

Abstract
Serotonin N-acetyltransferase is the enzyme responsible for the diurnal rhythm of melatonin production in the pineal gland of animals and humans. Inhibitors of this enzyme active in cell culture have not been reported previously. The compound N-bromoacetyltryptamine was shown to be a potent inhibitor of this enzyme in vitro and in a pineal cell culture assay (IC50 ≈ 500 nM). The mechanism of inhibition is suggested to involve a serotonin N-acetyltransferase-catalyzed alkylation reaction between N-bromoacetyltryptamine and reduced CoA, resulting in the production of a tight-binding bisubstrate analog inhibitor. This alkyltransferase activity is apparently catalyzed at a functionally distinct site compared with the acetyltransferase activity active site on serotonin N-acetyltransferase. Such active site plasticity is suggested to result from a subtle conformational alteration in the protein. This plasticity allows for an unusual form of mechanism-based inhibition with multiple turnovers, resulting in “molecular fratricide.” N-bromoacetyltryptamine should serve as a useful tool for dissecting the role of melatonin in circadian rhythm as well as a potential lead compound for therapeutic use in mood and sleep disorders.
Keywords: pineal, acetyltransferase, circadian rhythm
Melatonin (N-acetyl-5-methoxytryptamine) is a pineal hormone that contributes to physiologic timekeeping in a wide variety of organisms, including humans (1–3). Serotonin (5-hydroxytryptamine) N-acetyltransferase (arylalkylamine N-acetyltransferase, EC 2.3.1.87; AANAT), the melatonin rhythm enzyme, is light regulated and is responsible for the circadian cycle of melatonin production in the pineal gland (4–7). AANAT catalyzes the conversion of serotonin to N-acetylserotonin and in most cases is the rate-limiting step in melatonin production (7). Nocturnal increases in the level of this enzyme drive nocturnal increases in melatonin production. Conversion of N-acetylserotonin to melatonin is catalyzed by hydroxyindole O-methyltransferase, and this step is generally constitutive and not rate limiting for melatonin production. Inhibitors of AANAT would be useful for delineating the role of melatonin in circadian rhythms and its other proposed but more controversial effects on aging, cancer, cardiovascular function, and the immune system (1–3). No AANAT inhibitor with potent activity in vivo has been reported. Specific inhibitors of AANAT could potentially serve as therapeutic agents for alleviating sleep and mood disorders, both by raising pineal serotonin and reducing melatonin production (8, 9).
The recent molecular cloning of AANAT has led to an enhanced understanding of its regulation and enzymatic mechanism (10–12). AANAT is a ≈24-kDa protein and a member of a relatively large but poorly conserved acetyltransferase superfamily that includes enzymes important in antibiotic resistance such as the aminoglycoside acetyltransferases, and gene regulation, like some histone acetyltransferases (13, 14). The three-dimensional structures of members of this superfamily show significant similarity despite the low sequence homology, and it is likely that the catalytic mechanisms of these enzymes will also display conservation (10, 12, 13–17). Steady-state kinetics were used to show AANAT follows an Ordered BiBi kinetic scheme in which acetyl-CoA binding precedes serotonin (or the alternative substrate tryptamine), and the chemical step occurs after formation of the ternary complex (10). On the basis of this scheme, a bisubstrate analog (1), which links tryptamine and CoA via an acetyl bridge, was synthesized and shown to be a very potent and specific AANAT inhibitor (Ki ≈ 50 nM; Km for acetyl-CoA ≈ 200 μM; Km for tryptamine ≈150 μM) (18). Although bisubstrate analog 1 works well in vitro, it is unlikely to enter cells (19) because it contains a cluster of negative charges and is thus unlikely to inhibit AANAT in vivo.
The second and final step in the chemical synthesis of 1 involved reaction of the N-bromoacetyltryptamine (2) and CoASH (Fig. 1B). N-bromoacetyltryptamine (2) is an uncharged and fairly hydrophobic compound and on the basis of Hansch analysis was predicted to be cell membrane permeable (20) and worthy of further consideration as an AANAT inhibitor. We postulated that N-bromoacetyltryptamine (2) might inhibit AANAT either as an “affinity label” or more interestingly as a precursor to 1 in an enzyme-catalyzed fashion, a form of mechanism-based inhibition. There are two previous reports proposing that CoA-dependent acetyltransferases display related “alkyltransferase” activities (21, 22), although full details of the chemical and kinetic mechanisms of these atypical reactions have not been described. Here we report that AANAT efficiently catalyzes the alkyl transfer reaction between CoASH and N-bromoacetyltryptamine (2) at a site that is functionally distinct from that promoting acetyltransferase activity, and that 2 displays potent AANAT inhibition in pineal cell culture.
Figure 1.

(A) Serotonin N-acetyltransferase-catalyzed reaction between serotonin and acetyl-CoA. (B) Alkylation reaction between CoASH and N-bromoacetyltryptamine (2).
Materials and Methods
General.
Chemical reagents were commercially available as reagent grade unless otherwise noted. 5-Bromotryptamine was purchased from Biosynth Technologies (Skokie, IL). N-bromoacetyltryptamine (2) and the bisubstrate analog 1 were synthesized as reported previously (18). 1H NMR spectra were recorded on a Bruker (Billerica, MA) 400 MHz machine; electrospray mass spectrometry measurements were carried out on a Perkin–Elmer API-100 Sciex instrument. CoASH-agarose was purchased from Sigma.
Preparation of AANAT.
Protein.
Sheep (pineal) AANAT cDNA (residues 29–197) was subcloned into a pGEX-6P-1 plasmid with BamHI (N-terminal) and XhoI (C-terminal) restriction sites (DNA sequencing was carried out to verify the absence of mutations). AANAT protein was overproduced in Escherichia coli and purified as the GST-fusion protein as described previously (except that isopropyl thiogalactoside induction was carried out at 16°C for 20 h) (10). The GST-fusion protein was cut with Precision (Pharmacia) protease, passed over a glutathione column, and further purified over a CoA-agarose column to afford >97% pure AANAT as judged by SDS/PAGE and native gel. Mass spectrometric measurements revealed that the protein had the correct molecular weight. Acetyltransferase activity (kcat and Km for substrates) was essentially identical (within 30%) to those reported previously for GST–AANAT and GST-free AANAT (10). The protein concentration was quantified by Bradford assay as well as spectrophotometrically in the denatured state. A typical yield of pure AANAT protein was 8 mg per liter of E. coli culture.
Preparation of H112Q Mutant AANAT.
This AANAT DNA was prepared by using PCR mutagenesis, and the single mutation was confirmed by DNA sequencing. The H112Q AANAT protein was expressed as the GST fusion protein as described above, but soluble protein production was significantly reduced (≈10-fold). This mutant was studied only as the GST-fusion protein, which was 40–50% pure by SDS/PAGE (this purity was factored into estimates of protein concentration determined by Bradford assay). Previous studies showed that the GST-AANAT and GST-free AANAT display nearly identical acetyltransferase kinetic constants (10).
Inhibition of AANAT by 2.
Preincubation assay.
A solution of AANAT (1–10 μM) in assay buffer (pH 6.8/0.05 M sodium phosphate/500 mM NaCl/1 mM EDTA/0.05 mg/ml BSA; see ref. 10) was treated with 50 or 500 μM N-bromoacetyltryptamine (2) and 500 μM desulfo-CoA for 20 min at 30°C [a control incubation carried out in parallel lacked N-bromoacetyltryptamine (2)]. Inclusion of desulfo-CoA was based on previous studies that showed AANAT follows an ordered BiBi kinetic mechanism with acetyl-CoA binding preceding tryptamine (10). To measure residual acetyltransferase activity, the mixture was diluted 100-fold (50 μM 2) or 1,000-fold (500 μM 2) into assay buffer containing 1 mM acetyl-CoA and 1 mM tryptamine, allowed to react for 2 min before quenching with guanidinium-HCl, and the CoASH product was quantified spectrophotometrically by reduction of 5,5′-dithiobis(2-nitrobenzoic acid), as described previously (10).
Dialysis recovery.
This experiment was performed as described for the preincubation of 2 (50 μM or 0 μM as a control) with AANAT (1 μM), except that before the acetyltransferase activity assay, the mixture was dialyzed (membrane with 10- to 12-kDa cutoff) for 2–3 h at 4°C against assay buffer without inhibitor. Acetyltransferase activity was then measured after dilution as described above.
Standard inhibition assay.
AANAT activity in the presence of N-bromoacetyltryptamine (2) (0.2–4 μM) was measured in duplicate without the preincubation step (or desulfo-CoA addition) during a 2-min interval under the conditions described above. An IC50 value for 2 was determined graphically and is reported in the text.
Slow-Binding Inhibition by 1.
The time course of inhibition of AANAT acetyltransferase activity by 1 (200–800 nM) was measured at fixed acetyl-CoA (0.8 mM) and fixed tryptamine (0.8 mM) concentrations by using previously described methods (18) except that aliquots were taken at 20-s intervals up to 2 min and every minute thereafter. All measurements were made at least twice with less than 10% turnover of the limiting substrate. It should be pointed out that curvature in the zero inhibitor time course was negligible. Data were fit to Eq. 1 to obtain a family of v0 and vS as a function of the concentration of 1 (23) (v0, initial velocity; vS, steady-state velocity; k, apparent first-order rate constant to reach steady state; Ki, initial equilibrium binding constant; Ki*, steady-state equilibrium binding constant; P, product; t, time.) A replot of 1/v0 and 1/vS was used to determine Ki and Ki*, assuming a competitive inhibition model (23). With use of Eq. 2 and an independently determined k6 (see below), k5 was estimated to be ≈0.046 s−1.
Dissociation of 1 from AANAT.
AANAT (3 μM) was incubated in assay buffer with a saturating concentration of 1 (6 μM) for 10 min. Aliquots were removed at the indicated time points (every 20–30 s) and diluted (500-fold) into assay buffer containing saturating concentrations of acetyl-CoA and tryptamine (2.0 mM each), and the acetylation product was quantified at the indicated time in Fig. 2B. The data were fit to Eq. 3 to obtain a k6 of ≈0.01 s−1.
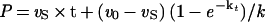 |
1 |
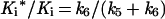 |
2 |
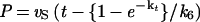 |
3 |
A control in the absence of inhibitor showed no detectable lag in product formation.
Figure 2.

Time course for inhibition of AANAT acetyltransferase activity by bisubstrate analog (1). (A) AANAT-catalyzed N-acetyltryptamine formation with respect to time in the presence of varying concentration of bisubstrate inhibitor (1). See Materials and Methods for details. (B) Recovery of AANAT acetyltransferase activity inhibited with bisubstrate inhibitor (1) after rapid dilution. The x-axis depicts the time after dilution into substrate mix during which product formation is measured. See Materials and Methods for details.
Alkyltransferase Assay.
Typical assays were carried out in Eppendorf tubes in assay buffer (50 mM sodium phosphate/500 mM NaCl/1 mM EDTA/50 μg/ml BSA, pH 6.8) at 30°C for 1–5 min with 0.3–5 μM AANAT/0–2 mM CoASH/0–8 mM N-bromoacetyltryptamine (2), in 0.08-ml reaction volumes. The reactions were quenched by dilution into aqueous guanidinium⋅HCl (pH 3) and held until HPLC injection. Separation and quantification by HPLC were carried out with a C-18 column (100 Å, 0.46 × 25 cm) and elution with a MeOH:50 mM aqueous potassium phosphate buffer (pH 4.5) gradient (0–3 min, 0% MeOH; 3- to 15-min linear increase to 65% MeOH; 15- to 20-min linear increase to 100% MeOH) (1 ml/min) and UV monitoring with a dual wavelength detector (set at 260 and 280 nm). Under these conditions, the retention times were: CoASH, 11 min; 1, 15 min; 2, 19 min. The bisubstrate analog 1 was identified and quantified by automated integration by using chemically synthesized material as a standard. Identity of the enzymatically-formed 1 was further confirmed by electrospray mass spectroscopic analysis. It proved necessary to include up to 5% vol/vol propylene glycol or MeOH as cosolvent to prevent precipitation of 2. It was established that the alkyltransferase reaction rates were identical with either cosolvent, and furthermore that these cosolvents did not affect AANAT acetyltransferase (<20%) at the concentrations used. No effect on alkyltransferase activity was observed by incubation of AANAT with 2 and desulfo-CoA for up to 30 min before alkyltransferase assay (suggesting that direct alkylation of AANAT by 2 did not contribute to observed rates). It was shown in control experiments that omission of the phosphate (and replacement with other buffering agents), high salt concentration, BSA, or EDTA did not significantly affect alkyltransferase catalysis. Measured alkyltransferase rates in the presence of AANAT were typically at least 5-fold higher than in the absence of AANAT, and the latter rates were subtracted in all cases.
Alkyltransferase activity was linear with respect to time and enzyme concentration in the ranges under investigation, as well as up to 10% turnover of the limiting substrate. Km (apparent) measurements for CoASH were made at concentrations from 0.05–2 mM ([N-bromoacetyltryptamine] (2) = 2 mM) and for N-bromoacetyltryptamine (2), 0.5–8 mM ([CoASH] = 2 mM). Data were fitted to the Michaelis–Menten equation by using Kaleidograph, and Km (apparent) and kcat values are shown in the text. All experiments were performed at least twice, and duplicate runs agreed within 20%.
Synthesis of Bisubstrate Analogs and Data.
Compounds were synthesized, purified, and spectroscopically characterized by 1H NMR, 31P NMR, and MS as described in the supplementary material (see www.pnas.org).
Inhibition of AANAT by Analogs.
Acetyltransferase Inhibition.
Assays were carried out in duplicate as previously described (18) with fixed substrate acetyl-CoA (0.3 mM) and tryptamine (0.3 mM) concentrations, and a range of at least five inhibitor concentrations varied around the Ki. Enzyme reactions were initiated with enzyme, quenched at 2 min, and product was analyzed. Kinetics were analyzed by using Dixon plots that were linear in each case. Ki values were estimated assuming a competitive inhibition model vs. acetyl-CoA (and noncompetitive vs. tryptamine) (10). Although these Ki values are only approximate because of the slow-binding behavior of bisubstrate analogs, previous work on compound 1 shows that there is good agreement between the Ki obtained with this analysis and the Ki* obtained from more extensive time-course measurements described above.
Alkyltransferase inhibition.
Assays were carried out in duplicate under the conditions outlined above with fixed CoASH (0.5 mM) and N-bromoacetyltryptamine (2 mM) concentrations and a range of at least five inhibitor concentrations varied around the Ki value. Enzyme was incubated with bisubstrate analog inhibitor for 10 min before the reaction was started to assure equilibrium binding between the slow-binding inhibitor and AANAT. The data were analyzed by Dixon plots, which were linear in each case. Ki values were estimated assuming a competitive inhibition model vs. acetyl-CoA. No change in inhibition of alkyltransferase (or acetyltransferase) activity was detected by prolonged equilibration between AANAT and inhibitor (up to 2 days), which provided evidence against a very slow conformational change in AANAT being responsible for two active sites.
Pineal Cell Culture AANAT Inhibition Assays.
Rat pinealocytes were grown in culture as described previously (24). Melatonin, cAMP, and AANAT activity levels were measured as reported previously (24). AANAT was stimulated in pinealocytes (35,000 cells) by adding norepinephrine (10 μM) to the media (0.3 ml) for 6 h in the presence of various concentrations of inhibitor in vehicle (60% vol/vol propylene glycol:water; the final propylene glycol concentration was 0.5%, which had no effect). Melatonin levels and AANAT acetyltransferase activity (after washing the cells, lysing them, and diluting the extracts in assay buffer) were measured at the end of this period. As a control experiment, pinealocytes were first treated with 2 (1 μg/ml) for 18 h, after which the cells were washed and induced with norepinephrine as described above. The levels of cAMP, melatonin, and AANAT activity reached levels of induction identical to control (data not shown). It was also determined that 2 has a similar IC50 (500 nM) for rat AANAT in vitro to sheep AANAT, which are ≈80% identical at the amino acid level.
Results and Discussion
To test the possibility that N-bromoacetyltryptamine (2) might be an affinity label of the enzyme, 1 μM AANAT was first incubated with 50 μM 2 for 20 min in the presence of 0.5 mM desulfo-CoA. The mixture was then diluted (100-fold) to reduce reversible antagonism effects by 2, and the residual acetyltransferase was measured. The activity of the enzyme was reduced by 60% with this treatment compared with control. A similar 60% rate reduction was observed starting with 10 μM AANAT and 500 μM 2, followed by a 1,000-fold dilution of enzyme. Further studies documented that this rate reduction could be recapitulated by omitting the preincubation step altogether but adding 0.5 μM N-bromoacetyltryptamine (2) (the final concentration of the above experiments) at the start of the 2-min activity assay. Analysis of a more complete concentration range (0.2–4 μM) of 2 at fixed substrate concentrations confirmed that 2 exhibited an IC50 of ≈0.5 μM and showed >90% inhibition at 5 μM. The lack of enhanced inhibition of AANAT by preincubation with large concentrations of 2 was evidence against covalent modification of the enzyme by 2. It seemed unlikely that this high potency was caused by standard reversible competitive inhibition from 2, because N-acetyltryptamine and N-trifluoroacetyltryptamine, structural analogs of 2, showed only modest inhibition (IC50 ≥ 200 μM for both; unpublished results). Rather, we thought that N-bromoacetyltryptamine (2) might undergo reaction with the small amount of CoASH present in the reaction mixture, and that the bisubstrate analog 1 once formed would be principally responsible for the enzyme inhibition. If such an inhibitory mechanism were operative, the loss of acetyltransferase activity should be reversed by removal of small molecule components by dialysis. Indeed, dialysis of the inhibited enzyme for 2 h, previously treated with 50 μM N-bromacetyltryptamine (2) and 0.5 mM desulfo-CoA, led to the nearly full recovery (>90%) of acetyltransferase activity.
Assuming that bisubstrate analog 1 formation was responsible for inhibition by N-bromacetyltryptamine (2), the intriguing question at this stage was whether AANAT was actually catalyzing this alkylation reaction (as shown in Fig. 1) or whether the spontaneous nonenzymatic alkylation velocity could account for the rate of formation of 1. We therefore designed an HPLC assay to measure the rate of bisubstrate analog 1 formation in the presence and absence of AANAT. The bisubstrate analog product 1 was well separated from CoASH, N-bromoacetyltryptamine (2) and other reaction mixture components as assessed by C-18 reversed phase HPLC. In the absence of enzyme at pH 6.8, bisubstrate analog 1 formation, measured at fixed N-bromoacetyltryptamine (2) concentration (2 mM), was linear with respect to time and first order with respect to CoASH concentration up to 2 mM. The apparent first order rate constant was 8.3 × 10−5 s−1 under these conditions. In a separate experiment, the reaction rate was first order with respect to N-bromoacetyltryptamine (2) concentration, and the second order rate constant was 0.042 M−1 s−1. The presence of AANAT enhanced significantly the formation rate of bisubstrate analog 1 compared with the nonenzymatic rate. The estimated kcat/kuncat was 3.3 × 104. The AANAT-catalyzed alkyltransferase activity showed Michaelis–Menten kinetics with Km for CoASH = 0.15 mM, Km for N-bromoacetyltryptamine = 3.6 mM, and kcat of 2.7 s−1. The kcat/Km(CoASH) = 1.8 × 104 M−1 s−1 (4.3 × 105-fold greater than the nonenzymatic second order rate constant).§ These steady-state rate constants are in a range not atypical for some enzymes, such as glutathione S-transferases (25), which catalyze alkyltransferase reactions physiologically.
The working assumption was that the alkyltransferase activity and the acetyltransferase activity were catalyzed at an identical active site on AANAT. The first clue that this might not be the case came from the surprising observation that AANAT-catalyzed formation of 1 displayed a linear rate despite the generation of relatively high concentrations (200 μM) of 1 (Ki = ≈50 nM for acetyltransferase reaction catalyzed by AANAT). Given the low Ki, a concentration greater than 10 μM of bisubstrate analog 1 would be expected to abolish completely (>98% inhibition) the acetyltransferase activity.
Because 1 presumably must dissociate from AANAT for each alkyl transfer reaction catalyzed, the alkyltransferase kcat (a steady-state parameter) should be less than or equal to the dissociation rate constant for 1 from AANAT. To gain further insight into the nature of the inhibition of the AANAT acetyltransferase activity by 1, we carried out a detailed kinetic analysis designed to reveal “slow-binding” kinetics. Technical improvements (including better mixing) in the early phase of the assay (20 and 40 s) allowed us to uncover subtle time-dependent features of the inhibition that were not detected in the original report (18). Thus a series of presteady-state measurements were carried out, and the results are shown in Fig. 2. Fig. 2A shows that AANAT-catalyzed acetyl transfer in the presence of 1 is slightly nonlinear for the first minute and then reaches a linear steady-state phase. Fig. 2B reveals that dissociation of 1 from AANAT has a half-life of approximately 60 s. These are characteristics of slow-binding/tight-binding inhibitors that follow an inhibition scheme shown in Eq. 4. Note that k6 (0.01 s−1) corresponds to the dissociation rate constant of 1 from AANAT and is ≈270-fold lower than the kcat (2.7 s−1) for the alkylation reaction.
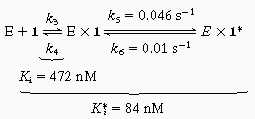 |
4 |
Competitive inhibition studies showed that the Ki values (>5 mM for both) for desulfo-CoA and acetyl-CoA were more than 5-fold greater than those previously measured (10) for the acetyltransferase reaction (Table 1). In addition, the Ki estimated for the bisubstrate analog 1 by Dixon analysis was 33 μM for alkyltransferase inhibition, which is approximately 700-fold higher than that (48 nM) measured for the acetyltransferase reaction (Table 1). In fact, it was demonstrated directly, by partitioning of the same AANAT preparation, that complete blockade (>99% inhibition) of the acetyltransferase activity could be achieved with 1 at a concentration (7.5 μM) that had little effect (<20% inhibition) on alkyltransferase activity. Taken together with the rate constant data discussed above, these findings argue persuasively that the acetyltransferase and alkyltransferase reactions are catalyzed at distinct active sites.
Table 1.
Inhibition of AANAT acetyltransferase or alkyltransferase sites by analogs
| Compound | Ki for acetyltransferase site, μM | Ki for alkyltransferase site, μM | Affinity ratio of acetyltransferase to alkyltransferase |
|---|---|---|---|
| Acetyl-CoA | 500* | >5,000 | >10 |
| Desulfo-CoA | 1000* | >5,000 | >5 |
| CoASH | – | 150† | – |
| 1 | 0.048 | 33 | 688 |
| 2 | – | 3,600† | – |
| 3 | 0.067 | 35 | 522 |
| 4 | 0.34 | 200 | 588 |
| 5 | 6.5 | >1,000 | – |
| 6 | 0.20 | 58 | 290 |
| 7 | <0.017 | 2.6 | – |
| 8 | 0.022 | 12.3 | 559 |
| 9 | 0.051 | 17.4 | 341 |
| 10 | 0.060 | 60 | 1,000 |
| 11 | 24 | >1,000 | – |
| 12 | 300 | – | – |
Values determined by Dixon Analysis (standard error approximately 20%; see Materials and Methods) (†, Km; *, taken from ref. 10).
Although we considered the possibility that the alkyltransferase catalysis was caused by a contaminating protein, this was shown to be unlikely for several reasons. The ratio of the two specific activities was constant throughout the purification of the enzyme and was very consistent among different preparations. Two additional chromatographic steps, cation exchange chromatography and gel filtration, showed complete coelution of the two activities. Furthermore, given that desulfo-CoA and acetyl-CoA show much weaker apparent affinity for the alkyltransferase active site compared with the acetyltransferase site (Table 1), it should have been possible to achieve selective elution of a contaminant from a CoASH affinity column by using these ligands. In fact, the activity profiles of AANAT fractions, eluted with either acetyl-CoA or desulfo-CoA, showed identical ratios of acetyltransferase and alkyltransferase activities in every fraction. Moreover, a mutant AANAT protein (H112Q; ref. 7) was defective both in acetyltransferase and alkyltransferase activity (>30-fold reduction in both activities), establishing that the same polypeptide was responsible for both activities. (The structural basis for this loss of acetyltransferase/alkyltransferase activity with H112Q (7) is unknown because His-112 is far removed from the active site in the x-ray structure.)
A series of bisubstrate analogs were synthesized (Fig. 3) and tested as AANAT acetyltransferase and alkyltransferase inhibitors. As seen in Table 1, increasing or decreasing the tether length between CoA and tryptamine (inhibitors 3–6) either had no effect or decreased inhibition potency for both acetyltransferase and alkyltransferase reactions compared with 1. Two substituted inhibitors, 7 and 8, showed enhanced potency toward both sites. Deletion of the 3′-phosphate from the CoA moiety (as in 10) led to minor effects on acetyltransferase or alkyltransferase inhibition potency, whereas further removing AMP (11) or ADP (12) showed more massive effects on inhibition potency. Although these structure–activity relationships allow a more refined approach to acetyltransferase inhibitor design (E. Wolf, J.D., E.M.K., P.A.C., S. Burley, manuscript in preparation), they also provide strong evidence of the relatedness of the two active sites. For every bisubstrate analog inhibitor, the relative affinity for the acetyltransferase site was about 600-fold greater than for the alkyltransferase site, and the rank order of affinity was nearly identical for both. Thus our favored model is that the steady-state alkyltransferase activity is catalyzed in a subtly conformationally altered active site compared with the acetyltransferase active site on AANAT.
Figure 3.

Structures of synthetic bisubstrate analogs tested as inhibitors of AANAT.
There are at least three possibilities that can account for this unusual behavior. First, the two active sites could reside within the framework of an individual AANAT monomer and be completely physically separate. This possibility seemed unlikely in view of the small size of the protein, the presence of a single active site revealed in the x-ray crystal structure of AANAT (ref. 26; E. Wolf, J.D., E.M.K., P.A.C., S. Burley, manuscript in preparation) and the likely structural similarity of the two sites revealed by the bisubstrate inhibitory patterns discussed above. Second, the activities could represent two stable forms of free monomers of AANAT that are very slow or unable to interconvert and differ in conformation or covalent structure. Although this possibility cannot be formally ruled out, evidence against this proposal is the precise coelution of the activities in several chromatographic steps including the coenzyme A with free sulfhydryl group (CoASH) column, and a homogeneous appearance on nondenaturing gel electrophoresis. Very slow conformational changes between monomeric AANAT forms also appear unlikely (see Materials and Methods).
The third possibility is that these catalytic sites represent similar but nonidentical pockets present in an AANAT protein homodimer. Lack of structural and functional identity of protein active sites in protein homodimers has been reported in other systems (27–32). Efforts to establish definitive evidence for the oligomerization state of AANAT in the presence of 1 by analytical ultracentrifugation have been unsuccessful.¶ However, the x-ray structure of AANAT reveals two potential dimer interfaces related by centers of symmetry (E. Wolf, J.D., E.M.K., P.A.C., S. Burley, manuscript in preparation). Moreover, there are two well-defined conformations of the inhibitor present in the AANAT active site (manuscript in preparation). It is tempting to speculate that these two conformations may correspond to the superimposed alkyltransferase and acetyltransferase binding interactions of 1 in individual protomers of a dimer. However, further studies will be needed to resolve the detailed structural basis for the AANAT active site plasticity.
Precedents exist for at least two clinically used drugs effecting enzyme inactivation by undergoing covalent reaction with cofactors within the active site of enzymes–finasteride within the human enzyme 5α reductase (33), and isoniazid metabolite within the mycobacterial enzyme InhA (34). To our knowledge, this is the first proposal of a mechanism of inhibition in which inhibitor activation chemistry (promiscuous reactivity; ref. 35) is catalyzed in a site of the target enzyme distinct from the inhibitory site of action (Fig. 4). The potential impact of such a scheme is to allow unprecedented amplification of inhibition, because activation of the inhibitor by the target enzyme can undergo multiple turnovers after its physiologically relevant active site has been poisoned. In this way, one enzyme molecule can be chemically induced to inhibit related family members, a form of molecular fratricide.
Figure 4.

Activation of AANAT inhibitor 2 by alkyltransferase action: multiple turnover inhibition. Tryp, tryptamine.
It was of obvious interest to evaluate N-bromoacetyltryptamine (2) in an in vivo situation both to validate the in vitro findings and to provide a new tool for circadian rhythm studies. To this end, we measured the production of melatonin in norepinephrine-stimulated pinealocytes (24) as a function of N-bromoacetyltryptamine concentration (Table 2). It can be seen that there was >50% inhibition of melatonin production with as little as 0.1 μg/ml (400 nM) N-bromoacetyltryptamine (2) and that 1 μg/ml (4 μM) resulted in background melatonin levels. Furthermore, there was no evidence of generalized cellular toxicity from 2 (4 μM) on the basis of a variety of criteria: (i) incubation of pineal cells with 1 μg/ml of 2 for 18 h followed by removal of inhibitor by washing allowed a normal response to 10 μM norepinephrine including cAMP, AANAT activity, and melatonin induction; (ii) microscopic features of cells appeared normal after treatment with 2; and (iii) AANAT was induced normally by norepinephrine in the pineal cells in the presence of 2 (data not shown). Given its intrinsic reactivity, whether 2 itself will ultimately be useful in whole animal studies or clinical situations remains to be seen. The success of a thiol alkylating agent showing specific protein kinase inhibition (36) (now undergoing clinical trials) as well as the well-established antibiotic chloramphenicol (which contains α-halocarboxamide functionality) support the view that 2 could be useful as a lead compound. N-bromoacetyltryptamine (2) thus represents the most potent cell culture AANAT inhibitor yet reported and may have a variety of new applications in the study of circadian rhythms.
Table 2.
Inhibition of melatonin formation in stimulated pinealocytes by N-bromoacetyltryptamine (2)
| Treatment | Melatonin formation, pmol/105 cells |
|---|---|
| Control | 0.84 ± 0.06 |
| Norepinephrine (10 μM) | 11.0 ± 2.1 |
| Norepinephrine (10 μM) + 2 (1 μg/ml) | 1.04 ± 0.32 |
| Norepinephrine (10 μM) + 2 (0.3 μg/ml) | 4.25 ± 0.26 |
| Norepinephrine (10 μM) + 2 (0.1 μg/ml) | 4.52 ± 0.42 |
| Norepinephrine (10 μM) + 2 (0.03 μg/ml) | 5.57 ± 0.41 |
For all measurements, n = 3. See Materials and Methods for details.
Supplementary Material
Acknowledgments
We are grateful to S. Burley and E. Wolf (Rockefeller University, New York, NY) for helpful insights and for sharing reagents. We thank A. Ho (University of Alberta, Edmonton) for carrying out the pinealocyte melatonin inhibition studies. We thank the Burroughs Wellcome Fund and the National Institutes of Health (NIH) (National Research Service Award to E.M.K.), the National Institutes of Health Medical Scientist Training Program (M.I.), and the Ellison Medical Foundation (P.A.C.) for financial support.
Abbreviations
- AANAT
arylalkylamine N-acetyltransferase or serotonin N-acetyltransferase
- CoAsh
coenzyme A with free sulfhydryl group
Footnotes
The magnitude of enzyme inhibition in the 2-min assay is still approximately 10-fold faster than that predicted on the basis of the kcat/Km for the alkyltransferase reaction and bisubstrate analog formation. This discrepancy may arise from a rapid initial turnover of the alkyltransferase reaction compared with the steady-state rate (i.e., a burst phase). Resolution of this issue awaits a more detailed presteady-state kinetic analysis, which is beyond the scope of this initial study.
Analytical centrifugation experiments in the presence of bisubstrate analog 1 were complicated by the chromophoric inhibitor analog, which led to baseline correction problems. Gel filtration experiments were complicated by an apparent intrinsic affinity of AANAT for Sepharose, which made interpretation difficult. Dynamic light scattering experiments in the presence of excess 1 were irreproducible but sometimes showed evidence of a monodisperse dimer. Therefore, the solution phase oligomerization state of the protein is not definitively known at this time.
References
- 1.Arendt J. Melatonin and the Mammalian Pineal Gland. New York: Chapman & Hall; 1995. [Google Scholar]
- 2.Pierpaoli W, Regelson W, Colman C. The Melatonin Miracle. Nature’s Age-Reversing, Disease-Fighting, Sex-Enhancing Hormone. New York: Pocket Books; 1995. [Google Scholar]
- 3.Reppert S M, Weaver D R. Cell. 1995;83:1059–1062. doi: 10.1016/0092-8674(95)90131-0. [DOI] [PubMed] [Google Scholar]
- 4.Klein D C, Weller J L. Science. 1970;169:1093–1095. doi: 10.1126/science.169.3950.1093. [DOI] [PubMed] [Google Scholar]
- 5.Coon S L, Roseboom P H, Baler R, Weller J L, Namboodiri M A A, Koonin E V, Klein D C. Science. 1995;270:1681–1683. doi: 10.1126/science.270.5242.1681. [DOI] [PubMed] [Google Scholar]
- 6.Borjigin J, Wang M M, Snyder S H. Nature (London) 1995;378:783–785. doi: 10.1038/378783a0. [DOI] [PubMed] [Google Scholar]
- 7.Klein D C, Coon S L, Roseboom P H, Weller J L, Bernard M, Gastel J A, Zatz M, Iuvone P M, Rodriquez I R, Begay V, et al. Recent Prog Horm Res. 1997;52:307–357. [PubMed] [Google Scholar]
- 8.Lewy A J, Sack R L, Miller L S, Hoban T M. Science. 1987;235:352–354. doi: 10.1126/science.3798117. [DOI] [PubMed] [Google Scholar]
- 9.Bonhomme N, Esposito E. J Clin Psychopharmacol. 1998;18:447–454. doi: 10.1097/00004714-199812000-00005. [DOI] [PubMed] [Google Scholar]
- 10.De Angelis J, Gastel J, Klein D C, Cole P A. J Biol Chem. 1998;273:3045–3050. doi: 10.1074/jbc.273.5.3045. [DOI] [PubMed] [Google Scholar]
- 11.Gastel J A, Roseboom P H, Rinaldi P A, Weller J L, Klein D C. Science. 1998;279:1358–1360. doi: 10.1126/science.279.5355.1358. [DOI] [PubMed] [Google Scholar]
- 12.Khalil E M, De Angelis J, Cole P A. J Biol Chem. 1998;273:30321–30327. doi: 10.1074/jbc.273.46.30321. [DOI] [PubMed] [Google Scholar]
- 13.Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley S K. Cell. 1998;94:439–449. doi: 10.1016/s0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 14.Yang X-J, Ogrryzko V V, Nishikawa J, Howard B H, Nakatani Y. Nature (London) 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 15.Hickman A B, Klein D C, Dyda F. Mol Cell. 1999;3:23–32. doi: 10.1016/s1097-2765(00)80171-9. [DOI] [PubMed] [Google Scholar]
- 16.Wybenga-Groot L E, Draker K-a, Wright G D, Berghuis A M. Structure (London) 1999;7:497–507. doi: 10.1016/s0969-2126(99)80066-5. [DOI] [PubMed] [Google Scholar]
- 17.Tanner K G, Trievel R C, Kuo M-H, Howard R M, Berger S L, Allis C D, Marmorstein R, Denu J M. J Biol Chem. 1999;274:18157–18160. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- 18.Khalil E, Cole P A. J Am Chem Soc. 1998;120:6195–6196. [Google Scholar]
- 19.Robishaw J D, Neely J R. Am J Physiol. 1985;248:E1–E9. doi: 10.1152/ajpendo.1985.248.1.E1. [DOI] [PubMed] [Google Scholar]
- 20.Silverman R B. The Organic Chemistry of Drug Design and Drug Action. New York: Academic; 1992. [Google Scholar]
- 21.Chase J F A, Tubbs P K. Biochem J. 1969;111:225–235. doi: 10.1042/bj1110225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Northrup D B, Williams J W. J Antibiotics. 1979;32:1147–1154. doi: 10.7164/antibiotics.32.1147. [DOI] [PubMed] [Google Scholar]
- 23.Morrison J F, Walsh C T. Adv Enzymol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 24.Ho A K, Chik C L. Am J Phys. 1992;26:E481–E488. doi: 10.1152/ajpendo.1992.263.3.E481. [DOI] [PubMed] [Google Scholar]
- 25.Jakoby W B. Adv Enz. 1978;46:383–414. doi: 10.1002/9780470122914.ch6. [DOI] [PubMed] [Google Scholar]
- 26.Hickman A B, Namboodiri M A, Klein D C, Dyda F. Cell. 1999;97:361–369. doi: 10.1016/s0092-8674(00)80745-x. [DOI] [PubMed] [Google Scholar]
- 27.Somoza J R, Chin M S, Focia P J, Wang C C, Fletterick R J. Biochemistry. 1996;35:7032–7040. doi: 10.1021/bi953072p. [DOI] [PubMed] [Google Scholar]
- 28.Zhang E, Brewer J M, Minor W, Carreira L A, Lebioda L. Biochemistry. 1997;36:12526–12534. doi: 10.1021/bi9712450. [DOI] [PubMed] [Google Scholar]
- 29.Mande S C, Mainfroid V, Kalk K H, Goraj K, Martial J A, Hol W G. Protein Sci. 1994;3:810–820. doi: 10.1002/pro.5560030510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stout T J, Stroud R M. Structure (London) 1996;4:67–77. doi: 10.1016/s0969-2126(96)00010-x. [DOI] [PubMed] [Google Scholar]
- 31.Phillips C, Dohnalek J, Gover S, Barrett M P, Adams M J. J Mol Biol. 1998;282:667–681. doi: 10.1006/jmbi.1998.2059. [DOI] [PubMed] [Google Scholar]
- 32.Dev I K, Dallas W S, Ferone R, Hanlon M, McKee D D, Yates B B. J Biol Chem. 1994;269:1873–1882. [PubMed] [Google Scholar]
- 33.Bull H G, Garcia-Calvo M, Andersson S, Baginsky W F, Chan H K, Ellsworth D E, Miller R R, Stearns R A, Bakshi R K, Rassmusson G H, et al. J Am Chem Soc. 1996;118:2359–2365. [Google Scholar]
- 34.Rozwarski D A, Grant G A, Barton D H R, Jacobs W R, Sacchetini J C. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 35.O’Brien P J, Herschlag D. Chem Biol. 1999;6:R91–R105. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 36.Fry D W. Pharmacol Ther. 1999;82:207–218. doi: 10.1016/s0163-7258(98)00050-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.