

Abstract
Adenovirus (Ad) types 2 and 5 encode at least five proteins within the E3 transcription unit that help the virus evade the immune system. Two such proteins, RIDα (formerly E3-10.4K) and RIDβ (formerly E3-14.5K), form the RID (receptor internalization and degradation) complex (formerly E3-10.4K/14.5K). RID mediates clearance from the cell surface and lysosomal degradation of a number of important members in the tumor necrosis factor receptor (TNFR) superfamily and the receptor tyrosine kinase receptor family. Affected receptors include Fas, TRAIL (TNF-related apoptosis-inducing ligand) receptor 1 (TR1), TR2, and epidermal growth factor receptor (EGFR). Degradation of Fas and TRAIL receptors protects Ad-infected cells from apoptosis. To investigate the mechanism of action of RIDα, 14 mutant RIDα proteins, each containing a three- to five-amino-acid deletion, were constructed and then expressed from the E3 region of a replication-competent recombinant Ad in the same context as wild-type RIDα. Each mutant protein was characterized with regard to five physical properties associated with wild-type RIDα, namely, protein stability, proteolytic cleavage, insertion into the membrane, complex formation with RIDβ, and transport to the cell surface. Additionally, the mutant proteins were tested for their ability to mediate internalization and degradation of EGFR and Fas and to protect cells from Fas-mediated apoptosis. The majority of mutant RIDα proteins (8 out of 14) were physically similar to wild-type RIDα. With regard to functional characteristics, the cytoplasmic domain of RIDα is largely unimportant for receptor internalization and degradation and the extracellular domain of RIDα is important for down-regulation of EGFR but not Fas.
Many viruses encode proteins that protect infected cells from the host immune system (3, 36). These proteins enable enhanced virus replication and/or provide an environment conducive to viral latency by delaying or preventing elimination of infected cells by the immune system (e.g., see the review by Abendroth and Arvin [1]). Since virus infection elicits a multifaceted attack by the immune system, numerous different virus-encoded proteins that counteract different aspects of the host immune response have been discovered (3, 36). DNA viruses such as adenoviruses (Ads) (8, 19, 30, 51), herpesviruses (12, 48), and poxviruses (15, 33) encode multiple proteins involved in immune evasion that target different facets of the immune response.
The early region 3 (E3) transcription unit of Ad types 2 and 5 encodes at least seven proteins, five of which are known to have immunomodulatory functions that help protect infected cells from the host immune response. The five proteins are E3-gp19K (35), E3-14.7K (47, 49), RIDα (formerly E3-10.4K) (44), RIDβ (formerly E3-14.5K) (43), and E3-6.7K (50). The mechanisms of action of these proteins are of interest for the insights they can provide into cellular processes, for the development of drugs that counteract the action of these proteins, and for the potential use of these proteins to treat human disease. The mechanisms of action of E3-gp19K and E3-14.7K have been reviewed recently (29).
RIDα and RIDβ subunits comprise the RID complex (formerly E3-10.4K/14.5K) (43-45). RIDβ is a type I integral membrane protein whose signal sequence is cleaved (22) and whose cytoplasmic domain is O glycosylated (24) and phosphorylated on serine 116 (25, 28). RIDα is also a type I integral membrane protein (22). The signal sequence is removed from only about 50% of the molecules so that RIDα exists as a full-length form and a form that is cleaved between amino acids 22 and 23 (22). Evidence indicates that these two versions of RIDα form a dimer through an intermolecular disulfide bond at cysteine 31 (18). Indirect immunofluorescence shows that RID is localized predominantly to the cell surface (18, 40, 41). However, when expressed individually, neither subunit localizes to the cell surface and instead both proteins display an intracellular localization pattern, with RIDα predominantly in the Golgi and RIDβ predominantly in the endoplasmic reticulum (ER) and Golgi (40, 41).
RID specifically down-regulates the cell surface expression of a number of receptor proteins, several of which are known to be important mediators of the immune response. Included among these receptors are three so-called death receptors, Fas (14, 38, 41), TRAIL receptor 1 (TR1) (4, 46), and TR2 (4), all of which belong to the tumor necrosis factor (TNF) receptor (TNFR) superfamily. Removal of these receptors from the cell surface can protect infected cells from the immune system by blocking apoptosis initiated by Fas ligand (14, 38, 41) and TRAIL (4, 46). In addition, McNees et al. recently reported that RID is able to internalize and degrade Fas in some lymphocyte cell lines (31). Also down-regulated are certain members of the receptor tyrosine kinase family, such as epidermal growth factor receptor (EGFR) (9, 45). The significance of EGFR clearance is not known. RID-mediated down-regulation of receptors is specific in that receptors such as transferrin receptor (9, 41), CD13, CD46, CD40 (14), and Her2 (26) are not cleared from the cell surface. The mechanism by which RID distinguishes among receptors that can and cannot be down-regulated is not understood. In addition, how RID is able to affect receptors from two unrelated families, which have little if any amino acid homology, has not yet been investigated.
Recent evidence clearly shows that E3-6.7K is required for RID-mediated clearance of TR2 (4). However, conflicting reports regarding the requirement of E3-6.7K for clearance of TR1 have appeared. One report indicates that E3-6.7K is not required at all for TR1 clearance (46), while another study found that E3-6.7K is needed for optimal clearance of TR1 (4). E3-6.7K has also been shown to independently inhibit apoptosis induced by a Fas agonist antibody, TNF, TRAIL, or thapsigargin (32). These authors suggested that E3-6.7K may act by maintaining ER Ca2+ homeostasis (32).
The means by which RID performs its function have only recently been addressed in detail, particularly with regard to the down-regulation of Fas. RID does not inhibit the de novo synthesis of Fas mRNA (14, 41) or protein (38) but instead acts by driving the degradation of existing Fas molecules (14, 38, 41). Immunofluorescence and confocal microscopy experiments show that Fas internalized from the cell surface is located in vesicles, which were identified as lysosomes by coimmunofluorescence with the lysosomal marker LAMP-1 (41) or LAMP-2 (14). The use of inhibitors of lysosomal function such as bafilomycin A1, ammonium chloride, and chloroquine confirmed that Fas is degraded in lysosomes (14, 41). RID targets receptors for degradation, but RID itself recycles back to the cell surface, as suggested by pulse-chase experiments (22, 40). However, it is not yet clear at what point RID returns to the cell surface. Put together, these data suggest a model whereby specific cell surface receptors are internalized via the endocytic pathway, where they are ultimately degraded in lysosomes. At some point along the endocytic pathway RID drops off its target molecules and recycles back to the cell surface.
Little is known about the role that each subunit of RID plays in forming the active RID complex. Recently, it was reported that RIDβ contains a tyrosine residue within the cytoplasmic domain that is critical for the ability of RID to degrade Fas and EGFR and to protect cells from apoptosis induced by TRAIL or a Fas agonist antibody (28). It was suggested that the tyrosine residue forms part of a YXXΦ (Y is tyrosine, X is any amino acid, and Φ is a hydrophobic amino acid with a bulky side chain) sorting signal because of the amino acid context of the tyrosine residue and because RID functions within the endocytic pathway. To begin the process of unraveling the molecular mechanism of action of RIDα, a series of small deletions (three to five codons) was introduced into the Ad2 RIDα gene. Each mutant protein was expressed from a recombinant Ad in the same context as wild-type RIDα and was assessed with regard to its physical and functional properties. The majority of mutant RIDα proteins behaved similarly to wild-type RIDα with regard to all physical attributes. In addition, the cytoplasmic tail of RIDα was not important for degradation of Fas or EGFR, but the extracellular domain contains sequences important for EGFR, but not Fas, degradation. Furthermore, mutant proteins that do not reach the cell surface were defective for EGFR and Fas degradation, lending strong support to the idea that RID functions at the cell surface.
MATERIALS AND METHODS
Cells and viruses.
A549 cells were maintained in Dulbecco's minimal essential medium with 10% (vol/vol) fetal bovine serum. HT-29.14S cells were maintained in McCoy's medium supplemented with 10% fetal bovine serum. Suspension cultures of human KB cells were maintained in Joklik modified minimal essential medium containing 5% (vol/vol) horse serum. All media were supplemented with penicillin (100 U/ml) and streptomycin (100 μg/ml). Virus stocks were prepared in suspension cultures of KB cells, purified by banding in CsCl, and titered on A549 cells as described previously (42).
The virus sub765, the parental virus for all of the RIDα deletion mutants used in this study, is a derivative of in723, an Ad5-Ad2-Ad5 recombinant that overproduces E3 mRNA f resulting in overproduction of RIDα and RIDβ, which are encoded by mRNA f (6). To construct sub765, a duplex consisting of oligonucleotides 1 and 2 (see below) was cloned in place of the 26-bp BamHI-to-XhoI fragment of the plasmid pED(in723) to create the plasmid pED(10.4-NH2). This plasmid was partially digested with EcoRI, and a duplex consisting of oligonucleotides 3 and 4 (see below) was inserted into the EcoRI site located near the 3′ end of the RIDα gene to create the plasmid pED(NH2-COOH). Mutations in the RIDα gene were generated by M13 site-specific mutagenesis of the BamHI fragment of pED(NH2-COOH) using the version 2 oligonucleotide-directed mutagenesis kit (Amersham Biosciences, Piscataway, N.J.). Following replacement of the BamHI fragment of pED(NH2-COOH) with the mutagenized BamHI fragment, recombinant Ads were constructed and plaque purified as described previously (17, 52).
The following oligonucleotide duplexes were used in the construction of pED(NH2-COOH): 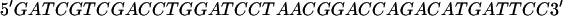



The RIDα deletion mutant viruses are named dl765.1 through dl765.14 as shown in Fig. 1. The nucleotide sequence flanking the deletion site (dash) for each mutation is shown below. The nucleotide numbering system is described by Cladaras and Wold (10), where the RIDα gene comprises nucleotides 2173 to 2489. The underlined nucleotides are mutated relative to the wild-type Ad2 sequence, resulting in amino acid substitutions as shown in Fig. 1. dl765.1, (2182)CGAGGA—TCCCTT(2205); dl765.2, (2227)TCT—GCT(2247); dl765.3, (2239)GCGCTCGCATCC—GAT(2262); dl765.4, (2266)ATC—TAC(2286); dl765.5, (2281)GTT—TTT(2301); dl765.6, (2300)TCC—ATC(2319); dl765.7, (2314)CTC—GTC(2340); dl765.8, (2335)GTA—CAG(2355); dl765.9, (2350)ATT—GTT(2370); dl765.10, (2372)GTTAAC—CTC(2391); dl765.11, (2389)CTC—CAA(2406); dl765.12, (2401)CCG—AGG(2418); dl765.13, (2419)ATC—CTT(2433); dl765.14, (2433)TAA (stop codon). The sequence of the RIDα gene of each mutant virus was confirmed by DNA sequence analysis using BigDye terminators and an ABI 377 sequence detection system (Applied Biosystems Inc., Foster City, Calif.). The viruses dl753 and dl748 were used as control viruses in some experiments. Neither dl753 nor dl748 synthesizes RIDα, but the level of RIDβ produced by dl753 is lower, and that produced by dl748 is higher, than the level produced by wild-type Ad (5).
FIG. 1.

Ad2 RIDα mutations used in this study. The wild-type Ad2 RIDα amino acid sequence that is present in the parental virus (sub765) is shown on the top line of each segment. For each mutant protein, only that portion of the amino acid sequence that contains the deletion is shown. Boldface, amino acids that differ from the wild-type (WT) sequence; dashes, deleted amino acids. The domain structure of RIDα is indicated above the sequence, and the amino acid numbers are shown below the sequence.
Western blot analysis.
Cells were infected with Ad mutants at 100 to 200 PFU per cell. For the analysis of RID in the membrane fraction, membranes were isolated as previously described (13). Membrane pellets were solubilized in 2× Laemmli buffer, subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to Immobilon-P membranes (Millipore, Bedford, Mass.). For analysis of Fas degradation, proteins solubilized at 18 h postinfection (p.i.) in radioimmunoprecipitation assay buffer were transferred to Immobilon-P. Rabbit antiserum P77-91, which was raised against a synthetic peptide corresponding to amino acids 77 to 91 of RIDα, was used at a dilution of 1:200. Rabbit antiserum P118-132, which was raised against a synthetic peptide corresponding to amino acids 118 to 132 of RIDβ, was used at a dilution of 1:400. A rabbit antibody directed against Fas (antibody C-20; Santa Cruz Biotechnology, Santa Cruz, Calif.) was used at a dilution of 1:1,000. Goat anti-rabbit immunoglobulin G (IgG) conjugated to horseradish peroxidase (Cappel/ICN, Costa Mesa, Calif.) was used as the secondary antibody at a dilution of 1:4,000. Proteins were visualized with ECL reagents (Amersham, Piscataway, N.J.).
Flow cytometry.
A549 cells were mock infected or infected with Ad that expresses wild-type or mutant forms of RIDα. At 24 h p.i. cells were removed from their dishes with phosphate-buffered saline containing EDTA (0.025%) and stained for fluorescence-activated cell sorter (FACS) analysis as described previously (46). The primary antibodies used were anti-human Fas antibody UB1 (Panvera, Madison, Wis.), anti-EGFR (528; Santa Cruz Biotechnology), and mouse IgG (as a negative control). The secondary antibody was a goat anti-mouse antibody conjugated to fluorescein isothiocyanate (FITC; Cappel/ICN). Stained cells were analyzed on a FACScaliber flow cytometer (Becton Dickinson, Palo Alto, Calif.) with CellQuest software (Becton Dickinson).
Metabolic labeling and immunoprecipitation.
KB and A549 cells were infected with 100 PFU of virus per cell for 18 h. Where indicated in the figure legends, KB cells were metabolically labeled with [35S]cysteine (Perkin-Elmer Life Sciences, Boston, Mass.) as described previously (44). Cells were washed twice with phosphate-buffered saline and lysed as previously described (44). RIDα or RIDβ was immunoprecipitated from lysates with rabbit antiserum P77-91 (anti-RIDα) or P118-132 (anti-RIDβ) and protein A-Sepharose beads (Sigma, St. Louis, Mo.). Immunoprecipitated proteins were washed as described previously (44) and then separated by SDS-PAGE. The gels were analyzed either by fluorography of the dried gel or by Western blotting.
Indirect immunofluorescence microscopy.
A549 cells were plated onto glass coverslips in growth medium. For analysis of RID expression, cells were either mock infected or infected with viruses in the presence of 1-β-d-arabinofuranosylcytosine and then were fixed at 22 h p.i. as previously described (40). Cells were stained with the P118-132 antiserum specific for RIDβ at a dilution of 1:250, followed by goat anti-rabbit IgG conjugated to FITC (Cappel/ICN). For examination of EGFR down-regulation, cells were fixed and then stained at 7 h p.i. with a rabbit anti-EGFR antibody (113368) at a dilution of 1:400 followed by goat anti-rabbit IgG conjugated to FITC (Cappel/ICN) at a dilution of 1:50. Cells were viewed with a Nikon Optiphot microscope equipped with epifluorescence and photographed as described previously (40).
Protein extraction with Triton X-114.
A549 cells were infected as described above and lysed with Triton X-114 extraction buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-114). The proteins were subjected to phase separation as previously described (23). Briefly, after removal of the particulate matter by centrifugation, the sample was layered over a 6% (wt/vol) sucrose cushion, warmed to 30°C to separate the phases, and centrifuged at 300 × g at room temperature. The detergent phase (an oily droplet at the bottom of the tube) was diluted, and proteins were separated by SDS-PAGE. RIDα was visualized by immunoblotting as described above.
Immune complex kinase assay.
KB cells were mock infected or infected with 150 PFU of virus per cell. Cells were harvested at 24 h p.i. and lysed with detergent as previously described (45). EGFR was immunoprecipitated from cell lysates with an EGFR-specific antipeptide antiserum (40) bound to protein A-Sepharose beads, and autophosphorylation of EGFR with [γ-32P]ATP was carried out as previously described (45). Proteins were dissociated from the Sepharose beads by boiling in Laemmli buffer and separated by SDS-PAGE on 7.5% gels. Phosphorylated EGFR was visualized by autoradiography.
Cell death assay.
HT-29.14S cells grown in 48-well plates were infected with Ads at 75 PFU per cell. At 17 h p.i., cells were treated for 28 h with cycloheximide (25 μg/ml) and the CH-11 anti-Fas agonist monoclonal antibody at various concentrations (0.5, 5.0, 50.0, and 500.0 ng/ml). Cytotoxicity resulting from agonist antibody-induced apoptosis was measured by a lactate dehyrogenase (LDH) release assay (Cytotox 96 kit; Promega, Madison, Wis.) in accordance with the manufacturer's instructions. Samples were assayed in triplicate, and results were read on an EL340 microplate reader (BioTec Instruments, Winooski, Vt.) at 490 nm. The percentage of specific lysis was determined with the following formula: [(experimental LDH release − spontaneous release)/(maximum release − spontaneous release)] × 100.
RESULTS
Construction of mutants.
The Ad-encoded RID complex effects the internalization of specific cell surface receptors, mediates their passage through the endocytic pathway to lysosomes, where the receptors are degraded, and presumably recycles back to the cell surface. This process has been described for members of two unrelated groups of receptors, the TNFR superfamily and the receptor tyrosine kinase family (4, 9, 14, 26, 38, 41). Since the mechanism of action of RID has scarcely been addressed, the present study was undertaken to examine three important issues: (i) which amino acid sequences within RIDα are required for RID-mediated receptor internalization and degradation, (ii) whether there are distinct steps in the process of RID-mediated receptor internalization and degradation that can be mapped to separate regions of the RIDα amino acid sequence, and (iii) whether different segments of RIDα are required for RID to function on the two different families of receptors.
To investigate these questions, a set of 14 nonoverlapping mutations in the RIDα gene was constructed. To avoid mutations that might grossly disrupt the structure of this small integral membrane protein, in-frame deletions that are only three to five codons long were introduced into the RIDα coding sequence (Fig. 1). Because of the mutagenesis strategy, some mutant proteins also contain one or more amino acid substitutions flanking the deletion (e.g., see sub765.3 in Fig. 1). The mutant genes were then introduced in place of the wild-type RIDα gene present in the virus sub765. The parental virus, sub765, was engineered to overexpress E3 mRNA f, which encodes RIDα and RIDβ (see Materials and Methods). The mutant viruses are named sub765.1, sub765.2, etc., but are referred to as 1, 2, etc. for simplicity, and the associated proteins are referred to as mutant proteins 1, 2, etc. Each mutation fell into one of four broad categories depending on which domain of RIDα contained the deletion, the signal sequence (mutant proteins 1 and 2), the extracellular domain (mutant 3 to 5), the transmembrane domain (mutant proteins 6 to 8), or the cytoplasmic domain (mutant proteins 9 to 14).
Stable expression of mutant RIDα proteins.
To address the stability and processing of each mutant protein, metabolically labeled RIDα was immunoprecipitated from infected cells with an anti-RIDα rabbit antiserum that is directed against a peptide corresponding to amino acids 77 to 91. The immunoprecipitates were analyzed by SDS-PAGE and fluorography. As expected, the parental virus that expresses wild-type RIDα directed the stable expression of two proteins that correspond to the two known forms of RIDα, the uncleaved form (upper band) and the cleaved form, in which the signal peptide has been removed (lower band) (Fig. 2, lane a) (22). Nearly all of the mutant RIDα proteins appeared to be stably expressed (Fig. 2, lanes c to k). Mutant protein 1 showed a low level of RIDα expression, which may have resulted from instability of the mutant protein or decreased expression (Fig. 2, lane b). Mutant proteins 13 and 14 were not detected (Fig. 2, lanes l and m), most probably because in both of them the portion of RIDα against which the antipeptide antiserum is directed is partially deleted.
FIG. 2.

Immunoprecipitation of wild-type and mutant RIDα proteins. KB cells were infected with parental virus that expresses wild-type (WT) RIDα (sub765) or viruses that express mutant forms of RIDα. Metabolically labeled proteins that were immunoprecipitated from cell lysates with an antiserum directed against RIDα were separated by SDS-PAGE. A fluorograph of the dried gel is shown. The sizes of the molecular weight standards (in thousands) are indicated to the left of the gel. The virus used for the infection is shown above the each lane. For viruses that express mutant RIDα proteins, the amino acids deleted from each particular mutant are also shown. The migration position of RIDα is indicated to the right.
Normally about 50% of wild-type RIDα is cleaved between amino acids 22 and 23, giving rise to two forms of RIDα (22). Surprisingly, mutant protein 2, which contains a deletion overlapping the cleavage site, was still efficiently cleaved (Fig. 2, lane c). The size of the lower band of mutant protein 2 compared to that of the corresponding wild-type band suggested that the cleavage occurred at nearly the same position as for wild-type RIDα (compare lanes a and c of Fig. 2). Mutant proteins 3 and 4 showed inefficient cleavage, if any at all (Fig. 2, lanes d and e). In addition, mutant proteins 6 to 8 showed predominantly one band, which migrated similarly to the cleaved form of wild-type RIDα (Fig. 2, lanes g to i). Note, however, that in subsequent experiments mutant proteins 7 and 8 exhibited equal amounts of cleaved and uncleaved forms, indicating that these two proteins are similar to wild-type RIDα with respect to cleavage (see Fig. 3 to 5). Of the mutant proteins examined in this experiment, only one (mutant protein 1) appeared to be unstable while several (mutant proteins 2 to 4, and 6) exhibited an altered cleavage pattern.
FIG. 3.

Mutant RIDα proteins interact with RIDβ. A549 cells were mock-infected or infected with parental virus that expresses wild-type (WT) RIDα (sub765), a virus that does not express RIDα (dl753), or viruses that express mutant forms of RIDα. Proteins were immunoprecipitated from cell lysates with an antibody directed against RIDβ. The immunoprecipitated proteins were split in two parts, separated by SDS-PAGE on two different gels, and transferred to Immobilon-P membranes. One membrane was probed with an anti-RIDα antiserum to detect coimmunoprecipitation of RIDα (A), while the other membrane was probed with an anti-RIDβ antiserum to check the amount of RIDβ immunoprecipitated (B). The virus used for the infection is shown above each lane. Arrows, RIDα-specific (A) and RIDβ-specific (B) bands.
FIG. 5.

Mutant RIDα proteins are integral membrane proteins. A549 cells were infected with parental virus that expresses wild-type (WT) RIDα (sub765), a virus that does not express RIDα (dl753), or viruses that express mutant forms of RIDα. Cell lysates were prepared with Triton X-114 extraction buffer. Proteins in the detergent phase were separated by SDS-PAGE and analyzed by Western blotting. The blot was probed with an anti-RIDα antiserum. RIDα-specific bands are indicated to the left. The virus used for the infections is shown above each lane.
Mutant RIDα proteins coimmunoprecipitate with RIDβ.
Formation of the RID complex is critically important for RID function since neither RID subunit by itself can internalize and degrade cell surface receptors (40, 45). Coimmunoprecipitation of RIDα by an antibody against RIDβ was used to assay for complex formation. Lysates from cells infected with the parental virus or viruses that express a mutant RIDα protein were subjected to immunoprecipitation with an antiserum against RIDβ. The immune complexes were then split in two parts and analyzed by Western blotting using an anti-RIDα antiserum (Fig. 3A) or an anti-RIDβ antiserum (Fig. 3B). Both RIDα bands were coimmunoprecipitated with RIDβ from the sub765-infected cells (Fig. 3A, lane b). A virus mutant that does not express RIDα (dl753) did not show any RIDα-specific bands (Fig. 3A, lane c). Nearly all of the mutant RIDα proteins were coimmunoprecipitated with RIDβ (Fig. 3A, lanes d to q). Note that mutant proteins 9 and 10, which were not analyzed in Fig. 2, were stable and properly processed (Fig. 3A, lanes l and m). Mutant proteins 12 to 14 appeared to be coimmunoprecipitated either poorly or not at all (Fig. 3A, lanes o to q). This is most likely a result of poor recognition of these mutant proteins in the Western blot and not a result of the inability of these proteins to be coimmunoprecipitated. Recall that the anti-RIDα antiserum used in the Western blot recognizes epitopes that overlap the amino acids that were deleted in these proteins. Importantly, when metabolically labeled proteins were used in a coimmunoprecipitation experiment, both forms of mutant proteins 12 to 14 were clearly visible, indicating that these proteins are stable and able to form a complex with RIDβ (data not shown). Figure 3B demonstrates that RIDβ was immunoprecipitated nearly equally well from all cell lysates.
Mutant RIDα proteins are localized to membranes.
Both the cleaved and full-length forms of RIDα are integral membrane proteins (22). Therefore, each mutant protein was assessed for its ability to associate with membranes. Lysates were prepared from uninfected and virus-infected cells. Following removal of nuclei from the cell lysates by low-speed centrifugation, membranes were pelleted by high-speed centrifugation. The membrane fraction was analyzed for the presence of RIDα by Western blot analysis. As expected, two RIDα bands were detected in the membrane fraction from parental virus-infected cells but no RIDα bands were seen in the dl753-infected sample since this virus does not express RIDα (Fig. 4). Mutant RIDα proteins 2 through 11 (Fig. 4, lanes e to n) were present abundantly in the membrane fraction, whereas mutant protein 1 was present at a low level (Fig. 4, lane d). Mutant proteins 12 to 14 were not detected, most likely because the P77-91 antiserum was used to detect RIDα (Fig. 4, lanes o to q). This experiment indicates that most of the mutant proteins were associated with membranes.
FIG. 4.

Localization of mutant RIDα proteins in the membrane fraction. A549 cells were mock infected or infected with parental virus that expresses wild-type (WT) RIDα (sub765), a virus that does not express RIDα (dl753), or viruses that express mutant forms of RIDα. The total membrane fraction was isolated from cell lysates (see Materials and Methods). Proteins extracted from the membranes were separated by SDS-PAGE and then analyzed by Western blotting. The blot was probed with an anti-RIDα antiserum. RIDα-specific bands are indicated to the left. The virus used for the infection is shown above each lane.
The previous experiment could not distinguish between peripheral association of RIDα with the membrane and integration of RIDα into the membrane, so a Triton X-114 extraction technique was performed on lysates of mock- and virus-infected cells. This technique partitions integral membrane proteins into the detergent phase while cytoplasmic and peripheral membrane proteins remain in the aqueous phase (23). Following Triton X-114 extraction, samples of the detergent phase were subjected to Western blot analysis to detect RIDα. Both forms of wild-type RIDα partitioned into the detergent phase (Fig. 5, lane a). Nearly all of the mutant proteins were also present in the detergent phase, indicating that these proteins were not just associated with the membrane but were inserted into it (Fig. 5, lanes c to n and p). The one exception was mutant protein 13, which did not show any bands, most likely due to the antiserum that was used to detect RIDα (Fig. 5, lane o). Mutant proteins 5 to 8 each have a transmembrane domain that is reduced to about 21 or 22 amino acids long, which should still be long enough to span the membrane (2, 27). These data indicate that nearly all of the mutant proteins are integral membrane proteins.
Subcellular localization of mutant RIDα proteins.
The subcellular localization of each mutant protein was analyzed by indirect immunofluorescence microscopy. This is a key issue since the probable site of action of the RID complex is the plasma membrane. Because the anti-RIDα serum does not efficiently react with RIDα when it is complexed with RIDβ, cell surface localization of RIDβ was used as a surrogate for the presence of RIDα at the plasma membrane. Recall that neither individual RID subunit reaches the plasma membrane unless the RID complex is formed. Mock-infected cells and cells infected with a mutant virus that does not express RIDβ showed a very low level of background staining for RIDβ (data not shown). Infection with sub765, which expresses wild-type RIDα and RIDβ, shows staining at the plasma membrane and the Golgi (Fig. 6A). As an example of what RIDβ staining looks like in the absence of RIDα, a virus mutant (dl748) that expresses RIDβ but not RIDα showed RIDβ staining in the ER and Golgi but not at the cell surface (Fig. 6B). Mutant protein 14 was similar to wild-type RIDα in that this mutant protein showed strong RIDβ cell surface staining (Fig. 6P). Mutant proteins 1 and 6 to 8 showed little if any cell surface staining but instead showed a RIDβ staining pattern similar to that of dl748 (Fig. 6C and H to J). The remaining mutant proteins (proteins 2 to 5 and 9 to 13) all showed weak cell surface staining, suggesting that transport of RID to the cell surface is impaired but not eliminated (Fig. 6D to G and K to O). In other experiments, cell surface staining for RIDβ with the mutant RIDα proteins 3 to 5, 9, 10, and 12 was more clearly visible. A number of the mutants showed punctate cytoplasmic staining, which may represent a defect in cell surface transport and/or RID complexes that have been internalized from the cell surface. These data indicate that the majority of mutant RIDα proteins are able to support translocation of the RID complex to the cell surface.
FIG. 6.

Subcellular localization of RID as determined by detection of RIDβ by indirect immunofluorescence microscopy. A549 cells plated on coverslips were infected with parental virus that expresses wild-type (WT) RIDα (sub765), a virus that does not express RIDα (dl748), or viruses that express mutant forms of RIDα. Cells were fixed and then stained for RIDβ with a rabbit anti-RIDβ antiserum followed by FITC-conjugated goat anti-rabbit IgG. The virus used for the infection is shown above each panel.
Internalization of Fas and EGFR.
To study the ability of the virus mutants to clear receptors from the cell surface, mock- and Ad-infected A549 cells were stained with an antibody against either Fas or EGFR and then subjected to FACS analysis. The mock-infected cells showed bright staining for Fas and EGFR (Fig. 7). Parental virus cleared both receptors from the cell surface, while a mutant (dl748) with the RIDα gene deleted was unable to clear either receptor. With respect to clearance of EGFR, mutant proteins 1 through 9 were unable to down-regulate EGFR, mutant proteins 11 and 12 were similar to wild-type RIDα, and mutant proteins 10, 13, and 14 only partially cleared EGFR from the cell surface (Fig. 7). When clearance of Fas was examined, a slightly different pattern of functional mutant proteins was observed. Similar to the case with EGFR, mutant proteins 1 and 6 to 9 were unable to clear Fas from the cell surface, mutant proteins 11 and 12 behaved like wild-type RIDα, and mutant proteins 10, 13, and 14 showed a partial-clearance phenotype. The only difference between the internalization of EGFR and Fas was that mutant proteins 2 to 5 were partially able to clear Fas, whereas these mutant proteins had no activity in clearing EGFR.
FIG. 7.

Internalization of EGFR and Fas from the cell surface. A549 cells were mock-infected or infected with parental virus (sub765; wild type), a virus that does not express RIDα (dl748; RIDα −), or the viruses that express mutant RIDα proteins (1 to 14). Cells harvested at 24 h p.i. were incubated with antisera against EGFR or Fas followed by a FITC-conjugated secondary antibody and then analyzed by flow cytometry.
To investigate the capacity of each mutant protein to direct internalization of EGFR into endosomes and lysosomes, mock-infected and virus-infected A549 cells were examined by indirect immunofluorescence microscopy. In mock-infected cells, EGFR was visualized at the plasma membrane and in the perinuclear region, which is indicative of the Golgi (Fig. 8A). When cells were infected with the parental virus, which expresses wild-type RIDα, EGFR was no longer seen at the cell surface but was located intracellularly in vesicles (Fig. 8B). Mutant proteins 11 and 12 showed an EGFR staining pattern similar to that observed with wild-type RIDα (Fig. 8I and J), while mutant proteins 13 and 14 showed intracellular vesicles as well as slight staining at the cell surface (Fig. 8K and L). The remaining mutants shown in Fig. 8 exhibited a staining pattern similar to that of mock-infected cells, indicating that these mutant proteins were defective for internalization of EGFR (Fig. 2C to H). In other experiments, mutant proteins 2, 6, 9, and 10 appeared to be defective for formation of EGFR vesicles (data not shown).
FIG. 8.

Subcellular localization of EGFR by indirect immunofluorescence. A549 cells were mock infected or infected with parental virus (sub765; wild type [WT]) or viruses that express mutant RIDα proteins. Cells were fixed at 7 h p.i. and then stained for EGFR with a rabbit anti-EGFR antiserum and FITC-conjugated secondary antibody. Shown above each panel is the virus used for infection.
Degradation of EGFR and Fas.
Following RID-mediated internalization, receptors are targeted for lysosomal degradation (4, 14, 41, 46). The degradation of EGFR was examined by an immune complex kinase assay in which EGFR was immunoprecipitated from cell lysates and the resulting immune complexes were incubated with [γ-32P]ATP to examine EGFR-catalyzed, tyrosine-specific autophosphorylation. The 32P-labeled EGFR was then analyzed by SDS-PAGE and autoradiography. As shown in Fig. 9A and as reported previously (45), a strong EGFR band was obtained from mock-infected cells, while very little EGFR was detected in sub765-infected cells expressing wild-type RIDα (Fig. 9A, lanes a and b). This indicates that EGFR was almost completely degraded in sub765-infected cells. Most of the RIDα mutant proteins were defective for degradation of EGFR, as evidenced by the strong EGFR bands in these samples (Fig. 9A, lanes c to e and g to m). The signal was greatly reduced with mutant proteins 11 and 12, indicating significant degradation of EGFR (Fig. 9A, lanes n and o), while mutant proteins 13 and 14 showed partial EGFR degradation (Fig. 9A, lanes p and q). Mutant protein 3 also showed some ability to degrade EGFR despite the lack of EGFR internalization seen in the FACS and immunofluorescence experiments (Fig. 9A, lane f). Overall, the data from the immune complex kinase assay agreed with the data obtained by FACS analysis and immunofluorescence (Fig. 7 and 8), indicating that the ability of the mutant RIDα proteins to clear EGFR from the cell surface correlates well with the ability to degrade EGFR.
FIG. 9.

RID-mediated degradation of EGFR and Fas. (A) KB cells were mock infected or infected with parental virus (sub765), a virus that does not express RIDα (dl753), or viruses that express mutant forms of RIDα at 150 PFU per cell. At 24 h p.i., cells were lysed with NP-40, EGFR was immunoprecipitated, and a kinase reaction was carried out in the immune complex by adding [γ-32P]ATP. Proteins in the immune complex were separated by SDS-PAGE, and EGFR was visualized by autoradiography. WT, wild type. (B and C) A549 cells were mock infected or infected with parental (sub765) virus or viruses that express mutant forms of RIDα at 200 PFU per cell. At 24 h p.i., cells were lysed and analyzed for the presence of Fas (B) or E1B-19K (C) by immunoblotting.
Degradation of Fas was also examined (Fig. 9B). Lysates prepared from mock-infected and virus-infected cells were subjected to SDS-PAGE and Western blot analysis with an anti-Fas antibody. A single band representing Fas was abundantly present in mock-infected cells; upon infection with the sub765 parental virus, the band was greatly diminished in intensity (Fig. 9B, lane b). A significant amount of Fas degradation occurred with mutant proteins 2 to 5 and 11 to 14 (Fig. 9B, lanes e to h and n to q), while Fas levels were unaffected with the remaining mutant proteins (Fig. 9B, lanes c, d, and i to m). Thus, as seen for EGFR, the ability of mutant RIDα proteins to degrade Fas corresponded well with the ability of the mutant proteins to internalize the receptor from the cell surface. For the Fas degradation experiment, the adenovirus E1B-19K protein was present at nearly the same level in all infected cell lysates, demonstrating that all infections were equivalent (Fig. 9C).
Inhibition of Fas agonist-induced apoptosis.
To determine if the ability to internalize and degrade Fas was biologically significant, a cell death assay that assesses the ability of mutant RIDα proteins to protect cells from Fas-mediated apoptosis was performed. HT-29.14S cells were used in this experiment because the E1B-19K protein does not inhibit apoptosis through the Fas pathway in these cells (38). Virus-infected HT-29.14S cells were treated with a Fas agonist antibody to induce apoptosis. After 28 h of treatment, the cell medium was assayed for LDH release as a measure of apoptosis. The parental virus (sub765) was able to protect cells from Fas agonist-induced apoptosis, consistent with the finding that the RID protein expressed by this virus mediates the degradation of nearly all Fas (Fig. 10). In contrast, cells infected with a mutant virus (dl748) that does not expresses any RIDα undergo a significant amount of apoptosis since RID is not functional (Fig. 10). Mutant proteins 2 to 5 (Fig. 10A) and 11, 12, and 14 (Fig. 10C) all provided good protection from lysis induced by the anti-Fas antibody, while mutant proteins 1 (Fig. 10A) and 6 to 8 (Fig. 10B) showed little if any protection from Fas-mediated apoptosis. Partial protection from apoptosis was seen for mutant proteins 9 (Fig. 10B) and 10 and 13 (Fig. 10C). In general, the ability to protect cells from Fas-agonist induced apoptosis correlated with the ability to internalize and degrade Fas, providing further evidence that down-regulation of Fas from the cell surface is the mechanism by which RID inhibits Fas-mediated apoptosis.
FIG. 10.

Inhibition of Fas agonist-induced cell death. HT-29.14S cells were mock infected or infected with parental virus (sub765), a virus that does not express RIDα (dl748), or viruses that express mutant forms of RIDα at 75 PFU per cell. Cell lysis was measured by an LDH release assay as described in Materials and Methods. The results are representative of at least two independent experiments. The results shown in panels A and B are from the same experiment but are divided into two panels for clarity. Panel C represents a separate experiment.
DISCUSSION
To discover what portions of RIDα are important for RID-mediated receptor internalization and degradation, a panel of small deletion mutations in the RIDα gene was created and the mutant genes were inserted in place of the wild-type Ad2 RIDα gene in a virus that was engineered to overexpress RIDα and RIDβ. Each mutant RIDα protein was assessed for five different physical characteristics and six functional properties associated with wild-type RIDα. The results, summarized in Fig. 11, reveal that 8 out of the 14 mutant proteins (proteins 2, 5, and 9 to 14) were similar to wild-type RIDα with regard to all five physical characteristics examined, namely, stable expression, proper proteolytic processing, insertion into the membrane, formation of a complex with RIDβ, and proper cellular localization. The difficulty in assessing all the physical characteristics of mutant proteins 13 and 14 likely stems from the fact that both proteins contain a deletion that overlaps the peptide against which the anti-RIDα antiserum was made, thus making it difficult to detect these two mutant proteins. Because of the extreme difficulty in detecting mutant protein 13, its physical properties were inferred to be similar to those of wild-type RIDα since the mutant protein was capable of reaching the cell surface and directing the degradation of EGFR and Fas. Overall, these data indicate that several regions of RIDα can tolerate small deletions without grossly affecting the stability, processing, and cell surface localization of the protein.
FIG. 11.

Summary diagram. The domain structure of RIDα is shown at the top. The location of each mutation is shown underneath along with the biochemical properties of each mutant protein and the functional properties with regard to EGFR and Fas cell surface clearance, internalization, and degradation and protection from Fas-mediated apoptosis. + and −, presence or absence of each biochemical property for each mutant protein, respectively (superscript “a” denotes properties that were inferred to be similar to those of wild-type RIDα). Red, yellow, and green shading indicate defective, partially active, and fully functional phenotypes, respectively.
Defects in one or more physical properties were noted for several mutant proteins. Most notably, mutant protein 1 appeared to be unstable since this protein was detected at only a very low level in most analyses and was defective in all functional assays. Mutant protein 2 was efficiently cleaved despite the fact that it lacks the normal cleavage site between residues 22 and 23 (22). Clearly, a new cleavage site is being utilized in this mutant protein. Mutant proteins 3 and 4, in each of which a portion of the extracellular domain that is C-terminal to the cleavage site is deleted, were cleaved inefficiently at best. This suggests that some aspect of the extracellular domain, beyond the primary amino acid sequence at the cleavage site, is important in order for cleavage to occur. Although mutant proteins 6 to 8 were capable of forming a complex with RIDβ, they were unable to support transport of the RID complex to the cell surface, suggesting that the transmembrane domain of RIDα may be important for this property. However, only weak RIDβ staining was detected at the cell surface with most of the other mutant RIDα proteins (proteins 2 to 5 and 9 to 13; Fig. 6D to G, and K to O), indicating that even small deletions elsewhere in RIDα result in less-efficient transport of the RID complex to the cell surface.
RID-mediated receptor internalization and degradation were investigated by assessing the ability of mutant RIDα proteins to down-regulate receptors representative of the TNFR superfamily (Fas) and the receptor tyrosine kinase family (EGFR). The results, summarized in Fig. 11, suggest that distinct portions of RIDα are required for down-regulation of Fas and EGFR. Mutations within the extracellular domain (mutant proteins 2 to 5) produced different phenotypes with respect to Fas and EGFR down-regulation, but mutations within the signal sequence, transmembrane domain, and cytoplasmic domain produced the same phenotype for Fas and EGFR down-regulation. Mutant proteins 2 through 5 were able to clear and degrade Fas and protect cells from Fas-mediated killing, suggesting that this region of RIDα does not contain sequences critical for down-regulation of Fas. In contrast, these same mutant proteins were generally defective for clearance and degradation of EGFR, suggesting an important role for this portion of RIDα in down-regulating this receptor. It is possible that these mutant proteins are not entirely defective for EGFR degradation but rather may simply have delayed kinetics for EGFR clearance. This idea was suggested by the ability of mutant protein 3 to partially degrade EGFR (Fig. 9A) despite showing no ability to clear or internalize EGFR (Fig. 7 and 8). A time course of EGFR degradation for each of these mutant proteins might resolve this issue. Regardless of the exact phenotype, however, mutations within the extracellular domain of RIDα clearly had differential effects on the ability to down-regulate Fas and EGFR. It would be of interest to determine if the distinct phenotypes with mutant proteins 2 through 5 are sustained for other members of the TNFR superfamily and receptor tyrosine kinase family upon which RID acts. If so, future studies on the mechanism of RID down-regulation of receptor tyrosine kinase family members should focus on this region of RIDα.
Previous hypotheses on the mechanism of action of RID-mediated receptor down-regulation have focused on the possible interaction between RID and the adaptor protein 2 (AP-2) complex, which connects the cellular sorting machinery with receptors targeted for endocytosis in clathrin-coated pits (20). In this regard it has been noted that the cytoplasmic tails of both RIDα and RIDβ contain potential cell sorting motifs (7, 14, 51). A YXXΦ motif, which binds to the mu subunits of AP complexes (34, 39), has been identified in RIDβ (28), and two potential dileucine motifs, which bind to the AP complex beta chain (37), have been recognized in RIDα (7, 14, 51). It was, therefore, of interest that mutant protein 14, in which both potential dileucine motifs were deleted, was still capable of Fas and EGFR internalization and degradation (Fig. 7 to 10). This suggests that the dileucine motifs in RIDα are not essential for RID-mediated receptor down-regulation, although these motifs may be required for full activity since mutant protein 14 shows partial activity for internalization and degradation of EGFR (Fig. 7, 8, and 9A). This, in turn, suggests the possibility that the YXXΦ motif in RIDβ compensates for the lack of dileucine motifs present in mutant protein 14.
Since RID internalizes and degrades cell surface receptors (4, 9, 14, 41, 46) and is localized to the plasma membrane (18, 40), it was hypothesized that RID acts at the cell surface and not in intracellular membranes (9, 14, 18, 40). It is also possible that RID functions within the cell in addition to or instead of functioning at the plasma membrane, as was suggested recently (11). The data obtained with mutant proteins 6 to 8, each of which contains a deletion within the transmembrane region of RIDα, lend support to the idea that RID functions at the plasma membrane and not internally. These mutant proteins do not support RID complex localization to the cell surface despite behaving like wild-type RIDα in most other ways (among these three mutant proteins the only other defect is in proteolytic processing by mutant protein 6). In accordance with these observations, mutant proteins 6 to 8 were all defective for internalization of both Fas and EGFR (Fig. 7 and 8). More interestingly, these mutant proteins were also unable to support RID-mediated degradation of Fas or EGFR (Fig. 9), strengthening the idea that RID acts only on receptors that have reached the cell surface.
A goal of this study was to determine if specific RIDα amino acid sequences that are important for individual steps within the degradation pathway could be identified. Nearly all of the mutant proteins that were defective for cell surface clearance of the receptors were also defective for receptor entry into endosomes and lysosomes and degradation. The one possible exception was mutant protein 10. This mutant protein displayed a partial phenotype for cell surface clearance of Fas and EGFR (Fig. 7) but was defective for internalization and degradation of both receptors (Fig. 8 and 9). The most likely explanation for the behavior of this mutant protein is that internalization and degradation of both receptors are kinetically delayed. Thus, these results suggest a model wherein RID may be important only in the initial step(s) in the pathway. For example, the function of RID may be to drive target receptors into clathrin-coated pits, forcing entry of the receptors into the endocytic pathway. However, once the receptors are within the coated pits, RID may not be involved in the subsequent steps that lead to receptor degradation.
In addition to its role in receptor degradation, RID inhibits TNF-induced translocation of cytosolic phospholipase A2 (cPLA2) from the cytosol to membranes (13), release of arachidonic acid (21), and activation of NF-κB (16). It has been suggested that at early times p.i. the inhibition of NF-κB activation is not due to RID-mediated degradation of TNFR1 since the level of this receptor at the cell surface was reduced only 5 to 10% after infection with a virus that expresses RID (16). In addition, inhibition of cPLA2 translocation and arachidonic acid release occurs before significant degradation of TNFR1 (Dimitrov et al., unpublished). These results suggest that these functions of RID are independent of its ability to degrade TNFR1. It would be of interest to use this panel of mutant proteins to determine if these TNF-related functions map to a region of RIDα different from the region required for the receptor degradation function.
Acknowledgments
We thank Chris Wells for technical assistance and Dawn Schwartz for help in preparing the manuscript.
This research was supported by grant CA58538 from the National Institutes of Health.
REFERENCES
- 1.Abendroth, A., and A. M. Arvin. 2001. Immune evasion as a pathogenic mechanism of varicella zoster virus. Semin. Immunol. 13:27-39. [DOI] [PubMed] [Google Scholar]
- 2.Adams, G. A., and J. K. Rose. 1985. Structural requirements of a membrane-spanning domain for protein anchoring and cell surface transport. Cell 41:1007-1015. [DOI] [PubMed] [Google Scholar]
- 3.Alcami, A., and U. H. Koszinowski. 2000. Viral mechanisms of immune evasion. Immunol. Today 21:447-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benedict, C., P. Norris, T. Prigozy, J. L. Bodmer, J. A. Mahr, C. Garnett, F. Martinon, J. Tschopp, L. R. Gooding, and C. F. Ware. 2001. Three adenovirus E3 proteins cooperate to evade apoptosis by tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and -2. J. Biol. Chem. 276:3270-3278. [DOI] [PubMed] [Google Scholar]
- 5.Brady, H. A., and W. S. M. Wold. 1987. Identification of a novel sequence that governs both polyadenylation and alternative splicing in region E3 of adenovirus. Nucleic Acids Res. 15:9397-9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brady, H. A., and W. S. M. Wold. 1988. Competition between splicing and polyadenylation reactions determines which adenovirus region E3 mRNAs are synthesized. Mol. Cell. Biol. 8:3291-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgert, H. G., and J. H. Blusch. 2000. Immunomodulatory functions encoded by the E3 transcription unit of adenoviruses. Virus Genes 21:13-25. [PubMed] [Google Scholar]
- 8.Burgert, H. G., Z. Ruzsics, S. Obermeier, A. Hilgendorf, M. Windheim, and A. Elsing. 2002. Subversion of host defense mechanisms by adenoviruses. Curr. Top. Microbiol. Immunol. 269:273-318. [DOI] [PubMed] [Google Scholar]
- 9.Carlin, C. R., A. E. Tollefson, H. A. Brady, B. L. Hoffman, and W. S. M. Wold. 1989. Epidermal growth factor receptor is down-regulated by a 10,400 MW protein encoded by the E3 region of adenovirus. Cell 57:135-144. [DOI] [PubMed] [Google Scholar]
- 10.Cladaras, C., and W. S. Wold. 1985. DNA sequence of the early E3 transcription unit of adenovirus 5. Virology 140:28-43. [DOI] [PubMed] [Google Scholar]
- 11.Crooks, D., S. J. Kil, J. M. McCaffery, and C. Carlin. 2000. E3-13.7 integral membrane proteins encoded by human adenoviruses alter epidermal growth factor receptor trafficking by interacting directly with receptors in early endosomes. Mol. Biol. Cell 11:3559-3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derfuss, T., and E. Meinl. 2002. Herpesviral proteins regulating apoptosis. Curr. Top. Microbiol. Immunol. 269:257-272. [DOI] [PubMed] [Google Scholar]
- 13.Dimitrov, T., P. Krajcsi, T. W. Hermiston, A. E. Tollefson, M. Hannink, and W. S. M. Wold. 1997. Adenovirus E3-10.4K/14.5K protein complex inhibits tumor necrosis factor-induced translocation of cytosolic phospholipase A2 to membranes. J. Virol. 71:2830-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elsing, A., and H.-G. Burgert. 1998. The adenovirus E3/10.4K-14.5K proteins down-modulate the apoptosis receptor Fas/Apo-1 by inducing its internalization. Proc. Natl. Acad. Sci. USA 95:10072-10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett, H., and G. McFadden. 2002. Poxviruses and apoptosis: a time to die. Curr. Opin. Microbiol. 5:395-402. [DOI] [PubMed] [Google Scholar]
- 16.Friedman, J. M., and M. S. Horwitz. 2002. Inhibition of tumor necrosis factor alpha-induced NF-κB activation by the adenovirus E3-10.4/14.5K complex. J. Virol. 76:5515-5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hermiston, T. W., A. E. Tollefson, and W. S. M. Wold. 1998. Construction of mutations in the adenovirus early region 3 (E3) transcription unit, p. 11-24. In W. S. M. Wold (ed.), Adenovirus methods and protocols. Humana Press, Totowa, N.J.
- 18.Hoffman, P., M. B. Yaffe, B. L. Hoffman, S. Yei, W. S. M. Wold, and C. Carlin. 1992. Characterization of the adenovirus E3 protein that down-regulates the epidermal growth factor receptor. Evidence for intermolecular disulfide bonding and plasma membrane localization. J. Biol. Chem. 267:13480-13487. [PubMed] [Google Scholar]
- 19.Horwitz, M. 2001. Adenovirus immunoregulatory genes and their cellular targets. Virology 279:1-8. [DOI] [PubMed] [Google Scholar]
- 20.Kirchhausen, T. 1999. Adaptors for clathrin-mediated traffic. Annu. Rev. Cell Dev. Biol. 15:705-732. [DOI] [PubMed] [Google Scholar]
- 21.Krajcsi, P., T. Dimitrov, T. W. Hermiston, A. E. Tollefson, T. S. Ranheim, S. B. Vande Pol, A. H. Stephenson, and W. S. M. Wold. 1996. The adenovirus E3-14.7K protein and the E3-10.4K/14.5K complex of proteins, which independently inhibit tumor necrosis factor (TNF)-induced apoptosis, also independently inhibit TNF-induced release of arachidonic acid. J. Virol. 70:4904-4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krajcsi, P., A. E. Tollefson, C. W. Anderson, A. R. Stewart, C. R. Carlin, and W. S. M. Wold. 1992. The E3-10.4K protein of adenovirus is an integral membrane protein that is partially cleaved between Ala22 and Ala23 and has a Ccyt orientation. Virology 187:131-144. [DOI] [PubMed] [Google Scholar]
- 23.Krajcsi, P., A. E. Tollefson, C. W. Anderson, and W. S. M. Wold. 1992. The adenovirus E3 14.5-kilodalton protein, which is required for down-regulation of the epidermal growth factor receptor and prevention of tumor necrosis factor cytolysis, is an integral membrane protein oriented with its C terminus in the cytoplasm. J. Virol. 66:1665-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krajcsi, P., A. E. Tollefson, and W. S. M. Wold. 1992. The E3-14.5K integral membrane protein of adenovirus that is required for down-regulation of the EGF receptor and for prevention of TNF cytolysis is O-glycosylated but not N-glycosylated. Virology 188:570-579. [DOI] [PubMed] [Google Scholar]
- 25.Krajcsi, P., and W. S. M. Wold. 1992. The adenovirus E3-14.5K protein which is required for prevention of TNF cytolysis and for down-regulation of the EGF receptor contains phosphoserine. Virology 187:492-498. [DOI] [PubMed] [Google Scholar]
- 26.Kuivinen, E., B. L. Hoffman, P. A. Hoffman, and C. R. Carlin. 1993. Structurally related class I and class II receptor protein tyrosine kinases are down-regulated by the same E3 protein coded for by human group C adenoviruses. J. Cell Biol. 120:1271-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuroiwa, T., M. Sakaguchi, K. Mihara, and T. Omura. 1991. Systematic analysis of stop-transfer sequence for microsomal membrane. J. Biol. Chem. 266:9251-9255. [PubMed] [Google Scholar]
- 28.Lichtenstein, D. L., P. Krajcsi, D. J. Esteban, A. E. Tollefson, and W. S. M. Wold. 2002. The adenovirus RIDβ subunit contains a tyrosine residue that is critical for RID-mediated receptor internalization and inhibition of Fas- and TRAIL-induced apoptosis. J. Virol. 76:11329-11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lichtenstein, D. L., K. Toth, K. Doronin, A. E. Tollefson, and W. S. Wold. Functions and mechanisms of action of the adenovirus E3 proteins. Int. Rev. Immunol., in press. [DOI] [PubMed]
- 30.Mahr, J. A., and L. R. Gooding. 1999. Immune evasion by adenoviruses. Immunol. Rev. 168:121-130. [DOI] [PubMed] [Google Scholar]
- 31.McNees, A. L., C. T. Garnett, and L. R. Gooding. 2002. The adenovirus E3 RID complex protects some cultured human T and B lymphocytes from Fas-induced apoptosis. J. Virol. 76:9716-9723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moise, A. R., J. R. Grant, T. Z. Vitalis, and W. A. Jefferies. 2002. Adenovirus E3-6.7K maintains calcium homeostasis and prevents apoptosis and arachidonic acid release. J. Virol. 76:1578-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss, B., and J. L. Shisler. 2001. Immunology 101 at poxvirus U: immune evasion genes. Semin. Immunol. 13:59-66. [DOI] [PubMed] [Google Scholar]
- 34.Ohno, H., J. Stewart, M. C. Fournier, H. Bosshart, I. Rhee, S. Miyatake, T. Saito, A. Gallusser, T. Kirchhausen, and J. S. Bonifacino. 1995. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269:1872-1875. [DOI] [PubMed] [Google Scholar]
- 35.Persson, H., H. Jornvall, and J. Zabielski. 1980. Multiple mRNA species for the precursor to an adenovirus-encoded glycoprotein: identification and structure of the signal sequence. Proc. Natl. Acad. Sci. USA 77:6349-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ploegh, H. L. 1998. Viral strategies of immune evasion. Science 280:248-253. [DOI] [PubMed] [Google Scholar]
- 37.Rapoport, I., Y. C. Chen, P. Cupers, S. E. Shoelson, and T. Kirchhausen. 1998. Dileucine-based sorting signals bind to the beta chain of AP-1 at a site distinct and regulated differently from the tyrosine-based motif-binding site. EMBO J. 17:2148-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shisler, J., C. Yang, B. Walter, C. F. Ware, and L. R. Gooding. 1997. The adenovirus E3-10.4K/14.5K complex mediates loss of cell surface Fas (CD95) and resistance to Fas-induced apoptosis. J. Virol. 71:8299-8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorkin, A., and G. Carpenter. 1993. Interaction of activated EGF receptors with coated pit adaptins. Science 261:612-615. [DOI] [PubMed] [Google Scholar]
- 40.Stewart, A. R., A. E. Tollefson, P. Krajcsi, S. P. Yei, and W. S. M. Wold. 1995. The adenovirus E3 10.4K and 14.5K proteins, which function to prevent cytolysis by tumor necrosis factor and to down-regulate the epidermal growth factor receptor, are localized in the plasma membrane. J. Virol. 69:172-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tollefson, A. E., T. W. Hermiston, D. L. Lichtenstein, C. F. Colle, R. A. Tripp, T. Dimitrov, K. Toth, C. E. Wells, P. C. Doherty, and W. S. M. Wold. 1998. Forced degradation of Fas inhibits apoptosis in adenovirus-infected cells. Nature 392:726-730. [DOI] [PubMed] [Google Scholar]
- 42.Tollefson, A. E., T. W. Hermiston, and W. S. M. Wold. 1998. Preparation and titration of CsCl-banded adenovirus stocks, p. 1-9. In W. S. M. Wold (ed.), Adenovirus methods and protocols. Humana Press, Totowa, N.J. [DOI] [PubMed]
- 43.Tollefson, A. E., P. Krajcsi, M. H. Pursley, L. R. Gooding, and W. S. M. Wold. 1990. A 14,500 MW protein is coded by region E3 of group C human adenoviruses. Virology 175:19-29. [DOI] [PubMed] [Google Scholar]
- 44.Tollefson, A. E., P. Krajcsi, S. P. Yei, C. R. Carlin, and W. S. M. Wold. 1990. A 10,400-molecular-weight membrane protein is coded by region E3 of adenovirus. J. Virol. 64:794-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tollefson, A. E., A. R. Stewart, S. P. Yei, S. K. Saha, and W. S. M. Wold. 1991. The 10,400- and 14,500-dalton proteins encoded by region E3 of adenovirus form a complex and function together to down-regulate the epidermal growth factor receptor. J. Virol. 65:3095-3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tollefson, A. E., K. Toth, K. Doronin, M. Kuppuswamy, O. A. Doronina, D. L. Lichtenstein, T. W. Hermiston, C. A. Smith, and W. S. M. Wold. 2001. Inhibition of TRAIL-induced apoptosis and forced internalization of TRAIL receptor 1 by adenovirus proteins. J. Virol. 75:8875-8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tollefson, A. E., and W. S. M. Wold. 1988. Identification and gene mapping of a 14,700-molecular-weight protein encoded by region E3 of group C adenoviruses. J. Virol. 62:33-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vossen, M. T., E. M. Westerhout, C. Soderberg-Naucler, and E. J. Wiertz. 2002. Viral immune evasion: a masterpiece of evolution. Immunogenetics 54:527-542. [DOI] [PubMed] [Google Scholar]
- 49.Wang, E. W., M. O. Scott, and R. P. Ricciardi. 1988. An adenovirus mRNA which encodes a 14,700-Mr protein that maps to the last open reading frame of region E3 is expressed during infection. J. Virol. 62:1456-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson-Rawls, J., S. K. Saha, P. Krajcsi, A. E. Tollefson, L. R. Gooding, and W. S. M. Wold. 1990. A 6700 MW membrane protein is encoded by region E3 of adenovirus type 2. Virology 178:204-212. [DOI] [PubMed] [Google Scholar]
- 51.Wold, W. S. M., and G. Chinnadurai. 2000. Adenovirus proteins that regulate apoptosis, p. 200-232. In A. J. Cann (ed.), DNA virus replication. Oxford University Press, Oxford, United Kingdom.
- 52.Wold, W. S. M., S. L. Deutscher, N. Takemori, B. M. Bhat, and S. C. Magie. 1986. Evidence that AGUAUAUGA and CCAAGAUGA initiate translation in the same mRNA region E3 of adenovirus. Virology 148:168-180. [DOI] [PubMed] [Google Scholar]