

Summary
Cowpea mosaic virus (CPMV) is a well-characterized nanoparticle that has been used for a variety of nanobiotechnology applications. CPMV interacts with several mammalian cell lines and tissues in vivo. To overcome natural CPMV targeting and re-direct CPMV particles to cells of interest, we attached a novel folic acid-PEG conjugate using the copper-catalyzed azide-alkyne cycloaddition reaction. PEGylation of CPMV completely eliminated background binding of the virus to tumor cells. The PEG-folate moiety allowed CPMV specific recognition of tumor cells bearing the folate receptor. In addition, by testing CPMV formulations with different amounts of the PEG-FA moiety displayed on the surface, we show that higher-density loading of targeting ligands on CPMV may not be necessary for efficient targeting to tumor cells. These studies help to define the requirements for efficiently targeting nanoparticles and protein cages to tumors.
Introduction
The ability to target tumors and deliver therapeutics to specific locations in the body is a primary goal in cancer medicine. Tumor-specific targeting has the potential to reduce adverse effects and increase the therapeutic benefit of highly toxic chemotherapies [1]. Tumor-targeting strategies include the use of various types of molecular and supramolecular scaffolds such as liposomes [2], iron oxide nanoparticles [3], silica-gold nanoshells [4], highly branched macromolecules such as dendrimers [5, 6], protein cages [7] and viruses [8–11]. Ligands capable of targeting tumors such as antibodies, peptides, or small-molecules are typically attached to the exterior surface. In many cases drugs or contrast agents can also be encapsulated inside the particles in order to combine targeted cell killing with intravital imaging [12–16].
Recently viruses and protein cages have been studied in the development of tumor-specific targeting strategies [8, 17]. These multivalent protein assemblies have the advantages of simple production, monodisperse size and structure, and surfaces that are compatible with circulation in vivo [10, 18–20]. One such particle is cowpea mosaic virus (CPMV), a plant virus that grows in the common cowpea plant (Vigna unguiculata) and forms a 31nm-diameter icosahedral structure [21, 22]. CPMV grows to very high yields in infected plants, and the purification is simple and inexpensive. In addition, CPMV is non-pathogenic for humans, and the products derived from plant virus culture are not contaminated with animal cells or viruses [23, 24]. CPMV particles are highly stable to extremes of temperature, pH and a variety of organic solvents such as DMSO [21, 25].
Surface modification of CPMV particles for targeting purposes has been performed by genetic and chemical means [26–31]. Chemical modification of CPMV surface lysine residues using fluorescent dye-labeled N-hydroxysuccinimide (NHS) esters or sulfhydryl groups using maleimide and haloacetamide reagents, has been extensively characterized [29, 32]. Surface carboxylate groups may also be modified for specific attachment [33]. Recently CPMV particles were shown to be stable to the conditions of the copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction, dramatically increasing the variety of ligands that may be efficiently conjugated to the capsid surface [34, 35].
Although it is a plant pathogen and does not replicate in animal cells, CPMV has been shown to interact with a variety of cell types both in vitro and in vivo [17, 36]. Fluorescently-labeled CPMV interacts with vascular endothelial cells when injected intravenously, allowing for a clear resolution of vascular structures in a variety of organs, while PEG-coated CPMV does not [36]. Interestingly, CPMV interacts specifically with a 54kD membrane protein found in mammalian cells, and this interaction is also blocked by PEG coating of the virus [37]. These studies demonstrate that CPMV has significant potential for use in targeting and imaging of both normal vasculature and tumors in vivo, but also underscores the need for cell-specific retargeting of the capsid.
One of the best-characterized ligands to be exploited for targeting tumor cells is the vitamin folic acid (FA). FA plays an essential role in human growth and development, and at the cellular level in cell division and DNA synthesis [38]. Uptake of FA into cells is mediated by the folate receptor (FR), and binding of FA to FR initiates receptor-mediated endocytosis and internalization of FA [39, 40]. Normally expression of FR on cells is low, however the demand for FA increases during cellular activation and proliferation. Indeed, FR expression is upregulated on a variety of human tumors, including cancers of the ovary, kidney, uterus, testis, brain, lung and myelocytic blood cells [12, 41–43]. Upregulation of FR has also been correlated with poor disease outcome [44]. Thus, FA has been studied as a promising targeting ligand for cancer detection, imaging, and treatment [45].
Several nanoparticle formulations have used FA-based targeting strategies. The derivatization of liposomes with FA has been the most intensively investigated thus far [13, 14, 46]. Other nanoparticle formulations employing FA-targeting include FA-coated gadolinium nanoparticles that were shown to increase uptake in tumors suggesting the possibility of neutron capture therapy [47], poly-amidoamine dendrimers (PAMAM) conjugated to FA [6, 48], and core-shell nanostructures decorated with FA [49]. The FA-targeting strategy has also been applied to gold nanoparticles [50], pegylated micelles [51], and adenoviruses [10] in an attempt to reach FR-expressing cells. PEG spacers have also been used to facilitate binding of the conjugated FA to cell-surface receptors [14, 51–54]. Nevertheless, precise control of attachment of targeting ligands at the nanoscale, as well as establishment of the relationship between ligand density and target cell recognition, has not been achieved.
Several attached large molecules such as peptides, antibody ligands, tumor receptor ligands, and transferrin, have been used to target viral nanoparticles and protein cages [29, 35, 55], but the efficiency of cellular targeting using capsid-arrayed small molecule ligands has been less well-studied. We report here the chemical attachment of FA, PEG, and PEG-conjugated FA to CPMV using the CuAAC reaction in order to direct the capsid specifically to tumor cells. The ability of the PEG molecules to inhibit natural CPMV-cell surface interactions and the ability of FA-modified CPMVs to bind to tumor cells was studied using confocal microscopy and flow cytometry-based binding assays. In addition, CPMV-based formulations that kept the amount of PEG constant and varied the loading of the FA ligand were evaluated for their ability to interact with FR-expressing cells in order to test the hypothesis that polyvalent binding might be advantageous for cell-specific targeting of CPMV.
Results
Previous studies have shown that CPMV recognizes and is taken up by a variety of mammalian cell types [36, 37]. To first ask whether CPMV conjugated directly to FA could redirect this natural targeting, an NHS ester of FA was conjugated to surface lysines on CPMV. A parallel reaction performed under identical conditions with fluorescein-NHS resulted in the attachment of 100±10 dyes per particle; we assume that the analogous folate conjugation gave a similar result [56, 57]. The natural uptake of CPMV is significant [36], and CPMV-FA particles showed no measurable increase in binding to FR-expressing KB cells in comparison to unmodified CPMV particles as measured by flow cytometry binding analysis and cell uptake studies (data not shown). Thus the natural cellular interactions of CPMV needed to be modified in order to achieve FR-specific targeting.
We previously showed that PEG (3400 Da) inhibits the natural targeting and uptake of CPMV [36]. In addition, the importance of a PEG spacer for efficient cellular recognition of FA-conjugated nanoparticles has also been previously noted [2]. To accommodate tailored loading of both PEG and FA on CPMV particles, a relatively short (500 Da) PEG chain was introduced as shown in Figure 1. A monodisperse azide-PEG-amine was coupled to the NHS ester of folic acid to give the FA-azide 2a. The corresponding polyvalent alkyne nanoparticle was obtained by reacting CPMV with a large excess of the alkynyl NHS ester reagent 1. This procedure has been shown to acylate 150–200 of the exposed lysine side chain amines of CPMV [56], and was chosen to provide an excess of available surface alkyne groups for subsequent attachment. The use of the CuAAC ligation reaction eliminates the need for protecting group manipulations and allows for reproducible attachments to be achieved with a minimum amount of material.
Figure 1. Synthesis of the PEG-folic acid conjugates.
FA derivatives are known to form aggregates at high concentrations [58], and there are some indications that too many FA molecules displayed on a surface can inhibit binding to the target receptor, perhaps because of such aggregation [59]. To address this phenomenon we limited the density of PEG-FAs on CPMV by using a relatively low concentration of azide derivatives in the CuAAC bioconjugation step. An estimate of the loading was obtained by performing a parallel reaction using N3-PEG-fluorescein (2b) with CPMV-alkyne under identical conditions, as we have previously described [57]. This transformation provided a value of 60±6 molecules per virus for the attachment of PEG-Fluorescein or PEG-FA, where a 1 mg/mL CPMV solution translates to a folic acid concentration of 10 µM.
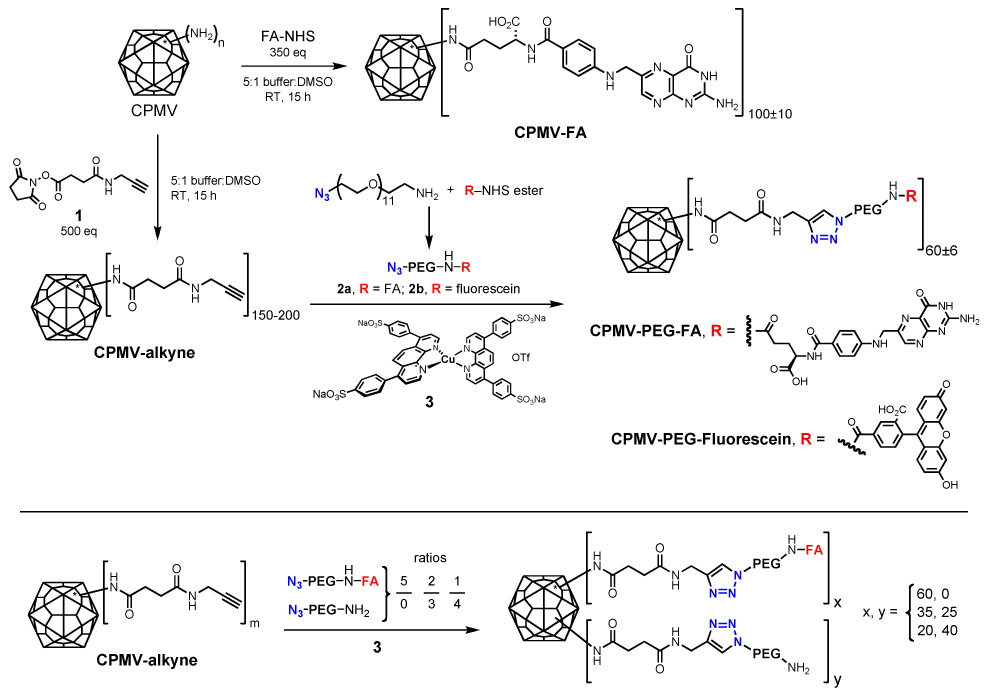
The integrity of the conjugated virus preparations was verified by analytical size exclusion chromatography (Superose 6), which also showed by a slightly smaller elution volume that the CPMV-(PEG-FA)60 conjugate appears to be slightly larger than unmodified CPMV (Figure 2 a). It is possible that the early elution of CPMV-(PEG-FA)60 could also be caused by altered interaction of PEG-FA moiety with the column material in comparison to the unmodified particle (designated CPMV-WT). The sharpness of the peaks for CPMV-(PEG-FA)60 in comparison to CPMV-WT suggests that the modification of the particles within the population is relatively uniform on a per-particle basis. This uniformity was supported by transmission electron microscopy (TEM) studies (Figure 2 e,f) of negatively-stained samples, which confirmed that the CPMV-(PEG-FA)60 particles were intact and showed no evidence of aggregation. Visual analysis of the samples showed that 100% of the CPMV-(PEG-FA)60 particles exhibited a halo-like structure on the surface of the particle that appeared to be uniform within the population, and which was absent in unmodified CPMV (Figure 2 e, f).
Figure 2. Virus integrity assessed by size exclusion chromatography and TEM, and Western blot.
A. Size exclusion FPLC analysis of CPMV-PEG-FA and WT-CPMV. B. Coomassie stain of WT-CPMV (1) and CPMV-(PEG-FA)60 (2), showing small (S) and large (L) capsid subunit proteins. C–D. Western blots of WT-CPMV (1) and CPMV-(PEG-FA)60 at higher concentration (2). Left panel, detection using rabbit anti-CPMV antibody. Right panel, detection using mouse anti-folic acid antibody. L: large subunit, and S: small subunit. E–F. Typical TEM image of WT-CPMV (e) and CPMV-(PEG-FA)60 (f), showing intact particles. The bar represents 200 nm.
Successful PEG-FA conjugation was further confirmed by gel electrophoresis and Western immunoblotting. Figure 2b shows the large (L) and small (S) capsid proteins of wild-type and PEG-FA-derivatized CPMV; unlike larger PEG chains used in previous studies [60], the small PEG-FA moiety used here shifted the positions of the fully denatured S or L bands only slightly toward higher molecular weight. Western immunoblots appear in Figures 2c,d, using higher concentrations of capsid protein. The use of anti-CPMV antibody showed the L and S subunits as well as minor amounts of higher molecular weight species which arise from incomplete denaturation of the durable CPMV capsid (Figure 2c). After attachment of PEG-FA, the higher molecular weight bands increased in intensity, presumably because the FA-derivatized protein subunits were more prone toward aggregation or resistant to denaturation (Figure 2c). Anti-FA antibody showed all of the bands from CPMV-(PEG-FA)60 to bear FA (Figure 2d). Of the five surface lysines found on CPMV, two dominate the acylation reactivity of the particle – one each on the S (lysine 38) and L (lysine 99) subunits [56, 61]. The average of 60 PEG-FA chains per particle may therefore be assumed to be distributed largely among the 120 sites provided by these two residues on the capsid exterior.
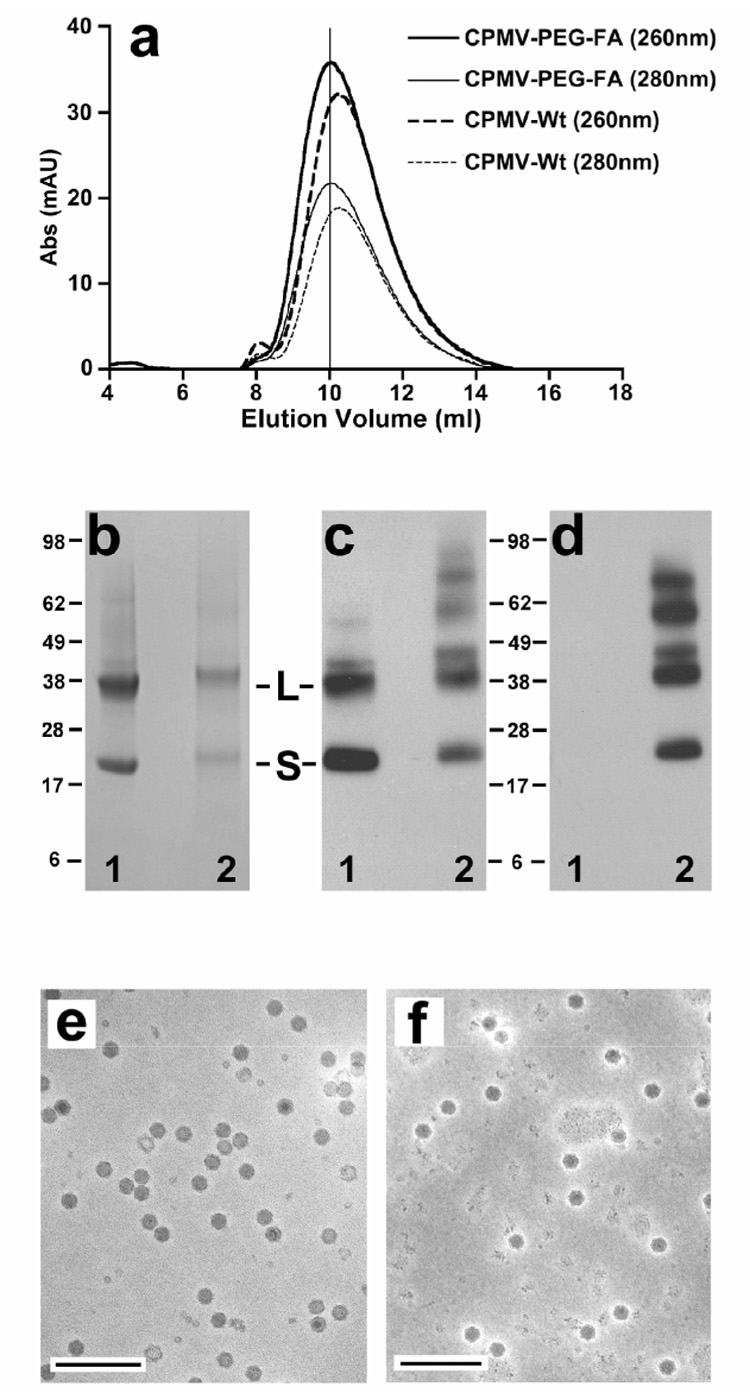
The ability of the relatively short (500 Da) PEG chains to inhibit the natural targeting of CPMV was assessed by flow cytometry. Two tumor cell lines, KB and HeLa, were used for these studies. KB, a human nasopharyngeal carcinoma cell line, has been widely used in tumor-targeting studies due to its high expression of FR and affinity for FA [42, 45, 48, 62–64]. HeLa cells, a well-established epitheloid cervical carcinoma, have been reported to express less FR than KB cells. The cellular uptake of CPMV particles in KB or HeLa cells after 2h incubation at 37°C was analyzed by fluorescence microscopy (Figure 3). As previously described, unmodified CPMV showed significant uptake in HeLa and KB cell lines (Figure 3 a,d), while the decoration of CPMV with PEG inhibited this phenomenon (Figure 3 b,e). Both types of cells also showed increased uptake of FA-conjugated viruses compared to unmodified CPMV (Figure 3 c,f). Interestingly, uptake of CPMV-(PEG-FA)60 in HeLa cells appeared as an evenly dispersed fluorescence in the cytoplasm, while CPMV uptake in HeLa cells showed a more punctate distribution of the virus throughout the cells. CPMV is naturally targeted to lysosomes [36], although the endocytic pathway used to reach this compartment is unknown. The uptake of FR in either cell type is not well-defined and has been shown to involve both clathrin-mediated and caveolin-mediated endocytic pathways. Differences between uptake patterns in each cell type could be due to differences in receptor expression level (e.g. saturation of the clathrin-mediated pathway in HeLa cells spilling into the caveolae pathway or vice-versa), or due to differences in the localization of endocytic vesicles or the trafficking pattern of vesicle recycling between the two cell types [63, 65, 66]. Nevertheless, our data indicate that the endocytic pathway used for uptake and trafficking of CPMV-PEG-FA is modified in comparison to CPMV-WT.
Figure 3. Fluorescent microscopy of monolayers of HeLa (top) and KB (bottom) cells.
Cells were incubated with the indicated virus particles (37°C, 2 h), followed by permeabilization and treatment with anti-CPMV antibody and then with a green fluorescent secondary antibody conjugate. Nuclei are stained with DAPI (blue). Scale bar, 10µm.
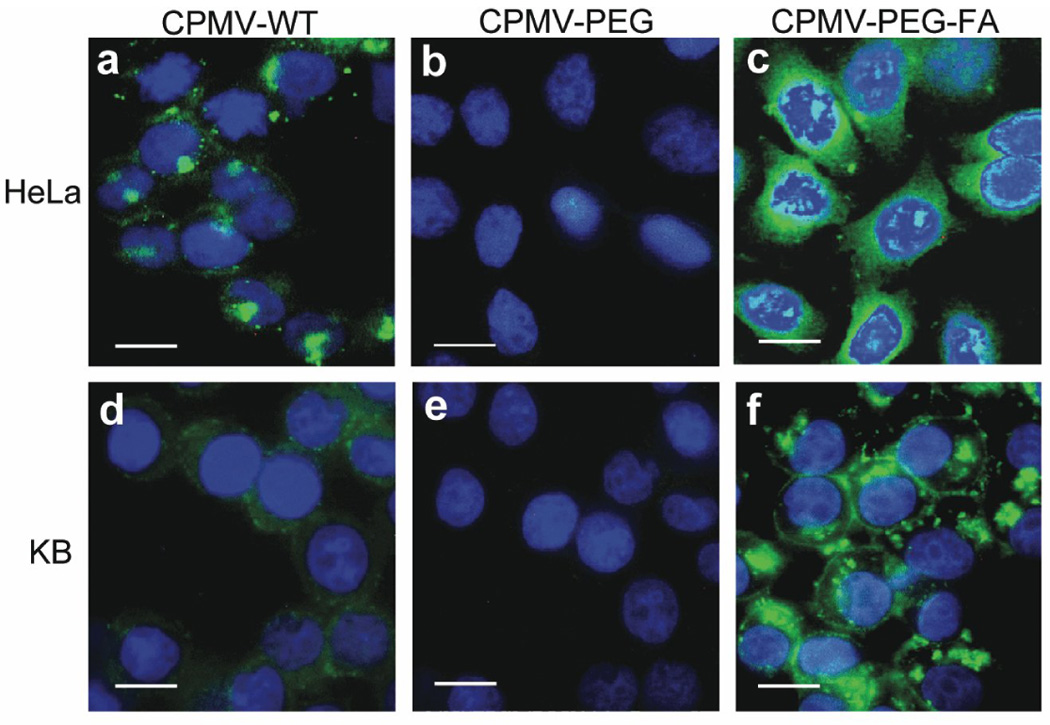
Differences in cell surface binding were measured by flow cytometry, where virus binding and subsequent steps were carried out at 4°C in order to minimize endocytosis. CPMV-(PEG-FA)60 bound to KB cells much better than to HeLa cells (94% and 22%, respectively, relative to cells alone; Figure 4 a,b, solid histograms), and both cell types interacted hardly at all with CPMV-PEG lacking the terminal FA moiety (1.6% and 0.7% to KB and HeLa cells, respectively; Figure 4 a,b). These results are consistent with the difference in the expression of FR receptors on the two cell types (approximately 2.8×105 receptors per cell for KB cells vs. 4-fold fewer receptors per cell for HeLa [67, 68]). Further details are provided in Supplemental Data and Figure S1.
Figure 4. Measurement of virus binding to tumor cells using flow cytometry.
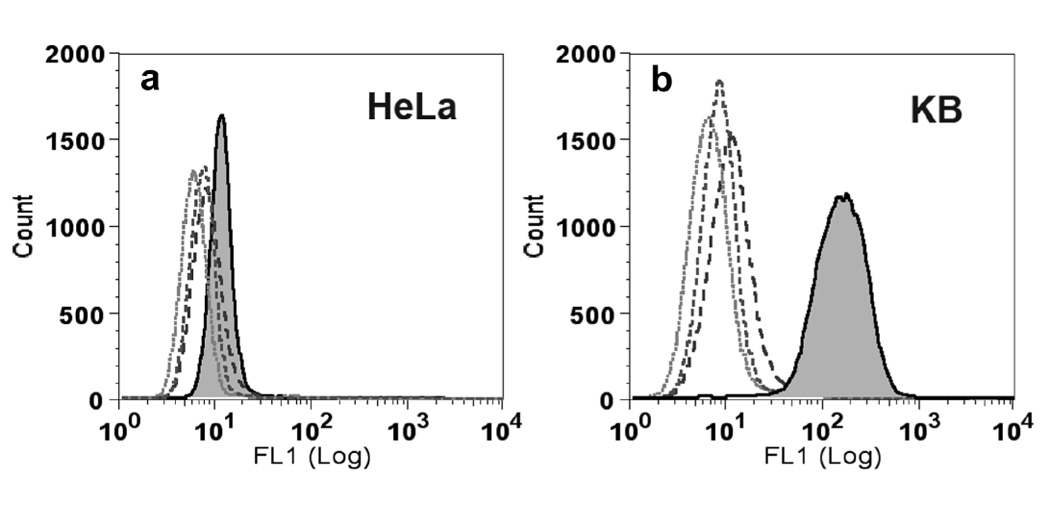
A. Binding of CPMVs to HeLa tumor cells. B. Binding of CPMV to KB tumor cells. The peaks from left to right are cells only (dotted histogram), CPMV-PEG (short dashed lines), WT-CPMV (long dashed lines), and CPMV-(PEG-FA)60 (filled histograms), respectively.
To further show that the interaction between CPMV-PEG-FA and tumor cells was due to specific interactions between the particles and FR on cells, the ability of free folic acid to compete for binding of CPMV-(PEG-FA)60 to KB cells was assessed by flow cytometry. In the experiment shown in Figure 5a, free folic acid at greater than 10 µM substantially inhibited CPMV-(PEG-FA)60 binding, and less than 1 µM folic acid had no effect. CPMV-(PEG-FA)60 was used at a concentration of 17 nM in particles, or 1 µM total FA. Nonlinear regression analysis (Prism, GraphPad Inc., San Diego CA) of a representative experiment revealed an IC50 value of 2.75µM. The average IC50 of four separate experiments was 4.9µM (Figure 5a). If each folate unit on CPMV acted as a free folate molecule and each virus-cell contact was made by a single FA-FR interaction, the IC50 value would, by definition, be equal to the virus-displayed FA concentration of 1 µM. Thus, FA displayed on the capsid surface binds with similar affinity on a per-folate basis than free folic acid. On a per-particle basis, the avidity of CPMV-(PEG-FA)60 for KB cells is much stronger (by approximately 300 times) than free FA.
Figure 5. Competition of CPMV-PEG-FA using free FA.
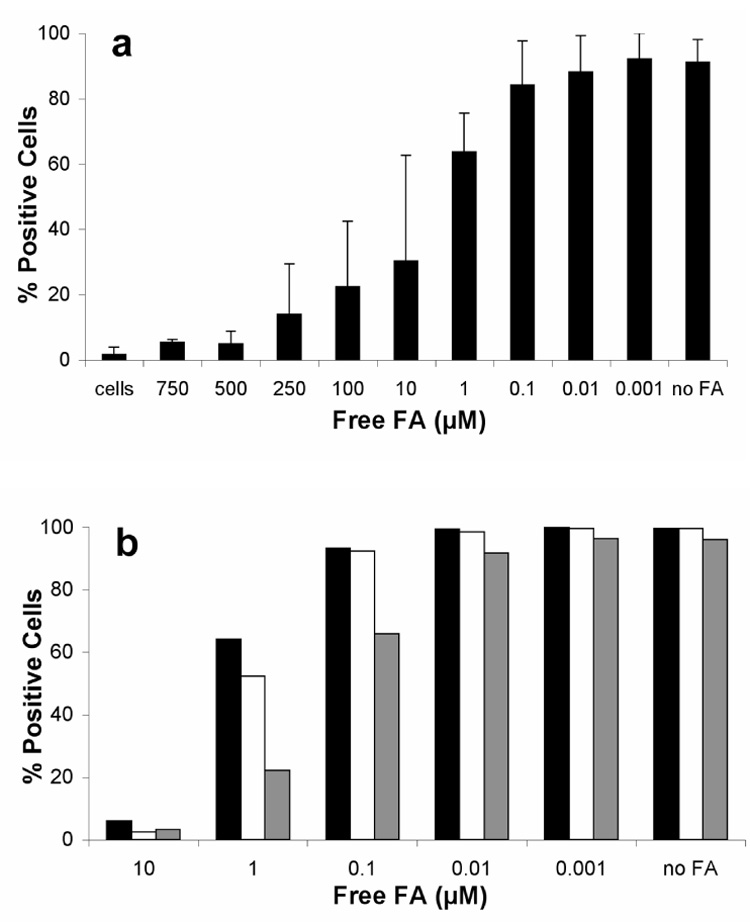
A. CPMV-PEG-FA bearing 60 FA/particle (1 µM FA) was incubated with KB cells in the presence of increasing amounts of FA as indicated. Error bars represent the mean +/− S.D. of triplicate experiments. B. Competition of CPMV-(PEG-FA)20 (grey), CPMV-(PEG-FA)35 (white bars) or CPMV-(PEG-FA)60 was performed using increasing amounts of FA as indicated. In each case, the concentration of virus particles was 17 nM, giving rise to the following concentrations of displayed FA: CPMV-(PEG-FA)20 = 0.33 µM FA; CPMV-(PEG-FA)35 = 0.6 µM FA; CPMV-(PEG-FA)60 = 1µM FA.
To further explore whether controlling FA loading at the nanoscale could influence interaction with FR, three different versions of CPMV-PEG-FA were formulated that had an equal loading of PEG, but varied in the number of FA units at the end of the PEG chains. Separate portions of CPMV-alkyne were reacted with azide-PEG-FA and two different combinations of azide-PEG-FA and azide-PEG-amine, giving rise to the attachment of 60, 35, and 20 PEG-FA units per particle (Figure 1, bottom). These are designated CPMV-(PEG-FA)60, CPMV-(PEG-FA)35 CPMV-(PEG-FA)20, respectively. Comparative western blots using identical amounts of virus particles confirmed the relative loading values using Spot Density Analysis (Alpha Innotech, San Leandro, CA; Supplemental figure S2a). While little difference in the cell uptake of each of these particles was observed at saturating conditions, titration of binding revealed a clear difference in binding that correlated with the loading of FA per particle (Supplemental Figure S2b). Moreover, in a competition assay using free FA to inhibit binding, in each case approximately the same concentration of free FA was required to disrupt binding of the particles to KB cells as was presented on the virus surface (Figure 5b). In other words, in no case was a substantial per-folate polyvalent effect observed, but in all cases the folate-decorated particles bound substantially better than free folic acid molecules alone.
Discussion
In this work, we demonstrate the specific binding and endocytosis of a FA-derivatized CPMV particle by cells bearing FR. Direct, controlled chemical conjugation of a monodisperse PEG-FA moiety to the CPMV surface by copper (I)-catalyzed azide-alkyne cycloaddition (CuAAC) was efficient and resulted in the attachment of a maximum of 60 ± 6 molecules of FA per particle. Wild-type CPMV exhibited significant binding, which was eliminated by the conjugation of the relatively short PEG chains to the virus surface. The addition of the FA moiety then allowed the particles to be recognized specifically by the tumor cell lines. Modulating the extent of FA loading using the CuAAC reaction demonstrated excellent control of particle modification at the nanoscale, and correlated with tumor cell recognition. These studies suggest that CPMV nanoparticles can be effectively redirected by surface conjugation to ligands of interest, allowing specific uptake into tumors while avoiding nonspecific uptake into normal cells.
At least two factors may contribute to the inability of HeLa or KB cells to specifically recognize FA directly conjugated to the CPMV capsid (as opposed to FA being displayed on the end of PEG chains). The simplest explanation is that endogenous and potentially multivalent CPMV-cell surface interactions are of higher affinity than the FA-FR interaction. Second, the conjugated FA units may simply be rendered inaccessible to FR by virtue of being attached to the rigid capsid structure by a short tether; previous reports have noted steric impediments to binding in other settings [14, 52, 53]. Alternatively, if endocytosis is initiated by multivalent binding, rather than a single-point contact (as has long been known for certain carbohydrate receptors [69, 70]), there may be a minimum distance that adjacent FAs need to achieve before such a process is initiated. The PEG linker can address both potential deficiencies, and studies are in progress to attempt to distinguish between these possibilities. It should be noted that the PEG linker used here is significantly shorter than has been previously employed for FA nanoparticles [52, 53, 64].
Our studies coupling 20–60 PEG-FA moieties to CPMV resulted in targeting that compares favorably to other FA-conjugated nanoparticle formulations. For example, conjugation of FA to PAMAM dendrimers resulted in loading of about 2.5 molecules of FA residues per dendrimer, due to an undesired reaction occurring between the carboxylic acid groups of FA and the amine groups of the dendrimer. In this case, no binding enhancement was observed: a concentration of 30 nM of free FA reduced by 50% the amount of association to KB cells reached by 30 nM of the dendrimer-FA conjugate [5, 48]. Coupling of FA to 10 nm Au nanoparticles has also been reported [50], but only 20% of the KB cells exposed to the nanoparticles actively took up the FA-conjugate. This uptake could be suppressed by 2 µM free FA.
PEGylation of CPMV, even using the small 500 Da material here, resulted in effective suppression of the particle’s natural cellular interactions. The strategy has been used previously in many other studies by our groups and others. For example, multifunctional PEGylated micelles conjugated to FA and carrying the antitumor drug adriamycin showed significant increase in cytotoxicity compared to non-targeted micelles. However, longer incubation times also induced significant cytotoxicity when non-targeted micelles were used, suggesting that PEGylation was not sufficient in blocking nonspecific binding of the micelles to tumor cells [51]. Attempts have also been made to retarget adenovirus particles to KB cells [10]. The adenovirus surface was modified with FA-PEG conjugate (24% occupancy), resulting in a ~40% increase in transfection efficiency over non-targeted pegylated virus. Nevertheless, the pegylated adenovirus was still able to transduce cells via its natural coxsackie-adenovirus receptor (CAR), suggesting that the degree of PEGylation was not sufficient to effectively retarget the virus.
One drawback in most chemical nanoparticle formulations is the lack of control over the spatial distribution on the outer shell of the ligand of interest. A sophisticated rational design of ligand orientation and stoichiometry would allow for the creation of a polyvalent scaffold that enhances the targeting properties of the nanoparticle and offers the possibility of cell targeting using lower concentrations of targeting molecules than would be possible for the monomeric form [71]. The increased binding of CPMV-PEG-FA over free FA (factor of approximately 2 per FA, or 120 per particle) is not sufficient to clearly indicate the existence of polyvalent (simultaneous multi-point) interactions [72] with FR, compared to other known examples using carbohydrates and cell surface lectins [73–77] or liposome-displayed anthrax peptides and receptors [78]. In part this may be due to the nature of the ligand-receptor pair, since more substantial avidity effects were observed using CPMV displaying carbohydrate moieties [28, 79](E. Kaltgrad and M.G. Finn, personal communication). Interestingly, a substantial polyvalent effect has recently been quantified by SPR measurements of binding affinity for FA displayed on dendrimers [78, 80]. While the more flexible nature of the dendrimer scaffold may better match cellular receptor patterns, as has been previously hypothesized [78], the target is also different (protein immobilized on SPR chips vs. cell-surface receptors), and competition with free monovalent folate was not reported in the dendrimer case [80]. For CPMV-PEG-FA, the roughly linear trend of free FA inhibition with variation of PEG-FA loading further suggests that monovalent FA-FR interactions are dominant, but also that local concentration of the FA ligand plays a role in binding of cells to the polyvalent scaffold. It seems likely that the effective off-rate of the particle is slowed by facile re-association with FR receptors brought about by the high local concentration of FA on the particle surface. Such effects may be all that is necessary to provide enough increased avidity and endocytosis to allow for effective tumor targeting [81].
Prior attempts to target tumors with displayed FA have yielded mixed results depending on the tumor site [47, 82, 83]. Endothelial cell targeting has also been widely studied in order to reach the tumor vasculature [15, 84]. Thus a combination of targeting moieties could enhance the specificity of nanoparticles to tumors in vivo. Toward these ends, the construction and in vivo testing of more sophisticated particles, taking advantage of the precise control of amino acid side-chain positioning and reactivity to install PEGs and tethered targeting moieties, is in progress in our laboratories.
Significance
The ability to specifically target therapeutics and imaging modalities to tumors is an important goal in biomedicine. Viruses and protein cage nanoparticles have proven utility in this area, however many of these materials have natural cellular interactions. CPMV in particular provides tremendous imaging resolution of normal and tumor vasculature, however the particles have significant affinity for cell-surface proteins. Thus any natural in vivo interaction of the nanoparticle must be masked and the particle retargeted to a tumor ligand of interest. In this article, we use copper (I) catalyzed azide-alkyne cycloaddition chemistry to precisely attach a defined number of folic acid tumor ligands, while redirecting the natural specificity of the particles by coating the remaining surface of the particles with low molecular-weight polyethylene glycol. The resulting particles are effectively retargeted specifically to the folic-acid receptor. The density of ligands may be controlled at the nanoscale, and competitive binding vs. free folic acid was shown to track linearly with the density of folate attached to the nanoparticle scaffold. The high local concentration of folic acid on the capsid surface enables these particles to be targeted efficiently to cell-surface FR receptors. To our knowledge this is the first time that small-molecule ligands have been successfully used to provide targeting specificity for virus-based nanoparticles.
Experimental Procedures
All reagents, unless otherwise specified, were purchased from commercial suppliers and used without further purification. Bifunctional N3-PEG-NH2 [N3(CH2CH2O)11CH2CH2NH2, MW 570, PDI = 1.0] was obtained from Polypure Inc. Copper(I) triflate was prepared according to literature procedures [85].
Propagation of CPMV in Plants
The primary leaves of Kentucky cowpea plants (Vigna unguiculata) were mechanically inoculated as 10-day old seedlings, bearing two primary leaves and with secondary leaves just beginning to show. Virus stocks were initiated from pCP1 and pCP2 plasmid cDNAs encoding full-length copies of the two RNA moieties of CPMV, RNA-1 and RNA-2, respectively. Carborundum was first dusted onto the leaves to aid in the wounding process. At approximately 3 weeks post inoculation, the symptomatic leaves were harvested, weighed and frozen at −70°C until virus purification. The virus was purified from the infected leaves by a method described elsewhere [86].
Synthesis of N3-PEG-FA (2)
FA-NHS ester was prepared from FA according to literature procedures [2]. FA-NHS (100 mg, 0.19 mmol) in anhydrous DMSO (10 mL) was treated with N3-PEG-NH2 (50 mg, 0.09 mmol) and the mixture was stirred while warming gently (ca. 35 °C) for 20 hours. DMSO was removed under high vacuum at RT. The residue was treated with CH2Cl2 and the solids were scraped gently from the walls of the flask. The mixture was sonicated to form a slurry, which was transferred to a fritted disk, and washed thoroughly with diethyl ether, CH2Cl2, and THF. The residual dark orange solid was dried and extracted with water until the aqueous extract was colorless. The aqueous fractions were combined and evaporated under vacuum to yield N3-PEG-FA (compound 2) as viscous orange oil (60 mg, 0.06 mmol, 66%). ¹H NMR (200 MHz, D2O): δ 8.59 (s, 1H), 7.52 (d, 2H), 6.64 (d, 2H), 4.48 (s, 1H), 4.15 (m, 2H), 3.58-3.35 (m, 40), 2.35-2.20 (m, 12H). An analogous procedure was employed for the synthesis of N3-PEG-fluorescein.
Preparation of CPMV-Alkyne
Compound 1 (50 mg, 0.20 mmol) was dissolved in DMSO (1 mL) and added to a solution of wild-type CPMV (5 mL, 7 mg/mL, 75 µM in protein subunits, 0.1 M phosphate buffer, pH 7.0). The mixture was gently agitated at RT for 15 hours and purified by sucrose gradient fractionation (10–40 % sucrose in 0.1 M pH 7.0 phosphate buffer, Beckman SW-28 Ti rotor, 28000 rpm, 3 hours). The intact virus was collected as a pale white band under intense illumination on a gradient fraction collector and subjected to ultracentrifugation (Beckman 50.2 Ti rotor, 42000 rpm, 3 hours) to form a colorless pellet. The solution was decanted and the colorless pellet was dissolved under N2 with sufficient buffer (Tris-Cl, 0.1 M, pH 8.0) to obtain a concentration of 7.3 mg/mL.
Conjugations to CPMV-Alkyne
The following reaction protocol was carried out in an inert atmosphere (N2) glove box with O2 level kept below 6 ppm until the final gel filtration step. For each of the three N3-PEG-FA loading levels, a 2 mL Eppendorf centrifuge tube was charged with CPMV-alkyne (7.3 mg/mL, 110 µL) and buffer (Tris-Cl, 0.1 M, pH 8.0, 260, 250, 220 µL, respectively). Degassed aqueous solutions of N3-PEG-FA (25 mM; 10, 20, 50 µL, respectively) and N3-PEG-NH2 (25 mM; 40, 30, 0 µL, respectively) were added simultaneously to the virus solution and mixed by gentle agitation. Thus, the total azide and PEG concentrations were kept constant for each of the three reactions, and the amount of FA vs. amine caps on the PEG chains were varied. A solution of copper(I) triflate (100 mM, CH3CN, 50 µL) was combined with a solution of sulfonated bathophenanthroline (100 mM in 100 mM Tris-Cl, pH 8.0, 100 µL) (We subsequently learned that Tris-Cl slightly inhibits the CuAAC reaction relative to HEPES buffer, and so the latter is now recommended) to form a catalyst of the formulation 3. An aliquot of the catalyst mixture (16 µL) was added to the tube containing the virus. The reaction mixture was immediately placed on a rotisserie for continuous agitation, and kept under N2 at RT for 15 hours. The product (designated CPMV-PEG-FA) was purified by three passages through size exclusion gel filtration columns (BioRad, p-100) which removed all residual catalyst and excess N3-PEG-FA. The integrity of the virus was verified by analytical size exclusion chromatography (Superose 6) and transmission electron microscopy (TEM, samples stained with 0.2% uranyl acetate; images acquired on a 100 Kv Philips Tecnai electron microscope). Conjugations of N3-PEG-NH2 and N3-PEG-Fluorescein to CPMV-alkyne were performed in the same manner using the following reagents: CPMV-alkyne (7.3 mg/mL, 110 µL), buffer (Tris-Cl, 0.1 M, pH 8.0, 250 µL), N3-PEG-NH2 or N3-PEG-Fluorescein (25 mM, 20µL). Virus concentrations were determined by UV-vis spectroscopy: at 0.1 mg/mL, CPMV gives a standard absorbance of 0.8 at 260 nm. The average molecular weight of the CPMV virion is 5.6×106.
Synthesis of CPMV-FA
FA-NHS (31 mg, 58 µmol) was dissolved in DMSO (1 mL) and added to a solution of wild-type CPMV (5 mL, 3 mg/mL, 32 µM in protein subunits, 0.1 M phosphate buffer, pH 7.0). The mixture was gently agitated at RT for 15 hours and purified by sucrose gradient fractionation (10–40 % sucrose in 0.1 M pH 7.0 phosphate buffer, Beckman SW-28 Ti rotor, 28000 rpm, 3 hours). The intact virus was collected as a pale white band under intense illumination on a gradient fraction collector and subjected to ultracentrifugation (Beckman 50.2 Ti rotor, 42000 rpm, 3 hours) to form a colorless pellet. The solution was decanted and the colorless pellet was dissolved with sufficient buffer (Tris-Cl, 0.1 M, pH 8.0) to obtain a concentration of 5.0 mg/mL. CPMV-FA was characterized in the manner described above for CPMV-alkyne + azide conjugates.
Western blotting
To detect CPMV, protein bands were visualized by Western blotting using anti-CPMV IgG (purified from rabbit polyclonal antisera on a protein G column; Amersham Pharmacia, Uppsala, Sweden) and diluted 1:1000 in blocking buffer (PBS, 5% normal goat serum, 0.05% Tween20). ImmunoPure Goat anti-Rabbit IgG, peroxidase conjugated, diluted 1:20,000 (Pierce, Rockford, IL) was used as secondary antibody. Folic acid conjugated to CPMV was detected by Western blotting using mouse anti-Folic acid monoclonal antibody (Sigma-Aldrich, St. Louis, MO) diluted 1:1000 in blocking buffer (PBS, 5% normal goat serum, 0.05% Tween20). Goat anti-mouse peroxidase conjugated, diluted 1:20,000 (Pierce, Rockford, IL) was used as secondary antibody. In both cases, detection of peroxidase was carried out using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL).
Cell Culture and Binding Studies
HeLa cells and KB cells, the latter a human nasopharyngeal epidermal carcinoma [62] were grown continuously as a monolayer using FA-free RPMI1640 medium (Gibco, Invitrogen, Carlsbad CA) containing 10% heat-inactivated fetal bovine serum (FBS), penicillin (50 units/mL), streptomycin (50 µg/mL), and 2 mM L-glutamine at 37°C in a 5% CO2/95% air humidified atmosphere. The concentration of folic acid was 5–6 nM in FA-free medium containing serum, close to the natural physiologic concentration (5–30 nM) [52, 87]. On the day before each experiment, the medium was replaced with FA-free RPMI 1640 containing all the supplements mentioned above, except 10% FBS.
Measurement of Virus Binding to HeLa and KB cells using Flow Cytometry
HeLa and KB cells, grown overnight in FA-depleted medium, were treated with trypsin, counted, and distributed in 100 µL portions into a 96-well V-bottom shaped plate at a concentration of 5×106 cells/mL. 10 µg of different virus preparations were added to each well and the cells were incubated on ice at 4°C for 1 hour. The cells were then washed 4 times using ice cold PBS buffer containing 1mM EDTA and 25 mM HEPES pH 7.5, centrifuged at 1600 rpm for 6 minutes at 4°C. Rabbit anti-CPMV primary antibody was then added to the cells in a 100 µL volume, and the cells were incubated on ice at 4°C for 30 minutes and washed as above. The same procedure was then repeated with goat anti-rabbit IgG AlexaFluor 488 conjugated antibody (Invitrogen, Carlsbad, CA), with incubation conducted in the dark. Finally, the cells were fixed using 2% formaldehyde in PBS buffer containing 1 mM EDTA and 25 mM HEPES pH 7.5. The samples were then analyzed using a FACS Calibur instrument (BD Biosciences, Franklin Lakes, NJ). Approximately 40,000 events were collected for each sample and the data were analyzed using FlowJo software (Tree Star, Inc, Ashland, OR). Binding and competition data were further analyzed by nonlinear regresson analysis (Prism, GraphPad Inc.). Independent control experiments established that the PEG-conjugated CPMV was recognized with approximately the same efficiency as native CPMV by the anti-CPMV antibody.
Cellular Uptake of the FA-Conjugated Virus in HeLa and KB Cells
Cells were seeded in a 12-well plate containing 12mm sterile glass cover slips at 1×105 cells/well and grown for 48 hours as previously described. On the day of the experiment, cells were washed once with FA-depleted medium, and 10 µg of different virus preparations were added to each well and the cells were incubated at 37°C in a 5% CO2/95% air humidified atmosphere for 2 hours. Cells were then washed four times using FA-depleted medium to remove unbound virus, on a rocker at room temperature for five minutes. Cells were then fixed using 4% paraformaldehyde in PBS for 20 minutes. After 4 washes with PBS buffer, cells were permeabilized using 0.1% Triton X-100 in PBS for 15 minutes. Non-specific binding was blocked by incubating the cells in 5% goat serum in PBS for 1 hour. Rabbit anti-CPMV antibody was added to the cells in 1% goat serum, 0.1% Triton X-100 in PBS, and cells were incubated at room temperature for 45 minutes with gentle agitation. Unbound antibody was then removed by washing four times with PBS. Goat anti-rabbit IgG AlexaFluor 488 conjugated antibody (Invitrogen) was added in 1% goat serum in PBS, and cells were gently agitated for a further 35 minutes. During the last five minutes of secondary antibody incubation, cell nuclei were stained by adding 100 µL of 4′,6-diamidino-2-phenylindole (DAPI 1:1000 dilution in water). Cells were then washed 4 times using PBS and cover slips covered with cells were mounted on slides using Vecta Shield mounting medium (Vector Laboratories). Cells were imaged using a Nikon Eclipse TS100 microscope, with a 100X oil objective.
Competition Assay
KB cells, grown overnight in FA-depleted medium, were treated with trypsin, counted, and distributed in 100 µL portions into a 96-well V-bottom shaped plate at a concentration of 5×106 cells/mL. 10 µg of CPMV-PEG-FA was added to the cells together with different concentrations of free FA, ranging from 750 µM to 1 nM. Cells were then treated as described above and the samples were analyzed using flow cytometry. The competition assay was also carried out using CPMV-PEG-FA that had been synthesized so that different amounts of PEG-FA conjugate were displayed on the surface of the virus.
Supplementary Material
Acknowledgments
We thank Dr. Eric Agner of Polypure, Inc. for a generous gift of the monodisperse azido-PEG-amine, and Kris Koudelka for assistance with flow cytometry and helpful discussions. This work was supported by National Institutes of Health grant R01CA112705.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 2.Lee RJ, L PS. Delivery of Liposomes into Cultured KB cells via Folate Receptor-mediated endocytosis. The Journal of Biological Chemistry. 1994;269:3198–3204. [PubMed] [Google Scholar]
- 3.Sonvico F, Mornet S, Vasseur S, Dubernet C, Jaillard D, Degrouard J, Hoebeke J, Duguet E, Colombo P, Couvreur P. Folate-conjugated iron oxide nanoparticles for solid tumor targeting as potential specific magnetic hyperthermia mediators: synthesis, physicochemical characterization, and in vitro experiments. Bioconjug Chem. 2005;16:1181–1188. doi: 10.1021/bc050050z. [DOI] [PubMed] [Google Scholar]
- 4.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci U S A. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi Y, B JRJ. Targeting cancer cells with DNA-assembled dendrimers: a mix and match strategy for cancer. Cell Cycle. 2005;4:669–671. doi: 10.4161/cc.4.5.1684. [DOI] [PubMed] [Google Scholar]
- 6.Shi X, Wang S, Meshinchi S, Van Antwerp ME, Bi X, Lee I, Baker JR., Jr Dendrimer-entrapped gold nanoparticles as a platform for cancer-cell targeting and imaging. Small. 2007;3:1245–1252. doi: 10.1002/smll.200700054. [DOI] [PubMed] [Google Scholar]
- 7.Flenniken ML, Willits DA, Harmsen AL, Liepold LO, Harmsen AG, Young MJ, Douglas T. Melanoma and lymphocyte cell-specific targeting incorporated into a heat shock protein cage architecture. Chem Biol. 2006;13:161–170. doi: 10.1016/j.chembiol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Douglas T, Young M. Viruses: making friends with old foes. Science. 2006;312:873–875. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- 9.Yamada T, Ueda M, Seno M, Kondo A, Tanizawa K, Kuroda S. Novel tissue and cell type-specific gene/drug delivery system using surface engineered hepatitis B virus nano-particles. Curr Drug Targets Infect Disord. 2004;4:163–167. doi: 10.2174/1568005043341037. [DOI] [PubMed] [Google Scholar]
- 10.Oh IK, Mok H, Park TG. Folate immobilized and PEGylated adenovirus for retargeting to tumor cells. Bioconjug Chem. 2006;17:721–727. doi: 10.1021/bc060030c. [DOI] [PubMed] [Google Scholar]
- 11.Singh P, Destito G, Schneemann A, Manchester M. Canine parvovirus-like particles, a novel nanomaterial for tumor targeting. J Nanobiotechnology. 2006;4:2. doi: 10.1186/1477-3155-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, L P. Folate-mediated targeting of antineoplastic drugs, imaging agents, and nucleic acids to cancer cells. Journal of Controlled Release. 1998;53:39–48. doi: 10.1016/s0168-3659(97)00236-8. [DOI] [PubMed] [Google Scholar]
- 13.Zhao XB, Lee RJ. Tumor-selective targeted delivery of genes and antisense oligodeoxyribonucleotides via the folate receptor. Adv Drug Deliv Rev. 2004;56:1193–1204. doi: 10.1016/j.addr.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Stephenson SM, L P, Lee RJ. Folate receptor-mediated targeting of liposomal drugs to cancer cells. Methods Enzymol. 2004;387:33–50. doi: 10.1016/S0076-6879(04)87003-4. [DOI] [PubMed] [Google Scholar]
- 15.Ruoslahti E. Drug targeting to specific vascular sites. Drug Discov Today. 2002;7:1138–1143. doi: 10.1016/s1359-6446(02)02501-1. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi H, Brechbiel MW. Dendrimer-based nanosized MRI contrast agents. Curr Pharm Biotechnol. 2004;5:539–549. doi: 10.2174/1389201043376571. [DOI] [PubMed] [Google Scholar]
- 17.Singh P, Gonzalez MJ, Manchester M. Viruses and their uses in nanotechnology. Drug Development Research. 2006;67:23–41. [Google Scholar]
- 18.Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 19.Ruoslahti E. Targeting tumor vasculature with homing peptides from phage display. Semin Cancer Biol. 2000;10:435–442. doi: 10.1006/scbi.2000.0334. [DOI] [PubMed] [Google Scholar]
- 20.Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh P, Thomas DM, Estrada MN, Powell E, Finn MG, Manchester M. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343:224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 21.Lomonossoff GP, Johnson JE. The synthesis and structure of comovirus capsids. Prog Biophys Mol Biol. 1991;55:107–137. doi: 10.1016/0079-6107(91)90003-b. [DOI] [PubMed] [Google Scholar]
- 22.Lin T, Chen Z, Usha R, Stauffacher CV, Dai JB, Schmidt T, Johnson JE. The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology. 1999;265:20–34. doi: 10.1006/viro.1999.0038. [DOI] [PubMed] [Google Scholar]
- 23.Brennan FR, Jones TD, Hamilton WD. Cowpea mosaic virus as a vaccine carrier of heterologous antigens. Mol. Biotechnol. 2001;17:15–26. doi: 10.1385/MB:17:1:15. [DOI] [PubMed] [Google Scholar]
- 24.Johnson J, Lin T, Lomonossoff G. Presentation of heterologous peptides on plant viruses: genetics, structure, and function. Annu Rev Phytopathol. 1997;35:67–86. doi: 10.1146/annurev.phyto.35.1.67. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q, Lin T, Tang L, Johnson J, Finn M. Icosahedral virus particles as addressable nanoscale building blocks. Angew. Chem. Int. Ed. 2002;41:459–462. doi: 10.1002/1521-3773(20020201)41:3<459::aid-anie459>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 26.Lomonossoff G, Rohll J, Spall V, Maule A, Loveland J, Porta C, Usha R, Johnson JE. Insertion of Foreign Antigenic Sites into the Plant Virus Cowpea Mosaic Virus. In: Goodenough P, editor. Proceedings of the Second AFRC Protein Engineering Conference. CPL Press; 1993. [Google Scholar]
- 27.Spall V, Porta C, Taylor K, Lin T, Johnson J, Lomonossoff G. Antigen expression on the surface of a plant virus for vaccine production. In: J N, P D, Shewry PR, editors. Engineering crops for industrial end uses. London: Portland Press; 1998. [Google Scholar]
- 28.Raja KS, Wang Q, Finn MG. Icosahedral virus particles as polyvalent carbohydrate display platforms. ChemBioChem. 2003;4:1348–1351. doi: 10.1002/cbic.200300759. [DOI] [PubMed] [Google Scholar]
- 29.Chatterji A, Ochoa W, Shamieh L, Salakian SP, Wong SM, Clingon G, Ghosh P, Lint T, Johnson J. Chemical conjugation of heterologous proteins on the surface of cowpea mosaic virus. Bioconjug. Chem. 2004;15:807–813. doi: 10.1021/bc0402888. [DOI] [PubMed] [Google Scholar]
- 30.Chatterji A, Burns LL, Taylor SS, Lomonossoff GP, Johnson JE, Lin T, Porta C. Cowpea mosaic virus: from the presentation of antigenic peptides to the display of active biomaterials. Intervirology. 2002;45:362–370. doi: 10.1159/000067929. [DOI] [PubMed] [Google Scholar]
- 31.Steinmetz NF, Lomonossoff GP, Evans DJ. Decoration of cowpea mosaic virus with multiple, redox-active, organometallic complexes. Small. 2006;2:530–533. doi: 10.1002/smll.200500453. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Lin T, Johnson J, Finn M. Natural supramolecular building blocks: cysteine-added mutants of cowpea mosaic virus. Chem. Biol. 2002;9:813–819. doi: 10.1016/s1074-5521(02)00166-7. [DOI] [PubMed] [Google Scholar]
- 33.Steinmetz NF, Lomonossoff GP, Evans DJ. Cowpea mosaic virus for material fabrication: addressable carboxylate groups on a programmable nanoscaffold. Langmuir. 2006;22:3488–3490. doi: 10.1021/la060078e. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. Bioconjugation by copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 35.Sen Gupta S, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Accelerated bioorthogonal conjugation: a practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconjug Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 36.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Viral nanoparticles as tools for intravital vascular imaging. Nat Med. 2006;12:354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koudelka KJ, Rae CS, Gonzalez MJ, Manchester M. Interaction between a 54-Kilodalton Mammalian Cell Surface Protein and Cowpea Mosaic Virus. J Virol. 2007;81:1632–1640. doi: 10.1128/JVI.00960-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massaro EJ, R JME. Folate and Human Development. Humana Press; 2002. p. 376. [Google Scholar]
- 39.Rijnboutt S, Jansen G, Posthuma G, Hynes JB, Schornagel JH, Strous GJ. Endocytosis of GPI-linked membrane folate receptor-alpha. J Cell Biol. 1996;132:35–47. doi: 10.1083/jcb.132.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birn H, Selhub J, Christensen EI. Internalization and intracellular transport of folate-binding protein in rat kidney proximal tubule. Am J Physiol. 1993;264:C302–C310. doi: 10.1152/ajpcell.1993.264.2.C302. [DOI] [PubMed] [Google Scholar]
- 41.Leamon C, Low PS. Membrane folate-binding proteins are responsible for folate-protein conjugate endocytosis into cultured cells. Biochem J. 1993;291:855–860. doi: 10.1042/bj2910855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy JA, L P. Folate-mediated targeting of therapeutic and imaging agents to cancers. Crit Rev Ther Drug Carrier Syst. 1998;15:587–627. [PubMed] [Google Scholar]
- 43.Lu Y, L PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Deliv. Reviews. 2002;54:675–693. doi: 10.1016/s0169-409x(02)00042-x. [DOI] [PubMed] [Google Scholar]
- 44.Hartmann LC, Keeney GL, Lingle WL, Christianson TJ, Varghese B, Hillman D, Oberg AL, Low PS. Folate receptor overexpression is associated with poor outcome in breast cancer. Int J Cancer. 2007;121:938–942. doi: 10.1002/ijc.22811. [DOI] [PubMed] [Google Scholar]
- 45.Reddy JA, Allagadda VM, Leamon CP. Targeting therapeutic and imaging agents to folate receptor positive tumors. Curr Pharm Biotechnol. 2005;6:131–150. doi: 10.2174/1389201053642376. [DOI] [PubMed] [Google Scholar]
- 46.Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94:2135–2146. doi: 10.1002/jps.20457. [DOI] [PubMed] [Google Scholar]
- 47.Oyewumi MO, Y R, Jay M, Coakley T, Mumper RJ. Comparison of cell uptake, biodistribution and tumor retention of folate-coated and PEG-coated gadolinium nanoparticles in tumor-bearing mice. Journal of Controlled Release. 2004;95:613–626. doi: 10.1016/j.jconrel.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Quintana A, Raczka E, Piehler L, Lee I, Myc A, Majoros I, Patri A, Thomas T, Mule J, Baker JJ. Design and function of a dendrimer-based therapeutic nanodevice targeted to tumor cells through the folate receptor. Pharm Res. 2002;19:1310–1316. doi: 10.1023/a:1020398624602. [DOI] [PubMed] [Google Scholar]
- 49.Pan D, Turner JL, Wooley KL. Folic acid-conjugated nanostructured materials designed for cancer cell targeting. Chem Commun (Camb) 2003;7:2400–2401. doi: 10.1039/b307878g. [DOI] [PubMed] [Google Scholar]
- 50.Dixit V, Van den Bossche J, Sherman DM, Thompson DH, Andres RP. Synthesis and grafting of thioctic acid-PEG-folate conjugates onto Au nanoparticles for selective targeting of folate receptor-positive tumor cells. Bioconjug Chem. 2006;17:603–609. doi: 10.1021/bc050335b. [DOI] [PubMed] [Google Scholar]
- 51.Bae Y, Jang WD, Nishiyama N, Fukushima S, Kataoka K. Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery. Mol Biosyst. 2005;1:242–250. doi: 10.1039/b500266d. [DOI] [PubMed] [Google Scholar]
- 52.Gabizon A, Horowitz AT, Goren D, Tzemach D, Mandelbaum-Shavit F, Qazen MM, Zalipsky S. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: in vitro studies. Bioconj. Chem. 1999;10:289–298. doi: 10.1021/bc9801124. [DOI] [PubMed] [Google Scholar]
- 53.Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv. Drug Deliv. Reviews. 2004;56:1177–1192. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 54.Salmaso S, Semenzato A, Caliceti P, Hoebeke J, Sonvico F, Dubernet C, Couvreur P. Specific antitumor targetable beta-cyclodextrin-poly(ethylene glycol)-folic acid drug delivery bioconjugate. Bioconjug Chem. 2004;15:997–1004. doi: 10.1021/bc034186d. [DOI] [PubMed] [Google Scholar]
- 55.Uchida M, Flenniken ML, Allen M, Willits DA, Crowley BE, Brumfield S, Willis AF, Jackiw L, Jutila M, Young MJ, Douglas T. Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles. J Am Chem Soc. 2006;128:16626–16633. doi: 10.1021/ja0655690. [DOI] [PubMed] [Google Scholar]
- 56.Wang Q, Kaltgrad E, Lin T, Johnson J, Finn M. Natural supramolecular building blocks: wild-type cowpea mosaic virus. Chem. Biol. 2002;9 doi: 10.1016/s1074-5521(02)00165-5. [DOI] [PubMed] [Google Scholar]
- 57.Kaltgrad E, Sen Gupta S, Punna S, Huang C, Chang A, Wong C, Finn MG, Blixt O. Anti-carbohydrate antibodies elicited by polyvalent display on a viral scaffold. ChemBioChem. 2007 doi: 10.1002/cbic.200700225. In Press. [DOI] [PubMed] [Google Scholar]
- 58.Ciuchi Federica, G DN, Franz Hermann, Gottarelli Giovanni, Mariani Paolo, Bossi Maria Grazia Ponzi, Spada Gian Piero. Self-Recognition and Self-Assembly of Folic Acid Salts: Columnar Liquid Crystalline Polymorphism and the Column Growth Process. J. Am. Chem. Soc. 1994;116:7064–7071. [Google Scholar]
- 59.Reddy JA, Abburi C, Hofland H, Howard SJ, Vlahov I, Wils P, Leamon CP. Folate-targeted, cationic liposome-mediated gene transfer into disseminated peritoneal tumors. Gene Ther. 2002;9:1542–1550. doi: 10.1038/sj.gt.3301833. [DOI] [PubMed] [Google Scholar]
- 60.Raja KS, Wang Q, Gonzalez MJ, Manchester M, Johnson JE, Finn MG. Hybrid virus-polymer materials. 1. Synthesis and properties of PEG-decorated cowpea mosaic virus. Biomacromolecules. 2003;4:472–476. doi: 10.1021/bm025740+. [DOI] [PubMed] [Google Scholar]
- 61.Chatterji A, Ochoa WF, Paine M, Ratna BR, Johnson JE, Lin T. New addresses on an addressable virus nanoblock; uniquely reactive Lys residues on cowpea mosaic virus. Chem Biol. 2004;11:855–863. doi: 10.1016/j.chembiol.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 62.Saikawa Y, Price K, Hance KW, Chen TY, Elwood PC. Structural and functional analysis of the human KB cells folate receptor gene P4 promoter:cooperation of three clustered Sp-1 binding sites with initiator region for basal promoter activity. Biochemistry. 1995;34:9951–9961. doi: 10.1021/bi00031a018. [DOI] [PubMed] [Google Scholar]
- 63.Sabharanjak S, Mayor S. Folate receptor endocytosis and trafficking. Adv Drug Deliv Rev. 2004;56:1099–1109. doi: 10.1016/j.addr.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 64.Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta. 1995;1233:134–144. doi: 10.1016/0005-2736(94)00235-h. [DOI] [PubMed] [Google Scholar]
- 65.Paulos CM, Reddy JA, Leamon CP, Turk MJ, Low PS. Ligand binding and kinetics of folate receptor recycling in vivo: impact on receptor-mediated drug delivery. Mol Pharmacol. 2004;66:1406–1414. doi: 10.1124/mol.104.003723. [DOI] [PubMed] [Google Scholar]
- 66.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 67.Saul JM, Annapragada A, Natarajan JV, Bellamkonda RV. Controlled targeting of liposomal doxorubicin via the folate receptor in vivo. Journal of Controlled Release. 2003;92:49–67. doi: 10.1016/s0168-3659(03)00295-5. [DOI] [PubMed] [Google Scholar]
- 68.Sonvico F, D C, Marsaud V, Appel M, Chacun H, Stella B, Renoir M, Colombo P, Couvreur P. Establishment of an in vitro model expressing the folate receptor for the investigation of targeted delivery systems. J. Drug Del. Sci. Tech. 2005;15:407–410. [Google Scholar]
- 69.Breitfeld PP, Simmons CF, Jr, Strous GJ, Geuze HJ, Schwartz AL. Cell biology of the asialoglycoprotein receptor system: a model of receptor-mediated endocytosis. Int Rev Cytol. 1985;97:47–95. doi: 10.1016/s0074-7696(08)62348-7. [DOI] [PubMed] [Google Scholar]
- 70.Kolb-Bachofen V. Hepatic receptor for asialo-glycoproteins. Ultrastructural demonstration of ligand-induced microaggregation of receptors. Biochim Biophys Acta. 1981;645:293–299. doi: 10.1016/0005-2736(81)90200-5. [DOI] [PubMed] [Google Scholar]
- 71.Wu P, Malkoch M, Hunt JN, Vestberg R, Kaltgrad E, Finn MG, Fokin VV, Sharpless KB, Hawker CJ. Multivalent, bifunctional dendrimers prepared by click chemistry. Chem Commun (Camb) 2005;14:5775–5777. doi: 10.1039/b512021g. [DOI] [PubMed] [Google Scholar]
- 72.Mathai Mammen S-KC, Whitesides George M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angewandte Chemie International Edition. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 73.Woller EK, Cloninger MJ. The lectin-binding properties of six generations of mannose-functionalized dendrimers. Org Lett. 2002;4:7–10. doi: 10.1021/ol016568+. [DOI] [PubMed] [Google Scholar]
- 74.Collins BE, Blixt O, Han S, Duong B, Li H, Nathan JK, Bovin N, Paulson JC. High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J Immunol. 2006;177:2994–3003. doi: 10.4049/jimmunol.177.5.2994. [DOI] [PubMed] [Google Scholar]
- 75.Collins BE, Paulson JC. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr Opin Chem Biol. 2004;8:617–625. doi: 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 76.Andre S, Kaltner H, Furuike T, Nishimura S, Gabius HJ. Persubstituted cyclodextrin-based glycoclusters as inhibitors of protein-carbohydrate recognition using purified plant and mammalian lectins and wild-type and lectin-gene-transfected tumor cells as targets. Bioconjug Chem. 2004;15:87–98. doi: 10.1021/bc0340666. [DOI] [PubMed] [Google Scholar]
- 77.Gestwicki JE, Kiessling LL. Inter-receptor communication through arrays of bacterial chemoreceptors. Nature. 2002;415:81–84. doi: 10.1038/415081a. [DOI] [PubMed] [Google Scholar]
- 78.Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S, Tao K, Mogridge J, Kane RS. Statistical pattern matching facilitates the design of polyvalent inhibitors of anthrax and cholera toxins. Nat Biotechnol. 2006;24:582–586. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- 79.Sen Gupta S, Raja KS, Kaltgrad E, Strable E, Finn MG. Virus-glycopolymer conjugates by copper(I) catalysis of atom transfer radical polymerization and azide-alkyne cycloaddition. Chem Commun (Camb) 2005;14:4315–4317. doi: 10.1039/b502444g. [DOI] [PubMed] [Google Scholar]
- 80.Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Jr, Banaszak Holl MM. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol. 2007;14:107–115. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 81.Weissleder R, Kelly K, Sun EY, Shtatland T, Josephson L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol. 2005;23:1418–1423. doi: 10.1038/nbt1159. [DOI] [PubMed] [Google Scholar]
- 82.Leamon CP, Cooper SR, Hardee GE. Folate-liposome-mediated antisense oligodeoxynucleotide targeting to cancer cells: evaluation in vitro and in vivo. Bioconjug Chem. 2003;14:738–747. doi: 10.1021/bc020089t. [DOI] [PubMed] [Google Scholar]
- 83.Muller C, Bruhlmeier M, Schubiger PA, Schibli R. Effects of antifolate drugs on the cellular uptake of radiofolates in vitro and in vivo. J Nucl Med. 2006;47:2057–2064. [PubMed] [Google Scholar]
- 84.Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, Ellerby HM, Bredesen DE, Pasqualini R, Ruoslahti E. Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci. USA. 2002;99:1527–1531. doi: 10.1073/pnas.241655998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kubas GJ. Copper(I) complexes. Inorg. Synth. 1979;19:90–92. [Google Scholar]
- 86.Dessens JT, Lomonossoff GP. Cauliflower mosaic virus 35S promoter-controlled DNA copies of cowpea mosaic virus RNAs are infectious on plants. J. Gen. Virol. 1993;74:889–892. doi: 10.1099/0022-1317-74-5-889. [DOI] [PubMed] [Google Scholar]
- 87.Antony AC. The biological chemistry of folate receptors. Blood. 1992;79:2807–2820. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.