

Abstract
We previously reported that the stimulation of monocyte-derived macrophages (MDM) by plate-bound intravenous immune globulins (IVIg) inhibits HIV-1 replication (Perez-Bercoff et al, J Virol, 2003, 77:4081). Here, we show that IgG immune complexes also suppress HIV-1 replication in MDMs and that activating receptors for the Fc portion of IgG - FcγRI, FcγRIIA and FcγRIII are responsible for the inhibition. MDM stimulation through FcγRs induces activation signals and the secretion of HIV-1 modulatory cytokines, such as M-CSF, TNF-α and MDC. However, none of these cytokines contribute to HIV-1 suppression. HIV-1 entry and post-integration steps of viral replication are not affected, whereas reduced levels of reverse transcription products and of integrated proviruses, as determined by real time PCR analysis, account for the suppression of HIV-1 gene expression in FcγR-activated MDMs. We found that FcγR-dependent activation of MDMs also inhibits the replication of HIV-2, SIVmac and SIVagm, suggesting a common control mechanism for primate immunodeficiency lentiviruses in activated macrophages.
Keywords: Animals, Cells, Cultured, DNA, Complementary, analysis, DNA, Viral, analysis, HIV-1, physiology, Humans, Lentivirus, physiology, Macrophage Activation, Macrophages, drug effects, immunology, virology, Primates, virology, Proviruses, isolation & purification, Receptors, IgG, agonists, genetics, Virus Replication
Keywords: HIV-1, macrophages, Fc Receptors, ell Activation, Lentiviruses
Introduction
Unlike other retroviruses, lentiviruses can integrate their DNA into the genome of non-dividing cells and can therefore replicate in monocytes and macrophages. HIV and SIV infection of monocytes and differentiated tissue-resident macrophages may play a major role in viral transmission, dissemination and persistence (1–4). The capacity of monocytes and macrophages to migrate in tissues makes them potential conveyors of HIV and SIV infections. Monocytes are thought to carry the virus to the central nervous system, and the expansion of subsets of activated monocytes has been associated with neurological diseases in AIDS (5, 6). Virions generated in infected macrophages are more efficient in establishing lymphocyte infection than cell-free virions (7). Macrophages may also favor cell-to-cell transmission to CD4 T cells by producing chemotactic cytokines and by interacting with cells during antigen presentation (8). In addition, HIV-1-infected macrophages can also induce the apoptosis of uninfected bystander T cells and neuronal cells (9). Finally, infected monocytes and macrophages may act as viral reservoirs for HIV and SIV and be the main source of virus production during the late stages of disease in pathogenic infections when the numbers of CD4 T cells are substantially reduced (10–14).
Macrophages play a major role in mounting innate and adaptive immune responses to pathogens. Macrophages react to HIV-1 infection by secreting cytokines, chemokines and other molecules having anti-viral activity or can directly control HIV-1 replication (15). However, HIV-1 infection may affect essential macrophage functions, such as antigen presentation, intracellular killing and phagocytosis (16). Therefore, the regulation of HIV-1 and related lentivirus replication in monocytes and macrophages might affect the host susceptibility to infection and could help to control viral dissemination and pathogenesis in infected individuals. In a SCID mouse model, virus spread and pathology was abolished by suppressing macrophage infection with anti-nerve growth factor antibodies (17).
We previously showed that the incubation of macrophages with IVIg bound on culture plates potently inhibit HIV-1 replication independently of viral tropism (18). Inhibition was not observed when macrophages were incubated with IVIg-F(ab′)2 fragments suggesting that it was mediated by receptor(s) for the Fc portion of IgG (FcγR) (18). FcγRs are a group of integral membrane proteins that bind to the Fc portion of IgG (19), and which can either activate or inhibit cell activation when engaged by IgG immune complexes. Activating FcγRs include the high-affinity receptor FcγRI (CD64), which can bind monomeric IgG, and the low-affinity receptors FcγRIIA/C (CD32) and FcγRIIIA (CD16), which do not bind monomeric IgG but bind IgG aggregates and antigen-antibody immune complexes (ICs) with a high avidity. Activating FcγRs possess immunoreceptor tyrosine-based activation motifs (ITAMs) that become phosphorylated upon FcγR clustering. ITAM phosphorylation promotes the recruitement of cytosolic protein tyrosine kinases. These kinases phosphorylate other proteins involved in signaling pathways, leading to the activation of PI-3K and MAP kinases (19). Inhibitory FcγRs consist of FcγRIIB, which contains an immunoreceptor tyrosine-based inhibition motif (ITIM). This motif enables FcγRIIB to negatively regulate cell activation triggered by ITAM-containing receptors when co-engaged with them.
In this study we aimed at identifying which FcγR(s) is(are) involved in viral inhibition. We quantitatively analyzed FcγR-mediated inhibition of HIV-1 replication using real time PCR. We also determined whether FcγR-mediated inhibition was limited to HIV-1 or was a general anti-retroviral mechanism by studying the effect of FcγR cross-linking on the replication of other primate lentiviruses in human macrophages.
Materials and Methods
Monocyte derived macrophages (MDM)
Human monocytes were isolated from buffy coats of healthy seronegative donors (Centre de Transfusion Sanguine Ile-de-France, Rungis and Hôpital de la Pitié-Salpêtrière, Paris, France) using lymphocyte separation medium (PAA laboratories GmbH, Haidmannweg) density gradient centrifugation and plastic adherence as previously described (18). Monocytes were then differentiated into macrophages by seven to 11 days culture in MDM medium (RPMI 1640 medium supplemented with 200 mM L-glutamine, 100 U penicillin, 100 μg streptomycin, 10 mM HEPES, 10 mM sodium pyruvate, 50 μM β-mercaptoethanol, 1% minimum essential medium vitamins, and 1% nonessential amino acids) supplemented with 15% of human AB serum in hydrophobic Teflon dishes (Lumox™. D Dutcher, Brumath, France) as previously described (18). Monocyte derived macrophages (MDM) were then harvested, washed and resuspended in MDM medium containing 10% heat-inactivated fetal calf serum (FCS) for experiments. The purity of CD14+ macrophages was usually more than 95% as assessed by immunofluorescent staining and flow cytometry analysis. FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16) were all expressed on the MDM surface but the proportion of cells expressing each receptor varied with different donors and MDM preparations.
LPS content in the media and all the reagents used for culture and stimulation of MDM was below the limit of detection of the QCL1000 Limulus amebocyte lysate test (LAL) (BioWhittaker, France).
Antibodies
Monoclonal Abs (MAbs) against FcγRs (CD64, clone 32.2; CD32, clones IV.3 and AT.10; CD16, clone 3G8) were purified from hybridoma supernatants. F(ab′)2 from MAbs was generated by pepsin digestion (ImmunoPure F(ab′)2 Preparation kit from PIERCE). Anti CD64 F(ab′)2 10.1 was from Ancell (COGER, France). Isotype-matched uncoupled or FITC- or PE-coupled irrelevant control MAbs or F(ab′)2 were from SIGMA (St Quentin Fallavier, France). All F(ab′)2 preparations used for stimulating MDM were passed through a polymixin B-column (Detoxi-Gel Endotoxin Removing Gel, PIERCE, Perbio Science France, Brebières, France) to eliminate potential endotoxin contamination and were then verified as LPS-free by the LAL test.
Human FcγRIIA-specific and FcγRIIB-specific polyclonal antibodies were generated in rabbits immunized with GST fusion proteins containing the intracytoplasmic domain of either human FcγRIIA (ICIIA) or FcγRIIB2 (ICIIB). Briefly, cDNAs encoding the intracytoplasmic domains of human FcγRIIA and FcγRIIB2 were amplified by PCR using the following primers: FcγRIIA, forward: CGCGGATCCGCGAATTCCACTGATCCTGTGAAG; reverse: CGGAATTCCGTTAGTTATTACTGTTGACATGGTC; FcγRIIB2, forward: GCGGATCCGCGAATCCCACTAATCCTGATGAG; reverse: CGGAATTCCCTAAATACGGTTCTGGTCATC.
Purified amplicons were ligated into the PGEX4T1 vector (Pharmacia Biotech 27-4580-01) and expressed in E. coli DH5α cells. GST-ICIIA and GST-ICIIB were purified on glutathion-agarose gel and were then used to immunize rabbits (one 200 μg injection in complete Freund’s adjuvant followed three weeks later by three 200 μg injections in incomplete Freund’s adjuvant every two weeks). Serum IgG were purified by affinity chromatography on Protein A-sepharose (Pharmacia). IgG from rabbits immunized with GST-ICIIA were absorbed by two passages through GST-ICIIB-coated sepharose 4B beads (Pharmacia), whereas IgG from rabbits immunized with GST-ICIIB were absorbed by passage through GST-ICIIA-coated sepharose 4B beads, to remove anti-GST and any possibly cross-reacting antibodies. Anti-FcγRIIA Abs recognized FcγRIIA but not FcγRIIB in rat basophilic leukemia (RBL) transfectants, whereas anti-FcγRIIB Abs recognized FcγRIIB but not FcγRIIA in the same transfectants, as assessed by western blotting analysis and intracellular immunofluorescence.
The above described FcγRIIA and FcγRIIB2 specific primers were used to analyze the receptor transcripts in RNA preparations from MDM by RT-PCR. RNA was extracted using RNeasy Kit (Qiagen, France). cDNA was synthesized from 0.5 μg total cell RNA using Taqman Reverse Transcription Reagents Kit (Applied Biosystem). 1/5, 1/50 or 1/500 of the volume of the reaction mixture were used for PCR amplification (30 cycles), using Taq (Invitrogen, ) in a GeneAmp PCR9700 (Applied Biosystems). The PCR products were analyzed by gel electrophoresis on 2% agarose gel.
Anti-TNF-α rabbit IgG (gift of J-M. Cavaillon, Institut Pasteur), anti-M-CSF goat IgG or anti-MDC chicken IgYs (both from R&D systems, Minneapolis, MN) were used for TNF-α, M-CSF or MDC neutralization respectively. Isotypic controls were rabbit, goat or chicken irrelevant Abs. Commercial Abs were detoxified by passage through polymixin B columns before utilization. TNF-α, M-CSF and MDC levels in culture supernatants were measured by using quantikine ELISAs (R&D systems).
The following Abs were also used: unconjugated Mouse anti-phosphotyrosine mAb 4G10: purified from hybridoma supernatant on Protein G-sepharose; Rabbit anti-phospho-PLC-gamma1(tyrosine-783) Abs: Santa Cruz (Santa Cruz, CA); Rabbit anti-erk1/2 and rabbit anti-phospho-erk1/2 (Thr202/tyr204) Abs: Cell Signaling (Beverly, MA); fluorochrome-conjugated CD11b-PE (clone Bear1) and CD4-PE (clone 13B8.2) (both from Beckman Coulter), CD14-FITC (clone Leu M3) and CD3-FITC (clone Leu3) (both from Becton Dickinson, San Jose, Calif.).
Flow cytometry analysis
Cells were stained either with FITC-conjugated or PE-conjugated mAbs or with unconjugatd mAbs or F(ab′)2 fragments followed by secondary FITC-goat anti-mouse IgG F(ab′)2 or FITC-goat anti-mouse Fab F(ab′)2 (Immunotech) and analyzed using a FACSCalibur flow cytometer (Beckman Coulter).
Immunoblotting
For tyrosine phosphorylation analysis, cells were lysed by three cycles of incubation for 1 min in liquid nitrogen followed by 1 min at 37°C in lysis buffer at pH 8.0 (50 mM Tris, pH8, 150 mM NaCl, 1% Tx100, 1 mM Na3VO4, 5 mM NaF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml pepstatin and 1mM PMSF). For FcγRII expression analysis, cells were lysed by boiling 5 min in 10 mM Tris pH 7.4, 1% SDS. Proteins were quantified using a Biorad protein assay (Hercules, CA). 40 μg proteins for tyrosine phosphorylation analysis or 10 μg proteins for FcγRII analysis were boiled in sample buffer, fractionated by SDS-PAGE and then transferred onto Immobilon-P membranes (Millipore, Bedford, MA). The membranes were saturated with either 5% BSA (Sigma Chemical Co) or 5% skimmed milk (Régilait, Saint-Martin-Belle-Roche, France) diluted in western buffer (150 mM NaCl, 10 mM Tris and 0.5% Tween 20 (Merk, Schuchardt, Germany) pH 7.4) and incubated with the indicated antibodies and then incubated with HRP-conjugated goat anti-rabbit or goat anti-mouse Ig antibodies. Labeled antibodies were detected using an ECL kit (Amersham Pharmacia biotech, Little Chalfont, Buckinghamshire). Blots for FcγRII analysis were first probed with the anti-FcγRIIB antibody, then stripped with stripping buffer (reblot+, Chemicon, Temecula,CA) as indicated by the manufacturer, and re-probed with the anti-FcγRIIA antibody.
MDM stimulation
MDM were stimulated using three different methods
Immobilized IVIg stimulation was carried out with hIgG for therapeutic use (IVIg) (Endobuline, BAXTER, Maurepas, France) (0.1 mg/ml in PBS) as previously described (18).
-
Stimulation with preformed immune complexes (ICs) was as follows: dinitrophenyl (DNP) groups were conjugated to LPS-free bovine serum albumin (BSA) (SIGMA) using dinitrobenzene sulfonate (DNBS, Eastman Kodak) in alkaline medium and then dialyzed against PBS. Culture plates were coated with 0.1 mg/ml DNP-BSA antigen by incubation for 2 hours at 37°C, washed with PBS, saturated by incubation with 1 mg/ml BSA in PBS for 30 minutes at 37°C and then incubated with 30 μg/ml rabbit anti-DNP antibodies (SIGMA) for 1 hour at 37°C to form ICs. MDMs were stimulated by plating on IC coated wells.
In some experiments 3 μm polystyrene beads (Polysciences Inc., France) opsonized with DNP-anti-DNP ICs were used for MDM stimulation. Polystyrene beads were absorbed with 400 μg/ml DNP-BSA in PBS according to manufacturer instructions, washed, and incubated with 100 μg/ml rabbit anti-DNP antibodies in PBS-BSA (IC-beads) or with PBS-BSA alone (Ag-beads). MDM were plated on 96-well plates and incubated with 100 μl medium containing IC-beads or Ag-beads at a bead:cell ratio of 10:1 and 30:1 and immediately infected.
Each FcγR was separately cross-linked on an MDM surface by incubating MDMs with the appropriate specific F(ab′)2 (5 μg F(ab′)2 per 106 MDM) for 30 min at 4°C. The MDMs were then washed with PBS and seeded on plates previously coated with 0.2 mg/ml goat anti-mouse Fab F(ab′)2 (SIGMA) and saturated with 1 mg/ml BSA. MDMs were incubated in parallel with irrelevant mouse F(ab′)2 as controls.
Cell viability was not affected in IgG or IC-stimulated MDM cultures, as evaluated by a WST-1-based colorimetric assay (not shown).
Viruses and MDM infection
HIV-1 infections
The following viral strains were used for productive infections: HIV-1Bal, HIV-2SBL, SIVmac251, SIVagmGril. Strains were propagated in PHA-activated human PBMCs (except SIVagm, propagated on SupT1 cells) and the culture supernatants were collected at times of peak p24 (HIV-1) or p27 (HIV-2, SIV) production. p24 and p27 were measured with commercial ELISA kits (Beckman Coulter, Paris, France). Viral stocks were titrated on PHA-activated human PBMCs except SIVagm, which was titrated on SupT1 cells. Multiplicities of infection (m.o.i.) used in this study were between 10−2 and 2 × 10−2.
For single-round infections, HIV-1 particles containing the luc reporter gene and pseudotyped with the VSV-G envelope protein (HIV-1VSV-G) that allows HIV-receptor independent entry into cells were used. HIV-1VSV-G virions were produced by transiently co-transfecting (SuperFect, Qiagen GmbH, Hilden, Germany) 293T cells with the proviral pNL-Luc-E−R+(20) vector and the VSV-G expression vector pCMV-G, as previously described (18). Supernatants were harvested 72 h after transfection, and p24gag levels were measured using a commercial ELISA kit (Beckman Coulter, Paris, France) with MDMs. Between 3 × 10−1 and 3 × 10−2 m.o.i. were used for MDM infection. Mock infections with equivalent amounts of p24 from supernatants from 293T cells transfected with pNL-Luc-E−R+ only were carried out in parallel as controls.
MDMs (0.8 × 105 – 1 × 105 cells/well in 96 well plates or 106 cells/well in 12 well plates) were infected either with viral strains or with pseudotyped particles by incubating cell with viral inoculum 1h at 37°C, or by a spinoculation protocol (1h centrifugation at room temperature at 1200 × g followed by 1h incubation at 37°C), to increase the efficiency of infection (21). MDMs were then washed with PBS and cultured in MDM medium.
In the experiments for detecting HIV DNA by PCR, HIV-1VSV-G preparations were previously treated with DNase I (Roche Diagnostics GmbH, Mannheim, Germany).
In cytokine/chemokine neutralization experiments, immobilized-IgG-stimulated or unstimulated MDMs were infected in triplicate with HIV-1Bal, and then cultured in 96 well-plates in the presence of neutralizing concentrations of the each specific Ab (anti-TNF-α, 1:150 dilution; anti-M-CSF, 7,5 μg/ml, anti-MDC, 10 μg/ml) or equal concentrations of the appropriate control Ab. Half culture supernatant was changed with fresh medium containing the appropriate concentrations of each Ab each 2 days.
WN virus infection: Production of WN virus strain IS-98-ST1 (GenBank accession number AF 481864) from mosquito Aedes pseudoscutellaris AP61 cell monolayers and virus titration on AP61 cells by focus immmunodetection assay (FAI) were performed as previously described (22). Infectivity titres were expressed as focus forming units (FFU). RPMI medium supplemented with 2% FCS was used for washing, infection and culture. MDM were washed three times and infected with 1 m.o.i of WN virus for 1 h at 37°C. MDM were then washed twice and incubated at 37°C for 72 hours, then cell culture supernatants were harvested and processed for viral titration. As a control for inhibition of WN virus replication, MDM were exposed to 10 IU/ml human recombinant IFN-α A/D (Biosource, France), during and after infection.
Measure of luciferase activity in cell lysates
At various times after infection with pseudotyped HIV-1 virions, each well of MDMs was lysed with 100 μl of luciferase cell culture lysis reagent (Promega France, Charbonnières, France). The luciferase activity was quantified in 20 μl of each lysate using the Promega Luciferase reporter 1000 Assay System and an LUMAT LB9501 luminometer (Berthold Technologies).
Real time PCR quantification of HIV-1 cDNA forms
At different times after infection, MDMs were washed in PBS and total DNA was extracted using the DNeasy Tissue Kit (Qiagen). The HIV-1 DNA forms R-U5, U5-Gag and 2-LTR were quantified using real time PCR with an ABI PRISM 7000 instrument (Applied Biosystems Applera France, Courtaboeuf, France). For all the real-time PCR, we used 100 ng of template DNA per reaction, corresponding to about 2 × 104 MDMs. DNA loading was controlled by concurrently amplifying the albumin gene by real-time PCR and quantifying with reference to a control human genomic DNA (Roche). The reaction mixture contained 1X Taqman Universal PCR master mix, 300 nM of each primer (except R-U5 primers, 200 nM) and 200 nM of the appropriate fluorogenic probe, in a final volume of 30 μl. PCR cycle conditions were: 50°C for 2 min, 95°C for 10 min and 40 cycles of 95°C for 15 sec and 60°C for 1 min. Copy numbers of R-U5 and U5-Gag were determined with reference to a standard curve prepared by concurrent amplification of serial dilutions of 8E5 cells containing one integrated copy of HIV-1 per cell (23). The copy number of 2-LTR was determined with reference to standard curves generated by serial dilutions of CEM cells infected with HIV-1NL4-3. The number of 2-LTR copies/per CEM cell was previously quantified against a standard curve generated by dilution of cloned DNA with matching sequences (pSLL-IIIb, gift of Audrey Brussel, Institut Pasteur, Paris, France) (24). R-U5 primers are described elsewhere (25), the probe was (FAM)-AGACGGGCACACACTA-(MGB). Primers and probes for U5-Gag (26), 2-LTR (27) and albumin (28) have been reported.
Integrated HIV-1 DNA was quantified by real time Alu-Gag nested PCR using primers and probes supplied by Norio Yamamoto, Tokyo Medical and Dental University, Tokyo, Japan. The first round of amplification was conducted on a Gene Amp PCR system 9700 (Applied Biosystems). Integrated HIV-1 sequences were amplified with the expand high fidelity kit (Roche) using an Alu primer (NY1F) and a Gag primer extended with an artificial tag sequence at the 5′ end of the oligonucleotide (NY1R). The reaction mixture contained 100 ng of DNA, 0.2 mM dNTP, 300 nM primers, 1X buffer with 1.5 mM MgCl2, and 1.05 U of polymerase. Reaction conditions were as follows: 95°C for 2 minutes, 15 cycles of 95°C for 15 seconds, 57°C for 30 seconds and 72°C for 3 minutes, and 72°C for 2 minutes. Real time nested PCR was carried out on the ABI PRISM 7000 system (Applied Biosystems) using 10 μl of 1/20 dilution of the first-round PCR product as a template with 300nM of LTR primer (NY2F), 300nM tag sequence primer (NY2R) and 200 nM Alu-LTR probe (NY2ALU) and with 1X Taqman Universal PCR master mix. The integrated HIV-1 DNA copy number was determined with reference to a standard curve generated by concurrent amplifications of a standard HeLa R7 Neo cell DNA. The HeLa R7 Neo cell line was generated as described (29). Briefly, Hela cells were infected with a VSV-G pseudotyped HIV-1 R7 Neo virus (gift from Audrey Brussel) containing a neomycin resistance gene and cultured for five weeks in the presence of G418. For each sample, a whole nested PCR procedure omitting the Alu primer in the first round PCR was carried out in parallel, showing a very low background. The number of integrated HIV-1 DNA copies was then adjusted by subtracting the copy number measured in the absence of the Alu primer in the first-round PCR from the copy number measured in the presence of Alu primer. Primers and probes for the Alu-Gag nested PCR are as follows:
NY1F: GGCTGAGGCAGGAGAATGG
NY1R: CAATATCATACGCCGAGAGTGCGCGCTTCAGCAAG
NY2F: AATAAAGCTTGCCTTGAGTGCTC
NY2R: CAATATCATACGCCGAGAGTGC
NY2ALU: (FAM)-AGTGTGTGCCCGTCTGTTGTGTGACTC-(TAMRA).
Results
Immune complexes inhibit HIV-1 replication in macrophages
We reported previously that HIV-1 replication is inhibited in macrophages stimulated with immobilized IVIg, but not by F(ab′)2 fragments of the same IVIg preparations (18). This observation suggested that Ag-Ab complexes might regulate viral infection by interacting with macrophage FcγRs. To confirm this interpretation, MDMs were plated onto immobilized immune complexes (ICs) made of DNP-BSA and IgG anti-DNP, and infected with HIV-1VSV-G pseudotype. We used HIV-1VSV-G pseudotyped viruses to assess the impact of IC-stimulation of MDM on a single cycle of viral replication. HIV-1 replication was dose-dependently inhibited in MDMs exposed to ICs, compared to unstimulated MDMs, but not in MDMs exposed to antigen or antibody alone (Fig. 1A). In some experiments, MDM were incubated with IC-opsonized polystyrene beads, which are readily phagocytosed by macrophages and may mimic antibody-opsonized bacteria or cell debris. IC-coated polystyrene beads, but not beads coated with antigen only, inhibited HIV-1VSV-G replication in a dose-dependent manner (Fig. 1B). However, inhibition induced by immobilized ICs was more reproducible (data not shown) and as efficient as when induced by immobilized IVIg (Fig. 1C). The levels of IC- or IVIg-induced viral inhibition vary in parallel in MDM preparations from different donors (Fig. 1C). Replication competent viruses, including HIV-1 Bal, were also inhibited by immobilized ICs (data not shown).
Figure 1.

IgG-immune complexes inhibit HIV-1 replication in MDMs. (A) MDMs infected with HIV-1VSV-G were plated on wells treated with BSA only (unstimulated, US) or coated with 30 μg/ml anti-DNP Abs (a-DNP) or with decreasing concentrations of DNP-BSA (10, 1, 0.1 μg/well) followed or not by 30 μg/ml anti-DNP Abs; (B) MDMs were plated and incubated with medium (US) or with 3μm polystyrene beads coated with DNP-BSA-anti DNP ICs (IC) or with DNP-BSA only (Ag) at a bead:cell ratio of 10:1 and 30:1 (10, 30) and infected with HIV-1VSV-G; (C) HIV-1VSV-G infected MDMs from three different donors were stimulated with either immobilized IVIg or ICs. Results are expressed as percentage of inhibition of luciferase activity (means and SD of three independent wells) found in unstimulated MDMs plated on BSA or DNP-BSA. (D) HIV-1VSV-G infected MDMs were plated on wells coated with 10 μg/well DNP-BSA or with complexes formed by DNP-BSA with 30 μg/ml of either anti-DNP IgG or anti-DNP F(ab′)2 and infected with HIV-1VSV-G. In all the experiments luciferase activity was measured in cell lysates 72 hours p.i. Results are expressed as means and SD of three independent wells.
When equivalent concentrations of anti-DNP F(ab′)2 fragments were used to form DNP-anti-DNP ICs, no inhibition of HIV-1 replication was observed (Fig. 1D), indicating that the Fc portion of IgG is required for inhibition. Since complement is not present in incubation medium, this result implies that HIV-1 inhibition induced by ICs is mediated by FcγR.
Inhibition of HIV-1 replication is mediated by activating FcγRs
Human macrophages express several FcγRs. To identify which FcγR(s) account(s) for IC-induced inhibition of HIV-1 replication, we investigated first the effect of engaging separately the three activating receptors known to be expressed on macrophages, and for which specific antibodies are available i.e. FcγRI, FcγRIIA and FcγRIIIA. MDMs were incubated with F(ab′)2 fragments of mAbs specific for each receptor or with irrelevant mouse F(ab′)2, and seeded onto wells coated with F(ab′)2 fragments of goat anti-mouse Fab Abs. MDMs were then infected with HIV-1 BaL, and viral replication was evaluated by measuring p24 production. The incubation of MDMs with each of the FcγR-specific F(ab′)2, but not with control F(ab′)2, decreased p24 production, compared with unstimulated MDMs. Anti-FcγRIIA IV.3 F(ab′)2 induced the strongest inhibition (Fig. 2). These results indicated that cross-linking activating receptors can induce an inhibition of HIV-1 replication. The level of viral suppression induced by F(ab′)2 fragments of antibodies against activating FcγRs was however generally lower than that induced by either IVIg or ICs ((Fig. 2 and data not shown).
Figure 2.

Cross-linking of activating FcγRs inhibit HIV-1 replication. MDMs were incubated with medium (unstimulated, US), with irrelevant F(ab′)2 (C) or with F(ab′)2 fragments of mAbs specific for FcγRI (32.2), FcγRIIA (IV.3) or FcγRIII (3G8), and plated on anti-mouse-Fab F(ab′)2-coated wells. As a control of HIV-1 inhibition, MDMs were stimulated in parallel with IVIg. MDMs were then infected with HIV-1 Bal. Results are expressed as means and SD of the percentage of infection in three independent wells (evaluated by p24 levels in supernatants) on day 8 p.i. with respect to unstimulated MDMs (p24 = 202 ng/ml). Comparisons among data sets were performed by independent sample t-test. A representative experiment of three performed with MDM from different donors is shown.
Human monocytes having been reported to express the inhibitory FcγRIIB, we investigated next the expression and the potential role of this receptor in HIV-1 inhibition. Because there are no available antibodies (Abs) that recognize specifically the extracellular domain of FcγRIIB, we generated FcγRIIB-specific and, as controls, FcγRIIA-specific polyclonal Abs by immunizing rabbits with peptides corresponding to the intracytoplasmic domain of each receptor. By Western blotting, anti-FcγRIIA Abs recognized proteins of the expected MW in lysates of cells stably transfected with cDNA encoding FcγRIIA but not in lysates of cells stably transfected with cDNA encoding FcγRIIB (Fig. 3A, left). Conversely, anti-FcγRIIB Abs recognized proteins of the expected MW in lysates of cells stably transfected with cDNA encoding FcγRIIB, but not in lysates of cells transfected with cDNA encoding FcγRIIA (Fig. 3A, left). We used these Abs to evaluate the expression of FcγRIIA and FcγRIIB in monocytes and macrophages. As a control, we also examined B lymphocytes which express FcγRIIB. As expected, FcγRIIB was readily detected in B lymphocytes by the anti-FcγRIIB Ab (Fig. 3A, right). In contrast, FcγRIIB was undetectable in monocyte or macrophage lysates at concentrations that showed very strong FcγRIIA signals (Fig. 3A, right). The same results were found with MDMs from four different donors. Thus, FcγRIIB was not detectably expressed in MDMs under our experimental conditions. When analyzed by RT-PCR with FcγRIIB or FcγRIIA specific primers, FcγRIIB transcripts were detected in MDM RNA, but in lower amount than FcγRIIA transcripts (Fig. 3B). Indeed, when analyzing three ten-fold dilutions of MDM cDNA, FcγRIIB transcripts were clearly detected in the first dilution and barely detected in the second dilution, whereas FcγRIIA transcripts were detected in all three dilutions (Fig. 3B). The same results were found with MDMs from two different donors. Altogether, WB and RT-PCR results indicate that FcγRIIA is the predominant FcγRII in MDMs. To assess whether, although undetected by Western blotting, FcγRIIB could modulate FcγRIIA-mediated inhibition of HIV-1 replication, we compared the effect of IV.3 F(ab′)2, which recognize the extracellular domain of FcγRIIA but not that of FcγRIIB, and the effect of AT10 F(ab′)2, which recognize the extracellular domains of FcγRIIA, FcγRIIB and FcγRIIC. Similar percentages of positive cells and similar mean fluorescence intensities were found when MDM were stained with either AT10 or IV.3 F(ab′)2 (Fig. 3C). AT10 and IV.3 F(ab′)2 induced comparable HIV-1 inhibition in infected MDM (Fig. 3D).
Figure 3.

FcγRIIB is undetectable by Western blotting in MDM and does not apparently regulate activating FcγR-mediated HIV-1 inhibition. (A) Left: RBL cells transfected with cDNA encoding FcγRIIA or anti-FcγRIIB were western blotted with polyclonal rabbit anti-FcγRIIA or anti-FcγRIIB. Right: B-lymphocytes, monocytes or MDM lysates (10 μg) were western blotted with polyclonal rabbit anti-FcγRIIA or anti-FcγRIIB. (B) RNA from MDMs was analyzed by RT-PCR for expression of FcγRIIA and FcγRIIB transcripts. 0.5 μg of RNA were reverse transcribed. PCR amplification with primers specific to each receptor was performed on sequential ten-fold dilutions of the cDNA mixture. (C) FACS analysis of MDMs stained with F(ab′)2 of mAbs directed against FcγRIIA (IV.3) (solid line) or against the FcγRs IIA, IIB and IIC (AT.10) (dashed line). (D) MDMs were incubated with medium (unstimulated, US), with ICs (IC), with 5μg/106 MDM of irrelevant F(ab′) (C) or with F(ab′)2 derived from IV.3 or AT10 mAbs at decreasing concentrations (5, 1, 0.2, 0.04 μg/106 MDM) and plated on anti-mouse-Fab F(ab′)2 coated wells to cross-link bound F(ab′)2. MDMs were infected with HIV-1 Bal. Results are expressed as percentage of infection (evaluated by p24 levels in supernatants) on day 6 p.i. with respect to unstimulated MDM (p24 = 658 ng/ml). Values are means and SD of three independent wells. Similar results were obtained in experiments performed with MDM from three different donors.
Both expression and functional analyses altogether suggest that activating, rather than inhibitory FcγRs account for the IC-induced inhibition of HIV-1 replication in MDM.
MDM stimulation through FcγR induces activation signals and cytokine secretion
We then investigated the signaling events induced by FcγRs in MDMs and their consequences on infection by HIV-1VSV-G. As expected, tyrosine phosphorylation of a number of intracellular proteins, including PLC-γ and Erk1/2, increased in IVIg-stimulated MDMs (Fig. 4A). PLC-γ phosphorylation was transient whereas Erk1/2 phosphorylation was sustained in non infected MDMs (Fig. 4A,C). HIV-1VSV-G infection did not detectably modify the phosphorylation patterns or kinetics (Fig. 4B), even when examined over an extended period of time (Fig. 4C).
Figure 4.
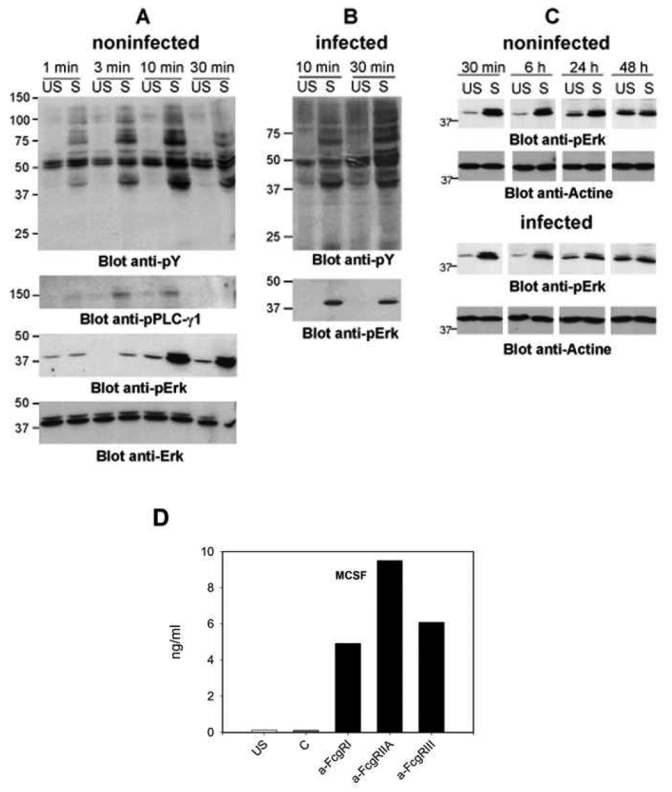
FcγR aggregation induces activation signals in MDMs and cytokine secretion (A–C) Non-infected or HIV-1VSV-G infected MDMs were stimulated by being plated onto IgG-coated (S) or non-coated wells (US) for the indicated times before being lysed. Proteins (40 μg) were electrophoresed and western blotted with anti-phosphotyrosine (anti-pY), anti-phospho-PLC-γ 1, or anti-phospho-Erk Abs. Anti-Erk Abs were used as loading controls. (D) M-CSF secretion after cross-linking of activating FcγRs. M-CSF secreted in the supernatants of unstimulated MDM (US), MDM incubated with irrelevant F(ab′)2 (C) or with F(ab′)2 specific for each activating FcγR and plated on anti-mouse-Fab F(ab′)2-coated wells was measured 48h after cell plating by ELISA (means of duplicates).
In order to assess the effect of blocking activating FcγR-mediated signaling on HIV-1 inhibition in IC-stimulated MDMs, we used piceatannol, reported as an inhibitor of the tyrosine kinase Syk that is recruited by phosphorylated ITAMs in FcγR aggregates (30). Piceatannol concentrations of 20–40 μM partially but significantly (p = 0.001) removed viral suppression in IC-stimulated MDM infected with HIV-1VSV-G (data not shown). However, piceatannol treatment also caused a dose-dependent inhibition of HIV-1 replication in unstimulated MDM, which was almost complete at concentrations higher than 50 μM (data not shown), possibly because of the inhibition of phosphorylation pathways involved in HIV-1 replication. Indeed piceatannol can inhibit not only Syk but also numerous tyrosine and serine-threonine kinases (31–33).
MDM stimulation by either IVIg or ICs induced chemokine and cytokine secretion, including M-CSF, MDC and TNF-α (Fig. S1 A and (18)). Cross-linking of activating FcγR with anti-FcγR F(ab′)2 on MDMs also induced the secretion of M-CSF (Fig. 4D) and other cytokines (not shown). Whether using IVIg or ICs or anti-FcγR F(ab′)2, in all cases the amounts of cytokines secreted by FcγR-activated MDMs, and particularly M-CSF, correlated with the magnitude of inhibition of HIV-1 replication (Figs. 2 & 4A, (18) and data not shown). However, we previously reported that HIV-1 suppression could not be induced by exposing MDMs to cytokine-containing supernatants from IVIG-stimulated MDMs and that MDC neutralization in IVIG-stimulated MDM cultures did not restore HIV-1 replication (18). These results indicate that neither MDC nor other secreted factors are responsible for FcγR-mediated HIV-1 inhibition. Supporting this conclusion, anti-M-CSF or anti-TNF-α neutralizing Abs reduced, rather than enhanced HIV-1 infection in unstimulated MDMs, and increased IVIG-induced inhibition (Fig. S1 B).
HIV-1 cDNA and integrated proviruses are decreased in FcγR-activated macrophages
To identify the steps in the HIV-1 replicative cycle that are inhibited in FcγR-activated MDMs, we measured the intermediate products of HIV-1 replication using real time PCR (rtPCR) in single-round infections with HIV-1VSV-G from entry to integration. We used primers and probes amplifying early (R-U5) and late (U5-Gag) products of reverse transcription (RT), 2-LTR circles (2-LTR), and integrated proviruses (Alu-LTR). We found similar HIV-1 replication inhibition profiles in MDMs activated either by IVIg or by ICs in MDMs from three different donors. A representative experiment is shown in Fig. 5. Luciferase activity in cell lysates was much lower in IC-activated MDMs (90% inhibition at 96h) than in unstimulated MDMs (Fig. 5A). Similar levels of R-U5 products were found by rtPCR in unstimulated and in IC-stimulated MDMs at 4 and 24 h post infection (p.i.) (Fig. 5B). At these early times, R-U5 products essentially reflect the input virus entered into the cells and the initial synthesis of the first products of retrotranscription. However, at later times p.i., the levels of both early and late RT products were decreased in IC-activated MDMs compared to unstimulated MDMs (Fig. 5B,C). Among the nuclear forms of HIV-1 cDNA, 2-LTR circles were only slightly less abundant in IC-activated MDMs than in unstimulated MDMs (Fig. 5D), whereas the number of integrated copies, determined from Alu-LTR levels, progressively decreased over time in IC-activated MDMs (64 and 76% inhibition at 48 and 96 hours p.i. respectively) (Fig. 5E). Accordingly, the ratio between 2-LTR circles and integrated forms was higher in IC-activated MDMs than in control MDMs and increased over time (2-LTR copies were at 6% and 2,6% of Alu-LTR copies at 48 h p.i. and at 38% and 15% at 96h p.i. in IC-activated and in unstimulated MDMs respectively) (Fig. 5F). This result suggests that unintegrated viral forms accumulate in FcγR-activated MDM while integrated forms decrease.
Figure 5.
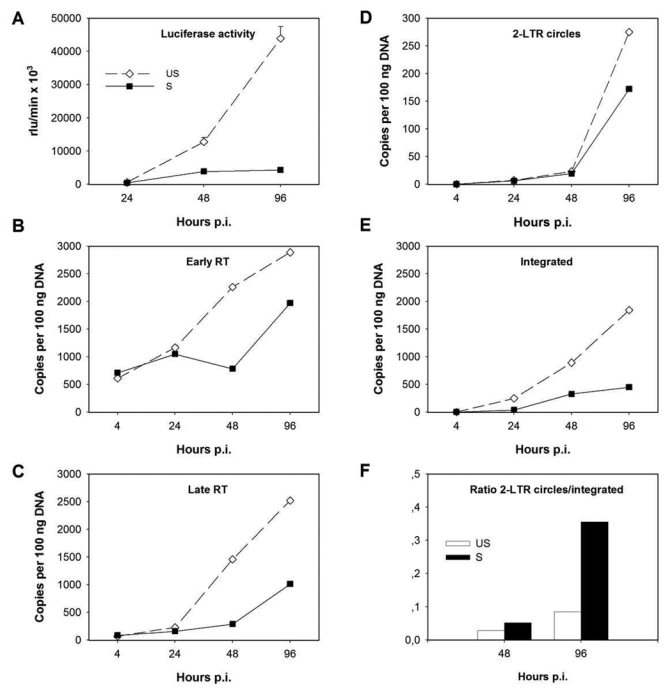
FcγR-mediated activation causes a reduction of HIV-1 retrotranscripts and of integrated proviruses in MDMs. MDMs were infected with DNAse treated HIV-1VSV-G and plated on DNP-BSA coated plates (unstimulated, US) or stimulated with ICs (S). Luciferase activity was monitored at 24, 48 and 96 h p.i. in MDM lysates (A). Early and late retrotranscription products and 2-LTR circles were analyzed by rtPCR using the appropriate primers (R-U5, U5-Gag, 2-LTR) and probes (B–D). Integrated copies were evaluated by Alu-LTR nested rtPCR. Values are means of duplicate measures at the indicated times p.i. (E). Ratios between 2-LTR circles and integrated nuclear forms of HIV-1 at 48 and 96 h p.i. as measured by rtPCR in unstimulated or IC-activated MDMs. Results from a representative experiment of three performed with MDM from different donors are shown.
Early post-integration steps are not inhibitedin FcγR-activated macrophages
We then determined whether FcγR-mediated activation of macrophages could affect HIV-1 post-integration steps, including transcription. MDMs were infected with HIV-1VSV-G and cells were kept in suspension for 72 or 96 hours before they were plated onto IVIg-coated or uncoated wells. Under these conditions, MDMs activation was triggered after most of the viral DNA should be integrated (Fig. 5 and results not shown). Forty eight hours after activation, similar levels of luciferase activity were found in lysates from unstimulated and from IVIg-stimulated MDMs (Fig. 6). By contrast, the same MDM preparation activated at the same time as infection showed a luciferase activity 82% lower in IVIg-stimulated MDMs than in unstimulated cells (Fig. 6). These results indicate that HIV-1 transcription and protein synthesis are not affected by FcγR-mediated activation in macrophages.
Figure 6.

FcγR-mediated activation of MDMs after viral integration does not affect HIV-1 gene expression. MDMs were infected with HIV-1VSV-G and immediately stimulated (0 h) with immobilized IgG or kept in suspension for 72 h or 96 h before stimulation. Luciferase activity in cell lysates was measured 72 h p.i. for MDM stimulated at time 0 and 48 h after activation for MDM stimulated 72 or 96 h p.i. Results are expressed as percentage of the luciferase activity (means and SD of 3 independent wells) found in unstimulated MDM. The experiment shown is representative of experiments on MDM from three different donors.
FcγR-mediated activation of macrophages inhibits the replication of primate lentiviruses
All lentiviruses can complete integration in non-dividing cells and can thus replicate in macrophages. We therefore wondered whether other primate lentiviruses would also be susceptible to FcγR-mediated inhibition in human macrophages. MDMs were infected with HIV-2sbl, SIVmac or SIVagm, plated onto wells coated with ICs, and viral p27 levels were measured in culture supernatants every three days for 23 days (Fig. 7A–C). All three lentiviruses replicated efficiently in control MDMs, with p27 production becoming greater than 1 μg/ml. p27 levels were markedly reduced in the cultures of MDMs infected with each of the three viruses and stimulated with ICs (Fig. 7A–C). FcγR-mediated inhibition of viral infection is therefore not limited to HIV-1 but affects other primate lentiviruses.
Figure 7.

FcγR-mediated activation inhibits HIV-2, SIVmac and SIVagm replication, but not WN virus replication in MDMs. (A–C) Unstimulated (dotted line) or IC-stimulated (solid line) MDMs were infected with HIV-2SBL, SIVmac251 or SIVagmGril and infection was monitored between seven and 23 days by p27 levels in culture supernatants. The results shown (means and SD of 3 independent wells) are representative of experiments on MDMs from three different donors. (D) MDMs were infected with WN virus. MDMs, unstimulated (US), stimulated with immobilized IgG (S), or treated by IFN-α were infected with IS-98-ST1 strain. Supernatants were harvested at 72 hours p.i. and virus infectivity was titrated by focus immunodetection assay (FIA). Data are expressed as means and SD of 3 independent wells.
FcγR-mediated inhibition affects HIV-1 reverse transcription and integration that are peculiar features of retroviral replication. We therefore investigated whether FcγR-mediated activation of MDMs could affect other viruses. For this experiment, we used the West Nile (WN) Virus, an unrelated macrophagotropic Flaviviridae virus. WN virus replication in unstimulated MDMs was compared with that in FcγR-stimulated MDMs (Fig. 7D). As a control for inhibition, MDMs were treated with IFN-α (34–36). MDMs were infected with WN virus IS-98-ST1 strain at 1 m.o.i. No cytopathic effect was observed in any condition (not shown). After 72 hours, titres of virus produced in cell culture supernatants were determined. No difference was observed in the virus titre between unstimulated and stimulated MDMs (Fig. 7D). However, as expected, virus titre was strongly decreased in IFN-α treated MDMs. These results suggest therefore that FcγR-mediated inhibition selectively affects lentiviruses.
Discussion
In the present study, we show 1) that the inhibition of HIV-1 replication that we previously reported in macrophages plated onto immobilized IVIg (18), can also be induced by IgG immune complexes; 2) that activating FcγRs account for HIV-1 inhibition, but that FcγR-induced cytokines are not responsible for HIV-1 inhibition; 3) that inhibition affects neither viral entry nor post integration steps, but causes a reduction of viral cDNA and blocks viral integration; 4) that FcγR-mediated suppression is not limited to HIV-1 but also affects other primate lentiviruses.
We showed that the three known ITAM-bearing FcγRs can mediate HIV-1 replication inhibition. Although the levels of HIV-1 inhibition varied depending on the MDM preparation, it was consistently higher upon FcγRIIA cross-linking than upon FcγRI or FcγRIIIA cross-linking (Fig. 2). Whether this difference is due to a higher expression of FcγRIIA on MDMs, to specific properties of this receptor, or to a higher affinity of the anti-FcγRIIA mAb used is not known. Inhibition of HIV-1 correlated with MDM activation, as judged by M-CSF and MDC secretion. Accordingly, the partial recovery of HIV-1 replication by piceatannol in IC-stimulated MDM may suggest that blocking signalling pathways downstream activating FcγRs can remove HIV-1 inhibition. However, the significance of this result was obscured by a direct inhibitory effect of piceatannol on HIV-1 replication. Both inhibition of HIV-1 and MDM activation were generally lower when induced by anti-FcγR F(ab′)2 than when induced by IVIg or ICs. This is consistent with previous studies showing that FcγR cross-linking with immobilized IgG induces TNF secretion by human monocytes, whereas cross-linking with anti-FcγR antibodies does not (37). It also indicates that ICs or IVIg are more efficient at aggregating FcγRs than anti-FcγR antibodies.
We found no evidence that FcγRIIB contributes to IC-induced HIV-1 inhibition in MDMs. On possible reason is the low expression of FcγRIIB in these cells. We did not detect FcγRIIB proteins by Western blotting, but we found relatively low levels of FcγRIIB transcripts by RT-PCR, in MDMs. These results are consistent with previous reports where FcγRIIB detection by Western blotting required very high amounts of monocytes (38, 39). FcγRIIB is highly regulated by culture conditions and the presence of cytokines, including IL-4 (38–40). Culture conditions used for macrophages differentiation, i.e. culture medium supplemented with human serum but with no added cytokines, may not be optimal for FcγRIIB expression. Our data, however, do not exclude that, if expressed at sufficient levels, FcγRIIB could negatively regulate HIV-1 replication inhibition by activated FcγRs. The coengagement of FcγRIIA/C and FcγRIIB by AT10 F(ab′)2, however, had the same effect on HIV-1 replication as the engagement of FcγRIIA alone by IV.3 F(ab′)2.
FcγRs have been involved in either enhancement or inhibition of HIV-1 infection when engaged by anti-HIV-1 antibodies. FcγRI has been suggested to contribute to the control of infection in HIV-1-infected patients by favoring the internalization of HIV-1-IgG complexes and the degradation of the virus in macrophages (41). Likewise, bispecific Abs that could target HIV-1 to macrophage activating FcγRs inhibited HIV-1 infection (42). On the contrary, FcγRI or FcγRIII have been suggested to enhance the entry of Ab-opsonized HIV-1 virions into macrophages (43–45). Whatever the effects of activating FcγRs when engaged by HIV-anti-HIV immune complexes, we consistently found that activating FcγRs inhibited HIV-1 infection when engaged by irrelevant IgG immune complexes. It was recently reported that FcγRIIA/IIIA polymorphisms which confer higher avidity binding to ICs are associated with protection against HIV infection, but, on the contrary, these same polymorphisms are associated with the likelihood of infection in HIV gp120-vaccinated individuals (46). Based on these data, one may speculate that in the presence of preexistent HIV-gp120 specific antibodies induced by vaccination, higher avidity for ICs may be deleterious favoring Ab-dependent enhancement of HIV infection. In contrast, in the absence of preexistent HIV-1 antibodies, higher avidity of FcγRs for circulating ICs may favor protection against incoming infection by limiting viral replication in IC-activated macrophages.
Macrophage activation by FcγRs affects the mechanisms that eventually lead to proviral integration. Previous qualitative PCR analysis of IVIg-stimulated MDMs infected with a replication competent virus detected a reduction in integrated proviral DNA but not in reverse transcription products (18). Using one-round infections, which avoid overlapping replication cycles, and quantitative PCR, we now show that, whereas reverse transcription was not affected at the earliest times p.i,, the levels of both early and late cDNA products were eventually reduced in activated MDMs (Fig. 5). Levels of integrated proviruses were further inhibited in either IVIg or IC-stimulated MDMs. By contrast, the ratio of circular 2-LTR forms to integrated forms was higher in activated macrophages than in controls (Fig. 5F). 2-LTR circles are unintegrated nuclear forms of HIV-1 DNA (47–49) and thus reflect the translocation of viral transcripts into the nucleus of infected cells. Our results suggest that unintegrated HIV-1 DNA accumulates because of an inhibition of integration, as observed with anti-integrase drugs (50). The level of HIV-1 integrated forms in FcγR-activated MDMs decreased to levels similar as viral replication inhibition levels, as shown by reduced luciferase activity (Fig. 5 and data not shown), suggesting that the postintegration steps of replication are unaffected. We confirmed this hypothesis by showing that FcγR-stimulation did not alter viral gene expression, once integration was achieved (Fig. 6).
The activation of PI3K or of the MAPK pathways has been shown to be essential for an efficient replication of HIV-1 (51–53). Therefore, one expects early signaling events triggered by activating FcγR cross-linking to favor HIV-1 replication rather than to exert an anti-viral effect. We thus suggest that FcγR-induced late signaling events, which need to be identified in future work, are involved in HIV-1 inhibition. These might include the mobilization, the neosynthesis or the suppression of molecules that are critical for reverse transcription and/or integration. FcγR-activated MDMs secrete several cytokines, such as M-CSF, TNF-α and MDC, which either up- or down-regulate HIV-1 infection in macrophages (54–56). Neutralization of these cytokines in MDM cultures showed that the inhibitory mechanisms induced by FcγR-aggregation overcome the enhancing effects of M-CSF and TNF-α on HIV-1 replication and are not linked to MDC (18) and data not shown). The pre-integration inhibition induced by FcγR stimulation is reminiscent of the inhibitory effects of IFN-α et β (57). IFN-α/β was, however, not detected in FcγR-activated MDM supernatant (18). In addition, West Nile virus infection, which is inhibited by IFN-α and β (Fig. 7B and (34, 35)), was not affected in FcγR-activated MDM. The participation of type I IFNs in HIV-1 inhibition in FcγR-activated MDM is therefore unlikely.
Two distinct inhibition mechanisms may be operating: one affecting the retrotranscription process, and another inhibiting viral integration after nuclear translocation of HIV-1 cDNA. A single mechanism may however inhibit both steps of the viral cycle. An increased degradation of reverse transcripts by endonucleases, as suggested for APOBEC3G (58), or of incoming viral proteins by the proteasome (59, 60), would reduce both the levels of reverse transcripts and the reverse transcription products available for integration. Alternatively, mechanisms that hinder HIV-1 integrase activity and/or affect the preintegration complex (PIC) stability would have a negative impact on both integration and reverse transcription. Indeed, although reverse transcription and integration occur in distinct cellular compartments, they take place in the same molecular environment formed by the PIC (61). Moreover, HIV-1 integrase was involved in different steps of the HIV-1 life cycle, including reverse transcription (62, 63).
Remarkably, FcγR-mediated anti-viral activity is not limited to HIV-1 as it also affects other primate lentiviruses. By contrast, unrelated macrophage-tropic viruses such as the West Nile Virus were not affected, suggesting that FcγR-mediated anti-viral activity is not a general anti-viral defense mechanism. If it targets highly conserved lentiviral proteins and/or their functions, such as reverse transcription and integration, one would expect inhibition to affect other HIV-1-related lentiviruses. It would be interesting to study the effect of FcγR-mediated activation of macrophages on the replication of lentiviruses in their natural hosts in more distant animal systems. This would be especially relevant for diseases caused by lentiviruses having a restricted tropism for macrophages, such as the caprine arthritis and encephalitis virus (CAEV) or the Maedi-Visna virus.
Supplementary Material
Acknowledgments
We are grateful to Audrey Brussel and Norio Yamamoto for reagents and methods for rtPCR, Emmanuelle Lenôtre (Applied Biosystems) for help in designing the R-U5 probe. We thank Hugues Sudry for his contribution to cytokine analysis and Philippe Despres for expert assistance in experiments of infection with the West Nile Virus. We thank Roger Legrand and Michael Ploquin for providing HIV-2, SIVmac and SIVagm strains.
This study was supported by the Agence Nationale de Recherches sur le SIDA (ANRS), France
References
- 1.Ignatius R, Tenner-Racz K, Messmer D, Gettie A, Blanchard J, Luckay A, Russo C, Smith S, Marx PA, Steinman RM, Racz P, Pope M. Increased macrophage infection upon subcutaneous inoculation of rhesus macaques with simian immunodeficiency virus-loaded dendritic cells or T cells but not with cell-free virus. J Virol. 2002;76:9787–9797. doi: 10.1128/JVI.76.19.9787-9797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gendelman HE, Orenstein JM, Baca LM, Weiser B, Burger H, Kalter DC, Meltzer MS. The macrophage in the persistence and pathogenesis of HIV infection. Aids. 1989;3:475–495. doi: 10.1097/00002030-198908000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Hirsch VM, Sharkey ME, Brown CR, Brichacek B, Goldstein S, Wakefield J, Byrum R, Elkins WR, Hahn BH, Lifson JD, Stevenson M. Vpx is required for dissemination and pathogenesis of SIV(SM) PBj: evidence of macrophage-dependent viral amplification. Nat Med. 1998;4:1401–1408. doi: 10.1038/3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharova N, Swingler C, Sharkey M, Stevenson M. Macrophages archive HIV-1 virions for dissemination in trans. Embo J. 2005;24:2481–2489. doi: 10.1038/sj.emboj.7600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams K, Westmoreland S, Greco J, Ratai E, Lentz M, Kim WK, Fuller RA, Kim JP, Autissier P, Sehgal PK, Schinazi RF, Bischofberger N, Piatak M, Jr, Lifson JD, Masliah E, Gonzalez RG. Magnetic resonance spectroscopy reveals that activated monocytes contribute to neuronal injury in SIV neuroAIDS. J Clin Invest. 2005;115:2534–2545. doi: 10.1172/JCI22953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995;25:3418–3424. doi: 10.1002/eji.1830251232. [DOI] [PubMed] [Google Scholar]
- 7.Carr JM, Hocking H, Li P, Burrell CJ. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology. 1999;265:319–329. doi: 10.1006/viro.1999.0047. [DOI] [PubMed] [Google Scholar]
- 8.Swingler S, Brichacek B, Jacque JM, Ulich C, Zhou J, Stevenson M. HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature. 2003;424:213–219. doi: 10.1038/nature01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahlknecht U, Herbein G. Macrophages and T-cell apoptosis in HIV infection: a leading role for accessory cells? Trends Immunol. 2001;22:256–260. doi: 10.1016/s1471-4906(01)01898-1. [DOI] [PubMed] [Google Scholar]
- 10.Gartner S, Markovits D, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTV-III/LAV infection. Science. 1986;233:215. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 11.Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, Hwangbo Y, Mullins JI, Corey L. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J Virol. 2002;76:707–716. doi: 10.1128/JVI.76.2.707-716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orenstein JM, Fox C, Wahl SM. Macrophages as a source of HIV during opportunistic infections. Science. 1997;276:1857–1861. doi: 10.1126/science.276.5320.1857. [DOI] [PubMed] [Google Scholar]
- 14.Igarashi T, Brown CR, Endo Y, Buckler-White A, Plishka R, Bischofberger N, Hirsch V, Martin MA. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): Implications for HIV-1 infections of humans. Proc Natl Acad Sci U S A. 2001;98:658–663. doi: 10.1073/pnas.021551798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verani A, Gras G, Pancino G. Macrophages and HIV-1: dangerous liaisons. Mol Immunol. 2005;42:195–212. doi: 10.1016/j.molimm.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 16.Kedzierska K, Azzam R, Ellery P, Mak J, Jaworowski A, Crowe SM. Defective phagocytosis by human monocyte/macrophages following HIV-1 infection: underlying mechanisms and modulation by adjunctive cytokine therapy. J Clin Virol. 2003;26:247–263. doi: 10.1016/s1386-6532(02)00123-3. [DOI] [PubMed] [Google Scholar]
- 17.Garaci E, Aquaro S, Lapenta C, Amendola A, Spada M, Covaceuszach S, Perno CF, Belardelli F. Anti-nerve growth factor Ab abrogates macrophage-mediated HIV-1 infection and depletion of CD4+ T lymphocytes in hu-SCID mice. Proc Natl Acad Sci U S A. 2003;100:8927–8932. doi: 10.1073/pnas.1332627100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Bercoff D, David A, Sudry H, Barre Sinoussi F, Pancino G. Fcγ-mediated suppression of human immunodeficiency virus type 1 replication in primary human macrophages. J Virol. 2003;77:4081–4094. doi: 10.1128/JVI.77.7.4081-4094.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daeron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 20.Connor RI, Benjamin KC, Choe S, Landau NR. Vpr Is Required for Efficient Replication of Human Immunodeficiency Virus Type-1 in Mononuclear Phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 21.O’Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000;74:10074–10080. doi: 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Despres P, Frenkiel MP, Deubel V. Differences between cell membrane fusion activities of two dengue type-1 isolates reflect modifications of viral structure. Virology. 1993;196:209–219. doi: 10.1006/viro.1993.1469. [DOI] [PubMed] [Google Scholar]
- 23.Folks T, Powell D, Lightfoote M, Koenig S, Fauci A, Benn S, Rabson A, Daugherty D, Gendelman H, Hoggan M. Biological and biochemical characterization of a cloned Leu-3- cell surviving infection with the acquired immune deficiency syndrome retrovirus. J Exp Med. 1986;164:280–290. doi: 10.1084/jem.164.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brussel A, Mathez D, Broche-Pierre S, Lancar R, Calvez T, Sonigo P, Leibowitch J. Longitudinal monitoring of 2-long terminal repeat circles in peripheral blood mononuclear cells from patients with chronic HIV-1 infection. Aids. 2003;17:645–652. doi: 10.1097/00002030-200303280-00001. [DOI] [PubMed] [Google Scholar]
- 25.Rouet F, Ekouevi DK, Chaix ML, Burgard M, Inwoley A, Tony TDA, Danel C, Anglaret X, Leroy V, Msellati P, Dabis F, Rouzioux C. Transfer and Evaluation of an Automated, Low-Cost Real-Time Reverse Transcription-PCR Test for Diagnosis and Monitoring of Human Immunodeficiency Virus Type 1 Infection in a West African Resource-Limited Setting. J Clin Microbiol. 2005;43:2709–2717. doi: 10.1128/JCM.43.6.2709-2717.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiu YL, V, Soros B, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki Y, Misawa N, Sato C, Ebina H, Masuda T, Yamamoto N, Koyanagi Y. Quantitative analysis of human immunodeficiency virus type 1 DNA dynamics by real-time PCR: integration efficiency in stimulated and unstimulated peripheral blood mononuclear cells. Virus Genes. 2003;27:177–188. doi: 10.1023/a:1025732728195. [DOI] [PubMed] [Google Scholar]
- 28.Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, Koup RA. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 29.Brussel A, Sonigo P. Analysis of early human immunodeficiency virus type 1 DNA synthesis by use of a new sensitive assay for quantifying integrated provirus. J Virol. 2003;77:10119–10124. doi: 10.1128/JVI.77.18.10119-10124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliver JM, Burg DL, Wilson BS, McLaughlin JL, Geahlen RL. Inhibition of mast cell Fc epsilon R1-mediated signaling and effector function by the Syk-selective inhibitor, piceatannol. J Biol Chem. 1994;269:29697–29703. [PubMed] [Google Scholar]
- 31.Geahlen RL, McLaughlin JL. Piceatannol (3,4,3′,5′-tetrahydroxy-trans-stilbene) is a naturally occurring protein-tyrosine kinase inhibitor. Biochem Biophys Res Commun. 1989;165:241–245. doi: 10.1016/0006-291x(89)91060-7. [DOI] [PubMed] [Google Scholar]
- 32.Wang BH, Lu ZX, Polya GM. Inhibition of eukaryote serine/threonine-specific protein kinases by piceatannol. Planta Med. 1998;64:195–199. doi: 10.1055/s-2006-957407. [DOI] [PubMed] [Google Scholar]
- 33.Ashikawa K, Majumdar S, Banerjee S, Bharti AC, Shishodia S, Aggarwal BB. Piceatannol inhibits TNF-induced NF-kappaB activation and NF-kappaB-mediated gene expression through suppression of IkappaBalpha kinase and p65 phosphorylation. J Immunol. 2002;169:6490–6497. doi: 10.4049/jimmunol.169.11.6490. [DOI] [PubMed] [Google Scholar]
- 34.Lucas M, Mashimo T, Frenkiel MP, Simon-Chazottes D, Montagutelli X, Ceccaldi PE, Guenet JL, Despres P. Infection of mouse neurones by West Nile virus is modulated by the interferon-inducible 2′-5′ oligoadenylate synthetase 1b protein. Immunol Cell Biol. 2003;81:230–236. doi: 10.1046/j.1440-1711.2003.01166.x. [DOI] [PubMed] [Google Scholar]
- 35.Anderson JF, Rahal JJ. Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg Infect Dis. 2002;8:107–108. doi: 10.3201/eid0801.010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Debets J, Van de Winkel J, Ceuppens J, Dieteren I, Buurman W. Cross-linking of both Fc gamma RI and Fc gamma RII induces secretion of tumor necrosis factor by human monocytes, requiring high affinity Fc-Fc gamma R interactions. Functional activation of Fc gamma RII by treatment with proteases or neuraminidase. J Immunol. 1990;144:1304–1310. [PubMed] [Google Scholar]
- 38.Pricop L, Redecha P, Teillaud JL, Frey J, Fridman WH, Sautes-Fridman C, Salmon JE. Differential modulation of stimulatory and inhibitory Fc gamma receptors on human monocytes by Th1 and Th2 cytokines. J Immunol. 2001;166:531–537. doi: 10.4049/jimmunol.166.1.531. [DOI] [PubMed] [Google Scholar]
- 39.Tridandapani S, Siefker K, Teillaud JL, Carter JE, Wewers MD, Anderson CL. Regulated expression and inhibitory function of Fcgamma RIIb in human monocytic cells. J Biol Chem. 2002;277:5082–5089. doi: 10.1074/jbc.M110277200. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Masuda E, Blank MC, Kirou KA, Gao X, Park MS, Pricop L. Cytokine-mediated regulation of activating and inhibitory Fc gamma receptors in human monocytes. J Leukoc Biol. 2005;77:767–776. doi: 10.1189/jlb.0904532. [DOI] [PubMed] [Google Scholar]
- 41.Holl V, Hemmerter S, Burrer R, Schmidt S, Bohbot A, Aubertin AM, Moog C. Involvement of Fc gamma RI (CD64) in the mechanism of HIV-1 inhibition by polyclonal IgG purified from infected patients in cultured monocyte-derived macrophages. J Immunol. 2004;173:6274–6283. doi: 10.4049/jimmunol.173.10.6274. [DOI] [PubMed] [Google Scholar]
- 42.Connor RI, Dinces NB, Howell AL, Romet-Lemonne JL, Pasquali JL, Fanger MW. Fc receptors for IgG (Fc gamma Rs) on human monocytes and macrophages are not infectivity receptors for human immunodeficiency virus type 1 (HIV-1): studies using bispecific antibodies to target HIV-1 to various myeloid cell surface molecules, including the Fc gamma R. Proc Natl Acad Sci USA. 1991;88:9593–9597. doi: 10.1073/pnas.88.21.9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeda A, Ennis FA. FcR-mediated enhancement of HIV-1 infection by antibody. AIDS Res Hum Retroviruses. 1990;6:999–1004. doi: 10.1089/aid.1990.6.999. [DOI] [PubMed] [Google Scholar]
- 44.Homsy J, Meyer M, Rateno M, Clarkson S, Levy JA. The Fc and not CD4 receptor mediated antibody enhancement of HIV infection in human cells. Science. 1989;244:1357–1360. doi: 10.1126/science.2786647. [DOI] [PubMed] [Google Scholar]
- 45.Trischmann H, Davis D, Lachmann PJ. Lymphocytotropic strains of HIV type 1 when complexed with enhancing antibodies can infect macrophages via Fc gamma RIII, independently of CD4. AIDS Res Hum Retroviruses. 1995;11:343–352. doi: 10.1089/aid.1995.11.343. [DOI] [PubMed] [Google Scholar]
- 46.Forthal D, Landucci G, Phan T, R H-T, P G. Fcg Receptor IIa and IIIa polymorphisms are associated with the risk of HIV infection. 13th Conference on Retroviruses and Opportunistic Infections; Denver, USA. 2006. [Google Scholar]
- 47.Kim SY, Byrn R, Groopman J, Baltimore D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: evidence for differential gene expression. J Virol. 1989;63:3708–3713. doi: 10.1128/jvi.63.9.3708-3713.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butler SL, Johnson EP, Bushman FD. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles. J Virol. 2002;76:3739–3747. doi: 10.1128/JVI.76.8.3739-3747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pierson TC, Kieffer TL, Ruff CT, Buck C, Gange SJ, Siliciano RF. Intrinsic stability of episomal circles formed during human immunodeficiency virus type 1 replication. J Virol. 2002;76:4138–4144. doi: 10.1128/JVI.76.8.4138-4144.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Gabuzda D. Regulation of Human Immunodeficiency Virus Type 1 Infectivity by the ERK Mitogen-Activated Protein Kinase Signaling Pathway. J Virol. 1999;73:3460–3466. doi: 10.1128/jvi.73.4.3460-3466.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Francois F, Klotman ME. Phosphatidylinositol 3-kinase regulates human immunodeficiency virus type 1 replication following viral entry in primary CD4+ T lymphocytes and macrophages. J Virol. 2003;77:2539–2549. doi: 10.1128/JVI.77.4.2539-2549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee C, Tomkowicz B, Freedman BD, Collman RG. HIV-1 gp120-induced TNF-{alpha} production by primary human macrophages is mediated by phosphatidylinositol-3 (PI-3) kinase and mitogen-activated protein (MAP) kinase pathways. J Leukoc Biol. 2005;78:1016–1023. doi: 10.1189/jlb.0105056. [DOI] [PubMed] [Google Scholar]
- 54.Kutza J, Crim L, Feldman S, Hayes MP, Gruber M, Beeler J, Clouse KA. Macrophage colony-stimulating factor antagonists inhibit replication of HIV-1 in human macrophages. J Immunol. 2000;164:4955–4960. doi: 10.4049/jimmunol.164.9.4955. [DOI] [PubMed] [Google Scholar]
- 55.Herbein G, Montaner LJ, Gordon S. Tumor necrosis factor alpha inhibits entry of human immunodeficiency virus ype 1 into primary human macrophages: a selective role for a 75-kilodalton receptor (published erratum appears in J. Virol. 1997 Mar; 71(3):1581) J Virol. 1996;70:7388–7397. doi: 10.1128/jvi.70.11.7388-7397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cota M, Mengozzi M, Vicenzi E, Panina-Bordignon P, Sinigaglia F, Transidico P, Sozzani S, Mantovani A, Poli G. Selective inhibition of HIV replication in primary macrophages but not T lymphocytes by macrophage-derived chemokine. Proc Natl Acad Sci U S A. 2000;97:9162–9167. doi: 10.1073/pnas.160359197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meylan PR, Guatelli JC, Munis JR, Richman DD, Kornbluth RS. Mechanisms for the inhibition of HIV replication by interferons-alpha, -beta, and -gamma in primary human macrophages. Virology. 1993;193:138–148. doi: 10.1006/viro.1993.1110. [DOI] [PubMed] [Google Scholar]
- 58.Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 59.Schwartz O, Marechal V, Friguet B, Arenzana-Seisdedos F, Heard JM. Antiviral activity of the proteasome on incoming human immunodeficiency virus type 1. J Virol. 1998;72:3845–3850. doi: 10.1128/jvi.72.5.3845-3850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei BL, Denton PW, O’Neill E, Luo T, Foster JL, Garcia JV. Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J Virol. 2005;79:5705–5712. doi: 10.1128/JVI.79.9.5705-5712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bukrinsky M, Sharova N, McDonald T, Pushkarskaya T, Tarpley W, Stevenson M. Association of Integrase, Matrix, and Reverse Transcriptase Antigens of Human Immunodeficiency Virus Type 1 with Viral Nucleic Acids Following Acute Infection. Proc Natl Acad Sci U S A. 1993;90:6125–6129. doi: 10.1073/pnas.90.13.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parrill AL. HIV-1 integrase inhibition: binding sites, structure activity relationships and future perspectives. Curr Med Chem. 2003;10:1811–1824. doi: 10.2174/0929867033457043. [DOI] [PubMed] [Google Scholar]
- 63.Zhu K, Dobard C, Chow SA. Requirement for integrase during reverse transcription of human immunodeficiency virus type 1 and the effect of cysteine mutations of integrase on its interactions with reverse transcriptase. J Virol. 2004;78:5045–5055. doi: 10.1128/JVI.78.10.5045-5055.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.