

Abstract
Neurexins mediate protein interactions at the synapse, playing an essential role in synaptic function. Extracellular domains of neurexins, and their fragments, bind a distinct profile of different proteins regulated by alternative splicing and Ca2+. The crystal structure of n1α_LNS#2 (the second LNS/LG domain of bovine neurexin 1α) reveals large structural differences compared with n1α_LNS#6 (or n1β_LNS), the only other LNS/LG domain for which a structure has been determined. The differences overlap the so-called hyper-variable surface, the putative protein interaction surface that is reshaped as a result of alternative splicing. A Ca2+-binding site is revealed at the center of the hyper-variable surface next to splice insertion sites. Isothermal titration calorimetry indicates that the Ca2+-binding site in n1α_LNS#2 has low affinity (Kd ~ 400 μm). Ca2+ binding ceases to be measurable when an 8- or 15-residue splice insert is present at the splice site SS#2 indicating that alternative splicing can affect Ca2+-binding sites of neurexin LNS/LG domains. Our studies initiate a framework for the putative protein interaction sites of neurexin LNS/LG domains. This framework is essential to understand how incorporation of alternative splice inserts expands the information from a limited set of neurexin genes to produce a large array of synaptic adhesion molecules with potentially very different synaptic function.
Neurexins are multidomain cell surface proteins found in the brain at the synapse (recently reviewed in Refs. 1–4). In mammals, the neurexin family consists of three genes that each encode an α- and a β-neurexin (5–8). Protruding into the synaptic cleft, the extracellular domain of α-neurexins consists of six LNS/LG2 domains, whereas β-neurexins contain only a single LNS/LG domain preceded by a short β-neurexin-specific sequence (nomenclature and abbreviations defined below and in Fig. 1). There is increased similarity between the first, third, and fifth LNS/LG domains as well as between the second, fourth, and sixth LNS/LG domains. Nevertheless the sequence identity is low between neurexin LNS/LG domains (20–25%).
FIGURE 1. Multimodular nature of the neurexin family.
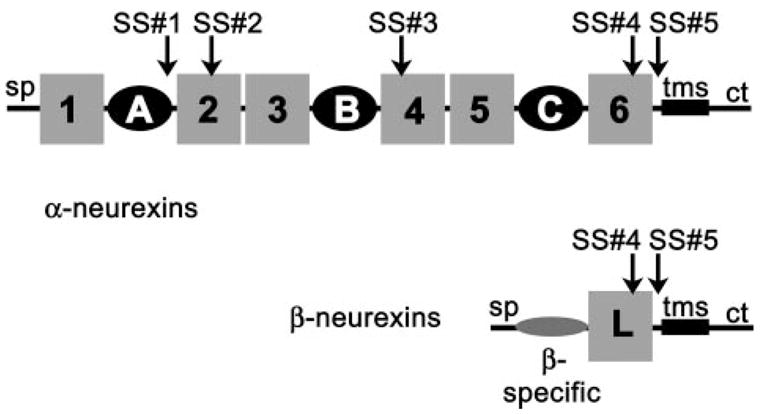
The extracellular domain of α-neurexins is composed of six LNS/LG domains (gray boxes) and three epidermal growth factor-like repeats (black circles). See text for the acronyms used in this study. The β-neurexin extracellular domain contains a single LNS/LG domain (gray box) that has the identical amino acid sequence to LNS#6 in the α-neurexin and a short β-neurexin-specific sequence (β-specific). In addition, neurexins contain a signal peptide (sp), a single transmembrane segment (tms), and a cytoplasmic tail (ct). Five alternative splice insert sites are found in the extracellular domain of neurexin 1α: SS#1, SS#2, SS#3, SS#4, and SS#5.
Neurexins facilitate synapse functioning and neurotransmitter release through protein-protein interactions (9–18). Neurexins interact inside the neuron with the cytomatrix of the active zone and outside the neuron with proteins at the synaptic cleft. Single LNS/LG domains of neurexins are sufficient to bind ligands (though not necessarily optimal), binding is Ca2+-dependent, and individual LNS/LG domains display distinct ligand-binding specificities (14, 17, 19–22). The second LNS/LG domain of neurexin 1α, neurexin 1α_LNS#2 (n1α_LNS#2), interacts with the extracellular matrix protein α-dystroglycan (22) and neurexophilin, a small neuropeptide-like protein (20, 23). LNS#6 in neurexin 1α (n1α_LNS#6) and the identical LNS domain in neurexin 1β (n1β_LNS) interact with neuroligins, a family of post-synaptic cell surface proteins (9, 14, 17, 24, 25), α-latrotoxin, a spider neurotoxin triggering massive neurotransmitter release (21), and α-dystroglycan as well (22). Neurexins may play an important adhesive role by forming trans-synaptic bridges with proteins on the post-synaptic membrane that align pre- and post-synaptic machineries to promote efficient neurotransmission (1, 3, 17, 26–29).
Neurexin gene transcripts in vertebrates undergo extensive alternative splicing to generate potentially over 2000 isoforms, encoding variant extracellular domains (Fig. 1) (7, 8, 30). Three splice insert sites localize to the LNS/LG domains: SS#2, which accommodates 0, 8, or 15 aa, SS#3, which accommodates a Gly residue or a 10-aa insert, and SS#4, which accommodates a splice insert of 0 or 30 aa (7, 8, 30). Relevant to the studies presented here, splice isoforms of bovine neurexin 1α_LNS#2 can contain no extra residues, a 8-aa splice insert HSGIGHAM, or a 15-aa splice insert HSGIGHAMVNKLHCS at SS#2. The splice insert sites are conserved between neurexin genes and between species, and the inserted polypeptide sequences display high sequence homology as well.
Splice isoforms are regulated developmentally and spatially, and they display distinct protein-binding characteristics. Alternative splicing at SS#2 in n1α_LNS#2 appears important; splice isoforms switch in a controlled fashion during developmental stages in vertebrate embryos (31, 32), and incorporation of an 8-or 15-aa splice insert at SS#2 prevents avid binding to α-dystroglycan (22). The physiological significance of the neurexin-α-dystroglycan interaction is not immediately apparent (12, 14), although it has been implicated in brain development (33). Alternative splicing of neurexins at SS#4 in n1α_LNS#6/n1β_LNS modulates interactions with neuroligin isoforms (17, 24, 25), α-latrotoxin (21), and α-dystroglycan as well (22). Thus a growing body of work demonstrates, at least in vitro, that alternative splicing of neurexin LNS/LG domains regulates protein-binding specificities. On an atomic level, however, it is completely unknown what the effects of incorporating alternative splice inserts into the neurexin extracellular domain are and how molecular properties are altered to regulate function.
The crystal structure of neurexin 1β_LNS (n1β_LNS) has revealed an immunoglobulin-like β-sandwich (34). Splice insert sites SS#2, SS#3, and SS#4 map to loops close together in space at one edge of the β-sandwich designated as the “hyper-variable” surface. Two proteins that are structurally but not functionally related to neurexin LNS/LG domains use surfaces analogous to the hyper-variable region to bind ligands: the laminin α2 LG4 domain, which binds α-dystroglycan, and the sex hormone-binding globulin, which binds steroids (35–37). It is therefore thought that neurexin LNS/LG domains contain special protein interaction surfaces that coincide with the hyper-variable surface that is regulated by alternative splicing (37).
Neurexins are crucial molecules at the synapse and bind key molecules involved in exocytosis of synaptic vesicles and adhesion of synaptic membranes. The neurexin family appears in a strategic position to influence synapse formation, functioning, and/or plasticity by incorporating isoforms with very distinct molecular properties into the vast protein network at the synapse (2, 38). Our studies aim to understand how alternative splicing creates and controls functional diversity within the neurexin family, one of the largest families of synaptic adhesion molecules found in higher vertebrates. To put the enormous array of neurexins isoforms into a framework, we are characterizing the molecular properties of neurexin LNS/LG domains and their isoforms. We have undertaken a structural and biophysical approach to examine the molecular properties of n1α_LNS#2. The crystal structure of bovine n1α_LNS#2 determined to a resolution of 2.1 Å reveals the presence of a Ca2+-binding site located exactly at the hyper-variable surface. The Ca2+-binding site is surrounded by a molecular surface that is highly specific to different neurexin LNS/LG domains in terms of sequence and structure. Calorimetric studies demonstrate that the Ca2+-binding site has low metal-binding affinity. The presence of splice inserts at splice site SS#2 abolishes measurable affinity for Ca2+, indicating that alternative splice inserts may control ligand binding not only directly but also indirectly through the regulation of Ca2+-binding sites. Our studies establish a paradigm for the hyper-variable surface of neurexin LNS/LG domains, the putative protein partner binding surface responsible for binding different protein partners in different LNS/LG domains.
EXPERIMENTAL PROCEDURES
Protein Overexpression and Purification
The cDNAs encoding bovine n1α_LNS#2 (residues 279–475, accession code nm_174404) with 0, 8, or 15 aa inserted at splice insertion site SS#2 (Fig. 1) were fused to glutathione S-transferase with standard molecular biology techniques using the pGEX-KG overexpression vector (39). The residue numbering scheme used here for bovine neurexin 1α starts with Met1 of the signal peptide and takes into account the presence of 20 residues in SS#1, 15 residues in SS#2, 10 residues in SS#3, and 30 residues in SS#4. The three native isoforms (with 0-, 8-, and 15-aa splice inserts) were expressed as thrombin-cleavable glutathione S-transferase fusion proteins in Escherichia coli BL21(DE3). Proteins were purified using glutathione-agarose beads, ion-exchange, and gel filtration using the procedure described in a previous study (34). Although n1α_LNS#2 isoforms did not bind either Source Q or Source S columns, contaminants were very efficiently removed. In addition, for structure determination purposes the selenomethionyl variant of n1α_LNS#2 with no splice insert present was overexpressed in E. coli B834(DE3) cells (Novagen) using specialized medium containing 25 mg/liter L-selenomethionine and a protocol obtained from Molecular Dimensions Ltd. (Athena Enzymes Systems). The selenomethionyl protein was purified with the same protocol as the wild-type proteins, except that 1 mm dithiothreitol was added to the buffers. Mass spectrometry confirmed that all four methionine residues per molecule were replaced efficiently with selenomethionine.
Crystallization and X-ray Data Collection
Crystals of selenomethionyl n1α_LNS#2 with no splice insert were grown by hanging drop vapor diffusion at 21 °C. Hanging drops contained 1 μl of protein (10 mg/ml in 20 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol) and 1 μl of the reservoir solution (10% polyethylene glycol 8000, 0.1 m Tris, pH 8.0, 5 mm CaCl2, 1% glycerol). Single crystals grew as thin plates with average dimensions of 0.5 × 0.3 × 0.04 mm. The crystals have the symmetry of space group P1 with cell dimensions a = 60.0 Å, b = 61.6 Å, c = 66.6Å, α= 78.5°, β = 78.7°, γ = 84.7°. Although the diffraction patterns could also be indexed in C2, merging the integrated intensities in C2 resulted in 64% of the reflections being rejected and an Rmerge of >50% for those remaining, indicating that the correct space group was in fact P1. The Matthews coefficient (40) indicated 4 molecules per asymmetric unit and 58% solvent content. Prior to data collection, crystals were cryoprotected with the reservoir solution containing 30% glycerol (v/v) and flash-cooled in liquid propane. Although crystals of native or selenomethionyl n1α_LNS#2 could be grown, splice isoforms of n1α_LNS#2 containing an 8- or 15-aa insert have so far proven resistant to crystallization.
A multiple wavelength anomalous dispersion experiment was carried out at the Cornell High Energy Synchrotron Source beam-line F2 on an ADSC Q210 detector. Three data sets were collected: one at the selenium absorption peak (data set FII, 12.665 keV, and 0.978952 Å), one at the inflection point (data set FI, 12.660 keV, and 0.979338 Å), and a third data set at a low energy remote wavelength (data set LR, 12.650 keV, and 0.980111 Å). Diffraction data were integrated and reduced with HKL2000 (41). Data were further scaled with programs from the CCP4 suite (42). Data collection statistics are summarized in Table 1.
TABLE 1.
Summary of the data collection phasing, and refinement statistics for selenomethionyl n1α_LNS#2
| Data seta | Resolutionb | Reflections (total/unique) | Completeness | Rmergec | I/σ | Phasing powerd | Rcullisd (iso/ano) |
|---|---|---|---|---|---|---|---|
| Å | % | ||||||
| FII | 2.1 (2.18–2.1) | 190,980/52,304 | 98.1 (96.6) | 8.9 (32.4) | 16.4 (2.7) | 2.13 | 0.77/0.76 |
| FI | 2.1 (2.18–2.1) | 191,448/52,276 | 98.2 (96.9) | 9.0 (29.1) | 18.0 (3.1) | 1.74 | 0.83/0.86 |
| LR | 2.1 (2.18–2.1) | 187,261/51,547 | 96.6 (85.9) | 7.2 (26.5) | 16.6 (2.9) | ||
| Figure of merit 10 to 3 Å: 0.619 for 16 selenium sites | |||||||
| Model | Value | ||||||
|
| |||||||
| Resolution (Å) | 20.0–2.1 (all reflections |F|/σ ≥ 0.0) | ||||||
| Protein atoms | 5,553 (727 residues) | ||||||
| Solvent atoms | 443 | ||||||
| Ligands | 4 Ca2+ ions, 4 glycerol molecules | ||||||
| Unique reflections | 51,492 | ||||||
| Working set/test set | 48,915/2,577 | ||||||
| Rwork (%) | 18.4 | ||||||
| Rfree (%) | 23.3 | ||||||
| r.m.s.d. bond lengths (Å) | 0.011 | ||||||
| r.m.s.d. bond angles (degrees) | 1.391 | ||||||
| r.m.s.d. between NCS-related molecules (Å) | 0.435 Å (181 Cα atoms) average over 4 molecules | ||||||
| Average B of main/side-chain atoms (Å2)e | 35.6 Å2 for 2,908 main-chain atoms/36.7 Å2 for 2,645 side-chain atoms | ||||||
| Average B of solvent atoms (Å2)e | 41.5 Å2 for 444 solvent molecules | ||||||
| Average B of Ca2+ ions (Å2)e | 26.0 Å2 for 4 ions | ||||||
FII (peak, 0.97895 Å), FI (inflection, 0.97934 Å), and LR (low energy remote 0.98011 Å).
Outer shell statistics are in parentheses.
Rmerge = Σ(|(I−〈Imean〉)|)/Σ(I).
Phasing power = 〈Fh〉/εand Rcullis = ε/δ iso, where ε is the lack of closure. The phasing power and Rcullis are calculated for data between 10 and 3 Å.
After TLS refinement by REFMAC.
Structure Determination
Sixteen selenium sites were identified with SOLVE (43) using the peak wavelength data set (FII) from the multiple wavelength anomalous dispersion experiment; however, the automated density modification and model building follow-up program RESOLVE (43) failed to generate interpretable density or a model for all four monomers in the asymmetric unit. By regarding our multiple wavelength anomalous dispersion experiment as a special case of multiple isomorphous replacement (44), anomalous and “isomorphous” contributions between the peak (FII), inflection point (FI), and low energy remote (LR) data sets were used to refine the selenium parameters and to calculate phases with MLPHARE (42). The phases were further improved through density modification, including 4-fold non-crystallographic symmetry (NCS) averaging with DM (45). In principle it should have been straightforward to derive NCS relationships between the four monomers by determining four sets of four selenium sites, with each set representing a single monomer. Because of a fortuitous arrangement of molecules in the asymmetric unit and their internal arrangement of selenium atoms, it was possible to group the 16 selenium sites into groups of 4 in numerous ways, each yielding a different set of NCS operators. None of the derived NCS relationships were able to improve the density of all four molecules simultaneously by 4-fold NCS averaging. We then derived new NCS relationships using the assumption that only three of four selenomethionyl residues per monomer were in the identical conformation in all four monomers and could be used to derive the NCS relationships. We systematically identified sets containing four groups of three selenium atoms (using FINDNCS, PROFESSS (42), and SITE2RT (46)) and tested the derived NCS operators for their ability to improve the electron density. Only one arrangement yielded operators able to improve the electron density for all four monomers simultaneously, through 4-fold averaging with DM. After completion of the atomic model for n1α_LNS#2, it became clear that, although Met321 and Met470 are highly ordered, the side chain of Met414 adopts somewhat different conformations in the four monomers (<2 Å between the most deviant selenium atoms); nevertheless these three methionine residues can be used to derive NCS operators. However, the side chain of Met442 adopts two very different conformations for two monomers, placing the selenium atoms apart by 2.7 Å. When Met442 was used together with the other three methionine residues, the operators derived from all four methionine positions were too inaccurate for density modification procedures.
Model Building, Refinement, and Validation
The initial model of n1α_LNS#2 was built with O (47) in 4-fold averaged electron density maps. Using all data from the low energy remote data set to 2.4 Å, the model was refined using CNS (48) with a protocol combining simulated annealing and conjugate gradient minimization, the combined maximum-likelihood and experimental phase target (MLHL), 4-fold NCS restraints, and a bulk solvent correction. The last two cycles of model building and refinement were carried out with REFMAC (49) using data to 2.1 Å, weak NCS restraints, TLS refinement with the four monomers as rigid bodies, and refinement of individual B-factors. The final model of four monomers in the asymmetric unit contains 727 residues and 443 well ordered solvent molecules, 4 Ca2+ ions, and 4 glycerol molecules (Table 1). The only residue in the disallowed region of the Ramachandran plot is Leu376. The figures were generated using Molscript (50) and Raster3D (51). The atomic model and x-ray data for bovine n1α_LNS#2 have been submitted to the Protein Data Bank (accession code 2H0B).
Superposition of n1α_LNS#2 and n1β_LNS
The four molecules of n1α_LNS#2 in the asymmetric unit of the crystal were superimposed using LSQMAN (52) (0.435-Å r.m.s.d. for 181 Cα atoms); the molecule that deviates the least among the four monomers was identified as the representative structure for n1α_LNS#2. The representative n1α_LNS#2 monomer (mol2) deviates only slightly from the other three monomers, at most in stretches of one or two residues containing Cα atoms deviating <1.5 Å. The eight molecules of n1β_LNS found in the asymmetric unit (pdb access code 1C4R) were superimposed (0.545-Å r.m.s.d. for 175 Cα atoms) and the representative n1β_LNS molecule selected as well. The largest deviations between the representative n1β_LNS structure and the seven other monomers confine to a three-residue stretch in loop β6–β7 containing Cα atoms that deviate at most by 2 Å, and to a five-residue stretch flanking SS#4 that contains Cα atoms deviating by no more than 3 Å (coincident with the arrow and label “loop β10–β11” in Fig. 3). The representative molecules of n1α_LNS#2 and n1β_LNS were subsequently superimposed with LSQMAN. Analysis of the superposition indicates that differences between n1α_LNS#2 and n1β_LNS are significant and not because of stretches structurally heterogeneous polypeptide or crystal packing artifacts affecting specific molecules.
FIGURE 3. Superposition of n1α_LNS#2 and n 1β_LNS.
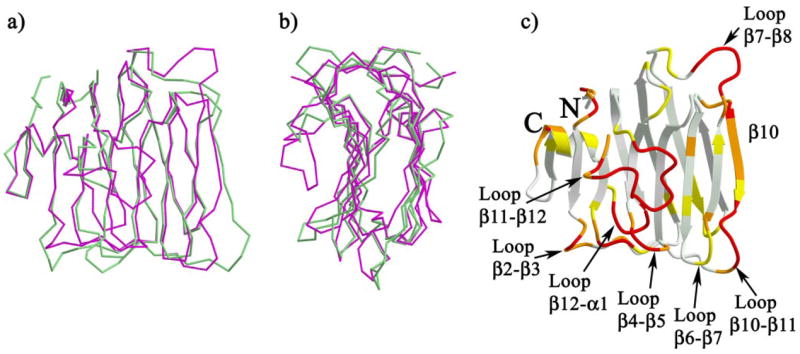
a and b, face and side views of the polypeptide Cα-trace superposition of n1α_LNS#2 (magenta) and n1β_LNS (green); c, ribbon diagram of n1α_LNS#2 colored according to the r.m.s.d. between pairs of structurally analogous Cα atoms from representative structures of n1α_LNS#2 and n1β_LNS: white (≤0.5 Å), light yellow-green (0.5 < r.m.s.d. ≤ 1.0 Å), yellow (1.0 < r.m.s.d. ≤ 1.5 Å), orange (1.5 < r.m.s.d. ≤ 3.0 Å), and red (>3.0 Å or not present in n1β_LNS). The N and C termini are indicated with N and C, respectively; loops and strands referred to in the text are indicated. Superposition and analyses were carried out using routines in LSQMAN (52) as described under “Experimental Procedures.”
Isothermal Titration Calorimetry
N1α_LNS#2 splice isoforms with 0, 8, or 15 amino acids at SS#2 were purified as described above and then rendered Ca2+-free by treating the protein in 20 mm HEPES, pH 8.5, 50 mm NaCl, 1 mm EDTA for 1 h at 4 °C. The protein samples were then buffer-exchanged using Amicon-10 concentrators (Millipore) five times with Ca2+-free buffer (20 mm HEPES, pH 8.5, 50 mm NaCl treated with CHELEX-100 resin) to remove the EDTA. For each protein sample, the flow-through from the final concentration step was collected and used as the experimental ITC “buffer” to prepare the 11.518 mm CaCl2 titrant solution and to determine the heat of dilution of titrant into buffer alone (i.e. in the absence of protein). ITC experiments were carried out with a VP-ITC MicroCal calorimeter. The experimental set-up was rendered Ca2+-free by treating the sample cell, automatic pipettor, and other reagent-handling aids like syringes, tubing, tubes, and stir bars with 1 mm EDTA for 2 min, followed by extensive washing with CHELEX-treated Millipore water, and finally with CHELEX-100 treated buffer (20 mm HEPES, pH 8.5, 50 mm NaCl). Calorimetric titrations were carried out by placing a protein solution (ranging from 0.35 to 0.5 mm) into the MicroCal sample cell, placing water in the reference cell, and titrating the protein with an 11.518 mm CaCl2 solution at 25 °C. CaCl2 was added to the sample cell in a series of 29 injections of 5 μl each, separated by 180 or 300 s. The raw ITC data were deconvoluted using the ORIGIN software provided by MicroCal to obtain least-square estimates of N (number of sites), ΔH° (heat change in calories/mol), and K (binding constant m−1). Before and after ITC runs with neurexins, a control ITC titration was performed with hen egg white lysozyme (Sigma) and N,N′,N″-triacetylchitotriose (Sigma) (Kd 2.6 μm (53)), to monitor the ITC machine performance. After each ITC run, the protein sample was removed from the sample cell and centrifuged at 10,000 rpm at 4 °C to monitor possible protein precipitation. All three n1α_LNS#2 splice isoforms exhibited a small amount of precipitation (estimated at <5%) after being subjected to an ITC run. In addition to performing calorimetric experiments on the isolated n1α_LNS#2 domains, calorimetric titration of n1α_LNS#2 with no insert at SS#2 and n1α_LNS#2 containing the eight-residue insert at SS#2 was also carried out on the N-terminally fused glutathione S-transferase fusion proteins, yielding similar binding isotherms.
RESULTS
Overall Structure of the Neurexin 1α LNS#2 Domain
The structure of neurexin 1α LNS#2 (n1α_LNS#2) was solved using crystals with space group P1 (cell dimensions a = 60.0 Å, b = 61.6 Å, c = 66.6 Å, α= 78.5°, β = 78.7°, γ = 84.7°) in a multi-anomalous dispersion experiment exploiting the anomalous signal of four selenium atoms per monomer (Table 1). Four independent monomers are found in the asymmetric unit. The structure of n1α_LNS#2 is composed of 13 β-strands and 1α-helix (Fig. 2). N1α_LNS#2 forms a β-sandwich with the dimensions 40 Å high, 36 Å wide, and 30 Å deep (Fig. 2, left-hand view). The β-sheet on the concave side of the molecule is formed by seven strands; the β-sheet on the convex side is formed by six β-strands. Loop β11–β12 protrudes extensively out in space filling the depression formed by the arching β-strands of the concave β-sheet.
FIGURE 2. Ribbon diagram of n1α_LNS#2.
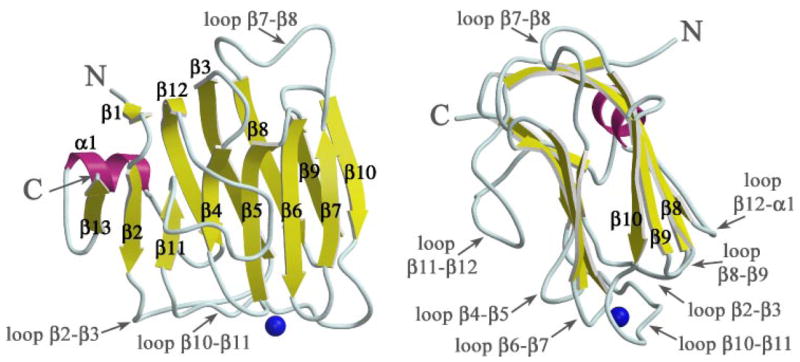
The β-sandwich is shown in a face view (left) and side view (right). β-Strands are depicted as yellow arrows, the single α-helix is shown in magenta, and the Ca2+ ion as a blue sphere. The N and C termini of the polypeptide chain are indicated with N and C, respectively.
Comparison of n1α_LNS#2 with n1β_LNS/n1α_LNS#6
The overall fold of n1α_LNS#2 is similar to n1β_LNS/n1α_LNS#6, even though the sequence identity between n1α_LNS#2 and n1β_LNS is only 20.4% for the 167 alignable Cα atoms residues out of 182 (Fig. 3, a and b). The regions of variation are located on one side of the molecule running along two edges of the β-sheet opposite to the N and C termini and in the polypeptide loop filling the concave side of the β-sandwich (Fig. 3c). The highly variable regions (>3 Å r.m.s.d. in Cα atom positions) involve predominantly loops: loop β2–β3, loop β4–β5, loop β7–β8, loop β10–β11, loop β11–β12, and loop β12–α1. The β-strand β10 is the only variable secondary structural element. The two biggest regions of contiguous variability are seen firstly along the edge of the β-sandwich encompassing part of loop β10–β11, the strand β10, and loop β7–β8, and secondly to the polypeptide loop filling the concave face of the molecule (loop β11–β12).
Calcium Binding Site in n1α_LNS#2
N1α_LNS#2 was crystallized in the presence of 5 mm CaCl2. Its x-ray structure reveals strong positive electron density peaks in three monomers and a weaker site in the fourth monomer (between 6.8 and 8.5 σ in composite simulated annealed omit maps calculated with SigmaA-weighted 2m|Fo| − D|Fc| coefficients and phases from a metal-free protein model). The density and the surrounding ligands are consistent with a Ca2+ ion bound to a Ca2+ -binding site (Fig. 4 and Table 2). A side-chain carboxyl (Asp329) and two main-chain carbonyl oxygens (Met414 and Leu346) provide protein ligands to the Ca2+ -binding site. The Ca2+ coordination (n = 6) is completed by three water molecules and adopts an approximate octahedral geometry. The Ca2+ to oxygen atom distances range from 2.2 to 2.5 Å, well within the range of 2.1–2.8 Å typically found for carboxylate and carbonyl ligands in proteins, and 2.3–2.9 Å for coordinating waters (54). N1α_LNS#2 contributes thus only three direct protein ligands to the Ca2+ -ion. The Ca2+ -binding site is located in the middle of the hyper-variable surface (Fig. 5, a and b). The protein region directly surrounding the Ca2+ -binding site is conserved between n1α_LNS#2 and n1β_LNS/n1α_LNS#6 both in terms of amino acid sequence (Fig. 5c) and three-dimensional structure (Fig. 5d). However, the loops encircling the Ca2+ -binding site display very little sequence conservation and great structural variation (Fig. 5, c and d). In other words there is a virtually complete breakdown of amino acid sequence conservation and structural conservation outside the immediate surroundings of the Ca2+ -binding site, indicating that the hyper-variable surfaces of different neurexin LNS/LG domains display highly specific surfaces able to provide domain specific protein partner recognition.
FIGURE 4. Ca2+-binding site in n1α_LNS#2.
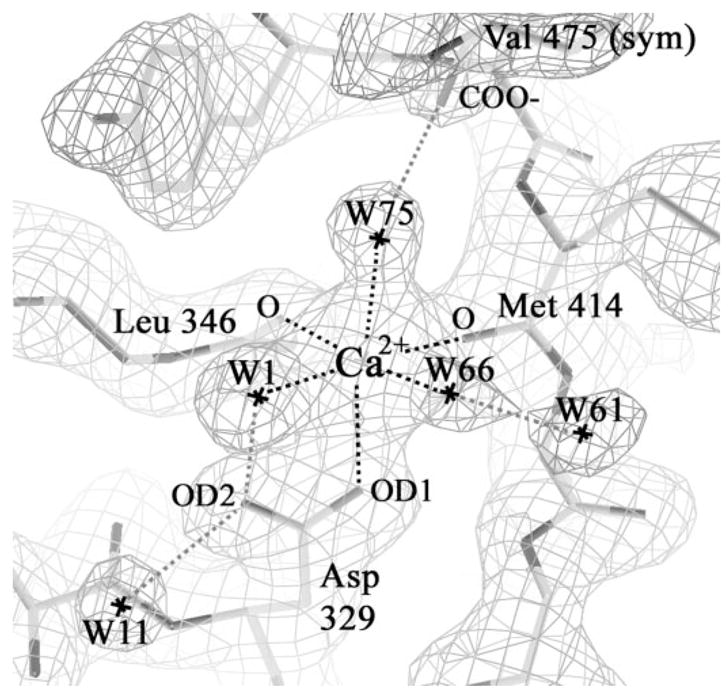
The SigmaA-weighted electron density 2m|Fo| − D|Fc| contoured at 1.2 σ and the atomic model are shown depicting the Ca2+-binding site found at the hyper-variable surface of n1α_LNS#2. Dotted lines indicate the interaction network between amino acid residues and solvent molecules chelating the Ca2+ ion. The binding site in Mol2 is depicted (see Table 2).
TABLE 2.
Ca2+-binding site in n1α_LNS#2
| Mol1 | Mol2 | Mol3 | Mol4 | |
|---|---|---|---|---|
| Å | ||||
| Met414 (C=O) | 2.2 | 2.3 | 2.3 | 2.3 |
| Leu346 (C=O) | 2.3 | 2.3 | 2.4 | 2.2 |
| Asp329 (Oδ1) | 2.4 | 2.4 | 2.4 | 2.4 |
| Wat1 | 2.3 | 2.5 | 2.3 | 2.3 |
| Wat66 | 2.3 | 2.4 | 2.2 | 2.4 |
| Wat75 | 2.5 | 2.5 | n.p.a | n.p. |
| Sym Glu356 (Oε1) | n.p. | n.p. | 2.3 | 2.4 |
| Sym Val475 (COO−) | p. | p. | n.p. | n.p. |
p. = present; n.p. = not present.
FIGURE 5. The hyper-variable surface in n1α_LNS#2.
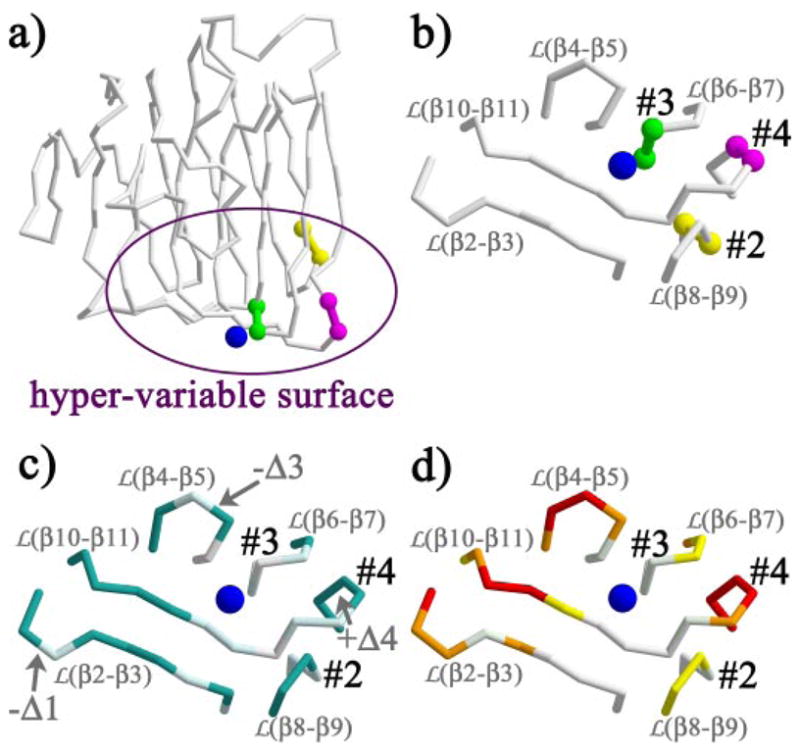
a, splice insert sites SS#2 (yellow), SS#3 (green), and SS#4 (magenta) visualized on the n1α_LNS#2 polypeptide backbone trace; b, the splice insert sites SS#2, SS#3, and SS#4 localize in a semicircle around the Ca2+-binding site (blue sphere); c, amino acid sequence conservation between n1α_LNS#2 and n1β_LNS/n1α_LNS#6 (accession code nm_174404); identical residues are shown in white, semi-conserved residues are in light cyan, and non-conserved residues are in dark aquamarine. Insertions or deletions of residues are indicated by +Δ or −Δ, respectively, followed by the number of residues present additionally or absent. Semi-conserved amino acids are defined as: (K/R/H), (P), (C), (F/Y/W/L), (D/E/Q/N), (V/I/M/A/L), (S/T/A), and (G/A); note that L and A are present in multiple groups; d, structural conservation between n1α_LNS#2 and n1β_LNS at the hyper-variable surface, coloring scheme as in Fig. 3c.
The Hyper-variable Surface: Modulation by Alternative Splicing
The three sites in LNS/LG domains that accommodate short polypeptide inserts as a result of alternative splicing were mapped onto the structure of n1α_LNS#2 (Fig. 5, a and b). The alternative splice insertion site SS#2 is found between the adjacent residues Gln378 and Val394 (using a residue numbering scheme accommodating a 15-residue splice insert). Alternative splicing at SS#2 inserts resi dues at the start of strand β9, just after a very tight two-residue loop between strand β8 and strand β9 (Figs. 2 and 5b). Sequence alignment of n1α_LNS#2, n1α_LNS#4, and n1α_LNS#6 indicates that splicing at SS#3 (taking place in n1α_LNS#4) inserts residues at a position equivalent to loop β6–β7 (Figs. 2 and 5b). The structures of n1α_LNS#2 and n1β_L/n1α_LNS#6 show that both loop β8–β9 and loop β6–β7 display very little structural variation (Fig. 5d), indicating that these loops do not readily adopt different conformations in the crystal structures and are potentially not very flexible. Hence, even though loop β8–β9 in n1α_LNS#2 domain can accommodate an extra 8- or 15-aa splice insert, in the splice free form it adopts the same conformation as its counterpart in n1β_LNS/n1α_LNS#6, which does not accommodate splice inserts. It is possible that a similar scenario is repeated with loop β6–β7, i.e. the loop can adopt the same spatial conformation in n1α_LNS#2, n1α_LNS#4, and n1β_LNS/n1α_LNS#6 as long as no splice insert is incorporated. Loop β10–β11, which houses SS#4 in n1β_LNS/n1α_LNS#6, incorporating 0 or 30 amino acids, adopts very different conformations between the struc tures of n1α_LNS#2 and n1β_LNS/n1α_LNS#6 revealing a lack of conformational restriction (Fig. 5d).
The Hyper-variable Surface: Presence of Splice Insert Sites and a Ca2+-binding site
The alternative splice insert sites SS#2, SS#3, and SS#4 found in neurexin LNS/LG domains localize in a semicircle around the Ca2+ ion (SS#3 4 Å, SS#4 11 Å, and SS#2 14 Å away from the Ca2+ ion, respectively) (Fig. 5b). We used ITC to measure the affinity of n1α_LNS#2 for Ca2+, and prompted by their proximity, we investigated whether the presence of alternative splice inserts would modulate Ca2+ binding. Titration of Ca2+ into a solution of n1α_LNS#2 with no insert present at SS#2 generates an exothermic reaction, releasing heat upon Ca2+ binding to protein. The raw ITC data were processed and fitted using a model for a single binding site. The Ca2+-binding site displays a low binding affinity with a Kd of ~400 μm (standard deviation, 48.1 μm) and a stoichiometry (n) of one Ca2+ per n1α_LNS#2 monomer. The values were derived from three independent experiments yielding Kd = 346 μm with n = 1.15, 442 μm with n = 0.94, and 400 μm with n = 1.1 (Fig. 6a). To test if the presence of an 8- or 15-aa insert at SS#2 affects Ca2+-binding affinity, we performed ITC experiments with the two splice isoforms as well. The presence of additional residues at SS#2 drastically alters the binding isotherm produced in the ITC reaction (Fig. 6, b and c, respectively). Titrating Ca2+ into a protein solution of the splice isoform n1α_LNS#2 + 8 aa or n1α_LNS#2 + 15 aa generates a very weak endothermic reaction from which no detectable Ca2+-binding affinity can be derived. Surprisingly, the presence of the 8-aa insert or the 15-aa insert at SS#2 has approximately the same effect on the enthalpy of the reaction.
FIGURE 6. ITC of n1α_LNS#2 splice isoforms.
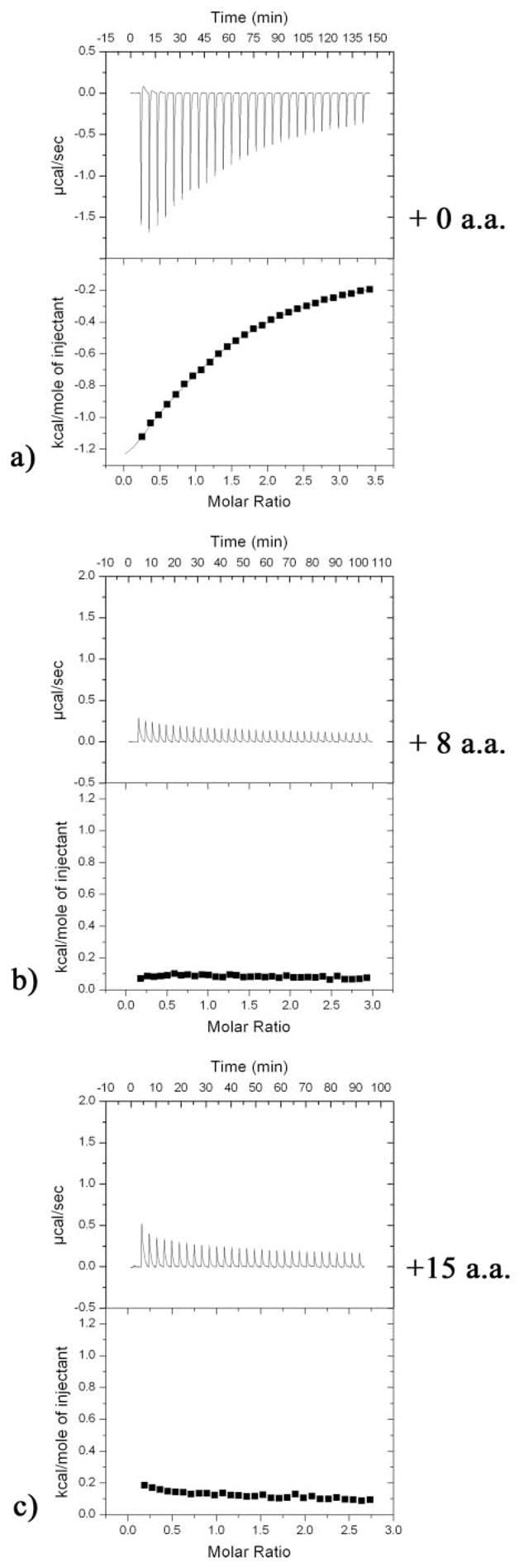
The titrations were carried out as described under “Experimental Procedures.” For each splice isoform: the top figure shows the calorimetric titration of 5-μl injections of a 11.518 mm CaCl2 solution into protein; the bottom figure shows the normalized binding isotherm displaying heat absorbed/released per kilocalorie/mol of CaCl2 injected as a function of molar ratio metal:protein (squares), with the best least-squares fit to a binding model for one site (solid line) superimposed. For each run the heat of dilution of the CaCl2 solution has been subtracted (measured with a titration of CaCl2 into flow-through buffer alone). a, n1α_LNS#2.
Our results establish a paradigm for hyper-variable surfaces in different neurexin LNS/LG domains as the putative protein interaction sites. We show that the hyper-variable surface contains 1) a conserved Ca2+-binding site, 2) specialized residues and three-dimensional structure surrounding the Ca2+-binding site that are poised to interact with the different protein partners recognized by the different domains, and 3) alternative splice insert sites that surround the Ca2+-binding site in close proximity (Fig. 5, b–d). The hyper-variable surface paradigm is extended with the finding that at least some alternative splice inserts may control protein partner binding indirectly by altering the affinities of the different neurexin LNS/LG domains for Ca2+.
DISCUSSION
Neurexin LNS/LG Domains as a Scaffold for Protein-Protein Interactions
To understand how different neurexin splice isoforms can impact synapse functioning on an atomic level, we are characterizing the molecular properties of different neurexin LNS/LG domains and their splice isoforms using structural, biochemical, and biophysical techniques. Previous studies have revealed that the structure of n1β_LNS/n1α_LNS#6 (solved in a metal-free form) contains a β-sandwich and that the three splice insertion sites found in neurexin LNS/LG domains map close together in space to form a hyper-variable surface (34). Our current studies reveal that, although n1α_LNS#2 adopts a similar overall fold to n1β_L/n1α_LNS#6, large differences in backbone conformation and sequence are observed at two edges of the β-sandwich (including the hyper-variable surface) and to the loop β11–β12 filling the depression formed by the concave curvature of the β-sandwich. The variable regions along the rim of the β-sandwich are found opposite the N and C termini, which tether LNS/LG domains in place in the full-length proteins. In plant lectins with a related fold, an analogous loop to loop β11–β12 is often involved in forming a carbohydrate-binding site (55), although in neurexins the functional significance for this loop has so far not been revealed. Hence, an increasing body of research points toward neurexin LNS/LG domains containing localized regions of variability that coincide with regions of functional significance; in the context of the full-length extracellular domains, these areas will be proffered to the solvent as interaction surfaces.
Neurexin LNS/LG Domains as Ca2+-binding Domains
The structure of n1α_LNS#2 crystallized in the presence of Ca2+ reveals a single metal-binding site located at the hyper-variable with no splice insert; b, n1α_LNS#2 with the 8-aa insert at SS#2; and c, n1α_LNS#2 with the 15-aa insert at SS#2. surface providing direct evidence that neurexin LNS/LG domains contain Ca2+-binding sites. The Ca2+ ion makes only three direct contacts to the neurexin polypeptide chain, using water molecules to complete an octahedral coordination. Our ITC studies indicate that the Ca2+-binding site in n1α_LNS#2 has only low affinity for Ca2+. Sugita and co-workers have shown that n1α_LNS#2 strictly requires Ca2+ to bind α-dystroglycan, and they were able to abolish α-dystroglycan binding by mutating Asp345 in n1α_LNS#2 (equivalent to Asp329 in our structure)3 that they suspected might be part of a Ca2+-binding site (22). The Ca2+-dependent nature of ligand binding can be explained if Ca2+ triggers conformational changes to neurexin LNS/LG domains that enable ligand binding. However, given the small number of residues in n1α_LNS#2 that directly coordinate the Ca2+ion, it is more conceivable that protein partners interact directly with the Ca2+ ion as they bind n1α_LNS#2, providing additional ligands to displace the water molecules observed in the crystal structure (Fig. 4). It will be important to determine if the Ca2+-binding properties of the other neurexin LNS/LG domains differ, and if properties characterized for the isolated n1α_LNS#2 are recapitulated in the context of the full-length protein.
Communication between Ca2+-binding Sites and Alternative Splice Insert Sites
The most striking observation from our studies is the close proximity of the Ca2+-binding site to SS#2, SS#3, and SS#4, strongly suggesting that incorporation of splice inserts can affect Ca2+ binding. In particular, extra residues inserted at SS#3 as a result of alternative splicing must be directly incorporated at the Ca2+-binding site and in a position to profoundly affect the Ca2+-binding environment (Fig. 5b). Using ITC, we demonstrate that the presence of splice inserts at SS#2 in n1α_LNS#2, though farther away from the Ca2+-binding site than SS#3, do indeed appear to alter properties of the Ca2+-binding site (Fig. 6). Because the outcome of a calorimetric reaction is the sum of the total heat released or absorbed in the sample cell, Ca2+ binding to the protein as well as any other changes triggered by Ca2+ addition contribute to the observed isotherm. For this reason there are two likely interpretations of the ITC data presented for n1α_LNS#2 (Fig. 6). The first interpretation is that the Ca2+-binding site in n1α_LNS#2 is obstructed if the 8- or 15-aa splice insert is present (or has very poor affinity), and Ca2+-binding is no longer measurable. The second interpretation is that Ca2+ still binds to splice isoforms containing the 8- or 15-aa splice insert but that Ca2+ triggers a conformational change in the protein or an alternative reaction that overshadows the exothermic energy produced upon Ca2+ binding to the protein. The effects of Ca2+ could be indirect; for example a conformational change to the protein might change the chemical environments of histidine residues present in both the 8- and 15-aa splice inserts (Fig. 1), shifting their pKa and altering their exchange of protons with the buffer. To evaluate what a binding isotherm would look like for a protein known not to bind Ca2+ even at molar concentrations (56), we titrated lysozyme with Ca2+ under conditions similar to those used to assay the n1α_LNS#2 isoforms, albeit using a different buffer and pH more suited to lysozyme. The calorimetric reaction resulted in a very small exothermic reaction on the same scale as the heat of dilution of the titrant, i.e. the baseline generated upon titrating Ca2+ into buffer alone (result not shown). Hence, the endothermic reaction observed upon titrating splice isoforms of n1α_LNS#2 with Ca2+, though small, may indicate that multiple events take place when Ca2+ is added to LNS/LG domains in solution (Fig. 6, b and c).
Significance of a Ca2+-binding Site in n1α_LNS#2
The low affinity of n1α_LNS#2 for Ca2+ (KD ~ 0.4 mm) comes as a surprise. Although no accurate measurements exist for the Ca2+concentration in the synaptic cleft, it is estimated to be ~1 mm and likely depletes more than 30–60% as pre-synaptic and post-synaptic Ca2+ channels open (57). The weak Ca2+-binding site found in n1α_LNS#2 could thus be subject to varying Ca2+ occupancies and hence varying abilities to interact with protein partners at the synapse. Incorporation of the 8- and 15-aa splice inserts appears to further decrease the affinity of Ca2+-binding site to the point where it likely would not be occupied even at a basal Ca2+ concentration. α-Dystroglycan, which binds n1α_LNS#2 only when Ca2+ is present (22), appears to selectively localize to a subset of inhibitory synapses (58). Our studies raise the question whether the fluctuating Ca2+concentrations at the synaptic cleft of these specialized synapses could regulate α-dystroglycan-neurexin interaction in vivo, influenced by the presence of alternative splice inserts at site SS#2.
Relation between LNS/LG Domains from Neurexins, Agrin, and Laminin
At first glance there are significant parallels between neurexin LNS/LG domains and the structurally related “G-domains” found in the large multidomain proteins agrin and laminin. These domains are all now known to bind Ca2+ (Fig. 4) (59, 60) at the rim of the β-sandwich, all three proteinsuseLNS/Lg domains to bind α-dystroglycan in aCa2+-dependent way (22, 61–64), and LNS/LG domains in agrin undergo alternative splicing (regulating their ability in neural cells to promote postsynaptic development at neuromuscular junctions, reviewed recently in Ref. 65). However, closer examination suggests differences on an atomic level that dictate how these domains employ structure to gain function.
N1α_LNS#2 and the agrin G3 domain implement splice inserts and a Ca2+-binding site in very different ways. The agrin G3 B-site (also called site Z) incorporates splice inserts in a loop equivalent to loop β2–β3 in n1α_LNS#2 (a loop that is not modified by splicing in neurexin LNS/LG domains but does localize close to the hyper-variable surface) (34, 60). Splice inserts at the agrin G3 B-site do not provide ligands to the Ca2+ ion and are flexible in the presence or absence of Ca2+, indicating little communication between the Ca2+ ion and the splice inserts (60). Strikingly, unlike apparently n1α_LNS#2, the agrin G3 affinity for Ca2+ is not radically affected by the presence of splice inserts (Kd ~ 0.6 mm for G3-B0 with no insert, versus Kd ~1mm for G3-B8 with an 8-aa insert) (60). Furthermore, in the case of neurexin LNS/LG domains, all three splice insertion sites arrange around the Ca2+-binding site, with loop β6 –β7 and loop β10 –β11 housing not only a splice insert but also providing a ligand to the Ca2+-binding site as well. Hence, neurexin LNS/LG domains appear to require specific interplay between Ca2+-binding and splice insert sites not seen in agrin G3.
No structural information is available yet for neurexin LNS/LG domains carrying splice inserts. Structural studies of the agrin G3 domain reveal that no significant rearrangements take place to accommodate splice inserts (0, 8, or 11 aa) at the B-site, extending splice inserts as flexible loops from the edge of the β-sandwich (60). Because two of three splice insert sites in neurexin LNS/LG domains map to regions that are highly structurally conserved between n1α_LNS#2 and n1β_L/n1α_LNS#6 (namely SS#2 and SS#3), it is possible that like agrin G3 the neurexin LNS/LG structural scaffold does not require major rearrangement to accommodate extra residues at these sites. However, the region encompassing splice site SS#4 (also found in agrin G2) is highly variable in structure between n1α_LNS#2 and n1β_LNS/n1α_LNS#6; this may indicate that the n1α_LNS#6/n1β_LNS domain has evolved a special scaffold able to accommodate the large 30-residue splice insert specific to SS#4.
Although laminin α2 G4 and neurexin n1α_LNS#2 are sufficient to bind α-dystroglycan, both in a Ca2+-dependent fashion, their crystal structures indicate that the binding surfaces are not conserved. Structure-based mutagenesis for the laminin α2 G4_G5 tandem has revealed the primary requirement of Arg2803 and the Ca2+ ion in G4; the structure shows that Arg2803 points toward the Ca2+ ion at very close distance (5.8 Å) (36). The hyper-variable surface of n1α_LNS#2 does not contain an analogue to laminin α2 G4 Arg2803. While a lysine (Lys326) is present at a similar place in the n1α_LNS#2 amino acid sequence, its side chain points away from the Ca2+ ion and the hyper-variable surface because loop β4–β5 (on which it resides) is two residues shorter, rearranging the loop conformation compared with laminin α2 G4. A possible alternative binding epitope for n1α_LNS#2 would be Arg377, which is 14 Å from the Ca2+ ion but directly precedes the splice insert site SS#2 (between Gln278 and Val294) incorporating the splice inserts known to regulate binding to α-dystroglycan. It is less clear for agrin whether single LNS/LG domains are sufficient to bind α-dystroglycan and which domains these might be, although alternative splicing of these domains does appear to regulate binding to α-dystroglycan (62–64). The emerging picture is that LNS/LG domains in neurexins, agrins, and laminins share a common protein fold, and even a common protein partner, but they diverge significantly in their atomic details, which are likely tailored to their different protein functions.
CONCLUSIONS
The neurexin family appears strategically placed to form an expansive set of synaptic building blocks. Alternative splicing, estimated to affect >74% of all multiexon genes in humans (66), is suspected to play a crucial role in the nervous system of higher organisms as a mechanism to derive higher complexity with a limited number of genes (67). Because of their potential to generate thousands of splice isoforms distributed in distinct spatial and temporal ways, neurexins are ideally suited to integrate into the vast protein network at synapses generating distinct and specialized functions. Systematic molecular characterization of different neurexin LNS/LG domains and their isoforms is a prerequisite to understanding how alternative splicing modulates the molecular properties of neurexin LNS/LG domains. The current studies indicate that molecular switches regulating protein partner binding (i.e. alternative splice inserts and Ca2+) locate next to each other on the molecular surface and work on each other. Our data provide the first evidence that alternative splice inserts in neurexin LNS/LG domains may regulate protein partner binding at the synapse not only directly but also indirectly as well by altering Ca2+-binding affinity at the hyper-variable surface. Future work will focus on further characterizing the effect of alternative splice inserts on neurexin structure and function.
Acknowledgments
We thank Prof. Johann Deisenhofer for support and encouragement during the initial stage of this project and Dr. Bruce Palfey for useful discussions regarding isothermal titration calorimetry.
Footnotes
This work was supported by the National Alliance for Research on Schizophrenia and Depression, the American Heart Association, and by the National Institute of Mental Health Grant R01MH077303. Synchrotron radiation at Cornell High Energy Synchrotron Source (CHESS) used for data collection was funded by the National Science Foundation under Grant DMR 0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which was supported by award RR-01646 from the National Institutes of Health, through its National Center for Research Resources.
The atomic coordinates and structure factors (code 2H0B) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The abbreviations used are: LNS, laminins, neurexins, and sex hormone-binding globulin; LG, laminin G domains; aa, amino acid(s); r.m.s.d., root mean square deviation; ITC, isothermal titration calorimetry; NCS, non-crystallographic symmetry.
S. Sugita, personal communication.
References
- 1.Cline H. Curr Biol. 2005;15:R203–R205. doi: 10.1016/j.cub.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Waites CL, Craig AM, Garner CC. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- 3.Craig AM, Graf ER, Linhoff MW. Trends Neurosci. 2006;29:8–20. doi: 10.1016/j.tins.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dean C, Dresbach T. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Ushkaryov YA, Petrenko AG, Geppert M, Sudhof TC. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- 6.Ushkaryov YA, Hata Y, Ichtchenko K, Moomaw C, Afendis S, Slaughter CA, Sudhof TC. J Biol Chem. 1994;269:11987–11992. [PubMed] [Google Scholar]
- 7.Rowen L, Young J, Birditt B, Kaur A, Madan A, Philipps DL, Qin S, Minx P, Wilson RK, Hood L, Graveley BR. Genomics. 2002;79:587–597. doi: 10.1006/geno.2002.6734. [DOI] [PubMed] [Google Scholar]
- 8.Tabuchi K, Sudhof TC. Genomics. 2002;79:849–859. doi: 10.1006/geno.2002.6780. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen T, Sudhof TC. J Biol Chem. 1997;272:26032–26039. doi: 10.1074/jbc.272.41.26032. [DOI] [PubMed] [Google Scholar]
- 10.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 11.Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Nat Neurosci. 2003;6:708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Sudhof TC. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- 13.Kattenstroth G, Tantalaki E, Sudhof TC, Gottmann K, Missler M. Proc Natl Acad Sci U S A. 2004;101:2607–2612. doi: 10.1073/pnas.0308626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Rohlmann A, Sargsyan V, Aramuni G, Hammer RE, Sudhof TC, Missler M. J Neurosci. 2005;25:4330–4342. doi: 10.1523/JNEUROSCI.0497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nam CI, Chen L. Proc Natl Acad Sci U S A. 2005;102:6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boucard AA, Chubykin AA, Comoletti D, Taylor P, Sudhof TC. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 18.Sons MS, Busche N, Strenzke N, Moser T, Ernsberger U, Mooren FC, Zhang W, Ahmad M, Steffens H, Schomburg ED, Plomp JJ, Missler M. Neuroscience. 2006;138:433–446. doi: 10.1016/j.neuroscience.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 19.Davletov BA, Krasnoperov V, Hata Y, Petrenko AG, Sudhof TC. J Biol Chem. 1995;270:23903–23905. doi: 10.1074/jbc.270.41.23903. [DOI] [PubMed] [Google Scholar]
- 20.Missler M, Hammer RE, Sudhof TC. J Biol Chem. 1998;273:34716–34723. doi: 10.1074/jbc.273.52.34716. [DOI] [PubMed] [Google Scholar]
- 21.Sugita S, Khvochtev M, Sudhof TC. Neuron. 1999;22:489–496. doi: 10.1016/s0896-6273(00)80704-7. [DOI] [PubMed] [Google Scholar]
- 22.Sugita S, Saito F, Tang J, Satz J, Campbell K, Sudhof TC. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrenko AG, Ullrich B, Missler M, Krasnoperov V, Rosahl TW, Sudhof TC. J Neurosci. 1996;16:4360–4369. doi: 10.1523/JNEUROSCI.16-14-04360.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof TC. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- 25.Ichtchenko K, Nguyen T, Sudhof TC. J Biol Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- 26.Littleton JT, Sheng M. Nature. 2003;423:931–932. doi: 10.1038/423931a. [DOI] [PubMed] [Google Scholar]
- 27.Cantallops I, Cline HT. Curr Biol. 2000;10:R620–R623. doi: 10.1016/s0960-9822(00)00663-1. [DOI] [PubMed] [Google Scholar]
- 28.Missler M. Trends Neurosci. 2003;26:176–178. doi: 10.1016/S0166-2236(03)00066-3. [DOI] [PubMed] [Google Scholar]
- 29.Rao A, Harms KJ, Craig AM. Nat Neurosci. 2000;3:747–749. doi: 10.1038/77636. [DOI] [PubMed] [Google Scholar]
- 30.Ullrich B, Ushkaryov YA, Sudhof TC. Neuron. 1995;14:497–507. doi: 10.1016/0896-6273(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 31.Zeng Z, Sharpe CR, Simons JP, Gorecki DC. Int J Dev Biol. 2006;50:39–46. doi: 10.1387/ijdb.052068zz. [DOI] [PubMed] [Google Scholar]
- 32.Patzke H, Ernsberger U. Mol Cell Neurosci. 2000;15:561–572. doi: 10.1006/mcne.2000.0853. [DOI] [PubMed] [Google Scholar]
- 33.Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 34.Rudenko G, Nguyen T, Chelliah Y, Sudhof TC, Deisenhofer J. Cell. 1999;99:93–101. doi: 10.1016/s0092-8674(00)80065-3. [DOI] [PubMed] [Google Scholar]
- 35.Grishkovskaya I, Avvakumov GV, Sklenar G, Dales D, Hammond GL, Muller YA. EMBO J. 2000;19:504–512. doi: 10.1093/emboj/19.4.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wizemann H, Garbe JH, Friedrich MV, Timpl R, Sasaki T, Hohenester E. J Mol Biol. 2003;332:635–642. doi: 10.1016/s0022-2836(03)00848-9. [DOI] [PubMed] [Google Scholar]
- 37.Rudenko G, Hohenester E, Muller YA. Trends Biochem Sci. 2001;26:363–368. doi: 10.1016/s0968-0004(01)01832-1. [DOI] [PubMed] [Google Scholar]
- 38.Grant SG. Biochem Soc Trans. 2006;34:59–63. doi: 10.1042/BST0340059. [DOI] [PubMed] [Google Scholar]
- 39.Guan KL, Dixon JE. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 40.Matthews BW. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Collaborative Computational Project Number 4. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:760–763. [Google Scholar]
- 43.Terwilliger TC. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 44.Ramakrishnan V, Biou V. Methods Enzymol. 1997;276:538–557. [PubMed] [Google Scholar]
- 45.Cowtan K. Joint CCP4 + ESF-EAMCB Newsletter on Protein. Crystallography. 1994;31:34–38. [Google Scholar]
- 46.Kleywegt GJ, Jones TA. In: From First Map to Final Model. Bailey S, Hubbard R, Waller D, editors. SERC Daresbury Laboratory; Warrington, UK: 1994. pp. 59–66. [Google Scholar]
- 47.Jones TA, Zou JY, Cowan SW, Kjeldgaard Acta Crystallogr Sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 48.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr Sect D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 49.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 50.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 51.Merritt EA, Bacon DJ. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 52.Kleywegt GJ, Jones TA. CCP4/ESF-EACBM Newsletter on Protein. Crystallography. 1994;31:9–14. [Google Scholar]
- 53.Cooper A, Johnson CM, Lakey JH, Nollmann M. Biophys Chem. 2001;93:215–230. doi: 10.1016/s0301-4622(01)00222-8. [DOI] [PubMed] [Google Scholar]
- 54.Dudev T, Lim C. Chem Rev. 2003;103:773–788. doi: 10.1021/cr020467n. [DOI] [PubMed] [Google Scholar]
- 55.Rini JM. Annu Rev Biophys Biomol Struct. 1995;24:551–577. doi: 10.1146/annurev.bb.24.060195.003003. [DOI] [PubMed] [Google Scholar]
- 56.Li G, Lee D, Wang L, Khvotchev M, Chiew SK, Arunachalam L, Collins T, Feng ZP, Sugita S. J Neurosci. 2005;25:10188–10197. doi: 10.1523/JNEUROSCI.3560-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith SM, Bergsman JB, Harata NC, Scheller RH, Tsien RW. Neuron. 2004;41:243–256. doi: 10.1016/s0896-6273(03)00837-7. [DOI] [PubMed] [Google Scholar]
- 58.Levi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM. J Neurosci. 2002;22:4274–4285. doi: 10.1523/JNEUROSCI.22-11-04274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tisi D, Talts JF, Timpl R, Hohenester E. EMBO J. 2000;19:1432–1440. doi: 10.1093/emboj/19.7.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stetefeld J, Alexandrescu AT, Maciejewski MW, Jenny M, Rathgeb-Szabo K, Schulthess T, Landwehr R, Frank S, Ruegg MA, Kammerer RA. Structure (Camb) 2004;12:503–515. doi: 10.1016/j.str.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 61.Talts JF, Andac Z, Gohring W, Brancaccio A, Timpl R. EMBO J. 1999;18:863–870. doi: 10.1093/emboj/18.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gesemann M, Cavalli V, Denzer AJ, Brancaccio A, Schumacher B, Ruegg MA. Neuron. 1996;16:755–767. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- 63.Hopf C, Hoch W. J Biol Chem. 1996;271:5231–5236. doi: 10.1074/jbc.271.9.5231. [DOI] [PubMed] [Google Scholar]
- 64.Campanelli JT, Gayer GG, Scheller RH. Development. 1996;122:1663–1672. doi: 10.1242/dev.122.5.1663. [DOI] [PubMed] [Google Scholar]
- 65.Kummer TT, Misgeld T, Sanes JR. Curr Opin Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 66.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 67.Graveley BR. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]