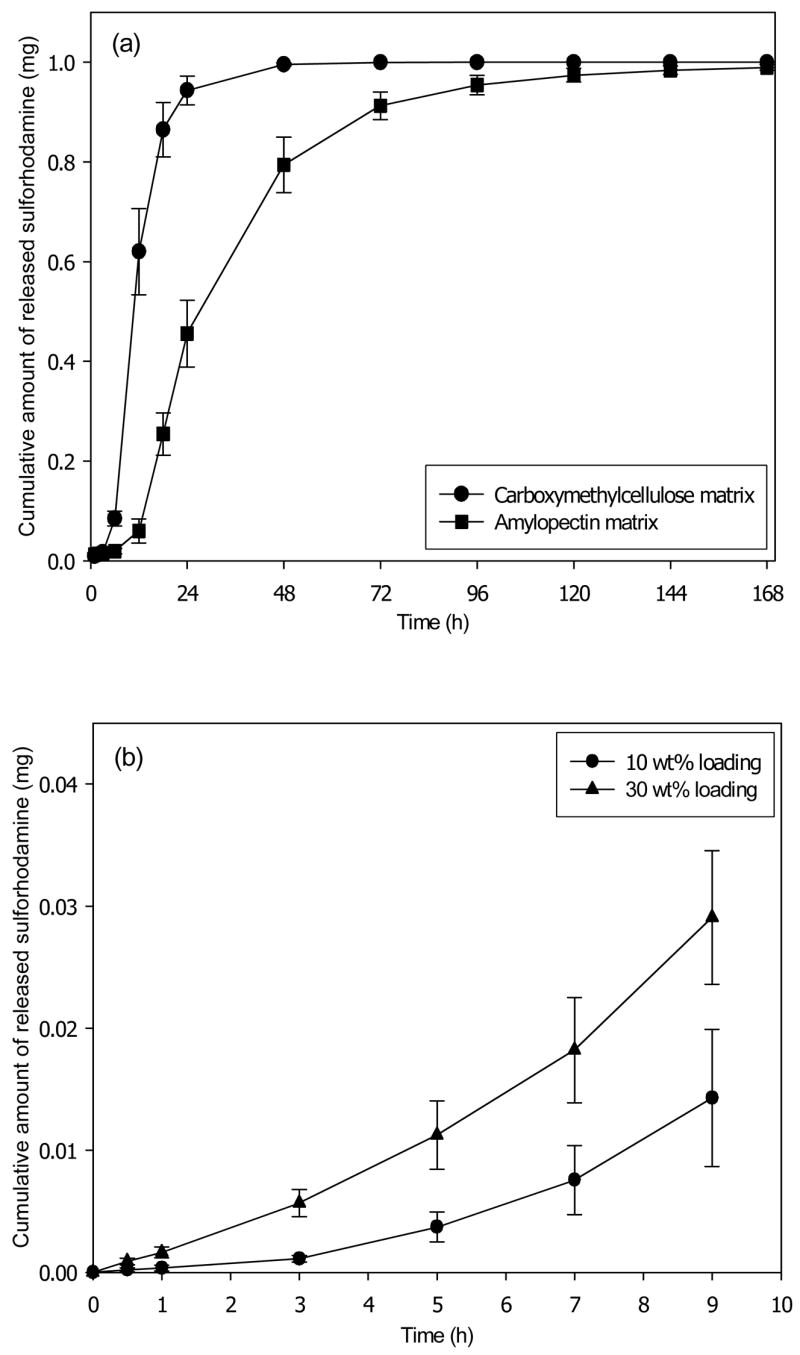
Transdermal release profile from dissolving microneedles patches. (a) Cumulative release of sulforhodamine encapsulated at 10 wt% in the pyramidal microneedles and the backing layer of patches made of CMC and amylopectin. (b) Cumulative release during the initial release period of sulforhodamine encapsulated at 0 wt% in the pyramidal microneedles and at 10 wt% or 30 wt% in the backing layer of CMC patches. Human cadaver epidermis was used. Average values are shown with standard error bars based on 3 replicate measurements.
These data validate the hypothesis that drug encapsulated within the backing layer of a microneedle patch can diffuse out of the patch and into skin. Moreover, they show that changing microneedle patch matrix material can alter release kinetics. It is important to be able to vary release kinetics based on patch design, because different drugs administered for different indications require different release patterns.
Release rate should also depend on sulforhodamine concentration in the patch. Consistent with this expectation, the drug release rate from a patch containing 30 wt% sulforhodamine was approximately three times greater than a patch containing 10 wt% sulforhodamine (Fig. 7b).
3.4 Protein stability after encapsulation in dissolving microneedles
Dissolving microneedles were designed to encapsulate sensitive biomolecules using a gentle fabrication process. To assess success of this design, we used lysozyme as a model protein and measured changes in secondary structure and enzymatic activity after encapsulation and storage in CMC microneedle patches.
Circular dichroism (CD) analysis of untreated lysozyme compared to lysozyme encapsulated within a microneedle patch and then released by dissolution in water showed no detectable change in protein secondary structure (Fig. 8a). Even after storage of microneedle patches containing lysozyme for 2 months at room temperature, protein structure was unchanged (Fig. 8a). As a positive control, the CD spectrum showed extensive degradation of secondary structure after thermal denaturation (Fig. 8a).
Figure 8.
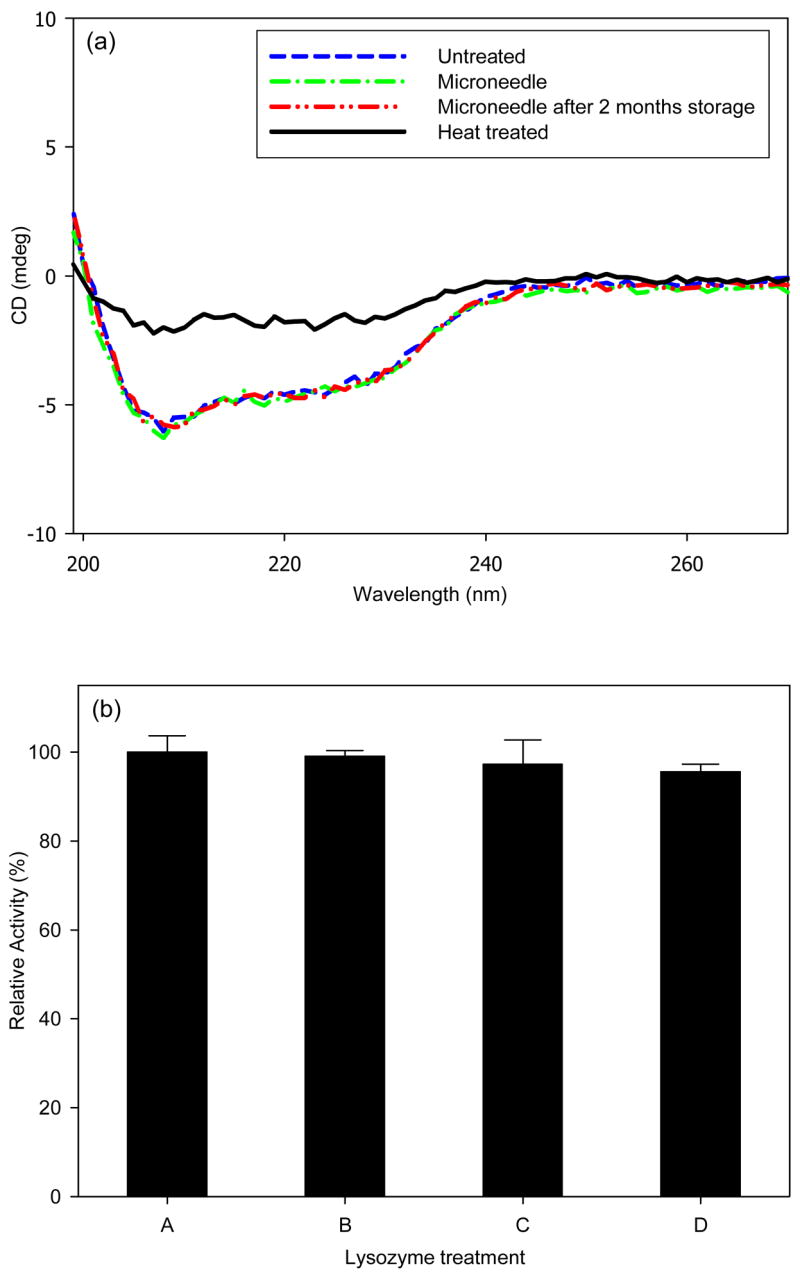
Protein stability after encapsulation and release from dissolving microneedles. (a) Circular dichroism spectrum of untreated lysozyme (negative control); lysozyme encapsulated in CMC microneedles and released by dissolution in PBS; lysozyme encapsulated in CMC microneedles and released by dissolution in PBS after 2 months storage at room temperature; and lysozyme denatured at 80ºC for 30 min (positive control). (b) Enzymatic activity of untreated lysozyme (A, negative control); lysozyme mixed with dissolved placebo CMC microneedles (B, negative control); lysozyme encapsulated in CMC microneedles and released by dissolution in PBS (C); lysozyme encapsulated in CMC microneedles and released by dissolution in PBS after 2 months storage at room temperature (D).
To further test lysozyme integrity, enzymatic activity of lysozyme was measured. To make sure that the presence of dissolved CMC after microneedle dissolution did not create an artifact, a CMC microneedle containing no lysozyme was dissolved in PBS and then mixed with untreated lysozyme. This resulted in no change in lysozyme activity (Student’s t-test, p=0.51). To test the effect of encapsulation, microneedles containing encapsulated lysozyme were dissolved in PBS and found to have no loss of enzymatic activity compared to untreated enzyme (Student’s t-test, p=0.28). After two months of storage, lysozyme released from microneedles retained 96% enzymatic activity, indicating a small loss of activity (Student’s t-test, p=0.03).
4. Discussion
4.1 Significance to drug delivery
Dissolving microneedles designed in this study may enable (1) bolus and sustained delivery of drugs into the skin, (2) self-administration of drugs that would otherwise require a hypodermic needle and (3) elimination of dangers associated with improper needle disposal and intentional re-use, especially in the developing world.
Previous studies that have developed microneedles that dissolve or degrade in the skin have either melted polymer into a mold at high temperature [8, 9], which can damage sensitive biomolecules, or have hand assembled individual needles of millimeter dimensions [24], which is a poorly controlled and non-manufacturable process. This study has developed a well-controlled, reproducible process for fabrication under gentle conditions that designed to maintain protein integrity.
Another feature of our dissolving microneedles is that the backing membrane to which microneedles are attached has been used, for the first time, as a drug reservoir for sustained drug delivery over many days. Previous approaches have encapsulated drug only within microneedle shafts [8, 9, 24] or, alternatively, coated drug onto the surface of microneedle shafts [7, 10]. These approaches have been limited to small (e.g., microgram) drug doses due to the inherently small volume and surface area of microneedles. Our use of the backing membrane as a drug reservoir may increase total doses to milligrams.
Previous studies have also pretreated skin by piercing it with microneedles and then applying a topical formulation or patch to deliver drug through the permeabilized skin [6, 25]. This two-step process may be cumbersome for patients and susceptible to mistakes. In this study, we mimicked this scenario, but accomplished it as a one-step process by using dissolving microneedles, which did not need to be removed after piercing the skin and were integrated onto the surface of a backing layer that served functionally as a transdermal patch. By controlling microneedle patch design, release kinetics was controlled over times ranging from minutes to days. Further design and formulation changes should be able to achieve a variety of different bolus and sustained release profiles.
Release from the backing layer reservoir probably occurs by a set of interacting phenomena. First, microneedles insert the skin and dissolve, which creates channels for drug transport into skin and for interstitial fluid transport out of skin. Although drug may diffuse through a dry backing layer, drug diffusion should be enhanced by hydrating the backing layer with interstitial fluid from skin. Consistent with this expectation, the backing layer of microneedle patches was seen to swell and soften over time during sustained release delivery experiments presented in Fig. 7. An example of this swelling is shown in Fig. 9, where a dissolving microneedle patch shows extensive swelling after 15 h on the skin (Fig. 9a). As a negative control, a patch backing layer fabricated without microneedles was also placed on skin, but did not swell after placement for the same time (Fig 9b). This suggests that the patch backing layer swelled by imbibing interstitial fluid from skin through channels created by microneedles. This observation is not only relevant to understanding drug delivery mechanisms, but also suggests uses to extract interstitial fluid for diagnostic applications, such as measuring glucose concentration in diabetics or monitoring industrial toxins in at-risk populations.
Figure 9.

Swelling of dissolving microneedle patch backing layer after insertion into skin using a Franz cell. (a) A patch of CMC pyramidal microneedles containing sulforhodamine inserted into skin for 15 h shows extensive swelling of the backing layer. (b) A backing layer of CMC that contains no microneedles (negative control) placed on the surface of skin for 15 h shows little swelling. Human epidermis was used. Imaging was by brightfield microscopy.
We found that a model enzyme, lysozyme, encapsulated in CMC microneedles was not substantially degraded after two months storage at room temperature (Fig. 8). This stability may be explained by the limited molecular mobility in the solid state that is known to enhance protein stability [26]. Additional studies are needed to determine if other proteins are similarly stable.
All of these findings are based on observations in vitro. Additional studies are needed to assess performance in vivo using animals and, ultimately, humans [27]. In addition, even though microneedles were fabricated using materials found in other FDA-approved formulations, dermatotoxicity and irritation have not been fully evaluated and needs to be addressed [28]. Finally, practical applications require robust and cost-effective manufacturing, which needs further study and development.
4.2 Microneedle design and fabrication
Selecting the right microneedle geometry and material were important to making functional dissolving microneedles. Most water-soluble materials are mechanically weaker than non-dissolving materials like metal or silicon. Given the many constraints, including water solubility, mechanical strength, processability, and established safety, we selected naturally occurring polysaccharides, i.e., CMC and amylopectin. However, polysaccharide microneedles having the same dimensions previously designed for metal and silicon microneedles were not strong enough. Therefore, mechanical strength was increased by reducing the microneedle aspect ratio, which permitted insertion into skin, although complete insertion of the microneedle shaft was not achieved.
The wider geometry of dissolving microneedles also increased microneedle volume, which can increase the amount of drug encapsulated per microneedle. The conical and pyramidal geometries used in this study had volumes of 8.4 and 18 nl, respectively. Based on a CMC specific gravity of 1.59 [29], a 0.1%, 1% or 10% drug loading (on a dry basis) corresponds to 0.028 μg, 0.28 μg or 2.8 μg of drug per pyramidal microneedle or 2.8 μg, 28 μg or 280 μg per 100-needle array. The maximum drug loading will depend largely on its impact on microneedle mechanical properties, which may be weakened, or possibly strengthened, by encapsulated drug and on drug stability. A microneedle made completely out of drug, such as the BSA microneedles shown in Fig. 1d, could contain 21.6 μg of drug per needle based on a BSA specific gravity of 1.2 [30].
5. Conclusion
This study presents a dissolving microneedle design involving fabrication under mild conditions that may be suitable for protein delivery and amenable to mass production. It was developed by selecting FDA-approved polysaccharides and modifying a casting method with centrifugation. By using a low aspect ratio and pyramidal geometry, dissolving microneedles were formulated to have sufficient mechanical strength to insert into skin. By selectively loading microneedle shafts, microneedle patches provided bolus release of a model drug upon the dissolution of the microneedle matrix inside skin. By loading the backing layer, microneedle patches provided sustained release probably due to drug diffusion and swelling of the backing layer over time. Model drug release rate was controlled by altering microneedle formulation. A model protein (i.e., lysozyme) maintained structural and functional integrity after encapsulation and release from dissolving microneedles, even after two months storage at room temperature. Overall, dissolving microneedles may be useful as a method for patients to self-administer drugs without the pain or hazards of hypodermic needles.
Acknowledgments
We thank Dr. Seong-O Choi for providing microneedle master structures and Prof. Jianmin Qu for the helpful discussions. This work was supported in part by the National Institutes of Health and carried out at the Institute for Bioengineering and Bioscience and the Center for Drug Design, Development, and Delivery at the Georgia Institute of Technology. Mark Prausnitz is the Emerson-Lewis Faculty Fellow at Georgia Tech.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Daugherty AL, Mrsny RJ. Emerging technologies that overcome biological barriers for therapeutic protein delivery. Expert Opinion on Biological Therapy. 2003;3(7):1071–1081. doi: 10.1517/14712598.3.7.1071. [DOI] [PubMed] [Google Scholar]
- 2.Nir Y, Paz A, Sabo E, Potasman I. Fear of injections in young adults: Prevalence and associations. American Journal of Tropical Medicine and Hygiene. 2003;68(3):341–344. [PubMed] [Google Scholar]
- 3.Simonsen L, Kane A, Lloyd J, Zaffran M, Kane M. Unsafe injections in the developing world and transmission of bloodborne pathogens: a review. Bulletin of the World Health Organization. 1999;77(10):789–800. [PMC free article] [PubMed] [Google Scholar]
- 4.Reed ML, Lye WK. Microsystems for drug and gene delivery. Proceedings of the IEEE. 2004;92(1):56–75. [Google Scholar]
- 5.Prausnitz MR. Microneedles for transdermal drug delivery. Advanced Drug Delivery Reviews. 2004;56(5):581–587. doi: 10.1016/j.addr.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 6.McAllister DV, Wang PM, Davis SP, Park JH, Canatella PJ, Allen MG, et al. Microfabricated needles for transdermal delivery of macromolecules and nanoparticles: Fabrication methods and transport studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24):13755–13760. doi: 10.1073/pnas.2331316100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matriano JA, Cormier M, Johnson J, Young WA, Buttery M, Nyam K, et al. Macroflux (R) microprojection array patch technology: A new and efficient approach for intracutaneous immunization. Pharmaceutical Research. 2002;19(1):63–70. doi: 10.1023/a:1013607400040. [DOI] [PubMed] [Google Scholar]
- 8.Miyano T, Tobinaga Y, Kanno T, Matsuzaki Y, Takeda H, Wakui M, et al. Sugar micro needles as transdermic drug delivery system. Biomedical Microdevices. 2005;7(3):185–188. doi: 10.1007/s10544-005-3024-7. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Allen MG, Prausnitz MR. Polymer microneedles for controlled-release drug delivery. Pharmaceutical Research. 2006;23(5):1008–1019. doi: 10.1007/s11095-006-0028-9. [DOI] [PubMed] [Google Scholar]
- 10.Gill HS, Prausnitz MR. Coated microneedles for transdermal delivery. Journal of Controlled Release. 2007;117(2):227–237. doi: 10.1016/j.jconrel.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JH, Yoon YK, Choi SO, Prausnitz MR, Allen MG. Tapered conical polymer microneedles fabricated using an integrated lens technique for transdermal drug delivery. IEEE Transactions on Biomedical Engineering. 2007;54(5):903–913. doi: 10.1109/TBME.2006.889173. [DOI] [PubMed] [Google Scholar]
- 12.Davis SP, Landis BJ, Adams ZH, Allen MG, Prausnitz MR. Insertion of microneedles into skin: measurement and prediction of insertion force and needle fracture force. Journal of Biomechanics. 2004;37(8):1155–1163. doi: 10.1016/j.jbiomech.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Smith WG. Analytic solutions for tapered column buckling. Computers & Structures. 1988;28(5):677–681. [Google Scholar]
- 14.Park JH, Allen MG, Prausnitz MR. Biodegradable polymer microneedles: Fabrication, mechanics and transdermal drug delivery. Journal of Controlled Release. 2005;104(1):51–66. doi: 10.1016/j.jconrel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Kligman AM, Christophel E. Preparation of isolated sheets of human stratum corneum. Archives of Dermatology. 1963;88(6):702. doi: 10.1001/archderm.1963.01590240026005. [DOI] [PubMed] [Google Scholar]
- 16.Bollag DM, Rozycki MD, Edelstein SJ. Protein Methods. 2. New York: Wiley-Liss; 1996. [Google Scholar]
- 17.Masschalck B, Van Houdt R, Van Haver EGR, Michiels CW. Inactivation of gram-negative bacteria by lysozyme, denatured lysozyme, and lysozyme-derived peptides under high hydrostatic pressure. Applied and Environmental Microbiology. 2001;67(1):339–344. doi: 10.1128/AEM.67.1.339-344.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Appel RA, Dmochowski RR, Herschorn S. Urethal injection for female stress incontinence. BJU International. 2006;98(s1):27–30. doi: 10.1111/j.1464-410X.2006.06305.x. [DOI] [PubMed] [Google Scholar]
- 19.Crookes BA, Cohn SM, Bonet H, Burton EA, Nelson J, Majetschak M, et al. Building a better fluid for emergency resuscitation of traumatic brain injury. Journal of Trauma-Injury Infection and Critical Care. 2004;57(3):547–554. doi: 10.1097/01.ta.0000135162.85859.4c. [DOI] [PubMed] [Google Scholar]
- 20.Feng XH, Pelton R, Leduc M. Mechanical properties of polyelectrolyte complex films based on polyvinylamine and carboxymethyl cellulose. Industrial & Engineering Chemistry Research. 2006;45(20):6665–6671. [Google Scholar]
- 21.Kalichevsky MT, Jaroszkiewicz EM, Ablett S, Blanshard JMV, Lillford PJ. The glass-transition of amylopectin measured by DSC, DMTA and NMR. Carbohydrate Polymers. 1992;18(2):77–88. [Google Scholar]
- 22.Dumitriu S. Polysaccharides in Medicinal Applications. New York: M. Dekker; 1996. [Google Scholar]
- 23.Martanto W, Moore JS, Couse T, Prausnitz MR. Mechanism of fluid infusion during microneedle insertion and retraction. Journal of Controlled Release. 2006;112(3):357–361. doi: 10.1016/j.jconrel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 24.Ito Y, Yoshimitsu JI, Shiroyama K, Sugioka N, Takada K. Self-dissolving microneedles for the percutaneous absorption of EPO in mice. Journal of Drug Targeting. 2006;14(5):255–261. doi: 10.1080/10611860600785080. [DOI] [PubMed] [Google Scholar]
- 25.Teo MAL, Shearwood C, Ng KC, Lu J, Moochhala S. In vitro and in vivo characterization of MEMS microneedles. Biomedical Microdevices. 2005;7(1):47–52. doi: 10.1007/s10544-005-6171-y. [DOI] [PubMed] [Google Scholar]
- 26.McNally EJ. In: Protein Formulation and Delivery. Eugene J McNally., editor. New York: M Dekker; 2000. [Google Scholar]
- 27.Godin B, Touitou E. Transdermal skin delivery: Predictions for humans from in vivo, ex vivo and animal models. Advanced Drug Delivery Reviews. 2007;59:1152–1161. doi: 10.1016/j.addr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Walters KA. Dermal absorption and toxicity assessment. New York: Marcel Dekker; 1998. [Google Scholar]
- 29.Material Safety Data Sheet provided by Sigma-Aldrich.
- 30.Chew NYK, Tang P, Chan HK, Raper JA. How much particle surface corrugation is sufficient to improve aerosol performance of powders? Pharmaceutical Research. 2005;22(1):148–152. doi: 10.1007/s11095-004-9020-4. [DOI] [PubMed] [Google Scholar]