

Abstract
TenA catalyzes the hydrolysis of 4-amino-5-aminomethyl-2-methylpyrimidine and participates in the salvage of base-degraded thiamin. Here we describe mutagenesis of the active site of TenA guided by structures of the enzyme complexed to a substrate analog and to the product. Catalytic roles for each of the active site residues are identified and a mechanism for the reaction is described.
1. Introduction
Thiamin pyrophosphate 1 is an essential cofactor in all living systems and is biosynthesized by a complex pathway 1, 2. Consequentially, many microorganisms also contain salvage pathways for thiamin fragments present in the environment and salvage pathways for thiamin alcohol 2,3 hydroxypyrimidine 3,4 thiazole 45 and base-degraded thiamin 56 have been identified.
The salvage of base-degraded thiamin is described in Figure 1. In this pathway, formyl aminopyrimidine 5, formed by base-mediated degradation of the thiazolium moiety of thiamin, is transported into the cell, deforymylated and the resulting aminopyrimidine 6 is converted to hydroxy-pyrimidine 3 by 4-amino-5-aminomethyl-2-methylpyrimidine hydrolase (TenA, thiaminase II).6 The salvaged hydroxy-pyrimidine (3) is then phosphorylated and incorporated into the de novo thiamin biosynthetic pathway.
Figure 1.
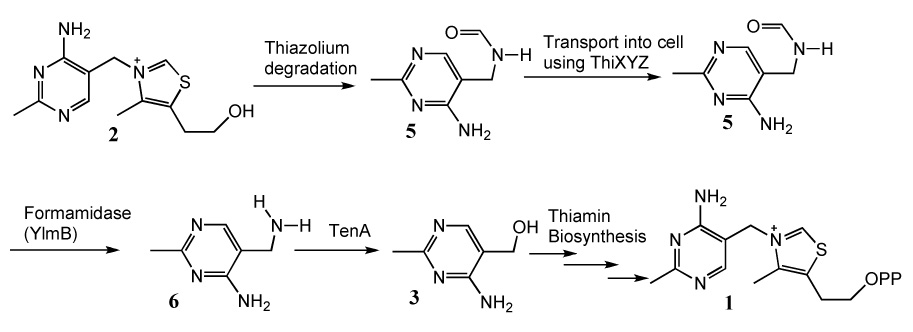
The salvage pathway for base-degraded thiamin.
The crystal structure of the amino-pyrimidine hydrolase (TenA), with hydroxy-pyrimidine 3 bound at the active site, has recently been reported.7 Here we describe a systematic mutagenesis study of the key active site residues identified from this structure. This information is used to propose a detailed mechanism for the TenA-catalyzed amino-pyrimidine hydrolysis.
2. Methods and Materials
Purification of TenA
TenA was cloned, overexpressed and purified as described previously7.
Preparation of TenA mutants
Standard methods were used for DNA manipulations. Plasmid DNA was purified with the Qiagen Miniprep kit. E. coli strain MachI (Invitrogen) was used as a recipient for transformations during plasmid construction and for plasmid propagation and storage. Site-directed mutagenesis was performed on the TenA overexpression plasmid (pBsTenA.XF1) by a standard PCR protocol using PfuTurbo DNA polymerase per the manufacturer’s instructions (Invitrogen) and DpnI (New England Biolabs) was used to digest the methylated parental DNA prior to transformation. Primers were designed to introduce or remove a diagnostic restriction enzyme site to facilitate screening for a mutated clone. Only clones producing the anticipated restriction pattern were sequenced. The mutagenesis primer pair consisted of the primer whose sequence is in Table 1 and its reverse complement.
Table 1.
Mutagenesis primers
| Mutant | Primer Sequence, top strand | Restriction Site |
|---|---|---|
| D44A | CCGTTTTAAATACTACGTACTTCAAGCTTCCTATTATTTAACGC | HindIII |
| C135A | GCGGCCCTGCTGCCCGCGTATTGGCTCTATTACGAGG | BstXI |
| E205A | GTCATCTCCAGCTACTATGCATATCAATTTTGGGGAATGG | NsiI |
| Y47F | CTTCAGGATTCCTATTTTCTGACGCATTTTGCAAAGG | HgaI |
| Y112F | CCTACGGCGTACTCTTTTACGTCCCATATGTACC | BsmFI |
Enzymatic assay
The enzymatic hydrolysis of amino-pyrimidine 6 (gift from Roche) was monitored using the glutamate dehydrogenase assay for ammonia.8 Each reaction mixture contained 5 units of glutamate dehydrogenase (unit is defined as the amount of glutamate dehydrogenase that will reduce 1 µmole of α-ketoglutarate to glutamate per min at pH 8.3 at 30°C), 5mM α-ketoglutarate, 0.1mM EDTA, 250uM NADPH, varing concentrations of amino-pyrimidine 6 and TenA, which was added last to initiate the reaction. The TenA concentration was determined from its calculated extinction coefficient at 280nm of 74,280M−1cm−1 (determined by the Edel-Hoch Method9). NADPH consumption was monitored by measuring the decrease in absorbance at 340nm. Km and kcat were obtained by fitting the initial rates, at varying substrate concentrations, (determined at less than 10% conversion) to the Michaelis-Menten equation using the program GraFit. These parameters for the native and mutated enzyme are shown in Table 2. The Km for Y112F and Y47F could not be directly measured because of the low activity of these mutants.
Table 2.
Kinetic parameters for TenA and its active site mutants.
| Protein | Km (µM) | kcat (min−1) | kcat/Km (min−1µM−1) | Relative kcat/Km |
|---|---|---|---|---|
| Native | 11.8 ± 1.6 | 22 ± 0.5 | 1.9 ± 0.3 | 1 |
| E205A | 251 ± 31.5 | 0.27 ± 0.006 | 0.001 ± 0.0001 | 0.0005 |
| D44A | 337 ± 94 | 0.09 ± 0.004 | 0.0003 ± 0.00008 | 0.0003 |
| Y112F | < 25 | 0.31 ± 0.02 | >0.012 ± 0.0007 | >0.01 |
| Y47F | < 25 | 0.75 ± 0.03 | >0.029 ± 0.0012 | >0.03 |
| C135A | Inactive | Inactive | Inactive | Inactive |
3. Results and Discussion
TenA catalyzes the substitution of the amino group of amino-pyrimidine 6 by water (Figure 1). This reaction is analogous to the well-studied thiaminase I-catalyzed substitution of the thiamin thiazole by a variety of nucleophiles which occurs by an addition elimination mechanism (Figure 2). In this mechanism, cysteine adds to C6 of the pyrimidine generating anion 7. Expulsion of the leaving group gives 8. Addition of the nucleophile (X-H), followed by expulsion of the active site cysteine completes the reaction. This mechanistic proposal is supported by the identification of the active site nucleophile as Cys113,10 the observation of ping-pong kinetics,11 retention of stereochemistry,12 and a structure of the enzyme with an analog of 7 bound at the active site.13
Figure 2.
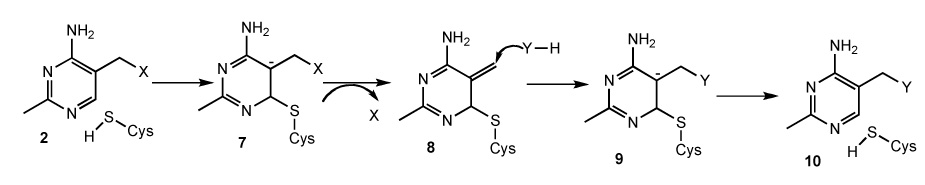
Mechanistic proposal for the Thiaminase I-catalyzed reaction. X represents the thiamin thiazole and Y represents a variety of nucleophiles.
The structure of TenA with bound product is shown in Figure 3A.7 The location of Cys135 close to C6 of the pyrimidine (3.2A) suggested that the mechanism of TenA was likely to be similar to the mechanism of thiaminase I. To test this, key residues surrounding the active site pyrimidine were altered by mutagenesis to probe their function.
Figure 3.
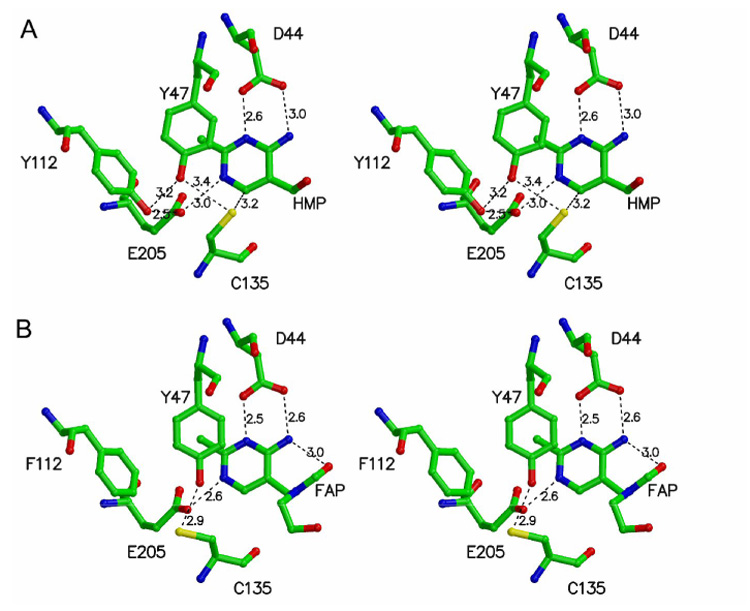
Active site structure of TenA. A) Native enzyme complexed with 4-amino-5-hydroxymethyl-2-methylpyrimidine. B) Y112F mutant
The results of these mutagenesis experiments are shown in Table 2 and are consistent with the mechanistic proposal outlined in Figure 4. In this mechanism, Cysteine 135 adds to the pyrimidine and the resulting pyrimidine anion is stabilized by protonation at N1 by Glutamic acid 205 to give 12. The observation that the C135A mutant is inactive and that the E205A mutant shows a 2000 fold reduction in catalytic activity is consistent with this. Cysteine 135 is activated by deprotonation using a catalytic triad consisting of Tyrosine 47, Tyrosine 112 and Glutamatic acid 205. Consistent with this, the Y112F mutant showed a 71 fold reduction in kcat, and the Y47F mutant showed a 29 fold reduction in kcat. Both mutants showed less than two fold increase in Km.
Figure 4.
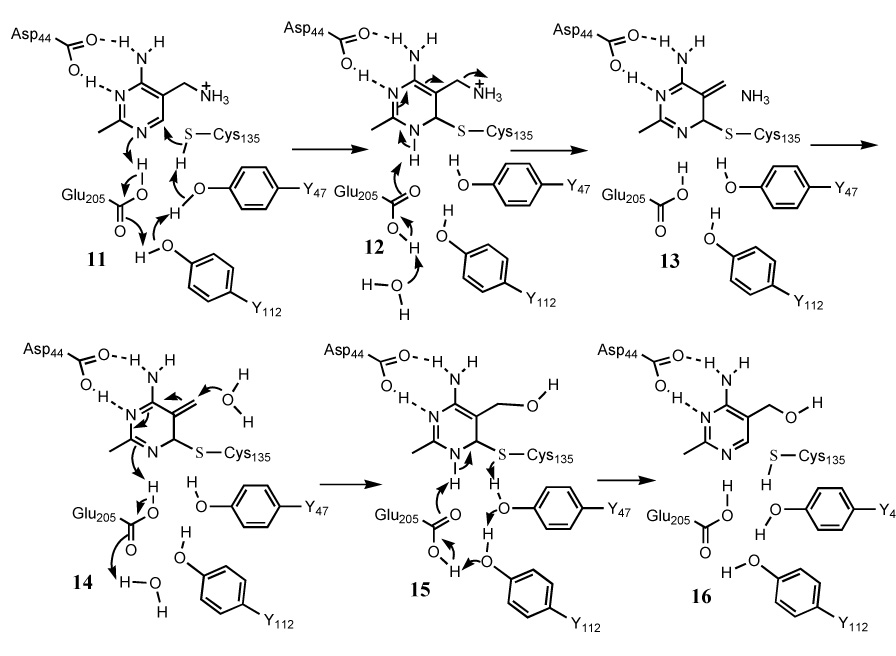
Mechanistic proposal for the TenA-catalyzed cleavage of aminopyrimidine 6.
The structure also suggests that Aspartic acid 44 anchors the substrate at the active site and further polarizes the pyrimidine for cysteine addition. The D44A mutant shows a 3,300 fold reduction in catalytic activity. Loss of ammonia from 12 gives 13. Surprisingly, the amine leaving group does not interact with any of the active site amino acid side chains. Reversal of these steps, using water as the nucleophile, completes the reaction (14 to 16).
TenA is involved in the salvage of the thiamin pyrimidine from base-degraded thiamin (Figure 1). Formyl aminopyrimideine 5 is not a substrate for TenA. To better understand the substrate selectivity of TenA, the structure of the Y112F mutant, complexed to 4-amino-5-(N-formyl-N-hydroxyethyl)aminomethyl-2-methylpyrimidine was determined (Figure 3B). This structure shows that this analog binds in a catalytically competent manner and that the enzyme also does not provide any acidic residues to stabilize an amide leaving group. Under these conditions, the substrate selectivity is most likely due to ammonia being a better leaving group than an amide anion (relative pKa values of 9.2 and 17 respectively).
Thiaminase I and TenA both catalyze pyrimidine substitution reactions at the C5' of the thiamin pyrimidine. These enzymes share no sequence or structural similarity,7 yet both enzymes utilize a similar addition-elimination mechanism. In contrast, thiamin phosphate synthase catalyzes the substitution of the pyrophosphate of 17 by thiazole phosphate using a dissociative mechanism involving a pyrimidine carbocation intermediate (Figure 5).14 A comparison of the mechanisms of these three enzymes suggests that substitution at C5' of the thiamin pyrimidine occurs by a dissociative mechanism when the leaving group is good (e.g. pyrophosphate) and by an addition elimination mechanism when the leaving group is poor (e.g. thiazole or ammonia). In contrast, benzylic substitution reactions proceed by associative (Sn2) and dissociative (Sn1) mechanisms depending on reaction conditions and on the nucleophile and leaving group. The addition elimination mechanism is not observed for benzylic substitution because of the high stability of the 6tt electron system in benzene which is disrupted by nucleophilic addition. The benzene ring also lacks the basic nitrogen atoms required to polarize the pyrimidine for nucleophilic addition.
Figure 5.
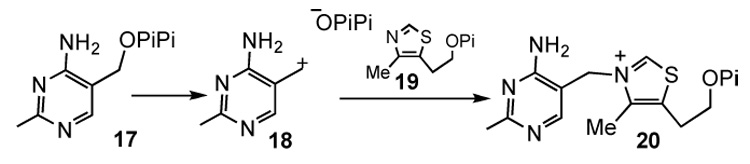
The thiamin phosphate synthase catalyzed reaction proceeds via a dissociative mechanism.
Structures.
Acknowledgments
We thank Dr. Cynthia Kinsland (Cornell Protein Expression Facility) for the preparation of the TenA mutants. This research was funded by grants from the NIH (DK44083 to TPB and DK067081 to SEE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Begley TP. Nat. Prod. Rep. 2006;33(1):15–25. doi: 10.1039/b207131m. [DOI] [PubMed] [Google Scholar]
- 2.Settembre EC, Begley TP, Ealick SE. Current Opinion in Structural Biology. 2003;13(6):739–747. doi: 10.1016/j.sbi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Melnick J, Lis E, Park J-H, Kinsland C, Mori H, Baba T, Perkins J, Schyns G, Vassieva O, Osterman A, Begley TP. J. Bacteriol. 2004;186(11):3660–3662. doi: 10.1128/JB.186.11.3660-3662.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park J-H, Burns K, Kinsland C, Begley TP. J. Bacteriol. 2004;186(5):1571–1573. doi: 10.1128/JB.186.5.1571-1573.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campobasso N, Mathews II, Begley TP, Ealick SE. Biochemistry. 2000;39(27):7868–7877. doi: 10.1021/bi0000061. [DOI] [PubMed] [Google Scholar]
- 6.Jenkins AHG, Schyns, Sebastien, Potot, Guangxing, Sun, Begley, Tadhg p. Nature Chemical Biology. 2007;3(8):492–497. doi: 10.1038/nchembio.2007.13. [DOI] [PubMed] [Google Scholar]
- 7.Toms AV, Haas AL, Park J-H, Begley TP, Ealick SE. Biochemistry. 2005;44(7):2319–2329. doi: 10.1021/bi0478648. [DOI] [PubMed] [Google Scholar]
- 8.Day N, Keillor JW. Anal. Biochem. 1999;274:141–144. doi: 10.1006/abio.1999.4255. [DOI] [PubMed] [Google Scholar]
- 9.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4(11):2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costello CA, Kelleher NL, Abe M, McLafferty FW, Begley TP. J Biol Chem. 1996;271(7):3445–3452. doi: 10.1074/jbc.271.7.3445. [DOI] [PubMed] [Google Scholar]
- 11.Puzach SSG, Ostrovskii ZV, Yu M. Biokhimiya. 1984;49(7):1178–1183. [Google Scholar]
- 12.Nicewonger R, Costello CA, Begley TP. J Org Chem. 1996;61(12):4172–4174. doi: 10.1021/jo960234u. [DOI] [PubMed] [Google Scholar]
- 13.Campobasso N, Costello CA, Kinsland C, Begley TP, Ealick SE. Biochemistry. 1998;37(45):15981–15989. doi: 10.1021/bi981673l. [DOI] [PubMed] [Google Scholar]
- 14.Hanes JW, Ealick SE, Begley TP. Journal of the American Chemical Society. 2007;129(16):4860–4861. doi: 10.1021/ja0679634. [DOI] [PMC free article] [PubMed] [Google Scholar]